

Abstract
The biological functions of the p53 related protein kinase (PRPK) remain unclear. We have previously demonstrated that PRPK is phosphorylated by the T-LAK cell-originated protein kinase (TOPK) and that phosphorylated PRPK (p-PRPK) promotes colon cancer metastasis. Here, we analyzed colon adenocarcinomas from 87 patients, and found that higher expression levels of p-PRPK were associated with later stages of metastatic dissemination (stages III or IV), as compared to earlier stages (stages I and II). Indeed, levels of p-PRPK were higher in metastatic versus malignant human colon adenocarcinomas. Knocking down PRPK expression attenuated colorectal liver and lung metastasis of colon cancer cells in vivo. An in vitro kinase assay indicated that active PRPK does not phosphorylate p53 directly. We found that PRPK phosphorylates survivin, a regulator of colon cancer metastasis. PRPK phosphorylates survivin at Thr34, which is important for survivin stability. Taken together, our data strongly suggest that the PRPK signaling pathway promotes colon cancer metastasis by modulating survivin stability, and that PRPK could be a new prognostic marker for the survival of colon cancer patients. In addition, we identified an FDA approved bacteriostatic antibiotic, fusidic acid sodium salt (fusidic acid or FA) as an inhibitor of PRPK, and show that FA combined with 5-fluorouracil (5-FU) inhibited PRPK activity and colon cancer metastasis to the lung in mice. We contend that the combination of FA with 5-FU could be an alternative therapeutic strategy to traditional chemotherapy for colon cancer patients with poor prognosis.
Keywords: phosphorylation, PRPK, survivin, metastasis, inhibition
Introduction
New research on molecules that contribute to metastasis, the pathways they control and the genes they regulate, is very important for understanding the prognosis and prevention of metastasis in the clinic. PRPK was first cloned from an interleukin-2-activated cytotoxic T-cell subtraction library and reported to up-regulate the transcriptional activity of p53 (1). This is where its acronym, “p53-related protein kinase”, was derived. The same authors identified a protein, CGI-121, as a binding partner of PRPK, which could inhibit the binding of PRPK to p53 (2). They also reported that the small Ras-like protein, GTPase protein Rab1c (Rab35), could act as a p53-modulating molecule by binding to p53-activating factors such as PRPK or other p53-related kinases (3). Human PRPK is a homolog to the yeast kinase piD261/Bud32 (Bud32) and could partially complement Bud32 deficiency (4). PRPK is phosphorylated at Ser250 by Akt/PBK (5) and by the T-LAK cell-originated protein kinase (TOPK) (6). The contribution of PRPK to DNA damage is associated with the G1checkpoint response (7). Drosophila PRPK is required for PI3-K/mTOR pathway-dependent growth and crucial for stimulating the basal protein biosynthetic machinery in response to insulin signaling (8). The biological functions of PRPK remain elusive, and are mostly based on screening data (7-10). The data regarding phosphorylation and regulation of p53 by PRPK are not clear and need to be evaluated. The authors, who first reported PRPK as a p53-related protein kinase (1), later concluded that PRPK does not directly phosphorylate p53. Pre-incubation with a COS-7 cell lysate was necessary for the in vitro kinase assay, and they concluded that the phosphorylation status of p53 was likely regulated not only by PRPK, but also by other kinases (3). For example, the p53 protein is also phosphorylated at Ser15 by ATR and ATM (11). The p53 protein is absent in yeast; however, the yeast PRPK homolog (piD261/BUD32) can phosphorylate p53 in vitro. Additionally, PRPK can partially complement the phenotype of yeast lacking the piD261/Bud32 gene, suggesting that PRPK could phosphorylate or interact with other Bud32 substrates/partners (4). Depletion of PRPK reportedly causes a p53-dependent and -independent increase in paclitaxel-induced caspase activation and p53 remains phosphorylated on Ser15 even after depletion of PRPK, suggesting that this is not a major role for PRPK in proliferating cells (10).
We have previously demonstrated that PRPK is a new substrate for TOPK and that the TOPK/PRPK signaling pathway promotes colorectal cancer metastasis (6). In this study, we found that the expression levels of phosphorylated PRPK (p-PRPK) are higher in human metastatic colorectal cancer tissues, as compared to normal colon tissues. Elevated levels of p-PRPK were correlated with more advanced pathological stages in colon cancer patients. In vitro kinase assay results indicated that active PRPK does not phosphorylate p53 directly. We investigated the role of PRPK in metastasis, and survivin, which stimulates metastasis, was identified as a substrate of PRPK. PRPK phosphorylates survivin at Thr 34, which is important for survivin protein stability. Thus, the PRPK signaling pathway promotes metastasis of colon cancer cells by modulating survivin stability. PRPK could be a novel anticancer target, indeed we found that the antibiotic FA, which inhibits PRPK activity, could suppress colon cancer metastasis to the lung, alone or in combination with 5-fluorouracil (5-FU).
Materials and Methods
Antibodies and reagents
Antibodies to detect PRPK (C-14), β-actin (C4), or survivin (D-8) and the protein A/G plus-agarose immunoprecipitation reagent were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-phosphor-survivin (Thr34; D2E11), phosphor-p53 (Ser15) (16G8) anti-mouse antibody were from Cell Signaling Technology, Inc. (Beverly, MA). Anti-Mutant p53 [Y5] antibody was from AbCam (Cambridge, MA). Alexa Fluor 488-goat anti-mouse IgG was from Life Technology (Carlsbad, CA). Anti-V5 was from Invitrogen (Carlsbad, CA). The GST-PRPK full-length recombinant protein was from Novus Biologicals (Littleton, CO). Anti-Flag, the antibiotic fusidic acid sodium salt (FA) and 5-fluorouracil (5-FU) were from Sigma (St Louis, MO). The phosphor-PRPK (Ser250) antibody was prepared by BioSynthesis, Inc. (Lewisville, TX; details in Materials and Methods). Active Chk1 for in vitro kinase assays was from Millipore (Billerica, MA). The JetPEI transfection reagent was purchased from Qbiogen, Inc. (Montreal, Quebec, Canada) and His-p53 recombinant protein was from ProSci. Inc. (Poway, CA).
Cell Culture
Human HCT116, HT29, HCT15, DLD1, WiDr and mouse CT26 colon cancer cells or CCD-18Co and HCEC normal colon cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) between 2009 and 2015. To eliminate the possibility of misidentification or cross-contamination of cells, ATCC performed several tests for authentication, including isoenzyme analysis to confirm human origin, DNA fingerprinting analysis of cell line-specific polymorphic markers, growth curve analysis to check doubling times, a microscope-based morphology analysis, and detection of mycoplasma. All cell lines were matched with their correct identity and were mycoplasma-free. Cells were maintained according to ATCC instructions before being frozen. Each vial of frozen cells was thawed and maintained for a maximum of 8 weeks.
The pSIN-Fluc vector expressing the firefly luciferase gene for generation of CT26 stable colorectal cancer cells (CT26-Luc) was a gift from Dr. Yasuhiro Ikeda (Mayo Clinic, Rochester, MN) (12). HCT116 cells were cultured in McCoy’s 5A medium and HT29 and HCT15 cells were cultured in DMEM/high glucose. DLD1 and CT26 cells were cultured in RPMI-1640 medium and WiDr, CCD-18Co and HCEC cells were cultured in MEM. All media were from Thermo Scientific Hyclone Laboratories, Inc. (Logan, UT) and contained 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 25 μM/ml gentamicin. Cells were grown in monolayers at 37°C in a 5% CO2 incubator.
The phosphorylated PRPK antibody
The phosphor-PRPK (Ser250) antibody (p-PRPK, anti-rabbit) was made by BioSynthesis, Inc. (Lewisville, TX). The antiserum was prepared using a synthesized 16-mer phosphopeptide consisting of an N-terminal cysteine (for coupling) and the sequence CLDEVRLRGRKR[pS250] MVG.
In vivo mouse models
All animal experiments were conducted in accordance with and approved by the University of Minnesota Institutional Animal Care and Use Committee (IACUC). Liver and lung metastasis mouse models were used (details about the animal experiments have been provided in “Supplementary Materials and Methods” under the headings “Pharmacokinetic studies” and “Detection of efficacy in vivo”).
