

Bohineust et al. establish that recently activated T cells exhibit a phase of unresponsiveness associated with a defect in calcium entry. This stage was essential to terminate priming, distracting T cells from APCs, and favoring their clonal expansion.
Abstract
T cells are primed in secondary lymphoid organs by establishing stable interactions with antigen-presenting cells (APCs). However, the cellular mechanisms underlying the termination of T cell priming and the initiation of clonal expansion remain largely unknown. Using intravital imaging, we observed that T cells typically divide without being associated to APCs. Supporting these findings, we demonstrate that recently activated T cells have an intrinsic defect in establishing stable contacts with APCs, a feature that was reflected by a blunted capacity to stop upon T cell receptor (TCR) engagement. T cell unresponsiveness was caused, in part, by a general block in extracellular calcium entry. Forcing TCR signals in activated T cells antagonized cell division, suggesting that T cell hyporesponsiveness acts as a safeguard mechanism against signals detrimental to mitosis. We propose that transient unresponsiveness represents an essential phase of T cell priming that promotes T cell disengagement from APCs and favors effective clonal expansion.
Graphical Abstract
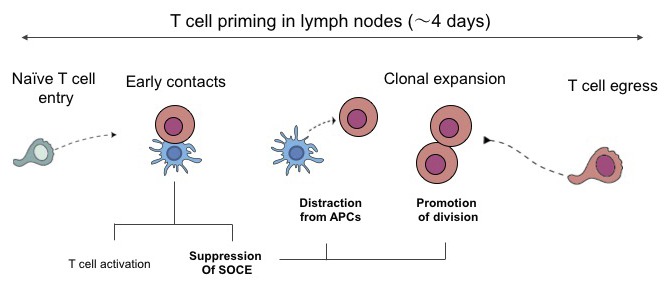
Introduction
T cell priming by dendritic cells (DCs) in the lymph node is a process that typically lasts for 3–4 d, at which point activated T cells egress and disseminate in the body. Intravital imaging has been instrumental to define the cellular orchestration of T cell priming (Miller et al., 2002; Bousso and Robey, 2003; Mempel et al., 2004; Bousso, 2008). In particular, one of the hallmarks of efficient priming is the establishment of hours-long T cell−DC interactions that are in some instances preceded by an early phase of transient contacts. By 48 h, many T cells have regained motility and clonal expansion is initiated.
The parameters regulating the formation of stable T cell–DC contacts have been extensively investigated. For example, peptides with a high binding stability on MHC molecules or displaying a high affinity for the TCR favor stable T cell–DC interactions (Skokos et al., 2007; Henrickson et al., 2008; Moreau et al., 2012; Pace et al., 2012; Ozga et al., 2016). Other factors, such as LFA-1–ICAM-1 interaction, promote tight contacts (Scholer et al., 2008). Conversely, the presence of regulatory T cells (Tadokoro et al., 2006; Tang et al., 2006; Pace et al., 2012) or expression of inhibitory receptors (CTLA-4, PD-1) can reduce the stability of T cell–DC interactions (Schneider et al., 2006; Fife et al., 2009).
Contrasting with the important knowledge acquired on the initiation of T cell activation, we critically lack information regarding the cellular mechanisms responsible for the termination of T cell priming. A first important question concerns the mechanism involved in T cell detachment from APCs after activation. One obvious possibility is that this reflects the progressive reduction in cognate peptide–MHC (pMHC) complexes at the surface of DCs. pMHC complexes with a half-life of a few hours may become limiting after 1–2 d (Henrickson et al., 2008). Active extraction of pMHC from the surface of DCs by T cells also reduces the number of TCR ligands over time (Kedl et al., 2002). T cell disengagement from APCs may also involve T cell–intrinsic mechanisms, including up-regulation of inhibitory receptors, down-regulation of TCR, or increased responsiveness to chemokines. Finally, DC death may provide a means for T cell to resume motility. A second key aspect of priming termination relates to the phase of clonal expansion. T cell division in contact to APCs has been observed in vitro (Oliaro et al., 2010) and is a proposed mechanism to drive asymmetric T cell division (Chang et al., 2007). However, whether T cells most often divide while in contact with DCs or after disengaging from APCs in vivo has yet to be fully resolved.
Here, we investigated the cellular mechanisms underlying the termination of T cell priming. Using functional reporters and intravital imaging, we uncovered a transient phase of T cell unresponsiveness after the initial activation that favors T cell disengagement from APCs. Finally, we provide evidence that such unresponsiveness protects T cells from receiving strong TCR stimulations that interfere with T cell division.
Results
Dynamics of T cell division during priming in lymph nodes
To study the termination of T cell priming, we first focused on the initiation of T cell clonal expansion in lymph nodes that typically starts after 48 h of activation (Miller et al., 2002; Beuneu et al., 2010). Using a synchronized system in which OT-I CD8+ T cells are stimulated by an intravenous injection of cognate peptide, we observed T cell proliferation in lymph nodes starting at 30–48 h after stimulation (Fig. 1 A). As illustrated in Fig. 1 (B–E) and Video 1, T cell divisions occurred in three phases: (1) rapidly migrating blastic T cells arrested and adopted a spherical shape (∼12 min), (2) T cells underwent mitosis (∼3 min), and (3) daughter T cells resumed their motility (∼8 min). Although the APCs were not visualized in this system, the observation that T cell motility preceded T cell division raises the possibility that T cell division does not proceed during interactions with APCs, but instead after detachment. We confirmed this idea using an experimental set-up in which both T cells and DCs are visualized. Specifically, we tracked the activation of CD8+ T cells bearing the OT-I TCR by transferred DCs pulsed with the cognate antigenic OVA peptide SIINFEKL (OVAp). In large agreement with previous studies, stable and long-lasting T cell–DC interactions dominated in the first hours (Video 2). By 24–48 h, many T cells had resumed motility. At this time, we also detected many events of T cell division (Fig. 1, F and G; and Video 3). Importantly, in virtually all the division events visualized, T cells exhibited a spherical shape and were not in contact with a cognate DC. On average, dividing T cells were located ∼50 µm away from the closest antigen-bearing DC (Fig. 1 G). Overall, our results obtained in two experimental set-ups support the idea that T cells divide, in most cases, while disengaged from antigen-bearing APCs.
Figure 1.

