

Abstract
The selective functionalization of remote C–H bonds via intramolecular hydrogen atom transfer (HAT) is transformative for organic synthesis. This radical-mediated strategy provides access to novel reactivity that is complementary to closed-shell pathways. As modern methods for mild generation of radicals are continually developed, inherent selectivity paradigms of HAT mechanisms offer unparalleled opportunities for developing new strategies for C–H functionalization. This review outlines the history, recent advances, and mechanistic underpinnings of intramolecular HAT as a guide to addressing ongoing challenges in this arena.
Keywords: hydrogen atom transfer, remote C–H functionalization, radicals, nitrogen-centered radicals, oxygen-centered radicals
Graphical Abstract
1 Introduction
In modern organic synthesis, C–H functionalization is the chemical equivalent of adding the final brush strokes to a painting. While it conveniently allows for blemishes to be swiftly corrected without restarting from a blank canvas, it also necessitates great care not to disrupt the overall beauty of a near-finished piece. Similarly, since the C–H bond is the most ubiquitous motif in organic chemistry, its modification within a complex molecule necessitates a high level of care and precision. Thus, the most important challenge of C–H functionalization is selectivity.1–4 In recent decades, metal-based C–H activation5–7 and insertion8,9 mechanisms have been employed with increasing frequency to address the challenges of chemo-, regio-, and stereoselectivity.
In contrast, radical-mediated C–H functionalization,10 while discovered earlier,11–13 has played a much less significant role in advancing the frontiers of selective C–H functionalization. This is somewhat surprising given that intra-molecular hydrogen atom transfer (HAT)14 of a C–H to a remote radical site15 is typically quite selective16–18 and exergonic.14 The latter feature suggests these processes may be promoted under milder reaction conditions than their metal-mediated counterparts. In our estimation, however, the traditionally harsh conditions employed for radical generation have precluded an objective evaluation of the full synthetic potential of open-shell pathways. For instance, radical initiation has historically been conducted with strong acid, refluxing peroxides, or high energy UV light. Fortunately, modern methods now allow mild access to radicals (e.g., iodonium reagents,19–21 photoredox catalysts22–24), thereby enabling synthetic chemists to more fully explore the inherent selectivity of C–H functionalizations mediated by the diverse chemistry25–28 of single-electron transfer (SET) pathways.
This review is intended to be more instructive than exhaustive. Therefore, its focus on intramolecular HAT will highlight the key discoveries in these areas, with emphasis on the development of new strategies to (i) generate unique radicals (ii) from tailored precursors and to (iii) trap these relayed radicals in novel ways. Our objective is to illustrate the innovations and general lessons from pioneering, and more recent, contributions in each of these areas, in order to inform the reader of the challenges and opportunities that lie ahead.
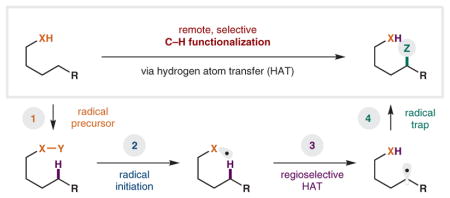
The general strategy of remote, directed C–H functionalization via HAT can be divided into four main components, as outlined above and detailed further in Scheme 1. Specifically, the mechanistic features that are common to all intramolecular HAT reactions include: (1) generation of a radical precursor; (2) initiation of the radical; (3) regioselective HAT; and (4) trapping of the relayed radical. A brief overview of the salient aspects of each elementary step is described below.
Scheme 1.

Fundamental mechanistic steps of intramolecular hydrogen atom transfer (HAT)
Radical precursor
The first, and perhaps most important, step in promoting an intramolecular HAT is the two-electron construction of a precursor that will enable access to the key, single-electron intermediate. The most common form of HAT-initiation is from N-, O-, or C-centered radicals,28 and this review is organized into three parts, each describing the reactivity of one of these radicals. As shown in Scheme 1, there is a range of related methods for accessing each radical type. For example, N-centered radicals are typically accessed via SET reduction or homolysis of a weak N–X bond, where X is a halide or N2 nucleofuge. Similarly, O-centered radicals are typically accessed via homolysis of a weak O–X bond (or via photoexcitation of a carbonyl). Historically, these weak heteroatom–halide bonds have been preformed, but they are now typically generated in situ.19 Finally, C-centered radicals (alkyl, aryl, and vinyl) can be accessed from weak C–X or C–N2 precursors.
Radical initiation
The next, key step in any HAT mechanism is the generation of the open-shell intermediate. Historically, this has been the most limiting aspect of HAT chemistry, as typical strategies employ strong acid, high temperatures, unstable peroxides, toxic organotin reagents, or high energy UV light. More recently, however, earth abundant metals (e.g., Fe, Cu, Ni), hypervalent iodine reagents, and visible-light-mediated photoredox catalysts enable mild access to radicals, harnessing inherent HAT selectivity pathways.
Regioselective HAT
Despite the rapid, exergonic nature of many intramolecular H-atom transfer events, H• abstraction frequently occurs with complete δ regioselectivity. The 1,5-HAT pathway is most typical thanks to a preorganized, six-membered, cyclic transition state, with nearly linear C–H–X geometry (153° for O•).16 For the C• mediated process, there is a strong enthalpic preference for 1,5-HAT over 1,4-HAT (ΔΔH: 6.6 kcal/mol).29 In contrast, 1,5-HAT over 1,6-HAT selectivity stems from a lower entropic barrier (ΔΔS: 8.3 eu, 2.5 kcal/mol) in the O• pathway.30 As a consequence, the rate of 1,5-HAT is at least 10 times faster (2.7 × 107 s−1) vs the 1,4 or 1,6 variants.31 Rare exceptions17 to this rule are cases where C5 lacks a H atom, or when adjacent C–H bonds are significantly weaker (e.g., benzylic, tertiary, α-oxy), or when geometry precludes 1,5-HAT. Otherwise, selective 1,5-HAT of an initiating radical typically offers access to a δ carbon radical, exclusively.
Radical trap
Last, but certainly not least, the overall transformation, and the specific identity of the group that is incorporated via C–H functionalization, relies on the trap that is used. In this regard, radical-mediated processes offer a wide array of synthetic complementarity to other metal-mediated approaches. For example, unlike the strong reliance of the latter on aryl halide or organometallic coupling partners, radical traps can range from caged radicals (X•, NO•, ON•), to weakly bonded main group molecules (N–X, Si–X, Sn–H, Sn–allyl), metal salts (CuX, CuSCN, CuN3), and π-systems (alkenes, arenes). This diversity of methods suitable for terminating HAT mechanisms allows for the widest scope of reactivity that is available for C–H functionalization via any single approach.
The following is a selected collection, showcasing the range of intramolecular HAT-mediated reactions, classified by the identity of the radical that initiates H-atom transfer (e.g., N, O, C).
2 Nitrogen-Centered Radicals
In the field of intramolecular HAT chemistry, N-centered radicals enjoy a prominent role.18 Historically, these radicals were the first used to initiate remote H-atom transfer. Additionally, they are the most tunable (via N-substitution), enabling fine modulation of their polarity,32 which is known to highly influence HAT reactivity. And thanks to their versatility, N-centered radicals are now accessible via the widest range of mild radical-initiation methods.33,34
2.1 sp3 N-Radical Initiation
The earliest example of selective C–H functionalization via HAT was reported by Hofmann in 1883 (Scheme 2).35 This N-centered radical reaction, now known as the Hofmann–Löffler–Freytag (HLF) reaction, arises from photolytic homolysis of a cationic N-haloamine.36,37 The resulting aminium radical cation is sufficiently polarized32 to enable the abstraction of a hydrogen atom from the δ carbon, producing a new C-centered radical. The selectivity of this 1,5-HAT is governed by a chairlike transition state. The ensuing δ C-radical is then trapped by recombination with a caged X• (or N–X) to generate a δ halide, which is then displaced via base-induced intramolecular cyclization. This strategy for accessing pyrrolidines directly from amines via δ C–H amination has enabled the streamlined synthesis of several natural products and their derivatives. For instance, in 1909 Löffler and Freytag employed this reaction in the synthesis of nicotine from the corresponding N-haloamine,38 and in 1958 Corey and Hertler employed this approach in the synthesis of a series of cycloaminated steroid derivatives.39 In 1979, Baldwin and Doll extended this method to the use of amides in the total synthesis of gelse-mincine.40 Overall, the HLF reaction has played a vital role in demonstrating HAT as a robust and selective radical-mediated C–H functionalization method.
Scheme 2.

δ C–H Amination via N-centered radicals by N–X homolysis
Hofmann’s seminal report inspired continued development of intramolecular HAT strategies for selective C–H functionalization, especially via N-centered radicals. In the original reports of the HLF reaction, the use of strong acid and high temperatures were required to generate the polarized N-radical, thereby prohibiting the use of acid-sensitive functional groups. Additionally, the need for N–X preformation limited the scope and efficiency of this method. In 1985, nearly a century later, Suárez and co-workers circumvented the need to preform the N-haloamine by generating N–I in situ (Scheme 3).41 By employing molecular iodine and a hypervalent iodine oxidant, PhI(OAc)2, N–I is generated from transiently formed AcOI,42 and the ensuing weak N–I bond is homolyzed by light to generate the analogous electrophilic nitrogen-centered radical. Furthermore, the need for strongly acidic media is circumvented by the use of electron-deficient protecting groups (e.g., NO2, CN) to polarize the N-radical. Through this modified HLF reaction mechanism, a series of pyrrolidines were generated, including steroid derivatives. Suárez and co-workers applied these milder reaction conditions to the synthesis of bicyclic lactams via transannular C–H lactamization,43 as well as to the δ C–H amination of carbohydrates at the anomeric carbon.44,45
Scheme 3.