Immunohistochemical analysis (IHC)
Commercially available patient tissue arrays from US Biomax Inc. (Rockville, MD), were used for IHC. US Biomax states, “All tissue is collected under the highest ethical standards with the donor being informed completely and with their consent. We make sure we follow standard medical care and protect the donors’ privacy. All human tissues are collected under HIPPA approved protocols. All animal tissues are collected under IACUC protocol. All samples have been tested negative for HIV and Hepatitis B or their counterparts in animals, and approved for commercial product development.”
The colon cancer tissue array (C0702) was comprised of malignant or metastatic adenocarcinomas and normal adjacent tissues (NAT; including TNM and pathology grade, 69 cases/69 cores). The colon adenocarcinoma tissue arrays (HCol-Ade180Sur-03) and (HCol-Ade180Sur-05) had survival data, including TNM and pathology grade, 90 cases and NAT, 90 cases. Immunostaining was performed on the tissue array using antibodies to detect p-PRPK (1:100) or mutant p53 (1:50). The biotinylated secondary antibody was rabbit anti-mouse IgG (1:200, 10% normal rabbit serum; Vector Laboratories, Burlingame, CA). The slides were developed in diaminobenzidine (DAB) and counter stained with hematoxylin, then dehydrated and mounted in permount. Images were captured and analyzed using the Image-Pro-Plus 6.2 software program.
Statistical Analysis
All quantitative results are expressed as mean values ± S.D. Statistically significant differences (p < 0.05) were obtained using the Student’s t test or one-way ANOVA. Additional detailed methods are described in Supplementary Materials and Methods.
Results
p-PRPK is overexpressed in metastatic colorectal cancer, and induces colon cancer cell migration or invasion
Total and p-PRPK protein levels are highly expressed in colon cancer cells, as compared to normal colon cells such as CCD-18Co or HCEC (Fig. 1A). To specifically detect p-PRPK levels, a phosphor-PRPK (Ser250) antibody was prepared, and the specificity of antibody was verified by peptide competition (Fig. 1B and Supplementary Fig. 1) using ICC staining of HCT116 colon cancer cells, and IHC staining of mouse lung cancer tissues from the control group of the xenograft model used in this study (see Supplementary Materials and Methods). Antibody staining was decreased by the specific peptide (LDEVRLRGRKR[pS250]MVG), indicating that this antibody can specifically detect the phosphorylation of PRPK at Ser250. The p-PRPK protein expression level (Fig. 1C) was significantly higher in metastatic colon adenocarcinoma tissue as compared to malignant colon adenocarcinoma tissues (p < 0.001), whereas neither protein is highly expressed in normal adjacent colon tissue. Matrigel assay results indicated that the invasion abilities of colon cancer cells correlated with the PRPK phosphorylation status in different colon cancer cell lines (Fig. 1D). Normal colon cells and HT29 colon cancer cells, which are not invasive, express low p-PRPK protein levels, whereas invasive colon cancer cell lines like HCT116 and HCT15, express high levels of p-PRPK. Next, we determined the effect of knocking down PRPK levels with sh-RNA on the invasion abilities of invasive cancer cell lines such as HCT116 and HCT15. We found that HCT116 and HCT15 colon cancer cells expressing shPRPK (Fig. 1E, upper panels) exhibited less migration compared with shMock cells (Fig. 1E, bottom panels). Cells expressing shPRPK exhibited attenuated invasion through Matrigel inserts compared with shMock cells (Fig. 1F). We also examined the growth of HCT116 and HCT15 cells expressing shPRPK or shMock (Supplementary Fig. 2A, B) and observed no differences in the growth rates of these cells in 10% serum. These results indicate that overexpression of PRPK is associated with increased migration and invasion, which is independent of cell proliferation.
Figure 1.
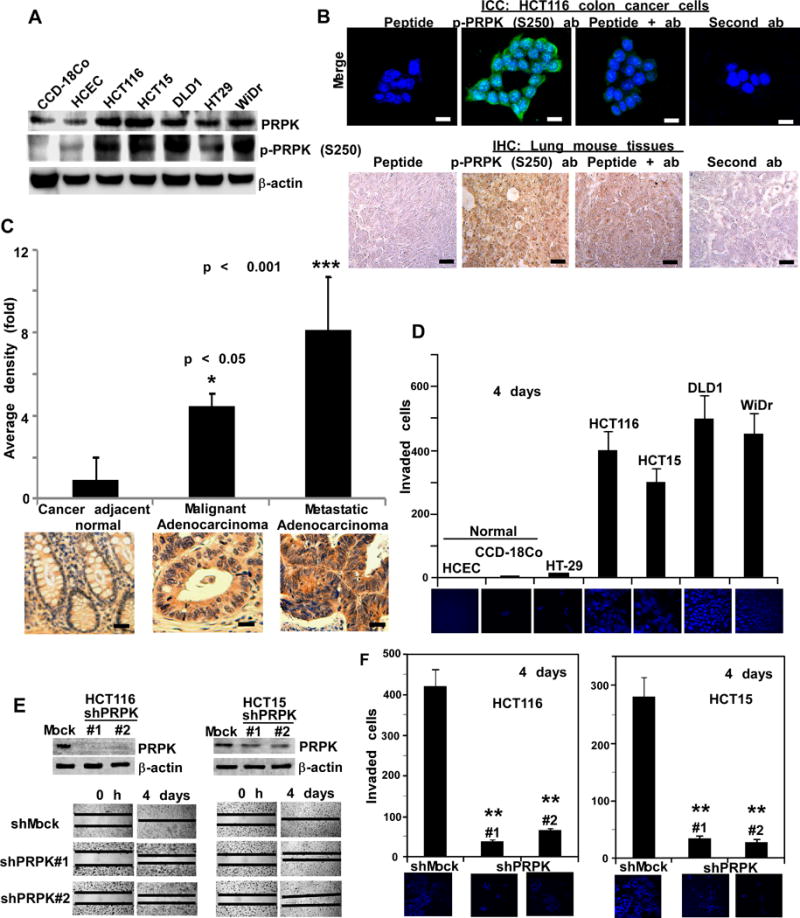
The p-PRPK protein is overexpressed in metastatic colorectal tissues. A, normal CCD-18Co or HCEC colon cells and 5 human colon cancer cell lines were screened to examine the expression level of total and p-PRPK proteins. B, determining the specificity of the p-PRPK (S250) antibody. For ICC (upper panels; scale bars = 20 μm), the green color indicates p-PRPK and DAPI is shown in blue; for IHC (lower panels; scale bars = 25 μm), the brown color indicates p-PRPK expression. C, expression of p-PRPK in malignant human colorectal adenocarcinomas (30 cases), metastatic adenocarcinoma tissues (30 cases), and matching normal colorectal tissue (10 cases). Representative photographs are shown (scale bars = 25 μm) and data are represented as mean values ± S.D. * Compared to normal adjacent, (p < 0.05), compared with malignant adenocarcinoma, (p < 0.001). D, Matrigel invasion assay of normal colon cells or colon cancer cells. E, efficiency of shPRPK in HCT116 or HCT15 cells (upper panels) is shown. Migration of shMock/HCT116 and shPRPK/HCT116 cells (left lower panels) or shMock/HCT15 cells and shPRPK/HCT15 cells (right lower panels) with a wound closure assay. F, invasive ability of shMock/HCT116 and shPRPK/HCT116 cells (left panels) or shMock/HCT15 and shPRPK/HCT115 cells (right panels) using a Matrigel invasion chamber. Invading cells were stained with DAPI (60X). ** Compared with cells expressing shMock (p < 0.001). For D and F, invading cells were counted on 2 membranes from 2 separate experiments.