Most dividing T cells during priming are not in contact with APCs. (A–E) Visualization of T cell division dynamics in the lymph node. GFP+ OT-I CD8+ T cells were adoptively transferred and stimulated by an i.v. injection of OVAp. At various time points, recipient mice were subjected to intravital imaging of the popliteal lymph node. (A) The graph shows the frequency of dividing T cells observed per hour of imaging. Two to four videos were analyzed for each time point. Data are shown as mean ± SD. (B) Time-lapse images showing the typical phases of T cell division (motility, arrest, mitosis, and recovery of motility). (C and D) Tracks (C) and velocity (D) for the dividing cell shown in B (black) and the two daughter cells (red and blue). (E) Timing for the indicated phases of T cell division measured after T cell arrest. 16 dividing T cells were tracked over time. Each dot represents the value for the different phases for each of these 16 division events. Mean values are shown in red. Results are representative of eight independent experiments. (F and G) Analysis of T cell division upon in vivo stimulation with DCs. Recipient mice were adoptively transferred with GFP-expressing OT-I CD8+ T cells and injected in the footpad with OVAp-pulsed CFP-expressing DCs. Intravital imaging of the draining popliteal lymph node was performed at 48 h. (F) The vast majority of dividing T cells (green) are not in contact with an antigen-bearing DC (cyan) at the time of mitosis. White arrows show the cell before division. Blue and red arrows show the daughter cells. (G) The graph shows the distance between dividing T cells and the closest DCs. T cells forming stable interaction (but not dividing) with DCs were scored similarly for comparison. Mean values are shown in red. Results are representative of three independent experiments. Significance testing was performed with the nonparametric Mann-Whitney test. ***, P < 0.001. Bars, 15 µm.
Recently activated T cells exhibit an intrinsic defect in binding to DCs
The fact that T cell division events were detected away from antigen-bearing DCs prompted us to investigate the ability of recently activated T cells to interact with APCs. Previous studies have shown that stable T cell–DC interactions are lost in the late phases of T cell activation and that T cells establish primarily transient contacts with DCs at this stage (Hugues et al., 2004; Mempel et al., 2004; Miller et al., 2004). However, it is unclear whether this observation originates from changes in DCs (in particular because of decreased antigen presentation) or from changes in T cells during the activation process. We tested the possible involvement of a T cell–intrinsic phenomenon in the progressive loss of T cell–DC contacts using two approaches in which we could relate T cell interactions to their activation status. In both settings, it is possible to compare the behavior of T cells that have reached different stages of activation when facing the same DCs in the same microenvironment. A first system was designed to distinguish undivided T cells from those that had undergone at least one round of cell division. For this, we transferred GFP-expressing OT-I CD8+ T cells labeled with a red vital dye (SNARF) into recipient mice challenged by peptide-pulsed DCs (Fig. 2 A). At 48 h, undivided T cells (GFP+SNARF+) could be readily discriminated from divided T cells (GFP+SNARFlow/−) by intravital imaging. Notably, we found that long-lasting contacts with DCs were largely restricted to undivided T cells (Fig. 2, B–D; and Video 4). In these settings, divided T cells typically interacted with DCs for <5 min (Fig. 2, C and D). To test whether such a defect in activated T cells appears before or after the first cell division, we used OT-I CD8+ T cells expressing a YFP reporter for IFN-γ gene activation (Yeti) and labeled with SNARF (Fig. 2 E). We have previously shown that YFP expression in T cells mirrored the level of T cell activation and is initially detected at 24 h, before the first T cell division (Beuneu et al., 2010). We found that T cells that were not or poorly activated (SNARF+YFP−) at the time of imaging could establish stable interactions with DCs (Fig. 2, F–H; and Video 5). In contrast, activated T cells (SNARF+YFP+) were highly motile, forming only short contacts upon encounters with DCs. Altogether, these experiments demonstrate that recently activated T cells exhibited a substantial defect in the formation of stable interactions with antigen-bearing DCs, implying that the reduction of stable contacts observed in the late phase of priming is largely a T cell–intrinsic phenomenon.
Figure 2.

Recently activated T cells exhibit an intrinsic defect in forming stable interactions with APCs. (A–D) Divided T cells do not form stable interactions with DCs. (A) Experimental set-up. GFP-expressing OT-I CD8+ T cells were labeled with SNARF and transferred into recipient mice. CFP-expressing DCs pulsed with OVAp were injected in the footpad. At 48 h, recipient mice were subjected to intravital imaging of the draining popliteal lymph node. (B) Representative time-lapse images showing that, in the same lymph node, undivided T cells (yellow) established long-lived interactions (arrowheads) with DCs (cyan), whereas divided T cells (green) failed to do so. Bar, 15 µm. (C) Duration of T cell–DC contacts from a representative video is graphed as a function of the GFP/SNARF ratio. Note that undivided T cells (low GFP/SNARF fluorescence ratio) formed interactions >10 min. Results are representative of three independent experiments. (D) The frequency of undivided or divided T cells forming interactions >5 min with DCs (stable contacts) is shown. Results are compiled from seven videos obtained in two independent experiments. (E–H) Recently activated T cells do not form stable interactions with DCs. (E) Experimental set-up. OT-I CD8+ T cells expressing a YFP reporter for IFN-γ (Yeti) were labeled with SNARF and transferred into recipient mice. GFP-expressing DCs pulsed with OVAp were injected in the footpad. At 24 h, recipient mice were subjected to intravital imaging of the draining popliteal lymph node. (F) Representative time-lapse images showing that, in the same lymph node, naive (red) but not activated (orange) T cells established long-lived interactions (arrowheads) with DCs (green). Bar, 15 µm. (G) Duration of T cell–DC contacts from a representative video is graphed as a function of the YFP/SNARF ratio. Note that only unactivated T cells (low YFP/SNARF fluorescence ratio) formed interactions >12 min. Representative of three independent experiments. (H) The frequency of unactivated (naive) or activated T cells forming interactions >5 min with DCs (stable contacts) is shown. Results are compiled from three videos obtained in a representative experiment out of three.
Recently activated T cells display a defect in TCR-induced stop signals
T cell dynamics are dictated by both stop and go signals promoting arrest and motility, respectively (Dustin, 2004). Thus, the defect of recently activated T cells in stably binding DCs could theoretically be caused by an increased ability to migrate (for instance, as a result of increased chemokine receptor expression) or by a decreased ability to stop upon TCR stimulation. We tested the latter possibility by assessing the capacity of recently activated T cells to undergo antigen-driven T cell stop upon systemic peptide delivery.