δ C–H Amination via in situ N–I formation and homolysis
In 2015, the Herrera46 and Muñiz47 groups separately explored further modifications of the HLF reaction (Scheme 4). Whereas the Suárez HLF reaction efficiently aminates weak C–H bonds (benzylic, tertiary, α-oxy), Herrera and coworkers investigated 1,5-HAT from primary C–H bonds. The challenge of this transformation arises from the high bond dissociation energy (BDE) of primary C–H bonds (>100 kcal/mol vs 90 kcal/mol, benzylic).48 Their solution involves adding PhI(OAc)2 oxidant to the reaction portionwise to prevent over-oxidized side products, in favor of desired reactivity. Alternatively, divergent reactivity to access lactams was accomplished using slow addition of I2, providing a wide scope of pyrrolidin-2-ones instead.
Scheme 4.

Modern improvements to Suarez’s δ C–H amination
Also in 2015, Martínez and Muñiz reported a catalytic variant of the Suárez reaction, using 10 mol% of I2 and a tailored hypervalent iodine oxidant.47 This represents the first catalytic HLF reaction. Its utility is best for weaker benzylic and tertiary C–H bonds. In 2017, the Muñiz and Reiher groups reported a dual-catalytic method for δ C–H amination in which an organic photoredox catalyst allows for catalytic reoxidation of the I− to the I2 co-catalyst. In this pathway, air serves as the terminal oxidant to reform I2 from the HI byproduct.49
Although many HLF modifications are iodine-mediated, excess I2 can generate undesired oxidized side-products, leading to poor product formation in cases such as the amination of secondary C–H bonds. In 2016, our group introduced a solution to this challenge that circumvents the build-up of excess I2 by generating it in situ via slow oxidation of NaI (Scheme 5).50 In this approach, triiodide (I3−) is generated in equilibrium upon combination of I2 and NaI, whereby I− scavenges I2 and limits side products derived from I2 oxidation. This triiodide-mediated approach is the first to enable δ C–H amination of amines with secondary C–H bonds, and serves to complement methods that are better suited for stronger C–H bonds. Notably, this reaction is selective for δ secondary C–H functionalization, even in the presence of weaker tertiary C–H bonds, and provides a broad scope of substituted pyrrolidines from these ubiquitous precursors. Interestingly, the use of NaCl or NaBr facilitates interception of two key intermediates in the proposed mechanism: the N–Cl amine, as well as the δ-Br amine.
Scheme 5.

Triiodide strategy for δ C–H amination of methylenes
Azides have been implemented as an alternative to haloamines as precursors to N-centered radicals, enabling similar reactivity (Scheme 6). Kim and co-workers generated N-radicals by combination of alkyl azides and Bu3Sn•, followed by loss of N2.51 The resultant, Sn-stabilized N-radical effectively performs a 1,5-HAT to yield the δ C-radical, which is then trapped by Bu3SnD affording selective δ-deuteration. Recently, metal-catalyzed variants have also been developed for conversion of azides into nitrenoids. Zhang and co-workers first employed this strategy using a Co(II) metalloporphyrin catalyst;52 mechanistic studies suggest the reaction proceeds through H-atom abstraction.53 Similarly, the Betley group reported a nitrene-mediated C–H amination of both activated and unactivated C–H bonds δ to azides, using high-spin Fe(II) catalysts.54 The mechanism was proposed to occur either via intramolecular HAT from the imido radical, or via a closed-shell C–H insertion pathway.
Scheme 6.

δ C–H Amination via azide-derived N-radicals and nitrenes
In addition to selective C–H aminations, N-centered radicals have also been employed to generate other functional groups from inert, distal C–H bonds. For example, intercepting the δ-halo intermediate of the HLF mechanism has been developed as an important method for installing a versatile halide group at the δ position via remote C–H functionalization (Scheme 7). Nikishin and co-workers reported the first δ chlorination in 1985 using stoichiometric CuCl2 and sodium persulfate.55 In 2015, Yu and Qin reported a modern variant to access δ chlorination using photoredox catalysis from the preformed N–Cl amine.56 While the I2-mediated HLF reaction typically results in rapid cyclization of the δ iodide, early reports in 1989 by Suárez included the observation of a small amount of δ iodide remaining, as well as trace δ diiodide byproduct.43 Togo and co-workers demonstrated that these multi-iodination products could be harnessed in the synthesis of saccharides via a triple C–H iodination of o-tosylamides at the benzylic position, followed by ring closure and hydrolysis.57
Scheme 7.

δ C–H Halogenation via halide radical traps
Selective C–H bromination was obtained by Corey and co-workers using in situ generation of a trifluoroacetamide N–Br via direct amide combination with acetyl hypobromite (AcOBr).58 Under these reaction conditions, HLF amination does not occur on the δ bromide. In 2017, J.-Q. Yu and co-workers introduced a Cu-catalyzed approach to δ bromination using Me3SiN3 and NBS to generate transient amidyl radicals and intercept their δ-C radicals.59 Completing the halogen series, Cook and co-workers reported the first δ C–H fluorination, enabled by Fe-catalyzed transposition of an N–F to a benzylic C–F with complete selectivity, derived from 1,5-HAT of an amidyl radical, even in the presence of other benzylic C–H bonds.60
In 2008, Baran and co-workers developed an HAT-based C–H halogenation strategy to access 1,3-diols from N-bromocarbamates (Scheme 8).61 Following photoinitiated homolysis of N–Br, 1,6-HAT occurs along with Br• trapping, affording the alkyl bromide. Although the HLF reaction typically undergoes 1,5-HAT, this carbamate tether promotes a seven-membered, cyclic transition state, affording the γ halide. The caveat is H• abstraction must be from a benzylic or tertiary C–H bond. The resulting alkyl bromide is then displaced, generating an iminocarbamate. This intermediate is hydrolyzed to unmask the 1,3-diol, providing access to a synthetically valuable motif in a one-pot synthesis from the N-bromocarbamate. Notably, this strategy enabled the total synthesis of several natural products, including rengyol and isorengyol. In 2017, Roizen and co-workers reported N-chlorosulfamates facilitate highly selective γ halogenation of secondary C–H bonds.62 This photoinitiated chlorination remains γ selective even in the presence of weaker C–H bonds that are tertiary or α to heteroatoms. It is expected this robust γ selectivity, dictated by sulfamate geometry, is extendable to other γ C–H functionalizations.
Scheme 8.

γ C–H Halogenation of alcohols via N–X homolysis
In 2015, J.-Q. Yu and co-workers utilized amidyl radicals to simultaneously functionalize both γ and δ C–H bonds in a single cascade reaction to generate iodolactams from alkyl amides (Scheme 9).63 This transformation begins with in situ conversion of an amide into its corresponding N–I intermediate by treatment with NIS. Following 1,5-HAT, iodination, and lactam formation, the mechanism then proceeds via an azido radical mediated β-scission of the C–N to form a terminal olefin. Subsequent 5-exo-trig cyclization and I• trapping yields the δ-iodo-γ-lactam. This selective dual-functionalization method converts two remote C–H bonds into vicinal functionalities in a single step.
Scheme 9.

γ,δ-Amino-iodination of amides via amination/scission cascade
The common feature in all of the previously described mechanisms is the SET reduction or homolysis of a weak N–X to generate an N• intermediate. Even the modern Suárez variant employs in situ N–X formation. In all these cases, there must be an X• or weak N–X present; and these are great traps for the δ radical. Thus, while there are several methods of accessing δ C–H functionalization with halides and heteroatoms, there is no possibility for accessing C–C formation via this mechanism.
In 2016, the Knowles64 and Rovis65 groups independently reported methods to construct distal C–C bonds via amidyl radicals and circumvented the need for pre-installation, or in situ generation, of an N–X bond (Scheme 10). This mechanism proceeds via a neutral amidyl radical, which is generated from oxidative proton-coupled electron transfer (PCET) by an excited iridium photocatalyst. As in the HLF reaction, the resulting amidyl radical undergoes a 1,5-HAT to form a carbon-centered radical. However, instead of undergoing typical X• trapping (since there is no halogen), the C-radical is trapped via 1,4-addition into a Michael acceptor, producing a new C–C bond from a tertiary δ C–H bond. Reduction of the newly formed α-EWG C-radical provides a stabilized anion and regenerates the Ir(III) catalyst. Upon protonation of the anion, the remote functionalized product is isolated. This first, interrupted HLF reaction represents the only method for δ C–H alkylation by HAT from an N-radical. Its success stems from avoiding the need for halogens (X2 or in situ N–X) to generate the N-radical, and thus non-halide based trapping mechanisms may now be accessed.
Scheme 10.

Photoredox-catalyzed γ or δ C–H alkylation via amidyl radicals
In 2017, Rovis and co-workers reported a related method, wherein C–H alkylation occurs on the carbonyl side chain of the amide (rather than the N-substituent).66 Since regioselectivity is again dictated by a 1,5-HAT mechanism from an amidyl radical, C–H alkylation is γ selective in this case.
2.2 sp2 N-Radical Initiation
An iminyl (sp2) N-centered radical exhibits complementary reactivity to aminyl (sp3) N-radicals. Extensive kinetic studies by Newcomb and co-workers67 have shown that iminyl radicals undergo faster addition to olefins and slower reduction by an intermolecular H-atom relative to neutral aminyl radicals. This distinct kinetic profile of the N(sp2)-centered radical allows access to different synthetic avenues. While pioneering work from Forrester,68 Zard,69 Narasaka,70 and Weinreb71 have shown the synthetic utility of iminyl radicals by their addition into π-systems, there are few reports on N(sp2) radical-based HAT. The harsh conditions typically employed to generate iminyl radicals (strong oxidants, elevated temperatures) have likely limited an extensive exploration of this reactivity.
An initial report by Forrester and co-workers in 1979 demonstrated iminyl radicals are capable of performing 1,5-HAT to form a radical δ to N, and γ to an imine (Scheme 11).72 These radicals were generated by decomposition of oximes bearing a pendant acid.73 Upon Cu-catalyzed, per-sulfate-mediated 1e− oxidation, and subsequent loss of CO2 and CO, the iminyl radical engages in several reactive pathways, including HAT. The resulting benzylic radical is oxidized under these conditions and trapped by the imine via intramolecular cyclization. Various tetralone derivatives were synthesized via this new iminyl-radical based approach, including pyridyl analogues.74,75
Scheme 11.