Knocking down PRPK expression attenuates colorectal liver or lung metastasis in vivo
Because gastrointestinal cancers, including colorectal and pancreatic cancers, commonly metastasize to the liver, we injected HCT116 or HCT15 colon cancer cells stably expressing shMock or shPRPK#1 into the spleen of athymic nude mice, and determined their abilities to metastasize to the liver. The area of liver metastasis from mice expressing shPRPK#1/HCT116 was significantly decreased compared to shMock (Fig. 2A and Supplementary Fig. 2C). The metastatic nodules were completely absent in livers from mice expressing shPRPK#1/HCT15 compared to shMock (Fig. 2B, bottom panel, and Supplementary Fig. 2D). Hematoxylin and Eosin (H&E) staining of livers is shown (Fig. 2A, B, right panels). Migration and invasion were inhibited in CT26 mouse colon cancer cells stably expressing shPRPK, as compared to shMock (Supplementary Fig. 3A, B, C). Because colorectal cancers can metastasize to the lung, HCT116 human and CT26 mouse colon cancer cells stably expressing shMock or shPRPK were injected into the tail of athymic nude mice (1×106/mouse) to evaluate metastasis to the lungs (Supplementary Fig. 3D, E). The area of lung metastasis from mice expressing shMock in HCT116 cells (28 days) was greater compared to shPRPK#1 (Fig. 2C, upper panels, circled areas). Lungs from mice expressing sh-Mock CT26 cells had 95% metastasis after 9 days, and lung metastasis from mice expressing shPRPK#4 was significantly decreased compared to shMock (Fig. 2D, left panels, circled areas). H&E staining of lungs with cells expressing shMock showed greater tumor metastasis compared with shPRPK (Fig. 2C, lower panels; Fig. 2D, right panels). These data confirmed that suppressing PRPK expression reduces liver or lung metastasis of human or mouse colon cancer cells. Kaplan-Meier survival analysis (Fig. 2E) demonstrated that mice inoculated with shMock (n = 9, red line) exhibit dramatically decreased survival time compared to mice inoculated with shPRPK#1 (p = 0.0015).
Figure 2.
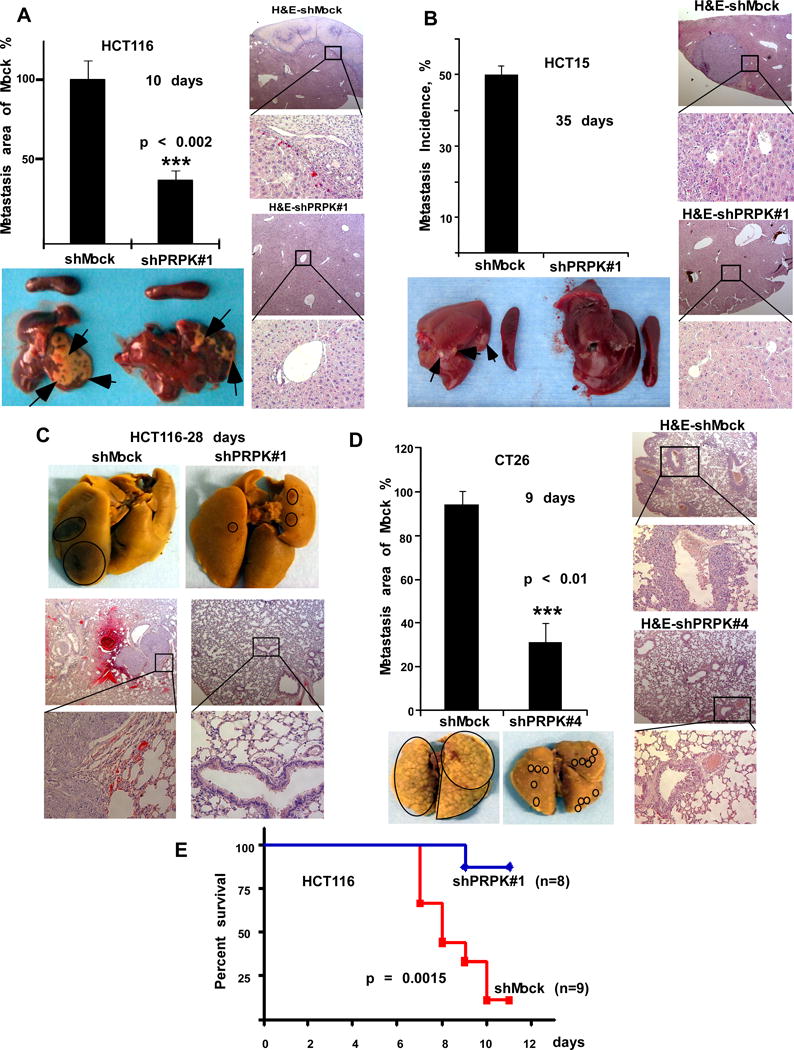
PRPK promotes liver or lung metastasis of human or mouse colon cancer cells. A, mouse liver metastasis after intrasplenic injection of shPRPK/HCT116 cells (n = 5 mice) or B, with shPRPK/HCT15 cells (n = 6 mice) was compared to livers from mice injected with shMock cells. Representative liver metastasis (arrows) is shown (1 representative liver/group). Compared with mice injected with shMock-expressing cells, (p < 0.002). C, lung metastasis in mice (n = 6) after tail vein injection of HCT116 cells expressing shMock or shPRPK (1 representative lung/group, circled areas). D, the area of lung metastasis (n = 5 mice) after tail injection of shPRPK/CT26 cells was decreased compared to metastasis in mice injected with shMock-expressing cells (circled areas, 1 representative lung/group). For A-D, H&E staining of lung tissues is shown (scale bar = 4 μm; 25X and 200X). E, Kaplan-Meier survival analysis with cells expressing shMock (n = 9, red line) and cells expressing shPRPK (n = 8, blue line; p = 0.0015).
Phosphorylation of survivin (Thr34) by PRPK, is required for stability of survivin
Survivin expression is up-regulated in human cancers, and is associated with resistance to chemotherapy and radiation therapy (13). Survivin is an activator of tumor cell motility and metastatic genes and plays an important role in colorectal tumorigenesis (14, 15). Using an in vitro kinase assay, we determined that PRPK phosphorylates survivin (Fig. 3A). By utilizing a specific p-survivin antibody, we determined that PRPK phosphorylated survivin at Thr34 (Fig. 3B, right panel), which is known to increase survivin’s stability (16). Mutation of survivin (Thr34Ala) can induce apoptosis and reduce angiogenesis, metastasis, and cell cycle progression (17). Wild-type and mutant His-survivin (Thr34Ala) proteins were analyzed for phosphorylation by PRPK. Audioradiography (Fig. 3B, left panel) and Western blotting (Fig. 3B, right panel) results indicated that mutant T34A abrogated phosphorylation of survivin, confirming that PRPK phosphorylates survivin at Thr34. Because the only reported physiological substrate of PRPK is p53, we used an in vitro kinase assay to compare the phosphorylation of p53 or survivin by PRPK (Fig. 3C, upper panel). Coomassie blue staining was used as the loading control (Fig. 3C, lower panel). Results indicate that PRPK does not phosphorylate p53 directly.
Figure 3.