We first characterized the changes in naive OT-I CD8+ T cell dynamics upon peptide injection (Fig. 3 A). In this system, T cells immediately stop upon peptide injection as reflected by velocity <2 µm/min, high arrest coefficient (>80%) and low straightness index (<0.3) (Fig. 3, B–D). A marked recovery of T cell motility was seen starting at 30 h and was even more prominent at 48 h (Fig. 3, B–D). To test whether recovery of T cell motility was simply due to antigen disappearance, we relied on a two-cohorts experiment (Fig. 3 E). A first cohort of OT-I CD8+ T cells was therefore injected and stimulated by i.v. peptide injection. A second cohort of differently labeled OT-I CD8+ T cells was injected 30 h later, and intravital lymph node imaging was performed 18 h after the second cohort injection. We noted that although most T cells from the second cohort were completely arrested, T cells from the first cohort in the same imaging area were largely motile (Fig. 3, F and G; Video 6), indicating that the recovery of motility is T cell–intrinsic and dependent on T cell activation state. Moreover, reinjection of a large amount of peptide resulted only in a modest deceleration of activated T cells, far from the complete arrest seen with naive T cells in the same conditions (Fig. 3, F and G; Fig. S1; Video 7). We conclude from these experiments that recently activated T cells do not effectively stop upon antigen recognition.
Figure 3.

Recently activated T cells do not effectively stop upon TCR engagement. (A–D) CD8+ T cell dynamics during the course of activation in vivo. (A) Experimental set-up. GFP+ OT-I CD8+ T cells were adoptively transferred, and recipients were stimulated by an i.v. injection of OVAp. At various time points, recipient mice were subjected to intravital imaging of the popliteal lymph node. (B–D) Graphs show the velocity (B), arrest coefficient (C), and straightness index (D) of individual cells at the indicated time point. Each dot represents an individual track, and the velocity represents the mean speed of the track. (E–G) CD8+ T cell arrest in response to the antigenic peptide is dependent on T cell activation status. (E) Experimental set-up. GFP+ OT-I CD8+ T cells were adoptively transferred, and recipients were stimulated by an i.v. injection of OVAp. At 30 h, a second cohort of SNARF-labeled OT-I CD8+ T cells was adoptively transferred. Intravital imaging of the popliteal lymph node was performed 18 h later. OVAp was reinjected during the imaging experiment. (F) T cell tracks (15 min long) of the first (green) and second cohort (red) of T cells obtained from a representative experiment are shown before and after OVAp reinjection. Bar, 15 µm. (G) Graphs show the velocity, arrest coefficient, and straightness index for individual T cells of the first and second cohorts before and after OVAp reinjection. Results are compiled from two videos obtained in one representative experiment out of three. Significance testing was performed with the nonparametric Mann-Whitney test. **, P < 0.01; and ***, P < 0.001. See also Fig. S1.
Role of TCR down-regulation and the PD-1 pathway in T cell stop defect
Several mechanisms could potentially explain the intrinsic defect of recently activated T cells in stopping upon TCR engagement. TCR down-regulation (Valitutti et al., 1995; Friedman et al., 2010) is a well-characterized process that occurs after strong stimulation and may prevent subsequent reattachment to DCs. Although we observed decreased expression of the CD3 complex on OT-I CD8+ T cells at 24 h after cognate peptide injection, levels were almost back to normal at 30 h (Fig. 4 A). Upon detailed examination of TCR chains, we noted however a reduction in Vβ5 expression on recently activated OT-I T cells at 48 h (Fig. 4 B), raising the possibility that TCR down-regulation contributes, at least partly, to limit T cell stop in recently activated T cells.
Figure 4.

Role of TCR downmodulation and of the PD-1 axis on T cell stop. (A and B) GFP+ OT-I CD8+ T cells were adoptively transferred, and recipients were stimulated by an i.v. injection of OVAp or left untreated as a control. (A) Kinetics of CD3ε (left) or PD-1 (right) surface expression on OT-I CD8+ T cells as determined by flow cytometry. Data are shown as mean ± SEM. (B) The surface expression of the indicated markers was assessed on GFP+ OT-I CD8+ T cells at 48 h in peptide-injected or untreated recipients. Data are representative of three independent experiments. (C) Anti–PD-1 treatment does not restore the ability of activated T cells to arrest upon antigenic peptide injection. GFP+ OT-I CD8+ T cells were adoptively transferred, and recipients were stimulated by an i.v. injection of OVAp. Mice were treated with anti–PD-1 or isotype control on day 1. Intravital imaging of the popliteal lymph node was performed on day 2. OVAp was reinjected during the imaging experiment. Velocity and arrest coefficient of individual T cells are shown in mice treated with anti–PD-1 or an isotype control before and after OVAp reinjection. Results are representative of two independent experiments. Each dot represents an individual track. Significance testing were performed with the nonparametric Mann-Whitney test. ***, P < 0.001. Also see Fig. S2.
As an additional hypothesis, we tested the involvement of the PD-1 pathway. Indeed, effector T cells in the skin were shown to progressively become irresponsive to TCR stimulation, a process that was reversed by PD-1 blockade (Honda et al., 2014). Because activated T cells rapidly up-regulate PD-1 expression (Fig. 4 A), we tested whether PD-1 blockade could restore the responsiveness of recently activated T cells to TCR stimulation. As shown in Fig. 4 C, PD-1 blockade did not restore the ability of activated T cells to fully arrest upon peptide reinjection. Of note, the lack of effect of the anti–PD-1 Ab was not cause by a failure of the Ab to diffuse in the lymph node because anti–PD-1 treatment increased T cell activation (Fig. S2) that could possibly even hasten the recovery of T cell motility (Fig. 4 C). Thus, inhibition via the PD-1 axis does not appear to explain the defect of recently activated T cells to stop.