γ C–H Functionalization via iminyl N(sp2) radicals
In 2011, the Chiba group reported a Cu-catalyzed benzylic oxygenation via iminyl radicals.76 The radical precursors were generated in situ by Grignard addition into nitriles to form aryl imines. Next, direct Cu-catalyzed oxidation under O2 atmosphere facilitated 1,5-HAT of the iminyl radical to form a benzylic radical, which is subsequently trapped by O2 to provide a 1,4-keto-imine, which upon hydrolysis affords the diketo product.
In 2017, Shu and Nevado reported a mild, photoredox-catalyzed method to access and harness iminyl radicals (Scheme 11).77 Employing acyl oximes as radical precursors, it was found that Ir photocatalysts could reduce the weak N–O bond of the oxime to generate an iminyl radical. Upon HAT, the δ C-radical could either result in C–N or C–C formation, depending on reaction conditions. In the former case, an oxidation event turns over the photocatalyst and allows for C–H amination via ring closure in the presence of DBU. On the other hand, C–H arylation is observed in a CH3CN/H2O mixture when the radical first adds into an arene before oxidation by the catalyst.
H. Fu and co-workers78 have similarly employed an oxime for photoinduced iminyl radical formation.79 However, by designing a more easily reduced (2,4-dinitrophenyl) nucleofuge, it was found that a metal catalyst is not needed. Instead, tertiary amines are able to form an excited state complex with the dinitrophenyl group to promote photoinduced fragmentation. In their system, the resultant iminyl radical undergoes 1,5-HAT to generate an α-amino radical. Oxidation of this species affords transient formation of an iminium, which is then trapped by the pendant imine. An additional oxidation under the reaction conditions provides the heterocyclic product.
Typically, N(sp2) radical precursors are generated by either condensation of hydroxylamine onto a ketone or acylation of an oxime. However, in 2017 the Nevado group employed a synthetically distinct route to generate an iminyl radical (Scheme 12),80 via Suzuki’s method of C• addition into vinyl azides.81 Upon decarboxylative radical formation and addition into vinyl azide, the intermediary α-azido radical rapidly expels N2 to form an iminyl radical that promotes HAT and subsequent γ-arylation.
Scheme 12.

C–H Arylation via vinyl azide-derived N(sp2) radicals
In 2012, the Chiba group reported the first use of an amidinyl radical in HAT (Scheme 13).82 These amidine radical precursors are readily derived from amines, and can be oxidized directly using a Cu catalyst and O2 as the oxidant. Upon HAT from tertiary or benzylic C–H bonds, the resulting radical (δ to the N, or β to the amidine) can be trapped in divergent ways to access either β-oxygenation or β-amination. In the former case, O2 serves as the trap, and an ensuing Cu-mediated fragmentation and cyclization affords oxazolines via β-oxygenation. In 2013, the Chiba group reported N-Ph amidines are converted into imidazolines via an alternate β-amination pathway that employs PhI(OAc)2 as the oxidant instead of O2.83 This powerful method of converting an amine into a vicinal diamine via β C–H amination was further extended by the Chiba group in 2014 through the development of a redox-neutral variant.84 The use of amidoximes, or the oxime variant of an amidine, as a radical precursor allows for an oxidant-free approach. In this case, the Cu serves as a radical initiator by reducing the weak N–O of the oxime, and the oxidized Cu later serves as a trap of the relayed radical, wherein oxidation allows for redox turnover of the Cu catalyst along with formation of the imidazoline.
Scheme 13.

β C–H Functionalization of amines via amidine radicals
In 2017, our group reported a complementary strategy to convert alcohols into β-amino alcohols by C–H amination via in situ generated imidate radicals (Scheme 14).85 To achieve the goal of simplified access to an N(sp2) radical from an abundant functional group (e.g., an alcohol), we envisioned imidates as radical precursors. They are easily prepared in situ via nitrile condensation with alcohols,86 and their closed-shell reactivity is well understood (e.g., the Overman rearrangement).87 However, imidate radicals were previously only employed in π-addition cyclizations.88 Fortunately, under our triiodide-mediated δ C–H amination conditions [i.e., NaI, PhI(OAc)2], it was observed that the β C–H amination of a range of alcohols was possible via this imidate-based strategy. In the mechanism, the imidate forms an N–I bond in situ, which is homolytically cleaved by visible light to generate an N(sp2) radical. Ensuing 1,5-HAT translocates the radical to the β carbon, which is subsequently trapped by I• and displaced intramolecularly to afford an oxazoline product. Notably, benzylic, allylic, secondary, and even primary C–H bonds are aminated in this approach. The identity of the imidate enables the amination of these bonds of varying strength. For example, tri-chloroacetimidate promotes benzylic and allylic amination, quantitatively, while benzimidates enable C–H amination of stronger primary and secondary C–H bonds. As an illustration of its synthetic utility, the oxazoline intermediate can be hydrolyzed to a free β-amino alcohol, or substituted by nucleophilic addition to access a family of β amines.
Scheme 14.

β C–H Amination of alcohols via imidate radicals
3 Oxygen-Centered Radicals
Another common initiator of intramolecular HAT is the oxygen-centered radical. The more electronegative oxygen atom (χ = 3.4, O vs 3.0, N or 2.5, C) provides a greater driving force for HAT due to the following two, related factors: (1) an open-shell is highly disfavored for this atom, whose electronegativity is second only to fluorine, thereby promoting HAT; and (2) the O–H that forms upon HAT is rather strong (BDE = 110 kcal/mol, O–H vs N–H, C–H, <100 kcal/mol). Thanks to these favorable driving forces, the remote radicals generated via HAT from O-centered radicals have become the most synthetically versatile, allowing for halogen, heteroatom, and even alkyl trapping of the intermediate carbon radical. The challenge for this mechanism ultimately lies in the generation of radicals on such an electronegative atom. The three most common pathways (carbonyl photo-excitation and alkoxy or non-alkoxy radical initiation) are described below.
3.1 Carbonyl Diradical Initiation
In the earliest example of HAT mediated by an O-centered radical, Norrish reported the photoinitiated generation of 1,4-diradicals from alkyl or aryl ketones bearing γ C–H bonds (Scheme 15).89 Upon photoexcitation of the ketone, the more reactive O-radical undergoes 1,5-HAT to generate a 1,4-diradical intermediate, with both C-radicals remaining. Depending on the nature of the photoexcited state, the diradical species will then either recombine to yield cyclobutanol (via triplet state, T1) or fragment via β-scission to form an enol and alkene (via singlet state, S1).90
Scheme 15.

Norrish photochemical strategy for C–H functionalization
Taking advantage of this mode of carbonyl reactivity in a landmark discovery, in 1973 Breslow and co-workers achieved selective abstraction of the C14 hydrogen atom within a steroid via the diradical of a benzophenone carbonyl, which was tethered to the C3 alcohol via esterification.91 In this case, a subsequent, oxidative HAT adjacent to the resulting C-radical furnishes a remote olefin. More recently in 2013, in the synthesis of ouabagenin Baran and co-workers utilized the C–C bond-forming Norrish reaction to construct the key C19 oxidation en route to the natural product from an acyclic, β-methyl ketone precursor.92
3.2 Alkoxy Radical Initiation
Since alcohols are one of the most common and versatile functional groups in synthesis, the formation of O-centered radicals from alcohols has enabled the development of a family of important C–H functionalization methods. The first example of HAT from an alcohol-derived alkoxy radical was discovered by Sir Derek Barton in 1960, and was eventually recognized with a Nobel Prize in 1969 (Scheme 16).93–95 Barton’s solution for generating the reactive O-radical was to first synthesize a precursor containing a weak N–O bond. This nitrite can be conveniently prepared via combination of an alcohol with nitrosyl chloride in dry pyridine. Photolytic homolysis forms both •NO and an alkoxy radical. Upon 1,5-HAT, the δ C-radical of the alcohol recombines with the •NO to form a δ-nitroso alcohol, which tautomerizes to form the δ-aminated oxime product. Barton and co-workers demonstrated the synthetic utility of this new C–H functionalization reaction within a variety of steroid cores, including aldosterone.96 There have been many important applications of this method for the remote C–H amination of hydrocarbon cores, such as the Corey group synthesis of histrionicotoxin.97 As an added example of the broad utility of this strategy for the incorporation of functionality at remote C–H bonds, the δ-amination of Me10-carborane was accomplished by Hawthorne and co-workers.98
Scheme 16.

The Barton reaction: δ C–H amination via O-centered radicals
Within a year of Barton’s discovery of an O-centered radical synthetic route via nitrite homolysis, the Smith99 and Walling100 groups developed alternate pathways to access alkoxy radical-mediated HAT via homolysis of O–X bonds (Scheme 17). In analogy to the N–Cl precursor of the HLF reaction, the O–Cl of hypochlorite precursors are readily homolyzed by photolysis to give alkoxy radicals. Upon HAT, the δ C-radical recombines with the •Cl to form a δ-chloro alcohol, which is readily cyclized with base to form tetrahydrofurans.101
Scheme 17.