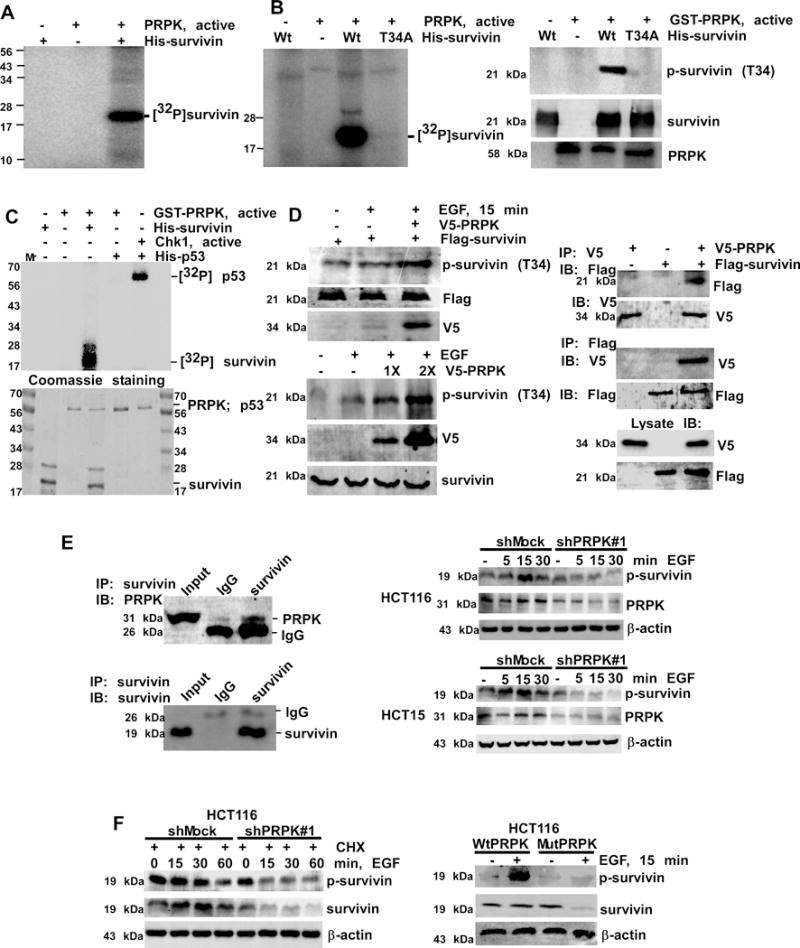
PRPK phosphorylates survivin and increases its stability. A, survivin is phosphorylated by PRPK in vitro. B, wild-type survivin (Wt) or a survivin Thr34Ala mutant (T34A) was used as a substrate for PRPK. Reactive products were visualized by autoradiography (left panel) or Western blotting (right panels). C, phosphorylation of survivin or p53 by PRPK in an in vitro kinase assay (upper panel). Phosphorylation of p53 by Chk1 is a positive control. Coomassie blue staining was used to verify equal loading of proteins (lower panel). D, left, PRPK phosphorylates survivin in cells treated with EGF. Equal amounts of pcDNA3-V5-PRPK and pcDNA-Flag-survivin (upper panels) or increasing amounts of pcDNA3-V5-PRPK (lower panels) were transiently transfected into HEK293 cells and phosphorylation of survivin was detected by Western blotting. D, right, PRPK binds with survivin in HEK293 cells. PcDNA3-V5-PRPK and pcDNA3-Flag-survivin were co-transfected into cells, immuno-precipitated with anti-V5 (upper panels) or anti-Flag (middle panels) to precipitate PRPK or survivin and probed with anti-Flag or anti-V5, respectively. Cell lysates were used to verify transfection efficiency of V5-PRPK and Flag-survivin (bottom panels). E, left, survivin binds with PRPK in HCT116 cells. Endogenous survivin was immunoprecipitated and probed with anti-PRPK. E, right, phosphorylation of survivin is decreased in shPRPK/ HCT116 (upper panels) or shPRPK/HCT15 (bottom panels) cells. The total levels of PRPK confirmed PRPK deficiency. F, cells were treated with CHX followed by EGF and then survivin stability was detected (left panels). The p-survivin or total survivin proteins were decreased in EGF-treated mutant (Mut) PRPK cells (right panels). β-Actin levels were used as loading controls.
We have previously shown that TOPK is required for EGF-induced PRPK phosphorylation (6), and we wanted to determine if phosphorylation of PRPK and survivin was also EGF-dependent. We found that this was indeed the case, as phosphorylation of PRPK (Ser250) and survivin (Thr34) was induced in a dose- and time-dependent manner by EGF (Supplementary Fig. 4A, B). HEK293 cells were co-transfected with pcDNA3-V5-PRPK and pcDNA3-Flag-survivin plasmids, and we observed that EGF increased the phosphorylation of survivin (Fig. 3D, left upper panels). Increasing the transfected amount of pcDNA3-V5-PRPK further increased the phosphorylation of survivin (Fig. 3D, left lower panels). V5-PRPK and Flag-survivin were transfected alone or together into HEK293 cells and the expression of each protein was confirmed by anti-V5 or anti-Flag, respectively (Fig. 3D, right lower). First, V5-tagged PRPK was immunoprecipitated and the Flag-tagged survivin protein was detected by Western blot analysis. Results confirmed that PRPK co-immunoprecipitated with survivin (Fig. 3D, right upper). Secondly, Flag-tagged survivin was immunoprecipitated and V5-tagged PRPK was detected by Western blotting. The results confirmed that survivin co-immunoprecipitated with PRPK (Fig. 3D, right middle). Finally, HCT116 cell extracts were immunoprecipitated with anti-survivin or control IgG. PRPK was detected in the immunoprecipitates obtained with anti-survivin, but not with control IgG (Fig. 3E), indicating that endogenous PRPK binds with survivin.
Survivin phosphorylation induced by EGF was decreased by knockdown of PRPK (shPRPK#1) in HCT116 or HCT15 cells (Fig. 3E, right panels, Supplementary Fig. 4C, D for shPRPK#2). These data suggest that PRPK is required for EGF-induced survivin phosphorylation. EGF regulates survivin stability (18) and survivin is stabilized by Thr34 phosphorylation (16). shMock/HCT116 or shPRPK#1/HCT116 cells were treated with cycloheximide (CHX) to block protein synthesis and then treated with EGF. Survivin stability in shPRPK#1 cells was decreased at 15 min following EGF treatment (Fig. 3F, left panels), suggesting that phosphorylation of survivin by PRPK enhances its stability. Phosphorylation of survivin was almost absent in MutPRPK cells (Fig. 3F, right panels). These results demonstrate that mutation of PRPK markedly attenuates its phosphorylation of survivin. Despite a high rate of drug resistance, 5-FU remains a common and widely used chemotherapeutic drug for the treatment of colon cancer (19). Treatment of colon cancer cells with 5-FU induces caspase-dependent apoptosis (20). Survivin is phosphorylated at Thr34 by Cdc2 and loss of Thr34 phosphorylation results in the dissociation of caspase-dependent apoptosis (16, 21). The mutation of survivin (Thr34Ala) induces apoptosis, thus we compared the level of cleaved caspase-3, a marker of apoptosis in cells expressing either mutant or wild-type PRPK (Supplementary Fig. 4E). After 48 h of treatment with 5-FU, the level of cleaved caspase-3 was increased in cells expressing mutant PRPK, compared to cells expressing WT PRPK. This indicated that overexpression of the mutant PRPK induces apoptosis, and has a similar biological effect to the survivin T34A mutation. Experiments were also performed with PRPK knockdown in HCT116 cells (Supplementary Figure 4F) and we did not observe dramatic differences in the levels of cleaved caspase 3 in cells with or without PRPK, thus apoptosis was not induced in the absence of 5-FU, which is consistent with our previous cell growth data. All these data confirm that PRPK interacts with survivin, phosphorylates survivin in vitro and ex vivo, and regulates survivin stability.