Recently activated T cells exhibit an intrinsic block in calcium signals
We and others have shown that calcium elevation is important for TCR-induced T cell arrest (Negulescu et al., 1996; Bhakta et al., 2005; Skokos et al., 2007; Waite et al., 2013; Moreau et al., 2015). In addition, we noted that the formation of stable conjugates between T cells and peptide-pulsed DCs required the presence of extracellular calcium (Fig. S3). We therefore sought to measure the calcium response of recently activated T cells to various stimuli ex vivo. We adoptively transferred GFP-expressing OT-I CD8+ T cells and injected (or not) recipient mice with OVAp. At 48 h, lymph node cells were prepared and stained with the calcium dye Indo-1. We compared the calcium responses of recently activated T cells (recovered from OVAp-treated mice) with naive T cells (recovered from nontreated recipients) by flow cytometry. Stimulation with the antigenic peptide or with the mitogen Concanavalin A (Con A) induced a calcium response in naive T cells, that was strongly reduced in recently activated T cells (Fig. 5, A and B). Similar observations were made by stimulating T cells with peptide-pulsed splenocytes (Fig. S4 A). To test whether such defect was caused solely by TCR proximal signaling (e.g., because of TCR down-regulation) or by a general block in store-operated calcium entry (SOCE), we treated T cells with thapsigargin, which provokes store depletion and activates SOCE. Thapsigargin successfully depleted ER calcium stores in both naive and activated T cells (Fig. S4 B) but only induced a robust calcium influx in naive T cells. Activated T cells failed to do so, pointing at a general defect in SOCE in these cells (Fig. 5, A and B). At 72 h, activated T cells had partly recovered their calcium responses, suggesting that the phase of unresponsiveness is transitory and maximal at 48 h (Fig. 5, A and B).
Figure 5.

Recently activated T cells have a defect in store-operated calcium entry. (A and B) GFP+ OT-I CD8+ T cells were adoptively transferred, and recipients were stimulated by an i.v. injection of OVAp or left unstimulated. After 48 or 72 h, lymph node cells were harvested and stained with the calcium-sensitive dye Indo-1. Cells were stimulated with OVAp, Con A, or thapsigargin, and calcium responses were analyzed by flow cytometry. (A) Kinetics of calcium responses are shown for activated T cells (48 or 72 h) or naive T cells isolated ex vivo. Results are representative of three independent experiments. (B) Maximal value from Indo-1 ratio is shown for naive and activated T cells stimulated with the indicated concentration of OVAp, Con A, or thapsigargin. Results are pooled from three independent experiments. Data are shown as mean ± SEM. Significance testing were performed by using an unpaired t test. *, P < 0.05; **, P < 0.01. (C) GFP+ OT-I CD8+ T cells were labeled with SNARF and transferred into recipient mice. DCs pulsed with OVAp were injected in the footpad at the same time. At 48 h, cells from the draining lymph node were stained with Indo-1 and stimulated by Con A or thapsigargin. Kinetics of calcium responses analyzed by flow cytometry are shown for undivided (SNARFhigh) and divided (SNARFlow) T cells recovered at 48 h. Results are representative of two independent experiments.
We next tested whether this defect in SOCE was also apparent during in vivo stimulation by peptide-pulsed DCs. Recipient mice were transferred i.v. with SNARF-labeled GFP+ OT-I CD8+ T cells and injected with OVAp-pulsed DCs. At 48 h, we compared ex vivo calcium responses of T cells based on their division status (Fig. 5 C). Robust calcium responses were detected in undivided OT-I T cells upon Con A and thapsigargin treatment. In contrast, only weak Ca2+ signals were detected in divided T cells present in the same lymph node. Of note, this defect in SOCE was not caused by a lower expression of either STIM1 or ORAI1 in activated T cells (Fig. S5). Given the importance of calcium signals for mediating T cell arrest, our results suggest that, in addition to previously described mechanisms limiting TCR signaling during activation, the suppression of SOCE in recently activated T cells likely contributes to their inability to stop upon TCR engagement.
Forcing TCR signals in recently activated T cells antagonizes cell division
Our results supported the idea that SOCE suppression in activated T cells promotes T cell disengagement from APCs and prevents activated T cells from reengaging new APCs. In cell lines, SOCE has been shown to be down-regulated during mitosis, a mechanism proposed to protect mitotic cells from detrimental Ca2+ influx (Preston et al., 1991; Smyth et al., 2009). We therefore asked whether suppression of SOCE during T cell activation represents an important mechanism for T cell division to proceed efficiently. To test this possibility, we stimulated OT-I CD8+ T cells in vivo and quantified T cell division at 48 h by intravital lymph node imaging. Because we have shown that an injection of a large amount (50 µg) of OVAp at 48 h triggers a partial deceleration in T cells (and thus partly bypass T cell hyporesponsiveness), we used this system to test whether forcing TCR stimulation at 48 h could be detrimental to T cell division. Using intravital imaging, we monitored T cell mitosis events in the popliteal lymph node 30 or 48 h after initial activation. We then reinjected OVAp and immediately imaged T cells in the exact same area. As shown in Fig. 6 (A, B, and D) and Video 8, the rate of T cell division rapidly dropped upon peptide injection. We repeated this experiment by activating T cells in vivo in the presence of anti–PD-1 (or an isotype control), a treatment that increases the rate of T cell division at 48 h. In these settings also, forcing TCR signaling by peptide injection strongly reduced the rate of T cell divisions (Fig. 6, C and E; and Video 9). Altogether, these results support the idea that TCR stimulation during the expansion phase antagonizes T cell division, suggesting that the suppression of SOCE during activation acts as a safeguard mechanism to limit external stimuli that would be deleterious for T cell division.
Figure 6.

Strong TCR stimulation in the late phase of priming antagonizes division. (A and B) LifeAct-GFP+ or GFP+ OT-I CD8+ T cells were adoptively transferred, and recipients were stimulated by an i.v. injection of OVAp. Intravital imaging of the popliteal lymph node was performed at 30 or 48 h. OVAp was reinjected during the imaging experiment. Mitosis events were analyzed before or immediately after OVAp reinjection. (A) Representative images before or after OVAp reinjection illustrating that T cell divisions are reduced upon stimulation. Bar, 30 µm. (B) Quantification of T cell division before and after OVAp reinjection. Each line represents one independent video, and each square corresponds to 1 min of imaging. Red squares indicate the occurrence of a mitotic event in the imaging field. (C) The same experiment was performed in the presence of anti–PD-1 treatment or isotype control at 48 h. In these conditions also, OVAp inhibited T cell mitosis. (D and E) Compilation of the number of T cell division per hour in the imaging field before and after OVAp reinjection for the indicated conditions and time points. Each pair of dots corresponds to one imaging area. Data are pooled from three (D) or two (E) independent experiments. Significance testing was performed by using a paired t test. *, P < 0.05; ***, P < 0.001.