δ C–H Halogenation via metal-mediated O–X cleavage
In the following decades, a tour de force of applications of this approach was developed by Čeković and co-workers.16 Among them, the use of hydroperoxides enables the isolation of these relatively more stable radical precursors. Iron-mediated cleavage of the peroxide O–O bond affords the δ C-radical intermediate, which can be trapped by copper(II) salts in rapid, radical-combination mechanisms reminiscent of those pioneered by the Kochi group.102,103 For example, the use of Cu(OAc)2 facilitates the subsequent single-electron transfer (SET) oxidation and elimination to form the δ-unsaturated alcohol.104,105 Alternatively, when other copper salts (CuX2, where X = SCN, N3, Cl, Br, I) are employed, substitution of the δ C with the X ligand of CuX2 is achieved, offering access to a range of δ-functionalized alcohols.106 A modern variant of this family of Čeković reactions includes the catalytic version reported by Ball and coworkers in 2010.107 In this protocol, the conversion of hydroperoxides to δ-chloro alcohols is achieved via the use of a sub-stoichiometric amount of a Cu catalyst for the first time, also in the presence of a terminal chloride source, NH4Cl, in excess. The use of a tridentate amine ligand appears vital for promoting catalytic peroxide reduction, likely due to the more reducing nature of this ATRP catalyst.
In 2014, Taniguchi and co-workers disclosed another interesting peroxide-based HAT, which is promoted by a sub-stoichiometric iron phthalocyanine catalyst.108 In their cascade mechanism, alkenes are converted into hydroperoxides by a combination of Fe(Pc), O2, and NaBH4. The ensuing O–O reduction is mediated by the Fe catalyst, whose turnover is again mediated by O2 and NaBH4, resulting in the formation of a 1,4-diol from the parent alkene.
In 1962, on the heels of Barton, Smith, and Walling’s discoveries of HAT reactions mediated by O-centered radicals (Scheme 18), Kalvoda and co-workers reported the first example of the direct generation of an alkoxy radical from an alcohol.109 The key to this solution is in situ formation of an O–X bond by the reagent combination of Pb(OAc)4 and I2. A hypoiodite, AcOI, is likely formed, which can combine with the alcohol to generate the weak O–I bond of the alcohol hypoiodite.42 Upon homolysis, HAT, and halide radical trap, the δ-iodo alcohol is formed in situ, which readily cyclizes to form the δ-oxygenated tetrahydrofuran product. Kalvoda and co-workers employed this approach in the synthesis of the pregnane steroids. In 1998, Ryu demonstrated the δ-carbon radical could be intercepted by CO to provide the carbonylated radical, which is oxidatively combined with the alcohol to provide a cyclic ester.110 This approach offers a valuable synthetic approach to access lactones directly from alcohols via δ C–H oxygenation.111
Scheme 18.

δ C–H Oxygenation of alcohols via in situ O–X formation
In 1969, Trahanovsky and co-workers demonstrated that ceric ammonium nitrate (CAN) could also be employed as an alternative to the Pb-based pathway.112 The intermediate radical may be trapped by the single-electron, Ce oxidant via either inner or outer sphere SET oxidation. Notably, in 1984, Suárez and co-workers made perhaps the most significant advance in this area by employing hypervalent iodine, PhI(OAc)2, to generate the alkoxy radical, in lieu of a Pb or Ce oxidant.113 This lead-free alternative offers a complementary pathway to access the weak O–I bond of the alcohol hypoiodite, and the ensuing etherification cascade pathway. Suárez and co-workers demonstrated the broad utility of this mild, photolytic method in the synthesis of a range of δ-oxygenated steroid and carbohydrate analogues.44,114
In addition to homolysis of O–X and O–O bonds, O-centered radicals may be accessed via scission of weak O–S or O–N bonds. For example, in 1997, Čeković reported that the O–SPh bond of benzenesulfenates is cleaved by photolytic generation of a thiophilic tin radical (Scheme 19).115–117 In this alternate approach to the typical homolysis pathway, the alkoxy radicals that are formed no longer have a radical counterpart and are available to interact with a range of traps following 1,5-HAT. Notably, these δ-carbon radicals combine rapidly with electron-deficient olefins (kadd = 3 × 105 M−1 s−1), even in the presence of Sn–H, which provides the terminal H-atom for this alkylation cascade. This strategy for multicomponent δ C–H alkylation, pioneered by Čeković and co-workers, has been the only known approach for HAT-mediated δ C–C bond formation, until the recent PCET-based method for N-centered radicals.64,65
Scheme 19.

δ C–H Alkylation via O–SPh or O–NPhth cleavage
As a bench-stable, isolable alternative to sulfenates, Kim and co-workers introduced N-alkoxy-phthalimides, whose weak O–N bond can also be cleaved by Sn radicals.118 In this case, Sn• combines with the imide carbonyl to form a captodatively stabilized C-radical, whose β-scission forms an alkoxy radical. When Sn–D is employed, δ-deuteration is observed, indicating the presence of an alkoxy-radical-mediated HAT mechanism. Sammis and co-workers employed these N-alkoxy-phthalimide precursors to enable intramolecular trapping of electronically neutral alkenes.119,120 Recently, a Sn-free alternative has been developed via the use of photoredox catalysis. In this case, a photoexcited Ir catalyst serves as a SET reductant capable of reducing the N–O bond generating a spectator phthalimide anion along with the alkoxy radical. In 2016, Chen and co-workers were able to trap the δ radical with allyl sulfones to enable δ C–H allylation.121 In the same year, Meggers and co-workers demonstrated that the Ir photoredox catalyzed SET reduction of the N–O could be coupled with a second catalytic cycle, wherein a chiral Rh complex could exhibit stereocontrol over the δ radical addition into enones.122 Indeed, this alkylation is the first and only method for enabling asymmetric termination of a radical resulting from an intramolecular HAT.
3.3 Non-alkoxy Radical Initiation
Alongside alkoxy radicals, the O-centered radicals that are substituted with adjacent heteroatoms (e.g., N–O•), are also synthetically useful, since they: (1) are typically easier to access via oxidation, and (2) afford distinct classes of products. For example, the Chiba group have shown that both oximes and hydrazones, which are each readily accessible by carbonyl condensation, are prone to facile oxidation when heated in the presence of TEMPO (Scheme 20).123 The heteroatom-substituted O• is stabilized by resonance, which facilitates its generation by mild oxidants, especially compared to alcohols, which are not as easily oxidized. Furthermore, the α-N influences the reactivity of the O• mediated HAT, as well as the ensuing cyclization to generate the isoxazoline product. With further aryl substitution, the Chiba group found that isoxazoles are also accessible. In a mechanistically similar vein, Pierce and co-workers have demonstrated that thiohydroxamic acids can be oxidized by DDQ to form an N–O• that effects HAT and subsequent oxidative cyclization to access oxathiazoles.124
Scheme 20.

δ C–H Oxygenation via in situ oxime-derived radicals
In a complementary pathway to SET reduction of the NO in N-alkoxy-phthalimides, the Oisaki and Kanai group have shown that oxidative conditions can leave the N–O intact and form an O-centered radical instead (Scheme 21). When this hydroxylamine radical is reversibly appended onto an alcohol, it can facilitate 1,6-HAT due to conformational constraints of the directing activator group. In 2016, they showed that Co catalysts under aerobic conditions can initiate radical generation and terminate the ensuing radical to afford γ-ketones on hydroxylamine-radical-appended alcohols.125 Also in 2016, the Oisaki and Kanai group showed that these radical precursor scaffolds can be combined with in situ generated NOx species to form γ-nitrated alcohols.126
Scheme 21.

δ C–H Oxygenation via in situ hydroxylamine radicals
A final class of O-centered radicals employed in HAT is generated from carboxylates (Scheme 22). Although it is easier to access O• from a carboxylate (1.4 V vs >2 V for alcohol)30 via SET oxidation using Ag, Pb, or I, the overriding challenge of this approach is that decarboxylation is quite facile (c.f. Kolbe, Hunsdiecker reactions). In 1983, Nikishin and co-workers discovered a method of bypassing this major byproduct-forming pathway by using a CuCl/Na2S2O8 oxidant.127 The ensuing HAT, Cl• trap, and chloride displacement affords γ-lactones via γ C–H oxygenation of acids. Interestingly, a NaCl/Na2S2O8 combination is also effective in promoting this transformation, thus the role of the copper remains unclear. In 2016, Du Bois and co-workers showed that phenylbutanoic acid derivatives bearing a benzylic γ C–H bond are also esterified by a Cu-catalyzed process.128 However, mechanistic experiments indicate that in contrast to non-benzyl carboxylates, intermolecular HAT of a benzyl C–H by sulfate radicals is more likely in this case.
Scheme 22.