Fusidic acid inhibits PRPK activity and reduces HCT116 migration and invasion
Betulinic acid (BA) is an inhibitor of prostate cancer growth that acts through the degradation of transcription factor specificity proteins (i.e., Sp1, Sp3 or Sp4), which regulate vascular endothelial growth factor and survivin, (22). We hypothesized that BA might reduce expression levels of survivin by attenuating PRPK activity. BA is not FDA approved, therefore we performed a shape similarity search between BA and a list of FDA-approved molecules (Supplementary Materials and Methods). We identified conformers that possessed a Tanimoto similarity coefficient higher than 0.7 when matched against the lowest energy conformation of BA. Schrödinger’s PHASE program was used to perform a shape similarity search and we found that the antibiotic FA and BA had a high Tanimoto score of 0.8 (Supplementary Fig. 4G). Even though the steroid portion differs by a 6–member ring, the molecules were similar in the location of the substituent that protruded from their rings. The location of the hydroxyl and the carboxylic acid groups provides both with hydrophilic groups in similar places at either end. Furthermore, the diene group on FA is also located in a similar area as the alkene group on BA. FA, licensed for use as sodium fusidate (Fig. 4A, inset), is effective primarily against gram-positive bacteria (23). Computer modeling showed that FA fits into the ATP-binding pocket of PRPK and formed hydrogen bonds with Lys40, Ala45 or Lys60 and hydrophobic interactions with Val47/Val58 or Ile179/Ile182 of PRPK (Fig. 4A). FA binds with PRPK in vitro and ex vivo (Fig. 4B) and inhibits PRPK activity in vitro (Fig. 4C, upper and middle). PRPK is phosphorylated by TOPK (6), therefore we checked whether TOPK activity was inhibited by FA. We found that FA did not inhibit TOPK activity (Fig. 4C, bottom). FA use has not been reported in cancer therapy and was nontoxic against HCT116 cells up to 100 μM (Fig. 4D). FA reduced cell migration (Fig. 4E, inset) and inhibited HCT116 colon cancer cell invasion dose-dependently (Fig. 4E). FA suppressed EGF-induced phosphorylation of survivin in HCT116 cells at a concentration similar to 5-FU (Fig. 4F, left), but was most effective when combined with 5-FU in CT26 cells (Fig. 4F, right). To evaluate whether the combination of 5-FU and FA is more effective than single compound treatment, we first determined the IC50 value for 5-FU and FA alone (Supplementary Fig. 4H). Based on these values, the Combination Index (CI) was analyzed from 16 different groups using CompuSyn software (24) with various concentrations of the two compounds (Supplementary Fig. 4I). Inhibition of cell viability (%) and CI were used to show that 5-FU and FA act synergistically when used together (Supplementary Fig. 4J).
Figure 4.
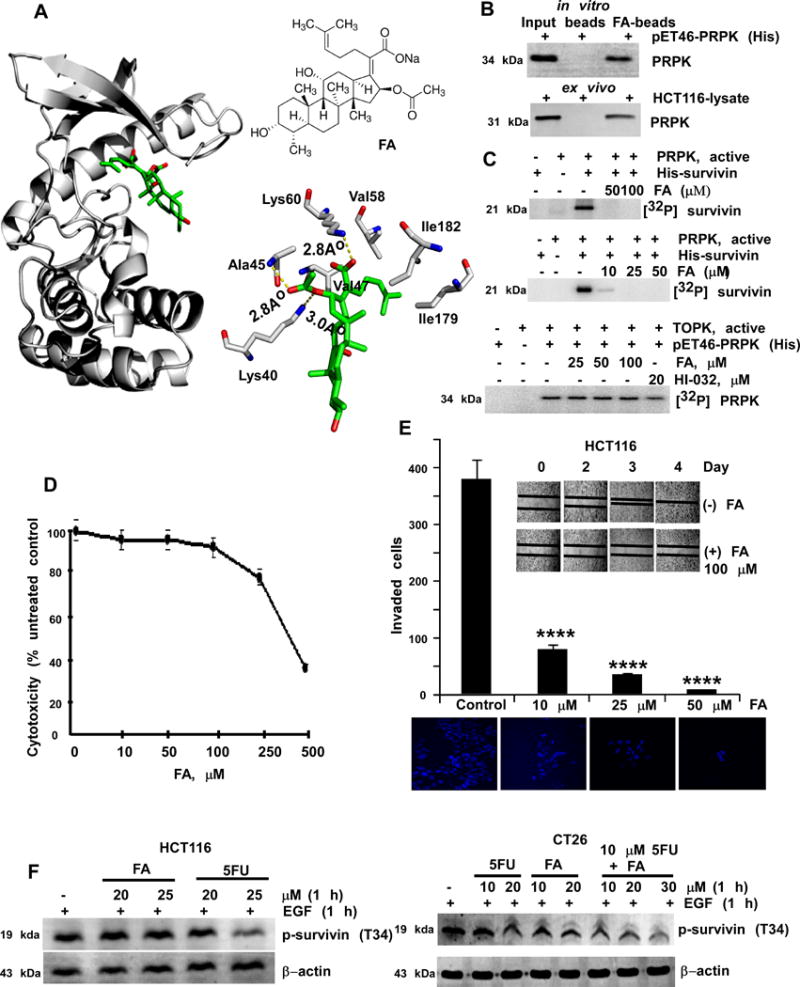
FA inhibits PRPK activity and reduces migration or invasion of HCT116 cells. A, docking model of FA (left) and the PRPK ATP-binding site. Chemical structure of FA (upper right). Two-dimensional illustration of the hydrogen bonding (yellow dotted lines, lower right) interactive network between FA (green) and PRPK. B, PRPK binds with FA. Recombinant PRPK (upper panel) or HCT116 cell lysate (lower panel) was incubated with FA-conjugated Sepharose 4B beads or Sepharose 4B beads-only. PRPK was detected by Western blotting. C, FA inhibits PRPK activity in vitro. D, FA is not cytotoxic to HCT116 cells up to 100 μM. E, FA inhibits migration (insert panels) and invasion (graph) of HCT116 cells. **** denote significant inhibition, as compared to the untreated controls, (p < 0.001). F, the effect on PRPK activity by 5-FU or FA (left panels) or combination of FA with 5-FU (right panels) was compared.
FA alone or in combination with 5-FU, suppresses colon lung metastasis in vivo
To detect colon cancer to lung metastasis, CT26 colon cancer cells expressing firefly luciferase (CT26-Luc) were used. The expression level of p-PRPK and growth of CT26 and CT26-Luc cells were similar (Supplementary Fig. 5A). CT26-Luc cells (1×106) exhibiting 3×106 luciferase activities (Supplementary Fig. 5B) were injected into the tail vein of mice.
We have shown that FA inhibited activity of PRPK in vitro and ex vivo. Mice were pre-treated with FA to try and achieve better experimental efficacy in terms of optimal PRPK inhibition. Previous studies have reported that elimination of FA from the blood is slow and accumulation of FA occurs (25-28). To confirm FA accumulation in our mouse model, FA was administered at 16 mg/kg/mouse (n=6 mice) one time daily for 10 days and FA levels in the serum were determined (Supplementary Figure 5C). We found that FA treated mice had FA levels of ~1 mg/ml in the serum, compared to untreated controls. Based on these results, we decided to pre-treat mice with FA for 10 days before inoculation of CT26-Luc cells in order to increase FA levels in the serum.
We conducted an in vivo experiment in which we evaluated the effect of FA pre-treatment on colon cancer metastasis. Mice were either pre-treated or not with different doses of FA and the effect of combining 5-FU was also determined (Supplementary Figure 5D). Results of in vivo imaging, which measured the bioluminescence of CT26-Luc cells, showed a significantly lower number of CT26-Luc cells detected in the lungs of treated groups compared to untreated controls (day 14 after tumor implantation; Fig. 5A). Results indicated that the area of lung metastasis in FA-treated mice was significantly decreased, especially in mice treated with a combination of FA and 5-FU (Fig. 5B, C) without changes in body weight (Fig. 5D). Thus, even though 5-FU or FA alone suppressed colon lung metastasis, combining FA with 5-FU strongly suppressed colon lung metastasis (Fig. 5C). Furthermore, the phosphorylated protein expression levels of survivin (i.e., substrate of PRPK) were analyzed in tissue slides of 3 lungs per group. Results showed that p-survivin was dramatically decreased in lung tissues from groups treated with 5-FU or FA, as compared to the untreated group (Fig. 5E).
Figure 5.