Discussion
In the present study, we investigated the cellular mechanisms underlying the termination of T cell priming, including T cell disengagement from DCs and initiation of clonal expansion. We found that the progressive loss of stable T cell–DC interactions during priming is a T cell–intrinsic rather than a DC-intrinsic mechanism. Specifically, recently activated T cells exhibited a defect in SOCE that was associated with a blunted capacity to stop upon TCR signals. Finally, we provide evidence that this block favors clonal expansion by preventing T cells from receiving late TCR signals that are detrimental to mitosis. Thus, our results identify an important mechanism for the termination of T cell priming.
Two-photon imaging has been instrumental in dissecting the dynamics of T cell priming in lymph nodes. In some systems, T cells initiate a first phase of dynamic contacts with DCs (phase 1) followed by stable interactions (phase 2) and later by the recovery of motility and progressive loss of contacts (phase 3; Hugues et al., 2004; Mempel et al., 2004; Miller et al., 2004). In other models, T cell priming starts directly with long-lasting contacts (phase 2; Shakhar et al., 2005; Celli et al., 2007; Moreau et al., 2012). The mechanisms driving these changes in cell dynamics are not fully understood, and it is unclear whether they reflect T cell– or DC-intrinsic mechanisms. For example, the transition from phase 1 to phase 2 has been proposed to reflect the progressive maturation of DCs, with fully mature DCs being more competent to form long-lasting conjugates than their immature counterparts (Hugues et al., 2004). In contrast, another study proposed that T cells progressively gain the ability to form stable contacts, suggestive of a T cell–intrinsic mechanism (Henrickson et al., 2008). The mechanisms driving the transition from phase 2 to phase 3 are even less understood. One obvious possibility is that the progressive loss of T cell–DC contacts during the late phases of T cell activation simply reflects the loss of pMHC complexes at the surface of DCs because the half-lives of pMHC can be in the range of hours. To test the possibility that the loss of T cell–DC contact originates, at least partly, from a T cell–intrinsic mechanism, we designed two experimental approaches to compare, in the same lymph node environment, the ability of T cells that have reached various stages of the priming process to bind DCs. By color-coding T cell division or T cell activation status, we could demonstrate that recently activated and divided T cells were less prone to bind to DCs compared to naive T cells, strongly suggesting that changes at the T cell level are essential to drive the transition from phase 2 to phase 3.
Further exploring this phenomenon, we showed that recently activated T cells were not responding to TCR-mediated stop signals. Several mechanisms could explain such T cell unresponsiveness. First, the PD-1 axis could limit T cell responsiveness over time, as observed in an effector phase in the skin (Honda et al., 2014). We did not find this mechanism to be at play during T cell priming in the lymph node because blocking the PD-1 pathway increased T cell activation status but did not prevent T cell unresponsiveness. Second, TCR internalization is typically observed in vitro and in vivo after strong TCR stimuli and could potentially limit TCR signaling in recently activated T cells (Valitutti et al., 1995). However, it is unlikely that diminished TCR levels solely explain the profound block observed in T cell responses to stimulation. Because elevation of intracellular calcium has been implicated in TCR-induced stop signal (Negulescu et al., 1996; Bhakta et al., 2005; Skokos et al., 2007; Waite et al., 2013; Moreau et al., 2015), we examined calcium mobilization capacity. We established that recently activated T cells exhibited a profound block in SOCE that paralleled their inability to stop upon TCR stimulation. Interestingly, a block in SOCE has previously been reported at the peak of expansion phase in a CD4+ T cell response suggesting that such mechanism may operate in various contexts and timings (Bikah et al., 2000). Previous in vitro studies have observed increased SOCE after T cell activation (Lioudyno et al., 2008; Thakur and Fomina, 2011). Differences in the timing of analysis and/or on the type of stimulation used (in vitro versus in vivo) may explain these discrepancies. Our study indicates that SOCE suppression occurs very rapidly during priming (within 48 h) providing a TCR-independent mechanism that may concur with other TCR signaling defects (Valitutti et al., 1995; Friedman et al., 2010; Best et al., 2013; Mayya and Dustin, 2016) to limit the late formation of T cell–DC contacts.
What is the physiological importance of such a phase of T cell unresponsiveness during priming? First, it prevents T cells from repeatedly engaging new DCs, a phenomenon that would limit their ability to egress and disseminate in the periphery. Beyond favoring the release of T cells from APCs, we provided evidence that such unresponsiveness is beneficial for effective clonal expansion because we showed that strong TCR signals antagonize cell division. Ca2+ has been proposed to participate in several processes during mitosis (Hepler, 1994), and it may be important to limit external perturbations and stimuli that would interfere with these processes (Arredouani et al., 2010). Consistent with this idea, SOCE has been shown to be specifically down-regulated in mitotic HeLa cells (Preston et al., 1991; Smyth et al., 2009). Future studies should help unravel the molecular basis for SOCE silencing in activated T cells.
In summary, the present work establishes that a phase of T cell unresponsiveness is integral to the process of T cell priming. We propose that this phase serves to temporally segregate two important cellular events during T cell priming: (1) the collection of activation signals during contact with DCs and (2) the clonal expansion away from the APCs. Whether similar mechanisms also operate to terminate effector T cell responses remains to be evaluated.
Materials and methods
Mice
Wild-type C57BL/6 (B6) mice were obtained from Charles River Laboratories; 6–12-wk-old mice were used. B6 transgenic mice expressing Rag1−/− OT-I TCR or UBC-GFP Rag1−/− OT-I TCR or LifeAct-GFP Rag1−/− OT-I TCR, CFP (CFP expressed under the actin promoter), mTomato, and B6-CD11c-YFP mice were bred in our animal facility. The knock-in bicistronic IFN-γ–YFP reporter mice (referred to as Yeti mice; Stetson et al., 2003) were a gift from R. Locksley (University of California, San Francisco, San Francisco, CA) and were crossed with the Rag1−/− OT-I TCR transgenic mice. All experiments were performed in agreement with relevant guidelines and regulations and approved by the Institut Pasteur committee on Animal Welfare (CETEA) under the protocol code of CETEA 2013–0089.