δ C–H Lactonization via in situ carboxylate O–H oxidation
4 Carbon-Centered Radicals
HAT mediated by C-centered radicals are rarer than their heteroatom counterparts. Unlike the N• and O• initiated mechanisms, in which C–H is exchanged for a stronger N–H or O–H bond,18 the C• pathway suffers from the challenge that both the HAT precursor and product similarly contain a C–H bond. Therefore, a necessary driving force is required in these transformations, such as the exchange of an C(sp2)-centered radical for a lower energy C(sp3) radical via HAT of an alkyl C–H to form a stronger aryl C–H bond. Although bond strength is not the only determinant of HAT-based chemical reactivity (since HAT reactions are often irreversible, exothermic processes with early transition states),14 BDE of reactants often serve as a useful guide for predicting HAT reactivity.129
4.1 sp2 C-Radical Initiation
The translocation of C• sites by intramolecular HAT between C–H bonds was first reported by the Curran group in 1988.130 This pioneering work made use of the energy difference between C(sp2) and C(sp3) radicals to promote HAT from an alkyl C–H to a vinyl radical (Scheme 23). In their early reports, the Curran group demonstrated that vinyl bromides are capable of serving as both radical precursor and intramolecular trap, to afford substituted cyclopentanes. In this mechanism, Bu3SnH also plays a dual role in chain propagation, via vinyl radical initiation as well as alkyl radical termination.131 A 2015 synthesis by Vellucci and Beaudry of the aspidosperma alkaloid, goniomitine, utilizes a vinyl bromide initiated HAT cascade.132 An excellent review by Dénès, Beaufils, and Renaud describes major developments in the synthesis of five-membered rings by this approach of translocation and cyclization of vinyl radicals.133
Scheme 23.

δ Alkylation via vinyl bromide derived C(sp2) radicals
An important application of this mechanism is the incorporation of aryl radical precursors as protecting groups on alcohols, as either benzyl or silyl ethers. An example of the latter includes use of an o-iodophenyl silyl ether as a radical precursor, which enables selective 1,5-HAT from an α-oxy C–H (Scheme 24).134,135 This stabilized radical was trapped by electrophilic olefins in an intramolecular fashion. Inspired by this radical translocation reaction, Gevorgyan and co-workers in 2016 developed a method to introduce an olefin functionality via Pd catalysis.136 In this mechanism, radical initiation and post-HAT elimination are both mediated by Pd in a mechanism that combines metal catalysis and intramolecular HAT.
Scheme 24.

α-Ether C–H functionalization via HAT from aryl radicals
Like alcohols, amines are also readily converted into radical precursors that can facilitate intramolecular HAT. The collaborating groups of Snieckus and Curran demonstrated that o-iodobenzamides are suitable radical precursors that provide streamlined access to α-amino radicals via HAT (Scheme 25).137 Upon tin-mediated abstraction of C(sp2)–I to form an aryl radical, HAT enables distal functionalizations via both intra- and intermolecular trapping of the α-amino radical. Applications of this α-amino C–H functionalization include cyclization, allylation, and deuteration.137–140 The mechanism of formation of the aryl radical by Bu3SnH has been studied extensively.141
Scheme 25.

C–H Functionalization of α-amides via HAT from aryl radicals
Ito and co-workers demonstrated amines (vs amides) also undergo HAT via an o-iodobenzyl precursor (Scheme 26).142 Employing SmI2 as a radical initiator, α-amino C–H abstraction is followed by ketone trapping to afford α C–H alkylated amines. The postulated, penultimate intermediate is an α-amino Sm species that readily combines with carbonyls.143 Undheim and co-workers extended this radical translocation of amine substrates to intermolecular α-amino C–H alkylation with alkene traps, such as acrylate.144,145 Using a 2-bromoindole as a precursor, Gribble and co-workers employed a C2-indole radical to effect HAT. Ensuing α-amido radical addition back into the indole trap forms indolines.146 As a Sn-free alternative, Murphy and coworkers developed a strategy that employs dialkylphosphine oxide as a radical initiator for iodide abstraction of o-iodoanilides. Following HAT, the α-acyl radical combines with the anilide to form indolone heterocycles via C–H arylation.147
Scheme 26.

C–H Alkylation of α-amines and α-amides via aryl radicals
Modern adaptations of these aryl radical mediated HAT reactions employ metal catalysts to serve the dual roles of radical initiation and termination. In 2010, the Nakamura group reported an iron-catalyzed α-amino arylation reaction, in which aryl Grignards are cross-coupled with the α-amino radical (Scheme 27).148 Similarly, Ni- and Ir-catalyzed variants of this class of reactions have been developed by the Kalyani149 and Xu150 groups, respectively. In these mechanistically, related reactions, pendant arenes serve as radical traps. A Mg-mediated version was also reported in 2016 by the Zeng group.151 Here, either 1e− reduction, or fluoride abstraction, generates the aryl radical species, which effects 1,5-HAT to yield a Mg-stabilized α-amido radical that is trapped by chlorosilanes to construct C–Si bonds, expanding the radical translocation method beyond the introduction of C–X, C–O, C–C, and C–N bonds.
Scheme 27.

Metal-catalyzed α C–H functionalization via aryl radicals
While aryl halides are most common, other substituted arenes can also be employed as radical precursors. For example, arenediazoniums, ArN2+, are synthetically useful aryl radical precursors152 that facilitate efficient C–C formation, such as in the Meerwein arylation of olefins.153 In 1954, Hey and Turpin demonstrated that the diazonium salts of o-aminobenzamides are demethylated upon diazonium decomposition.154 Building on this pioneering discovery, Weinreb and co-workers developed a general method for α-functionalization of benzamides (Scheme 28).155 This method involves Cu-catalyzed decomposition of the diazonium to an aryl radical, followed by 1,5-HAT of the α-amino C–H. Rapid oxidation of this electron-rich radical leads to transient iminium cation formation that is trapped by alcohols. In 2016, the Zhang and Qi groups developed metal-free versions of this transformation using ascorbic acid as the radical initiator, or organic photocatalysts.156,157 Additionally, the hemiaminal products were treated with a Lewis acid and either allylSiMe3 or Me3SiCN to afford a net C–H α-allylation or α-cyanation. Alternatively, Maulide and co-workers employed a hydrazine that serves two roles: reductant to generate the aryl radical, and nucleophile for addition into the transient iminium formed upon α-amino radical oxidation.158 The resultant cyclic aminals open to form stable hydrazones, which can be treated with a second (alkyne) nucleophile to provide ring-opened addition products.
Scheme 28.

α-Amino C–H functionalization via arenediazonium-derived radicals
In 2012, Baran and co-workers employed an aryltriazene as a radical precursor, since it serves as an arenediazonium equivalent upon loss of an amine (Scheme 29).159 In the presence of an acid or metal salt, the in situ generated diazonium is rapidly reduced and extrudes N2 to form an aryl radical. After a rare, 1,7-HAT (likely dictated by the sulfonate geometry), subsequent oxidation and elimination is facilitated by TEMPO to form remote alkenes. This remote desaturation is accessible for both alcohols and amines by use of the triazene-based precursor/protecting groups. In a related method, Ragains and co-workers employed an Ir photocatalyst to promote the same triazene sulfonate-mediated 1,7-HAT.160 However, the termination entails a catalyst-turnover oxidation, which affords a tertiary cation that is hydrolyzed with H2O to yield a net γ C–H hydroxylation.
Scheme 29.

Remote C–H functionalization via ArN2-derived radicals
H. Fu and co-workers incorporated these triazenes within amides to promote 1,6-HAT (dictated by formation of a tertiary radical). This remote C(sp3) radical is trapped by a pendant aryl group to form heterocycles.161 The further development of new pathways based on such orthogonal radical precursors may enable access to iterative HAT-based C–H functionalizations with different traps and selectivities.
4.2 sp3 C-Radical Initiation
The challenge associated with HAT initiated by C(sp3)-centered radicals is the lack of energetic difference between the initial C• and the resultant C• after HAT. A solution to this problem was developed by Crich and co-workers, wherein generation of a more stable α-oxy radical is the driving force (Scheme 30).162 In a pioneering example of sp3 C• initiated HAT, epimerization of α-mannoside to β-mannoside was accomplished, overcoming an anomeric stereo-preference, a challenge in its own right.
Scheme 30.

Anomeric radicals derived from alkyl bromides
In this radical-mediated epimerization, Sn• initiated reduction of an alkyl bromide affords a primary, sp3 C• that undergoes 1,5-HAT to generate a more stable, secondary, α-oxy radical, with added anomeric stabilization. A terminal, chain-propagating Sn–H reduction from the less sterically encumbered bottom face affords a 2:1 ratio of α/β mannoside, favoring the isomer not stabilized by the anomeric effect. The Ueno–Stork method was employed to rapidly install the sp3 C–Br, radical precursor via bromoetherification.163,164 After epimerization, this traceless ketal is hydrolyzed via acidic workup.
Another strategy for promoting HAT via an sp3 C• is to employ an α-EWG halide, whose C–X bond strength is weaker than the translocated, secondary C–X. In 1997, Masnyk reported the use of an α-sulfonyl iodide as a radical precursor to effect an HAT-induced cyclization to form cyclopentanes from acyclic alkanes via δ C–H functionalization (Scheme 31).165 In this case, radical initiation via thermal peroxide initiation or photolytic Sn–Sn cleavage precedes I• abstraction from a radical precursor that is easily prepared by α-iodination of the acidic alkyl sulfone. Upon 1,5-HAT, a secondary sp3 C• abstracts an iodide via radical recombination, or chain propagation, to afford an atom-transfer adduct. The resulting δ-iodo sulfone undergoes base-mediated intramolecular cyclization to generate cy-clopentanes, an all-carbon version of the HLF reaction.
Scheme 31.

δ C–H Alkylation via atom-transfer of an C(sp3)–I
Although these sp3 C• induced HAT mechanisms are rarest, and most challenging, there remains great opportunity to design new strategies that favor C• to C• translocation. For example, mechanistic studies by Wood and co-workers have demonstrated that captodative stabilization can override a kinetic isotope effect in dictating the fate of 1,5-HAT.166 In particular, it was observed that radical translocation favors formation of more stable, amino acid α-radicals over simply α-amino ones that lack the additional push-pull effects of captodative stabilization.167
In 2017, Gevorgyan and co-workers introduced a Pd-photo-catalyzed remote desaturation of aliphatic alcohols (Scheme 32).168 Employing (halomethyl)silanes as precursors (developed by Nishiyama and co-workers for radical addition to olefins),169 they demonstrated a hybrid Pd-radical mechanism facilitates SET reduction, regioselective HAT, and Pd-catalyzed β-hydrogen elimination to afford selective, remote desaturation of aliphatic alcohols.
Scheme 32.