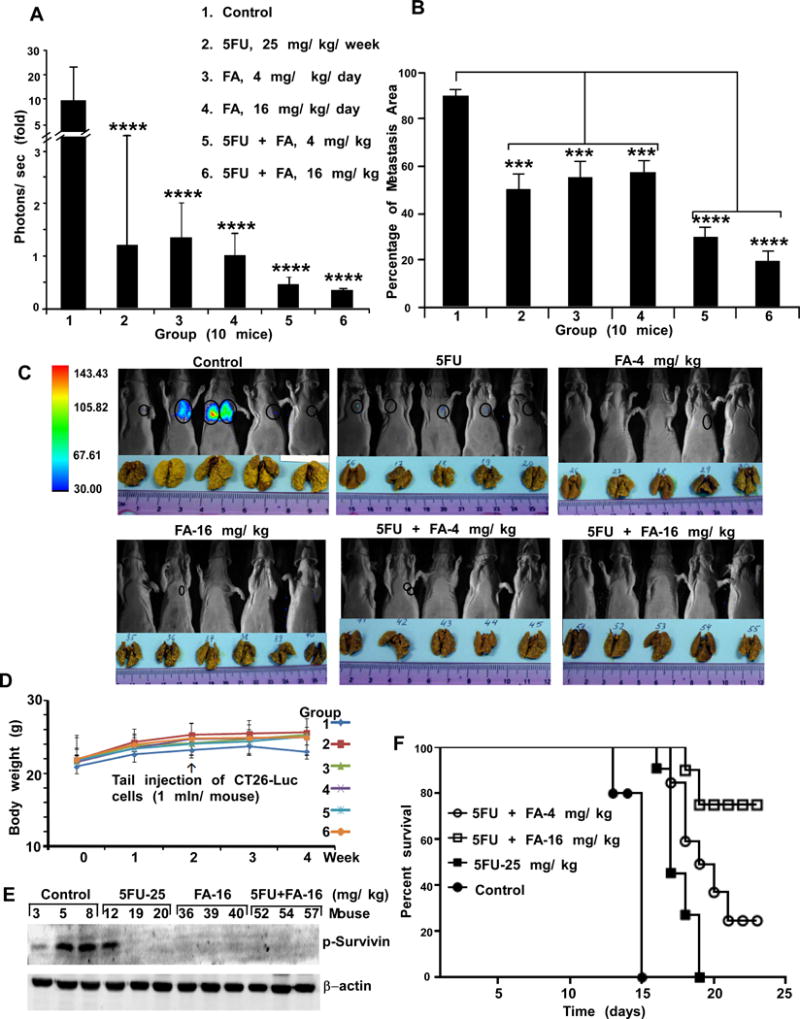
FA alone or combined with 5-FU suppresses colon lung metastasis in mice. A, CT26-Luc cells were injected into the tail vein (n = 10 mice/group). Lung tumors were observed by in vivo optical imaging with D-luciferin, which was injected intraperitoneally. Before inoculating CT26-Luc cells, groups 3-6 were intraperitoneally injected with FA for 10 days. The imaging was performed using the Xtreme Image system and bioluminescence was quantified using Bruker MI. The asterisks (****) indicate significantly less metastases (p < 0.001) in CT26-Luc cells in treated mice. B, lungs were fixed in Bouin’s solution to visualize metastatic nodules (yellow color) and data were analyzed using GraphPad Prism 5. The percentage of lung metastasis observed in treated groups was significantly decreased (***p < 0.01, ****p < 0.001) compared with untreated controls. The treated groups of mice have the same nomenclature as panel A. C, representative mice (5 mice/group) with bioluminescence signal (circled) and metastatic lungs (yellow color) are shown. D, FA or its combination with 5-FU does not affect mouse body weight. The treated groups of mice have the same nomenclature as panel A. E, the expression levels of phosphorylated survivin were analyzed in lung tissues (3 mice/group) by Western blotting. F, overall percent survival of animals (9 mice/group) with experimental lung metastasis of CT26-Luc colon cancer cells after treatment with 5-FU, FA or the combinations 5-FU and FA.
We also conducted an in vivo experiment in which mice were treated using a treatment schedule where there was no FA pre-treatment (Supplementary Figure 6A). Results of in vivo imaging, without pre-treatment with FA, also indicated a significantly lower number of CT26-Luc cells detected in the lungs of treated groups (except the group treated with a low dose of FA-4 mg/kg, alone) as compared to the untreated control group (Supplementary Figure 6B and C). The levels of p-survivin, the substrate of PRPK, were decreased in the lung tissues of the mouse experimental groups that were treated with 5-FU and FA alone. The levels of p-survivin were decreased dramatically when FA and 5-FU were used in combination (Supplementary Figure 6B, bottom). These results are similar to those obtained in the in vivo experiments with pre-treatment of FA (Figure 5E). We also examined the life-span of mice, in the different treatment groups. The survival curves showed that treatment by combination FA-16 mg/kg/mouse/day with 5-FU significantly increased life of mice compare with 5-FU mono therapy (Fig. 5 F).
The expression level of p-PRPK is associated with colon cancer stage
The 2016 survival rates for colon cancer, by stages, have been observed to decrease from 92% for patients with stage I, to 11% in patients with stage IV colon cancer (29). A colon cancer adenocarcinoma tissue array with 87 cases of tumors classified by clinical stage, and survival data was examined for p-PRPK protein levels. The 5-year survival after surgery was stage-dependent and correlated with the statistical data of the American Cancer Society (Fig. 6A). The p-PRPK protein levels in tumor tissues from patients with early stage I or II were increased, as compared to normal adjacent tissues (NAT). Levels of p-PRPK were dramatically increased with metastatic dissemination at stage III or IV, as compared to early stage I or II (Fig. 6B).
Figure 6.
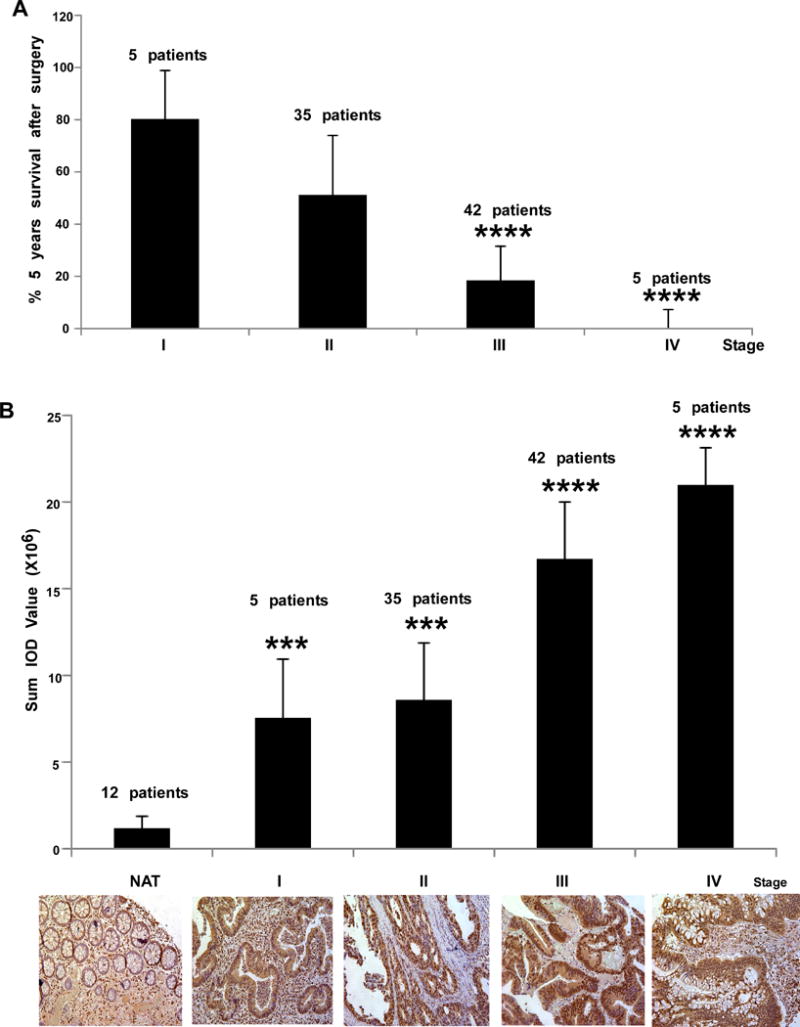
The expression levels of phosphorylated PRPK are associated with different colon cancer stages. A, the 5-year survival rate of patients was examined by tissue array (87 cases of colon adenocarcinoma) categorized by stage. Survival rate of patients with stage III or IV was significantly decreased (****p < 0.001) compared to stage I patients. B, the same tissue array was analyzed by IHC to determine expression levels of phosphorylated PRPK. Phosphorylated PRPK levels were significantly increased (***p < 0.001) in tissues at stages I and II compared with normal adjacent tissue (NAT) and at stages III and IV compared with stages I and II (****p < 0.001). Representative staining of lung tissues is shown (lower panels; 200X).