Flow cytometry and antibodies
Cell suspensions were Fc-blocked by using anti-CD16/32 Ab (93; BioLegend). Dead cells were labeled with Live/Dead fixable dead cell stains (Life Technologies). Stainings were performed with the following mAbs: CD3 (17A2; BioLegend), PD-1 (J43; eBiosciences), CD8α (53–6.7; BioLegend), CD62L (Mel-14; eBioscience), Vα2 (B20.1; BioLegend), Vβ5.1/5.2 (MR9-4; BioLegend), and CD25 (PC61, BioLegend). Polyclonal Ab to ORAI1 and STIM1 were purchased from GeneTex. Intracellular stainings were performed by using the PermWash buffer (BD Biosciences). Analyses were performed with an LSR/Fortessa cytometer (BD Biosciences) and analyzed with FlowJo software v.10.1 (Tree Star). For anti–PD-1 treatment experiment, recipient mice were injected at the indicated time with 250 µg blocking anti–PD-1 antibody (RMP1-14; Bio X Cell) or an IgG2Aκ isotype control (2A3, Bio X Cell).
In vivo T cell activation by peptide injection
OT-I CD8+ T cells were isolated from the lymph nodes of UBC-GFP Rag1−/− OT-I TCR transgenic mice, and 5 × 106 cells were adoptively transferred into B6 recipient mice by i.v. injection. 18 h later, the OVA257–264 peptide SIINFEKL (OVAp) was injected i.v. (50 µg). At different times after peptide injection (3, 24, 30, 48, and 72) lymph nodes were harvested and digested in RPMI with 1 mg/ml collagenase and 0.1 mg/ml DNase (Sigma) for 20 min at 37°C. Alternatively, mice were anesthetized and the popliteal lymph nodes prepared for intravital imaging as previously described (Moreau and Bousso, 2017). In some experiments, a second cohort of OT-I CD8+ T cells labeled with 2.5 µM SNARF (Invitrogen) was adoptively transferred into mice by i.v. injection 18 h before intravital imaging.
In vivo T cell activation by DCs
DCs were isolated from spleens of B6-CFP or B6-CD11c-YFP transgenic mice by positive selection with CD11c microbeads (Miltenyi Biotec). Purified DCs were pulsed with the indicated concentration of OVAp for 10 min at room temperature. 5–10 × 106 pulsed DCs were injected subcutaneously in the footpad of recipient B6 mice. OT-I CD8+ T cells were isolated from the lymph nodes of UBC-GFP Rag1−/− OT-I TCR transgenic mice and stained with 2.5 µM SNARF, and 0.5–3 × 106 T cells were injected i.v. in the same mice just after DC injection. Alternatively, OT-I CD8+ T cells were isolated from the lymph nodes of Yeti mice and labeled with SNARF, and 5 × 106 T cells were injected i.v. in the same mice. Intravital two-photon imaging of the draining popliteal lymph node was performed at the indicated time.
Ex vivo calcium measurements
OT-I CD8+ T cells were isolated from the lymph nodes of UBC-GFP Rag1−/− OT-I TCR transgenic mice, and 5 × 106 cells were adoptively transferred into B6 recipient mice by i.v. injection. 18 h later the OVA257-264 peptide SIINFEKL (OVAp) was injected i.v. or not (50 µg). At 48 and 72 h lymph nodes were harvested, crushed, and stained with Indo-1/AM (2.5 µM; Molecular Probes) for 40 min at 37°C. Cells were washed and concentrated at 2 × 106 cells/ml in complete RPMI (10% FCS) and kept at 37°C. Calcium responses were analyzed by using an LSR/Fortessa cytometer. A baseline Indo-1 fluorescence was recorded for 1 min, the stimulus was added, and acquisition was extended for 4 additional minutes. An Indo-1 ratio of fluorescence was calculated from signals emitted at 405 nm (Ca2+ bound dye) and 485 nm (Ca2+ free dye) and followed over time. A kinetic analysis was performed with FlowJo software, and the smoothed GeoMean of Indo-1 ratio was plotted. The following stimuli were used: OVAp (20 µg), Con A (100–200 mg/ml; Sigma), thapsigargin (1–10 µM; Sigma), or OVAp-pulsed splenocytes. When indicated, EGTA (5 µM) was added to chelate extracellular calcium.
Intravital two-photon imaging
We used an upright microscope (DM6000B, SP5; Leica Microsystems) with a 25×/1.05 NA dipping objective (Olympus). Excitation was provided by a Chameleon Ultra Ti/Sapphire (Coherent) tuned at 950 nm. The following filter set was used for imaging second harmonics generation (SHG)/GFP/SNARF: 483/32 BP, 509 LP, 520/35 BP, 605 LP, 641/75 BP. Alternatively, we used an upright microscope (FVMPE-RS; Olympus) with a 25×/1.05 NA dipping objective (Olympus). Excitation was provided by an Insight DS+ Dual laser (Spectra-Physics) tuned at 950 nm and 1,040 nm. The following filter set was used for imaging CFP/GFP/SNARF: 483/32 BP, 505 SP, 520/35 BP, 605 LP, 641/75 BP; (SHG)/GFP/YFP/SNARF: 483/32 BP, 505 SP, 520/35 BP, 520 LP, 542/27 BP, 605 LP, 641/75 BP. Typically, image fields of 512 × 512 pixels, from 8 to 12 z planes spaced 7 µm apart were collected every 30 s for 1 h. When indicated, recipient mice were injected i.v. with 50 µg OVAp, and a second 1-h video was acquired. Videos were processed and analyzed with Imaris software (Bitplane) or Fiji software (ImageJ 1.49 m). Only cell tracks with a duration >5 min were kept. The arrest coefficient was defined as the percentage of time during which instantaneous velocity was <3 µm/min. Straightness was calculated as the ratio of the distance from origin to the total distance traveled. Contact durations were determined by examining juxtaposition of T cell and DC fluorescent signals in individual z planes. The mean intensities of GFP, YFP, and SNARF fluorescence were measured with Fiji to determine the GFP/SNARF or YFP/SNARF ratio in individual T cells. The numbers of division events were counted for each video before and after peptide injection. For statistical purposes, only image fields with at least five division events before OVAp reinjection were considered. The distance between dividing T cells and the closest DCs was determined from the x, y, and z coordinates of the cell centers.