Pd-Catalyzed, remote desaturation via C(sp3)–I
5 Conclusions
Selective C–H functionalization via intramolecular HAT is a strategy that benefits from a mechanistically distinct pathway with complementary reactivity parameters to metal-based C–H activation routes. As new methods for mild radical-generation continually give way to new reaction development, there is also a sustained expansion of our understanding of the inherent chemo-, regio-, and stereo-selectivity rules for HAT mechanisms. While this mode of C–H functionalization is perhaps the oldest, its current revival of interest with new tools and methods offers ample opportunity for further, major contributions to the field of organic synthesis. For example, thanks to developments in the past year alone, δ C–C formation has been achieved for the first time (for N• initiation), asymmetric δ C–C formation is now possible with chiral catalysis (via O• initiation), and anilines offer orthogonal HAT initiation as diazonium surrogates (by C• initiation). In pursuit of solving ongoing challenges, new radical precursors will likely need to be designed, allowing for more creative methods of trapping these relayed radicals. Needless to say, with continued development of disruptive new methods,170 the future of C–H functionalization by radical translocation will continue to be exciting.
Acknowledgments
Funding Information
We thank the National Institutes of Health (R35 GM119812), National Science Foundation (CAREER 1654656), and American Chemical Society Petroleum Research Fund for financial support. L.M.S. is grateful for an NSF Graduate Research Fellowship.
We thank Sean Rafferty and Ethan Wappes for editing this manuscript.
Biography