PRPK regulates colon cancer metastasis independently of p53 status
To determine if PRPK regulates p53 function directly, we checked the status of p-p53 (Ser15) in HCT116 cells with shPRPK mediated knock-down. Results indicated that in PRPK depleted cells, there was no change in p-p53 (Ser15) levels following EGF treatment (Supplementary Figure 7A). Furthermore, in p53+/+/HCT116 or p53−/−/HCT116 cancer cells, there was no difference in the expression levels of p-PRPK and total PRPK (Supplementary Figure 7B). We also examined whether the p53 status of p53+/+/HCT116 or p53−/−/HCT116 cells had an effect on migration using a wound healing assay (Supplementary Figure 7C). No differences were observed in the migration of p53+/+/HCT116 or p53−/−/HCT116 cells, suggesting that the p53 status had no effect on the PRPK regulated migration of HCT116 cells. Also, the depletion of PRPK had no effect on the expression levels of p53 or p-p53 (Ser15) in liver or lung samples with metastasis from the in vivo experiments (Supplementary Figure 7D and E). Next, we examined p-p53 or mutant p53 expression levels in the same tissue array with different tumor grades that we had used for the analysis of p-PRPK levels (Fig. 6B). Our results showed that levels of mutated p53 levels in Stages I, II or III were significantly higher, as compared to normal colon tissues (Supplementary Figure 7F). However, levels of mutated p53 were decreased in Stages III and IV, as compared with Stage II. These results suggest that levels of mutated p53 are associated with colon cancer development in Stages I to II, but are not associated with colon metastasis (Stages III or IV). Furthermore, phosphorylated p53 levels were not significantly changed in each stage compared with normal colon tissues (Supplementary Figure 7G). In contrast, our result showed that phosphorylated PRPK levels were gradually increased in Stages III or IV, compared with Stages I or II in colon cancer tissues (Fig. 6B). Overall, our results indicate that phosphorylated PRPK protein levels are not directly associated with levels of either mutated or phosphorylated p53 in the different stages of colon cancer. Together, these findings strongly suggest that the role of PRPK in colon cancer metastasis is independent of p53.
Discussion
The biological role of PRPK has not been well-defined. We observed that the protein expression level of PRPK in colon cancer cell lines was high compared to normal colon cells. For PRPK, only one phosphorylation site, Ser250, which is required for PRPK activity, has been reported (5, 6). In human colon tissue array results, the p-PRPK protein levels were higher in metastatic adenocarcinoma tissues, as compared with malignant adenocarcinoma tissues. We found that PRPK was not directly associated with p53 phosphorylation as previously reported (1, 4, 5). The up-regulation of p53 activity suppresses cancer malignancy (30); however mutant p53 proteins not only lose their tumor suppressive activities, but often gain additional oncogenic functions (31). We analyzed p-PRPK levels in several colon cancer cell lines with different p53 status. The cell lines HCT116 and DLD-1 express wild-type p53 and HCT15, HT-29 and WiDr cells express a mutant p53. All cell lines expressed similar levels of the total PRPK protein, and only HT-29 exhibited low levels of p-PRPK. These results suggest that PRPK and p-PRPK protein levels in different colon cancer cell types are not associated with p53 status in these cells. Using an in vitro kinase assay, we found that PRPK does not phosphorylate p53 directly. Active Chk1, which phosphorylates p53 (32), was used as a positive control.
Metastasis is the formation of secondary tumor foci in one or more organs away from the primary lesion (33). Identifying new mechanisms contributing to metastasis is important for discovering novel chemotherapeutic targets. We explored the biological function of PRPK in colon cancer metastasis. For experiments, we used HCT116 cells, which express wild-type p53, and HCT15 cells, which express a mutant p53. HCT116 and HCT15 cells expressing shPRPK exhibited less migration and showed attenuated invasion through Matrigel inserts. Knocking down PRPK expression prevented colorectal liver or lung metastasis of HCT116 and HCT15 colon cancer cells in vivo, and dramatically increased survival of mice. Because the in vitro kinase assay showed that PRPK does not phosphorylate p53, we searched for other substrates for PRPK. Survivin is very important for regulation of metastasis in many cancers, including colon (13-15). Survivin is phosphorylated by Cdc2 at Thr34 (17), and this phosphorylation is important for survivin activity and stability (16, 17). We showed that PRPK binds with, and phosphorylates survivin at Thr34, and that this phosphorylation increases survivin stability.
Many mitogen-activated protein kinases (MAPKs) are involved in cell adhesion, migration and invasion (34). The PRPK/survivin interaction represents a new signaling axis that promotes colorectal cancer metastasis. PRPK promoted liver metastasis of HCT116 or HCT15 human colon cancer cells, or lung metastasis of CT26 mouse colon cancer cells, in a xenograft mouse model. PRPK is more highly expressed in metastatic colon adenocarcinoma tissues compared to malignant adenocarcinoma. Blocking expression of PRPK in human or mouse colon cancer cells reduced their migration and invasion capability and metastatic properties. Survivin is up-regulated in human cancer and associated with poor prognosis (13, 15). Survivin is overexpressed in colorectal cancer (14) and stimulates liver metastasis of colon cancer (21). The phosphorylation of survivin at Thr34 is important for its therapeutic potential and its molecular mechanism of action (17). The loss of Thr34 phosphorylation results in inhibition of metastasis (35). We found that PRPK phosphorylates survivin at Thr34, and increases the stability of survivin. Phosphorylated PRPK expression was significantly elevated in colon tumor tissues with higher pathological stages III or IV compared to I or II. Thus PRPK could serve as a diagnostic marker and potential therapeutic target for high-risk colon cancer. We found that FA inhibits PRPK activity and is effective in reducing colon cancer metastasis in lung similar to 5-FU; whereas a combination of FA and 5-FU had additional anti-metastatic efficacy in vivo. Combining both agents reduced colon metastasis to the lung compared to either agent alone. We believe that this combination is a promising therapeutic approach that could be applied to a wide range of human colon cancer malignancies. PRPK is thus a driver of colon cancer metastasis. The observation that FA targets PRPK, not TOPK and dramatically inhibits colon cancer metastasis suggests that PRPK could be a more direct target for reducing metastasis. Importantly, our study identified survivin, already known to be important for metastasis, as a substrate for PRPK. The association of p53 with PRPK was not supported by our study, thus the name “p53-related protein kinase” is likely a misnomer. Finally, the translation of FDA-approved FA into the clinic in combination with the standard treatment of colon cancer with 5-FU could occur quite rapidly.