Statistical analyses
All statistical tests were performed with Prism v.6.0g (GraphPad), and data are represented as mean ± SEM. We used alternatively unpaired t test, paired t test, or Mann-Whitney U test for two-group comparison. All p-values were calculated with two-tailed statistical tests and 95% confidence intervals. *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Online supplemental material
Fig. S1 shows the defect of recently activated T cells in stopping upon TCR engagement at various time points. Fig. S2 shows increased T cell activation upon anti–PD-1 treatment. Fig. S3 shows that extracellular calcium is required for efficient T cell–DC conjugation. Fig. S4 shows that recently activated T cells have defective calcium responses upon stimulation with peptide-pulsed splenocytes and that thapsigargin efficiently induces the depletion of intracellular calcium stores in both naive and activated T cells. Fig. S5 shows the expression of STIM1 and ORAI1 in naive and activated T cells. Video 1 shows that T cell motility precedes T cell division in the lymph node. Video 2 shows that T cells primarily establish stable and long-lasting interactions with DCs in the first hours of priming. Video 3 shows that most T cells divide away from DCs. Video 4 shows that long-lasting contacts with DCs are restricted to undivided T cells. Video 5 shows that recently activated T cells exhibit an intrinsic defect in establishing long-lasting contacts with DCs. Video 6 shows that T cell arrest in the lymph node is dependent on T cell activation status. Video 7 shows that recently activated T cells do not effectively stop upon antigen recognition. Videos 8 and 9 show that forcing TCR stimulation in the late phase of priming limits T cell divisions.
Supplementary Material
Acknowledgments
We thank the members of the Bousso laboratory for critical review of the manuscript. We acknowledge the Technology Core of the Center for Translational Science at Institut Pasteur for support in conducting this study and the specific contribution of S. Novault.
This work was supported by Institut Pasteur, Institut National de la Santé et de la Recherche Medicale, and a starting grant (Lymphocytecontact) from the European Research Council. A. Bohineust was funded by the European Research Council and by a fellowship from the Ligue Contre le Cancer.
The authors declare no competing financial interests.
Author contributions: A. Bohineust, Z. Garcia, H. Beuneu, and F. Lemaître conducted the experiments. P. Bousso and A. Bohineust designed the experiments, analyzed the data, and wrote the manuscript.
References
- Arredouani A., Yu F., Sun L., and Machaca K.. 2010. Regulation of store-operated Ca2+ entry during the cell cycle. J. Cell Sci. 123:2155–2162. 10.1242/jcs.069690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best J.A., Blair D.A., Knell J., Yang E., Mayya V., Doedens A., Dustin M.L., and Goldrath A.W.. Immunological Genome Project Consortium . 2013. Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat. Immunol. 14:404–412. 10.1038/ni.2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuneu H., Lemaître F., Deguine J., Moreau H.D., Bouvier I., Garcia Z., Albert M.L., and Bousso P.. 2010. Visualizing the functional diversification of CD8+ T cell responses in lymph nodes. Immunity. 33:412–423. 10.1016/j.immuni.2010.08.016 [DOI] [PubMed] [Google Scholar]
- Bhakta N.R., Oh D.Y., and Lewis R.S.. 2005. Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat. Immunol. 6:143–151. 10.1038/ni1161 [DOI] [PubMed] [Google Scholar]
- Bikah G., Pogue-Caley R.R., McHeyzer-Williams L.J., and McHeyzer-Williams M.G.. 2000. Regulating T helper cell immunity through antigen responsiveness and calcium entry. Nat. Immunol. 1:402–412. 10.1038/80841 [DOI] [PubMed] [Google Scholar]
- Bousso P. 2008. T-cell activation by dendritic cells in the lymph node: lessons from the movies. Nat. Rev. Immunol. 8:675–684. 10.1038/nri2379 [DOI] [PubMed] [Google Scholar]
- Bousso P., and Robey E.. 2003. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat. Immunol. 4:579–585. 10.1038/ni928 [DOI] [PubMed] [Google Scholar]
- Celli S., Lemaître F., and Bousso P.. 2007. Real-time manipulation of T cell-dendritic cell interactions in vivo reveals the importance of prolonged contacts for CD4+ T cell activation. Immunity. 27:625–634. 10.1016/j.immuni.2007.08.018 [DOI] [PubMed] [Google Scholar]
- Chang J.T., Palanivel V.R., Kinjyo I., Schambach F., Intlekofer A.M., Banerjee A., Longworth S.A., Vinup K.E., Mrass P., Oliaro J., et al. 2007. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 315:1687–1691. 10.1126/science.1139393 [DOI] [PubMed] [Google Scholar]
- Dustin M.L. 2004. Stop and go traffic to tune T cell responses. Immunity. 21:305–314. 10.1016/j.immuni.2004.08.016 [DOI] [PubMed] [Google Scholar]
- Fife B.T., Pauken K.E., Eagar T.N., Obu T., Wu J., Tang Q., Azuma M., Krummel M.F., and Bluestone J.A.. 2009. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 10:1185–1192. 10.1038/ni.1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R.S., Beemiller P., Sorensen C.M., Jacobelli J., and Krummel M.F.. 2010. Real-time analysis of T cell receptors in naive cells in vitro and in vivo reveals flexibility in synapse and signaling dynamics. J. Exp. Med. 207:2733–2749. 10.1084/jem.20091201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrickson S.E., Mempel T.R., Mazo I.B., Liu B., Artyomov M.N., Zheng H., Peixoto A., Flynn M.P., Senman B., Junt T., et al. 2008. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat. Immunol. 9:282–291. 10.1038/ni1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler P.K. 1994. The role of calcium in cell division. Cell Calcium. 16:322–330. 10.1016/0143-4160(94)90096-5 [DOI] [PubMed] [Google Scholar]
- Honda T., Egen J.G., Lämmermann T., Kastenmüller W., Torabi-Parizi P., and Germain R.N.. 2014. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity. 40:235–247. 10.1016/j.immuni.2013.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugues S., Fetler L., Bonifaz L., Helft J., Amblard F., and Amigorena S.. 2004. Distinct T cell dynamics in lymph nodes during the induction of tolerance and immunity. Nat. Immunol. 5:1235–1242. 10.1038/ni1134 [DOI] [PubMed] [Google Scholar]