(Left to right: Leah, Kohki, David) Leah Stateman (Orland Park, IL) earned a B.Sc. with honors at Illinois State University in 2015, where she studied the synthesis of carbaporphyrinoids with Prof. Timothy Lash. At OSU, she has pioneered our team’s development of catalytic, remote C–H functionalizations via hydrogen atom transfer. Leah is a 2016 National Science Foundation Graduate Research Fellow.
Kohki Nakafuku (Loveland, OH) earned a B.Sc. with honors at Duke University in 2014, where he studied the kinetics and mechanism of gold-catalyzed allene racemization with Prof. Ross Widenhoefer. At OSU, he has pioneered the development of an HAT-based radical relay chaperone strategy with applications towards directed C–H amination.
David Nagib (Philadelphia, PA) earned a B.Sc. with honors at Boston College in 2006, where he studied peptide-catalyzed de-symmetrization with Prof. Scott Miller. At Princeton University, he earned a Ph.D. in 2011 with Prof. David MacMillan, where he developed new trifluoromethylations via photoredox catalysis. As an NIH Postdoctoral Scholar at the University of California, Berkeley, he studied C–H activation via oxidative gold mechanisms with Prof. F. Dean Toste. Since 2014, David has been an Assistant Professor in the Department of Chemistry and Biochemistry at The Ohio State University, where his team’s research on radical-mediated C–H functionalization has been recognized by the American Chemical Society (PRF, 2015), National Institutes of Health (MIRA, 2016), National Science Foundation (CAREER, 2017), and Thieme Chemistry Journal Award (2017).
References
- 1.Bergman RG. Nature (London) 2007;446:391. doi: 10.1038/446391a. [DOI] [PubMed] [Google Scholar]
- 2.White MC. Science (Washington, D C) 2012;335:807. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem Int Ed. 2012;51:8960. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]
- 4.Wencel-Delord J, Glorius F. Nat Chem. 2013;5:369. doi: 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]
- 5.Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Chem Soc Rev. 2009;38:3242. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]
- 6.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 7.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies HML, Manning JR. Nature (London) 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roizen JL, Harvey ME, Du Bois J. Acc Chem Res. 2012;45:911. doi: 10.1021/ar200318q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi H, Zhang G, Wang H, Huang Z, Wang J, Singh AK, Lei A. Chem Rev. 2017;117:9016. doi: 10.1021/acs.chemrev.6b00620. [DOI] [PubMed] [Google Scholar]
- 11.Majetich G, Wheless K. Tetrahedron. 1995;51:7095. [Google Scholar]
- 12.Breslow R. Chem Soc Rev. 1972:553. [Google Scholar]
- 13.Heusler K, Kalvoda J. Angew Chem Int Ed. 1964;3:525. [Google Scholar]
- 14.Mayer JM. Acc Chem Res. 2011;44:36. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson J, Pillai J, Lush RK. Chem Soc Rev. 2001;30:94. [Google Scholar]
- 16.Čeković Ž. Tetrahedron. 2003;59:8073. [Google Scholar]
- 17.Nechab M, Mondal S, Bertrand MP. Chem Eur J. 2014;20:16034. doi: 10.1002/chem.201403951. [DOI] [PubMed] [Google Scholar]
- 18.Chiba S, Chen H. Org Biomol Chem. 2014;12:4051. doi: 10.1039/c4ob00469h. [DOI] [PubMed] [Google Scholar]
- 19.Togo H, Katohgi M. Synlett. 2001;565 [Google Scholar]
- 20.Merritt E, Olofsson B. Angew Chem Int Ed. 2009;48:9052. doi: 10.1002/anie.200904689. [DOI] [PubMed] [Google Scholar]
- 21.Zhdankin VV, Stang PJ. Chem Rev. 2008;108:5299. doi: 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]
- 23.Romero NA, Nicewicz DA. Chem Rev. 2016;116:10075. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- 24.Shaw MH, Twilton J, MacMillan DWC. J Org Chem. 2016;81:6898. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sibi MP, Manyem S, Zimmerman J. Chem Rev. 2003;103:3263. doi: 10.1021/cr020044l. [DOI] [PubMed] [Google Scholar]
- 26.Studer A, Curran DP. Angew Chem Int Ed. 2016;55:58. doi: 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]
- 27.Yan M, Lo JC, Edwards JT, Baran PS. J Am Chem Soc. 2016;138:12692. doi: 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taniguchi T. Synthesis. 2017;49:3511. [Google Scholar]
- 29.Huang XL, Dannenberg JJ. J Org Chem. 1991;56:5421. [Google Scholar]
- 30.Roth HG, Romero NA, Nicewicz DA. Synlett. 2016;27:714. [Google Scholar]
- 31.Horner JH, Choi SY, Newcomb M. Org Lett. 2000;2:3369. doi: 10.1021/ol006469g. [DOI] [PubMed] [Google Scholar]
- 32.Šakić D, Zipse H. Adv Synth Catal. 2016;358:3983. [Google Scholar]
- 33.Zard SZ. Chem Soc Rev. 2008;37:1603. doi: 10.1039/b613443m. [DOI] [PubMed] [Google Scholar]
- 34.Chen JR, Hu XQ, Lu LQ, Xiao WJ. Chem Soc Rev. 2016;45:2044. doi: 10.1039/c5cs00655d. [DOI] [PubMed] [Google Scholar]
- 35.Hofmann AW. Chem Ber. 1883;16:558. [Google Scholar]
- 36.Corey EJ, Hertler WR. J Am Chem Soc. 1960;82:1657. [Google Scholar]
- 37.Wolff ME. Chem Rev. 1963;63:55. [Google Scholar]
- 38.Löffler K, Freytag C. Chem Ber. 1909;42:3427. [Google Scholar]
- 39.Corey EJ, Hertler WR. J Am Chem Soc. 1958;80:2903. [Google Scholar]
- 40.Baldwin SW, Doll RJ. Tetrahedron Lett. 1979;20:3275. [Google Scholar]
- 41.De Armas P, Carrau R, Concepción JI, Francisco CG, Hernández R, Suárez E. Tetrahedron Lett. 1985;26:2493. [Google Scholar]
- 42.Courtneidge JL, Lusztyk J, Pagé D. Tetrahedron Lett. 1994;35:1003. [Google Scholar]
- 43.Dorta RL, Francisco CG, Suárez E. J Chem Soc, Chem Commun. 1989:1168. [Google Scholar]
- 44.Francisco CG, Herrera AJ, Kennedy AR, Melián D, Suárez E. Angew Chem Int Ed. 2002;41:856. doi: 10.1002/1521-3773(20020301)41:5<856::aid-anie856>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 45.Francisco CG, Herrera AJ, Suárez E. J Org Chem. 2003;68:1012. doi: 10.1021/jo026314h. [DOI] [PubMed] [Google Scholar]
- 46.Paz NR, Rodríguez-Sosa D, Valdés H, Marticorena R, Melián D, Copano MB, González CC, Herrera AJ. Org Lett. 2015;17:2370. doi: 10.1021/acs.orglett.5b00866. [DOI] [PubMed] [Google Scholar]
- 47.Martínez C, Muñiz K. Angew Chem Int Ed. 2015;54:8287. doi: 10.1002/anie.201501122. [DOI] [PubMed] [Google Scholar]
- 48.Blanksby SJ, Ellison GB. Acc Chem Res. 2003;36:255. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- 49.Becker P, Duhamel T, Stein CJ, Reiher M, Muñiz K. Angew Chem Int Ed. 2017;56:8004. [Google Scholar]
- 50.Wappes EA, Fosu SC, Chopko TC, Nagib DA. Angew Chem Int Ed. 2016;55:9974. doi: 10.1002/anie.201604704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim S, Yeon KM, Yoon KS. Tetrahedron Lett. 1997;38:3919. [Google Scholar]
- 52.Lu H, Jiang H, Wojtas L, Zhang XP. Angew Chem Int Ed. 2010;49:10192. doi: 10.1002/anie.201005552. [DOI] [PubMed] [Google Scholar]
- 53.Lyaskovskyy V, Suarez AIO, Lu H, Jiang H, Zhang XP, de Bruin B. J Am Chem Soc. 2011;133:12264. doi: 10.1021/ja204800a. [DOI] [PubMed] [Google Scholar]
- 54.Hennessy ET, Betley TA. Science (Washington, D C) 2013;340:591. doi: 10.1126/science.1233701. [DOI] [PubMed] [Google Scholar]
- 55.Nikishin GI, Troyansky EI, Lazareva MI. Tetrahedron Lett. 1985;26:3743. [Google Scholar]
- 56.Qin Q, Yu S. Org Lett. 2015;17:1894. doi: 10.1021/acs.orglett.5b00582. [DOI] [PubMed] [Google Scholar]
- 57.Katohgi M, Togo H, Yamaguchi K, Masataka Y. Tetrahedron. 1999;55:14885. [Google Scholar]
- 58.Reddy LR, Reddy BVS, Corey EJ. Org Lett. 2006;8:2819. doi: 10.1021/ol060952v. [DOI] [PubMed] [Google Scholar]
- 59.Liu T, Myers MC, Yu J-Q. Angew Chem Int Ed. 2017;56:306. doi: 10.1002/anie.201608210. [DOI] [PubMed] [Google Scholar]
- 60.Groendyke BJ, Abusalim DI, Cook SP. J Am Chem Soc. 2016;138:12771. doi: 10.1021/jacs.6b08171. [DOI] [PubMed] [Google Scholar]
- 61.Chen K, Richter JM, Baran PS. J Am Chem Soc. 2008;130:7247. doi: 10.1021/ja802491q. [DOI] [PubMed] [Google Scholar]
- 62.Short MA, Blackburn JM, Roizen JL. Angew Chem Int Ed. 2018;57:296. doi: 10.1002/anie.201710322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu T, Mei T-S, Yu J-Q. J Am Chem Soc. 2015;137:5871. doi: 10.1021/jacs.5b02065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Choi GJ, Zhu Q, Miller DC, Gu CJ, Knowles RR. Nature (London) 2016;539:268. doi: 10.1038/nature19811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chu JCK, Rovis T. Nature (London) 2016;539:272. doi: 10.1038/nature19810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen D-F, Chu JCK, Rovis T. J Am Chem Soc. 2017;139:14897. doi: 10.1021/jacs.7b09306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Homer JH, Musa OM, Bouvier A, Newcomb M. J Am Chem Soc. 1998;120:7738. [Google Scholar]
- 68.Forrester AR, Gill M, Sadd JS, Thomson RH. J Chem Soc, Chem Commun. 1975:677. [Google Scholar]
- 69.Boivin J, Fouquet E, Zard SZ. Tetrahedron Lett. 1991;32:4299. [Google Scholar]
- 70.Uchiyama K, Hayashi Y, Narasaka K. Chem Lett. 1998;27:1261. [Google Scholar]
- 71.Lin X, Stien D, Weinreb SM. Org Lett. 1999;1:637. doi: 10.1021/ol990720e. [DOI] [PubMed] [Google Scholar]
- 72.Forrester AR, Gill M, Thomson RH. J Chem Soc, Perkin Trans 1. 1979:621. doi: 10.1039/j39700001081. [DOI] [PubMed] [Google Scholar]
- 73.Forrester AR, Gill M, Meyer CJ, Sadd JS, Thomson RH. J Chem Soc, Perkin Trans 1. 1979:606. [Google Scholar]
- 74.Forrester AR, Gill M, Napier RJ, Thomson RH. J Chem Soc, Perkin Trans 1. 1979:632. [Google Scholar]
- 75.Forrester AR, Napier RJ, Thomson RH. J Chem Soc, Perkin Trans 1. 1981:984. doi: 10.1039/j39700001081. [DOI] [PubMed] [Google Scholar]
- 76.Zhang L, Ang GY, Chiba S. Org Lett. 2011;13:1622. doi: 10.1021/ol200425c. [DOI] [PubMed] [Google Scholar]
- 77.Shu W, Nevado C. Angew Chem Int Ed. 2017;56:1881. doi: 10.1002/anie.201609885. [DOI] [PubMed] [Google Scholar]
- 78.Li J, Zhang P, Jiang M, Yang H, Zhao Y, Fu H. Org Lett. 2017;19:1994. doi: 10.1021/acs.orglett.7b00533. [DOI] [PubMed] [Google Scholar]
- 79.Davies J, Booth SG, Essafi S, Dryfe RAW, Leonori D. Angew Chem Int Ed. 2015;54:14017. doi: 10.1002/anie.201507641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shu W, Lorente A, Gómez-Bengoa E, Nevado C, Booth RG. Nat Commun. 2017;8:13832. doi: 10.1038/ncomms13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Suzuki A, Tabata M, Ueda M. Tetrahedron Lett. 1975;16:2195. [Google Scholar]