Supplementary Material
Acknowledgments
The authors thank Dr. Lorenzo A. Pinna (Universita di Padova, Padova, Italy) for the pQE-81L-PRPK plasmid and Dr. Yasuhiro Ikeda (Mayo Clinic, Rochester, MN) for providing the pSIN-Fluc vector. We also thank Dr. Tia Rai for assistance in the editing and submission of this manuscript. This work was supported by donations from The Hormel Foundation (Z. Dong) and National Institutes of Health grants CA172457, CA187027 and CA196639 (Z. Dong)
Financial Support: This work was supported by donations from The Hormel Foundation (Z. Dong) and National Institutes of Health grants CA172457, CA187027 and CA196639 (Z. Dong)
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Abe Y, Matsumoto S, Wei S, Nezu K, Miyoshi A, Kito K, et al. Cloning and characterization of a p53-related protein kinase expressed in interleukin-2-activated cytotoxic T-cells, epithelial tumor cell lines, and the testes. J Biol Chem. 2001;276:44003–11. doi: 10.1074/jbc.M105669200. [DOI] [PubMed] [Google Scholar]
- 2.Miyoshi A, Kito K, Aramoto T, Abe Y, Kobayashi N, Ueda N. Identification of CGI-121, a novel PRPK (p53-related protein kinase)-binding protein. Biochem Biophys Res Commun. 2003;303:399–405. doi: 10.1016/s0006-291x(03)00333-4. [DOI] [PubMed] [Google Scholar]
- 3.Abe Y, Takeuchi T, Imai Y, Murase R, Kamei Y, Fujibuchi T, et al. A Small Ras-like protein Ray/Rab1c modulates the p53-regulating activity of PRPK. Biochem Biophys Res Commun. 2006;344:377–85. doi: 10.1016/j.bbrc.2006.03.071. [DOI] [PubMed] [Google Scholar]
- 4.Facchin S, Lopreiato R, Ruzzene M, Marin O, Sartori G, Gotz C, et al. Functional homology between yeast piD261/Bud32 and human PRPK: both phosphorylate p53 and PRPK partially complements piD261/Bud32 deficiency. FEBS Lett. 2003;549:63–6. doi: 10.1016/s0014-5793(03)00770-1. [DOI] [PubMed] [Google Scholar]
- 5.Facchin S, Ruzzene M, Peggion C, Sartori G, Carignani G, Marin O, et al. Phosphorylation and activation of the atypical kinase p53-related protein kinase (PRPK) by Akt/PKB. Cell Mol Life Sci. 2007;64:2680–9. doi: 10.1007/s00018-007-7179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zykova TA, Zhu F, Wang L, Li H, Bai R, Lim DY, et al. The T-LAK Cell-originated Protein Kinase Signal Pathway Promotes Colorectal Cancer Metastasis. EBioMedicine. 2017;18:73–82. doi: 10.1016/j.ebiom.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson E, Stockwell SR, Li H, Aherne W, Cuomo ME, Mittnacht S. Mechanism-based screen establishes signalling framework for DNA damage-associated G1 checkpoint response. PloS One. 2012;7:e31627. doi: 10.1371/journal.pone.0031627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ibar C, Cataldo VF, Vasquez-Doorman C, Olguin P, Glavic A. Drosophila p53-related protein kinase is required for PI3K/TOR pathway-dependent growth. Development. 2013;140:1282–91. doi: 10.1242/dev.086918. [DOI] [PubMed] [Google Scholar]
- 9.Hecker A, Graille M, Madec E, Gadelle D, Le Cam E, van Tilbergh H, et al. The universal Kae1 protein and the associated Bud32 kinase (PRPK), a mysterious protein couple probably essential for genome maintenance in Archaea and Eukarya. Biochem Soc Trans. 2009;37:29–35. doi: 10.1042/BST0370029. [DOI] [PubMed] [Google Scholar]
- 10.Peterson D, Lee J, Lei XC, Forrest WF, Davis DP, Jackson PK, et al. A chemosensitization screen identifies TP53RK, a kinase that restrains apoptosis after mitotic stress. Cancer Res. 2010;70:6325–35. doi: 10.1158/0008-5472.CAN-10-0015. [DOI] [PubMed] [Google Scholar]
- 11.Nakagawa K, Taya Y, Tamai K, Yamaizumi M. Requirement of ATM in phosphorylation of the human p53 protein at serine 15 following DNA double-strand breaks. Mol Cell Biol. 1999;19:2828–34. doi: 10.1128/mcb.19.4.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasegawa K, Nakamura T, Harvey M, Ikeda Y, Oberg A, Figini M, et al. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clin Cancer Res. 2006;12:6170–8. doi: 10.1158/1078-0432.CCR-06-0992. [DOI] [PubMed] [Google Scholar]
- 13.Cheung CH, Cheng L, Chang KY, Chen HH, Chang JY. Investigations of survivin: the past, present and future. Front Biosci. 2011;16:952–61. doi: 10.2741/3728. [DOI] [PubMed] [Google Scholar]
- 14.Chen WC, Liu Q, Fu JX, Kang SY. Expression of survivin and its significance in colorectal cancer. World J Gastroenterol. 2004;10:2886–9. doi: 10.3748/wjg.v10.i19.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawasaki H, Toyoda M, Shinohara H, Okuda J, Watanabe I, Yamamoto T, et al. Expression of survivin correlates with apoptosis, proliferation, and angiogenesis during human colorectal tumorigenesis. Cancer. 2001;91:2026–32. doi: 10.1002/1097-0142(20010601)91:11<2026::aid-cncr1228>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 16.O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, et al. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97:13103–7. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aspe JR, Wall NR. Survivin-T34A: molecular mechanism and therapeutic potential. Onco Targets Ther. 2010;3:247–54. doi: 10.2147/OTT.S15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Gambosova K, Cooper ZA, Holloway MP, Kassai A, Izquierdo D, et al. EGF regulates survivin stability through the Raf-1/ERK pathway in insulin-secreting pancreatic beta-cells. BMC Mol Biol. 2010;11:66. doi: 10.1186/1471-2199-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaferian S, Negahdari B, Eatemadi A. Colon cancer targeting using conjugates biomaterial 5-flurouracil. Biomed Pharmacother. 2016;84:780–8. doi: 10.1016/j.biopha.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Fu Y, Yang G, Zhu F, Peng C, Li W, Li H, et al. Antioxidants decrease the apoptotic effect of 5-Fu in colon cancer by regulating Src-dependent caspase-7 phosphorylation. Cell Death Dis. 2014;5:e983. doi: 10.1038/cddis.2013.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res. 2003;63:230–5. [PubMed] [Google Scholar]
- 22.Chintharlapalli S, Papineni S, Ramaiah SK, Safe S. Betulinic acid inhibits prostate cancer growth through inhibition of specificity protein transcription factors. Cancer Res. 2007;67:2816–23. doi: 10.1158/0008-5472.CAN-06-3735. [DOI] [PubMed] [Google Scholar]
- 23.Leclercq R, Bismuth R, Casin I, Cavallo JD, Croize J, Felten A, et al. In vitro activity of fusidic acid against streptococci isolated from skin and soft tissue infections. J Antimicrob Chemother. 2000;45:27–9. doi: 10.1093/jac/45.1.27. [DOI] [PubMed] [Google Scholar]
- 24.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 25.Bellahsene A, Forsgren A. Effect of fusidic acid on the immune response in mice. Infect Immun. 1980;29:873–8. doi: 10.1128/iai.29.3.873-878.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taburet AM, Guibert J, Kitzis MD, Sorensen H, Acar JF, Singlas E. Pharmacokinetics of sodium fusidate after single and repeated infusions and oral administration of a new formulation. J Antimicrob Chemother. 1990;25(Suppl B):23–31. doi: 10.1093/jac/25.suppl_b.23. [DOI] [PubMed] [Google Scholar]
- 27.Turnidge J. Fusidic acid pharmacology, pharmacokinetics and pharmacodynamics. Int J Antimicrob Agents. 1999;12(Suppl 2):S23–34. doi: 10.1016/s0924-8579(98)00071-5. [DOI] [PubMed] [Google Scholar]
- 28.Vaillant L, Machet L, Taburet AM, Sorensen H, Lorette G. Levels of fusidic acid in skin blister fluid and serum after repeated administration of two dosages (250 and 500 mg) Brit J Dermatol. 1992;126:591–5. doi: 10.1111/j.1365-2133.1992.tb00105.x. [DOI] [PubMed] [Google Scholar]
- 29.Society AC. Colorectal Cancer Fact Figures 2014-2016. Atlanta, GA: 2016. [Google Scholar]
- 30.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–70. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer. 2011;2:466–74. doi: 10.1177/1947601911408889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ou YH, Chung PH, Sun TP, Shieh SY. p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol Biol Cell. 2005;16:1684–95. doi: 10.1091/mbc.E04-08-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nature Reviews Genetics. 2007;8:341–52. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- 34.Reddy KB, Nabha SM, Atanaskova N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Reviews. 2003;22:395–403. doi: 10.1023/a:1023781114568. [DOI] [PubMed] [Google Scholar]
- 35.Peng XC, Yang L, Yang LP, Mao YQ, Yang HS, Liu JY, et al. Efficient inhibition of murine breast cancer growth and metastasis by gene transferred mouse survivin Thr34–>Ala mutant. J Exp Clin Cancer Res. 2008;27:46. doi: 10.1186/1756-9966-27-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.