- Kedl R.M., Schaefer B.C., Kappler J.W., and Marrack P.. 2002. T cells down-modulate peptide-MHC complexes on APCs in vivo. Nat. Immunol. 3:27–32. 10.1038/ni742 [DOI] [PubMed] [Google Scholar]
- Lioudyno M.I., Kozak J.A., Penna A., Safrina O., Zhang S.L., Sen D., Roos J., Stauderman K.A., and Cahalan M.D.. 2008. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc. Natl. Acad. Sci. USA. 105:2011–2016. 10.1073/pnas.0706122105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayya V., and Dustin M.L.. 2016. What scales the T cell response? Trends Immunol. 37:513–522. 10.1016/j.it.2016.06.005 [DOI] [PubMed] [Google Scholar]
- Mempel T.R., Henrickson S.E., and Von Andrian U.H.. 2004. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 427:154–159. 10.1038/nature02238 [DOI] [PubMed] [Google Scholar]
- Miller M.J., Wei S.H., Parker I., and Cahalan M.D.. 2002. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 296:1869–1873. 10.1126/science.1070051 [DOI] [PubMed] [Google Scholar]
- Miller M.J., Safrina O., Parker I., and Cahalan M.D.. 2004. Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J. Exp. Med. 200:847–856. 10.1084/jem.20041236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau H.D., and Bousso P.. 2017. In vivo imaging of T cell immunological synapses and kinapses in lymph nodes. Methods Mol. Biol. 1584:559–568. 10.1007/978-1-4939-6881-7_35 [DOI] [PubMed] [Google Scholar]
- Moreau H.D., Lemaître F., Terriac E., Azar G., Piel M., Lennon-Dumenil A.M., and Bousso P.. 2012. Dynamic in situ cytometry uncovers T cell receptor signaling during immunological synapses and kinapses in vivo. Immunity. 37:351–363. 10.1016/j.immuni.2012.05.014 [DOI] [PubMed] [Google Scholar]
- Moreau H.D., Lemaître F., Garrod K.R., Garcia Z., Lennon-Duménil A.M., and Bousso P.. 2015. Signal strength regulates antigen-mediated T-cell deceleration by distinct mechanisms to promote local exploration or arrest. Proc. Natl. Acad. Sci. USA. 112:12151–12156. 10.1073/pnas.1506654112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negulescu P.A., Krasieva T.B., Khan A., Kerschbaum H.H., and Cahalan M.D.. 1996. Polarity of T cell shape, motility, and sensitivity to antigen. Immunity. 4:421–430. 10.1016/S1074-7613(00)80409-4 [DOI] [PubMed] [Google Scholar]
- Oliaro J., Van Ham V., Sacirbegovic F., Pasam A., Bomzon Z., Pham K., Ludford-Menting M.J., Waterhouse N.J., Bots M., Hawkins E.D., et al. 2010. Asymmetric cell division of T cells upon antigen presentation uses multiple conserved mechanisms. J. Immunol. 185:367–375. 10.4049/jimmunol.0903627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozga A.J., Moalli F., Abe J., Swoger J., Sharpe J., Zehn D., Kreutzfeldt M., Merkler D., Ripoll J., and Stein J.V.. 2016. pMHC affinity controls duration of CD8+ T cell-DC interactions and imprints timing of effector differentiation versus expansion. J. Exp. Med. 213:2811–2829. 10.1084/jem.20160206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace L., Tempez A., Arnold-Schrauf C., Lemaitre F., Bousso P., Fetler L., Sparwasser T., and Amigorena S.. 2012. Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science. 338:532–536. 10.1126/science.1227049 [DOI] [PubMed] [Google Scholar]
- Preston S.F., Sha’afi R.I., and Berlin R.D.. 1991. Regulation of Ca2+ influx during mitosis: Ca2+ influx and depletion of intracellular Ca2+ stores are coupled in interphase but not mitosis. Cell Regul. 2:915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider H., Downey J., Smith A., Zinselmeyer B.H., Rush C., Brewer J.M., Wei B., Hogg N., Garside P., and Rudd C.E.. 2006. Reversal of the TCR stop signal by CTLA-4. Science. 313:1972–1975. 10.1126/science.1131078 [DOI] [PubMed] [Google Scholar]
- Scholer A., Hugues S., Boissonnas A., Fetler L., and Amigorena S.. 2008. Intercellular adhesion molecule-1-dependent stable interactions between T cells and dendritic cells determine CD8+ T cell memory. Immunity. 28:258–270. 10.1016/j.immuni.2007.12.016 [DOI] [PubMed] [Google Scholar]
- Shakhar G., Lindquist R.L., Skokos D., Dudziak D., Huang J.H., Nussenzweig M.C., and Dustin M.L.. 2005. Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nat. Immunol. 6:707–714. 10.1038/ni1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skokos D., Shakhar G., Varma R., Waite J.C., Cameron T.O., Lindquist R.L., Schwickert T., Nussenzweig M.C., and Dustin M.L.. 2007. Peptide-MHC potency governs dynamic interactions between T cells and dendritic cells in lymph nodes. Nat. Immunol. 8:835–844. 10.1038/ni1490 [DOI] [PubMed] [Google Scholar]
- Smyth J.T., Petranka J.G., Boyles R.R., DeHaven W.I., Fukushima M., Johnson K.L., Williams J.G., and Putney J.W. Jr. 2009. Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat. Cell Biol. 11:1465–1472. 10.1038/ncb1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson D.B., Mohrs M., Reinhardt R.L., Baron J.L., Wang Z.E., Gapin L., Kronenberg M., and Locksley R.M.. 2003. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J. Exp. Med. 198:1069–1076. 10.1084/jem.20030630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro C.E., Shakhar G., Shen S., Ding Y., Lino A.C., Maraver A., Lafaille J.J., and Dustin M.L.. 2006. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J. Exp. Med. 203:505–511. 10.1084/jem.20050783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q., Adams J.Y., Tooley A.J., Bi M., Fife B.T., Serra P., Santamaria P., Locksley R.M., Krummel M.F., and Bluestone J.A.. 2006. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 7:83–92. 10.1038/ni1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur P., and Fomina A.F.. 2011. Density of functional Ca2+ release-activated Ca2+ (CRAC) channels declines after T-cell activation. Channels (Austin). 5:510–517. 10.4161/chan.5.6.18222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valitutti S., Müller S., Cella M., Padovan E., and Lanzavecchia A.. 1995. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 375:148–151. 10.1038/375148a0 [DOI] [PubMed] [Google Scholar]
- Waite J.C., Vardhana S., Shaw P.J., Jang J.E., McCarl C.A., Cameron T.O., Feske S., and Dustin M.L.. 2013. Interference with Ca(2+) release activated Ca(2+) (CRAC) channel function delays T-cell arrest in vivo. Eur. J. Immunol. 43:3343–3354. 10.1002/eji.201243255 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.