- 82.Wang YF, Chen H, Zhu X, Chiba S. J Am Chem Soc. 2012;134:11980. doi: 10.1021/ja305833a. [DOI] [PubMed] [Google Scholar]
- 83.Chen H, Sanjaya S, Wang Y-F, Chiba S. Org Lett. 2013;15:212. doi: 10.1021/ol303302r. [DOI] [PubMed] [Google Scholar]
- 84.Chen H, Chiba S. Org Biomol Chem. 2014;12:42. doi: 10.1039/c3ob41871e. [DOI] [PubMed] [Google Scholar]
- 85.Wappes EA, Nakafuku KM, Nagib DA. J Am Chem Soc. 2017;139:10204. doi: 10.1021/jacs.7b05214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pinner A, Klein F. Chem Ber. 1877;10:1889. [Google Scholar]
- 87.Overman LE. J Am Chem Soc. 1976;98:2901. [Google Scholar]
- 88.Glover SA, Hammond GP, Hamnan DG, Mills JG, Rowbottom CA. Aust J Chem. 1993;46:1213. [Google Scholar]
- 89.Norrish RGW, Appleyard MES. J Chem Soc. 1934:874. [Google Scholar]
- 90.Lu Y, Carlson D, Morrison H. J Am Chem Soc. 1993;115:6446. [Google Scholar]
- 91.Breslow R, Baldwin S, Flechtner T, Kalicky P, Liu S, Washburn W. J Am Chem Soc. 1973;95:3251. doi: 10.1021/ja00791a031. [DOI] [PubMed] [Google Scholar]
- 92.Renata H, Zhou Q, Baran PS. Science (Washington, D C) 2013;339:59. doi: 10.1126/science.1230631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barton DHR, Beaton JM, Geller LE, Pechet MM. J Am Chem Soc. 1960;82:2640. [Google Scholar]
- 94.Barton DHR, Beaton JM, Geller LE, Pechet MM. J Am Chem Soc. 1961;83:4076. [Google Scholar]
- 95.Barton DHR, Hesse RH, Pechet MM, Smith LC. J Chem Soc, Perkin Trans 1. 1979:1159. [Google Scholar]
- 96.Barton DHR, Beaton JM. J Am Chem Soc. 1960;82:2641. [Google Scholar]
- 97.Corey EJ, Arnett JF, Widiger GN. J Am Chem Soc. 1975;97:430. doi: 10.1021/ja00835a039. [DOI] [PubMed] [Google Scholar]
- 98.Herzog A, Knobler CB, Hawthorne MF. Angew Chem Int Ed. 1998;37:1552. doi: 10.1002/(SICI)1521-3773(19980619)37:11<1552::AID-ANIE1552>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 99.Greene FD, Savitz ML, Lau HH, Osterholtz FD, Smith WN. J Am Chem Soc. 1961;83:2196. [Google Scholar]
- 100.Walling C, Padwa A. J Am Chem Soc. 1961;83:2207. [Google Scholar]
- 101.Walling C, Padwa A. J Am Chem Soc. 1963;85:1597. [Google Scholar]
- 102.Kochi JK, Bemis A, Jenkins CL. J Am Chem Soc. 1968;90:4616. [Google Scholar]
- 103.Kochi JK. Acc Chem Res. 1974;7:351. [Google Scholar]
- 104.Čeković Z, Green MM. J Am Chem Soc. 1974;96:3000. [Google Scholar]
- 105.Čeković Ž, Dimttruević L, Djokić G, Srnić T. Tetrahedron. 1979;35:2021. [Google Scholar]
- 106.Čeković Z, Cvetković M. Tetrahedron Lett. 1982;23:3791. [Google Scholar]
- 107.Kundu R, Ball ZT. Org Lett. 2010;12:2460. doi: 10.1021/ol100472t. [DOI] [PubMed] [Google Scholar]
- 108.Hashimoto T, Hirose D, Taniguchi T. Angew Chem Int Ed. 2014;53:2730. doi: 10.1002/anie.201308675. [DOI] [PubMed] [Google Scholar]
- 109.Meystre CH, Heusler K, Kalvoda J, Wieland P, Anner G, Wettstein A. Helv Chim Acta. 1962;45:1317. [Google Scholar]
- 110.Tsunoi S, Ryu I, Okuda T, Tanaka M, Komatsu M, Sonoda N. J Am Chem Soc. 1998;120:8692. [Google Scholar]
- 111.Ryu I. Chem Soc Rev. 2001;30:16. [Google Scholar]
- 112.Trahanovsky WS, Young MG, Nave PM. Tetrahedron Lett. 1969;10:2501. [Google Scholar]
- 113.Concepción JI, Francisco CG, Hernández R, Salazar JA, Suárez E. Tetrahedron Lett. 1984;25:1953. [Google Scholar]
- 114.Francisco CG, Herrera AJ, Suárez E. Tetrahedron Lett. 2000;41:7869. [Google Scholar]
- 115.Petrović G, Čeković Ž. Tetrahedron Lett. 1997;38:627. [Google Scholar]
- 116.Petrović G, Saičić RN, Čeković Ž. Tetrahedron Lett. 1997;38:7107. [Google Scholar]
- 117.Petrović G, Čeković Ž. Tetrahedron. 1999;55:1377. [Google Scholar]
- 118.Kim S, Lee TA, Song Y. Synlett. 1998:471. [Google Scholar]
- 119.Zhu H, Wickenden JG, Campbell NE, Leung JCT, Johnson KM, Sammis GM. Org Lett. 2009;11:2019. doi: 10.1021/ol900481e. [DOI] [PubMed] [Google Scholar]
- 120.Zhu H, Leung JCT, Sammis GM. J Org Chem. 2015;80:965. doi: 10.1021/jo502499a. [DOI] [PubMed] [Google Scholar]
- 121.Zhang J, Li Y, Zhang F, Hu C, Chen Y. Angew Chem Int Ed. 2016;55:1872. doi: 10.1002/anie.201510014. [DOI] [PubMed] [Google Scholar]
- 122.Wang C, Harms K, Meggers E. Angew Chem Int Ed. 2016;55:13495. doi: 10.1002/anie.201607305. [DOI] [PubMed] [Google Scholar]
- 123.Zhu X, Wang Y-F, Ren W, Zhang F-L, Chiba S. Org Lett. 2013;15:3214. doi: 10.1021/ol4014969. [DOI] [PubMed] [Google Scholar]
- 124.Lemercier BC, Pierce JG. Org Lett. 2015;17:4542. doi: 10.1021/acs.orglett.5b02256. [DOI] [PubMed] [Google Scholar]
- 125.Ozawa J, Tashiro M, Ni J, Oisaki K, Kanai M. Chem Sci. 2016;7:1904. doi: 10.1039/c5sc04476f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ni J, Ozawa J, Oisaki K, Kanai M. Org Biomol Chem. 2016;14:4378. doi: 10.1039/c6ob00678g. [DOI] [PubMed] [Google Scholar]
- 127.Nikishin GI, Svitanko IV, Troyansky EI. J Chem Soc, Perkin Trans 2. 1983:595. [Google Scholar]
- 128.Sathyamoorthi S, Du Bois J. Org Lett. 2016;18:6308. doi: 10.1021/acs.orglett.6b03176. [DOI] [PubMed] [Google Scholar]
- 129.Warren JJ, Tronic TA, Mayer JM. Chemical Reviews. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Curran DP, Kim D, Liu HT, Shen W. J Am Chem Soc. 1988;110:5900. [Google Scholar]
- 131.Curran DP, Shen W. J Am Chem Soc. 1993;115:6051. [Google Scholar]
- 132.Vellucci JK, Beaudry CM. Org Lett. 2015;17:4558. doi: 10.1021/acs.orglett.5b02277. [DOI] [PubMed] [Google Scholar]
- 133.Dénès F, Beaufils F, Renaud P. Synlett. 2008:2389. [Google Scholar]
- 134.Curran DP, Xu J. J Am Chem Soc. 1996;118:3142. [Google Scholar]
- 135.Curran DP, Yu H. Synthesis. 1992:123. [Google Scholar]
- 136.Parasram M, Chuentragool P, Sarkar D, Gevorgyan V. J Am Chem Soc. 2016;138:6340. doi: 10.1021/jacs.6b01628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Snieckus V, Cuevas JC, Sloan CP, Liu H, Curran DP. J Am Chem Soc. 1990;112:896. [Google Scholar]
- 138.Curran DP, Abraham AC, Liu H. J Org Chem. 1991;56:4335. [Google Scholar]
- 139.Curran DP, Abraham AC. Tetrahedron. 1993;49:4821. [Google Scholar]
- 140.Curran DP, Yu H, Liu H. Tetrahedron. 1994;50:7343. [Google Scholar]
- 141.Beckwith ALJ, Bowry VW, Bowman WR, Mann E, Parr J, Storey JMD. Angew Chem Int Ed. 2004;43:95. doi: 10.1002/anie.200352419. [DOI] [PubMed] [Google Scholar]
- 142.Murakami M, Hayashi M, Ito Y. J Org Chem. 1992;57:793. [Google Scholar]
- 143.Booth SE, Benneche T, Undheim K. Tetrahedron. 1995;51:3665. [Google Scholar]
- 144.Undheim K, Williams L. J Chem Soc, Chem Commun. 1994:883. [Google Scholar]
- 145.Williams L, Booth SE, Undheim K. Tetrahedron. 1994;50:13697. [Google Scholar]
- 146.Gribble GW, Fraser HL, Badenock JC. Chem Commun. 2001:805. [Google Scholar]
- 147.Khan TA, Tripoli R, Crawford JJ, Martin CG, Murphy JA. Org Lett. 2003;5:2971. doi: 10.1021/ol035173i. [DOI] [PubMed] [Google Scholar]
- 148.Yoshikai N, Mieczkowski A, Matsumoto A, Ilies L, Nakamura E. J Am Chem Soc. 2010;132:5568. doi: 10.1021/ja100651t. [DOI] [PubMed] [Google Scholar]
- 149.Wertjes WC, Wolfe LC, Waller PJ, Kalyani D. Org Lett. 2013;15:5986. doi: 10.1021/ol402869h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chen J-Q, Wei Y-L, Xu G-Q, Liang Y-M, Xu P-F. Chem Commun. 2016;52:6455. doi: 10.1039/c6cc02007k. [DOI] [PubMed] [Google Scholar]
- 151.Liu P, Tang J, Zeng X. Org Lett. 2016;18:5536. doi: 10.1021/acs.orglett.6b02784. [DOI] [PubMed] [Google Scholar]
- 152.Galli C. Chem Rev. 1988;88:765. [Google Scholar]
- 153.Meerwein H, Büchner E, van Emster K. J Prakt Chem. 1939;152:237. [Google Scholar]
- 154.Hey DH, Turpin DG. J Chem Soc. 1954:2471. [Google Scholar]
- 155.Han G, LaPorte MG, McIntosh MC, Weinreb SM, Parvez M. J Org Chem. 1996;61:9483. [Google Scholar]
- 156.Huang F-Q, Dong X, Qi L-W, Zhang B. Tetrahedron Lett. 2016;57:1600. [Google Scholar]
- 157.Huang F-Q, Zhou G-X, Dong X, Qi L-W, Zhang B. Asian J Org Chem. 2016;5:192. [Google Scholar]
- 158.Shaaban S, Oh J, Maulide N. Org Lett. 2016;18:345. doi: 10.1021/acs.orglett.5b03431. [DOI] [PubMed] [Google Scholar]
- 159.Voica A-F, Mendoza A, Gutekunst WR, Fraga JO, Baran PS. Nat Chem. 2012;4:629. doi: 10.1038/nchem.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hollister KA, Conner ES, Spell ML, Deveaux K, Maneval L, Beal MW, Ragains JR. Angew Chem Int Ed. 2015;54:7837. doi: 10.1002/anie.201500880. [DOI] [PubMed] [Google Scholar]
- 161.Tian H, Yang H, Zhu C, Fu H. Sci Rep. 2016;6:19931. doi: 10.1038/srep19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Crich D, Sun S, Brunckova J. J Org Chem. 1996;61:605. doi: 10.1021/jo951194h. [DOI] [PubMed] [Google Scholar]
- 163.Ueno Y, Chino K, Watanabe M, Moriya O, Okawara M. J Am Chem Soc. 1982;104:5564. [Google Scholar]
- 164.Stork G, Mook R, Biller SA, Rychnovsky SD. J Am Chem Soc. 1983;105:3741. [Google Scholar]
- 165.Masnyk M. Tetrahedron Lett. 1997;38:879. [Google Scholar]
- 166.Wood ME, Bissiriou S, Lowe C, Windeatt KM. Org Biomol Chem. 2013;11:2712. doi: 10.1039/c3ob40275d. [DOI] [PubMed] [Google Scholar]
- 167.Viehe HG, Janousek Z, Merenyi R, Stella L. Acc Chem Res. 1985;18:148. [Google Scholar]
- 168.Parasram M, Chuentragool P, Wang Y, Shi Y, Gevorgyan V. J Am Chem Soc. 2017;139:14857. doi: 10.1021/jacs.7b08459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nishiyama H, Kitajima T, Matsumoto M, Itoh K. J Org Chem. 1984;49:2298. [Google Scholar]
- 170.(a) Sathyamoorthi S, Banerjee S, Du Bois J, Burns NZ, Zare RN. Chem Sci. 2018;9:100. doi: 10.1039/c7sc04611a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ratushnyy M, Parasram M, Wang Y, Gevorgyan V. Angew Chem Int Ed doi. doi: 10.1002/anie.201712775. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chuentragool P, Parasram M, Shi Y, Gevorgyan V. J Am Chem Soc. 2018 doi: 10.1021/jacs.8b00488. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hu A, Guo JJ, Pan H, Tang H, Gao Z, Zuo Z. J Am Chem Soc. 2018;140:1612. doi: 10.1021/jacs.7b13131. [DOI] [PubMed] [Google Scholar]; (e) Dauncey EM, Morcillo SP, Douglas JJ, Sheikh NS. Angew Chem Int Ed. 2018;57:744. doi: 10.1002/anie.201710790. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Jiang H, Studer A. Angew Chem Int Ed. 2018;57:1692. doi: 10.1002/anie.201712066. [DOI] [PubMed] [Google Scholar]; (g) Zhou B, Sato H, Ilies L, Nakamura E. ACS Catal. 2018;8:8. [Google Scholar]