

Abstract
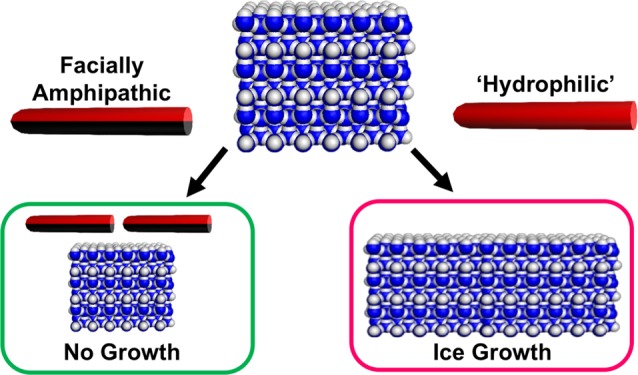
Antifreeze glycoproteins (AFGPs) from polar fish are the most potent ice recrystallization (growth) inhibitors known, and synthetic mimics are required for low-temperature applications such as cell cryopreservation. Here we introduce facially amphipathic glycopolymers that mimic the three-dimensional structure of AFGPs. Glycopolymers featuring segregated hydrophilic and hydrophobic faces were prepared by ring-opening metathesis polymerization, and their rigid conformation was confirmed by small-angle neutron scattering. Ice recrystallization inhibition (IRI) activity was reduced when a hydrophilic oxo-ether was installed on the glycan-opposing face, but significant activity was restored by incorporating a hydrophobic dimethylfulvene residue. This biomimetic strategy demonstrates that segregated domains of distinct hydrophilicity/hydrophobicity are a crucial motif to introduce IRI activity, which increases our understanding of the complex ice crystal inhibition processes.
Antifreeze glycoproteins (AFGPs) are found in the tissues and blood serum of extremophile fish species and act to modulate the growth of extracellular ice.1 A key property of AFGPs is ice recrystallization inhibition (IRI), which slows ice crystal growth (distinct from nucleation2).3 Ice recrystallization is a major cause of cell death during the freezing of cells and tissue for transfusion, fundamental biomedicine, and cell biology. Hence, AFGPs (or their mimics) have many potential applications.4 Cryopreservation with AFGPs (and non-glycosylated antifreeze proteins, AFPs)5 is limited, however, by their secondary property of dynamic ice shaping, whereby the AFGPs shape the ice into needle-like (spicular) morphologies, which can pierce cell membranes.3 AFGPs are also challenging to synthesize, requiring multistep procedures.6,7 Gibson and co-workers have developed synthetic polymers8,9 based upon poly(vinyl alcohol) and poly(ampholytes) which have been found to enhance the cryopreservation of blood10−12 and nucleated cells.4,13
In the case of AFPs, defined ice-binding faces have been identified using structural biology methods.14 Conversely, there is no crystal structure available for AFGPs, and the exact structural motifs required for IRI are unknown, although the glycan unit is essential for ice shaping.7 Solution NMR studies suggest that AFGPs form a polyproline II type of helix, with the glycans on one face and peptides on the opposite, forming a facially amphipathic structure.15 It is emerging that this segregated display of hydrophobic/hydrophilic groups, rather than a “binding site”, is the essential feature for IRI activity.8,16,17 Molecular modeling recently revealed that the hydrophobic face, not the glycans, of AFGPs interacts with the ice, and that the spatial segregation along the polyproline II helix is essential.18 Gibson and co-workers have shown that homopolyproline has a weak IRI,4 and that self-assembled metallohelicities with “patchy” amphipathy are potent IRIs,19 which supports a hypothesis that well-defined ice-binding domains are not essential for IRI.20 Amphipathy has also been seen to be important in ice nucleation.21 This evidence suggests that IRI, but not ice shaping,22 could be selectively introduced into new and emerging (bio)materials, if precise control over hydrophilic/hydrophobic domains is possible.
The design of polymers with solvent-exposed hydrophobic domains is, however, nontrivial. Block copolymeric amphiphiles spontaneously self-assemble into micelles/vesicles to reduce the hydrophobic domain’s contact with water, and hence only “water-loving” surfaces are exposed.23 Tew and co-workers have developed facially amphipathic cationic polymers, with opposing positive charges and lipophilic domains to mimic the function of antimicrobial peptides.24,25 A crucial design step was the use of ring-opening metathesis polymerization (ROMP), which introduces rigid alkene backbones, while balancing the hydrophobicity/hydrophilicity to maintain both solubility and the presentation of hydrophobic faces. These have shown particular potency as potential antimicrobials.26
Considering the above, we designed and synthesized locally rigid, facially amphipathic glycopolymers. A combination of ice binding assays, modeling, and small-angle neutron scattering (SANS) provides compelling evidence that local amphipathy is an essential motif for introducing IRI activity, providing design rules for new materials to mimic AFGP function.
Figure 1A shows the solution-state structure of AFGP,7 with the disaccharide units spatially segregated from the hydrophobic peptide backbone. Our approach was to use ROMP to introduce local rigidity,27 in contrast to flexible backbones obtained from radical polymerization. Four monomers were synthesized to give a range of amphipathies: M1 was prepared by acetylation of a commercial norbornenediol; M2 and M3 were synthesized by Koenigs–Knorr coupling of acetobromo-α-d-galactose with exo,exo-[oxo/fulvene]norborneneimide; and M4 was synthesized by substitution of monomethoxyhexaethylene glycol monotosylate (Figure 1B). The hydrophilic galactose and hydrophobic fulvene motifs were selected in particular due to their intrinsic rigidities, giving monomers with structurally distinct domains of opposing polarity. These monomers were polymerized using Grubbs’s third-generation catalyst, and acetate protecting groups were subsequently removed by treatment with sodium methoxide followed by ion exchange (Figure 1C). The panel of amphipathic polymers was characterized by size-exclusion chromatography (SEC; Đ < 1.4) and NMR (nuclear magnetic resonance) and IR (infrared) spectroscopy (see Supporting Information (SI) and Table 1).
Figure 1.
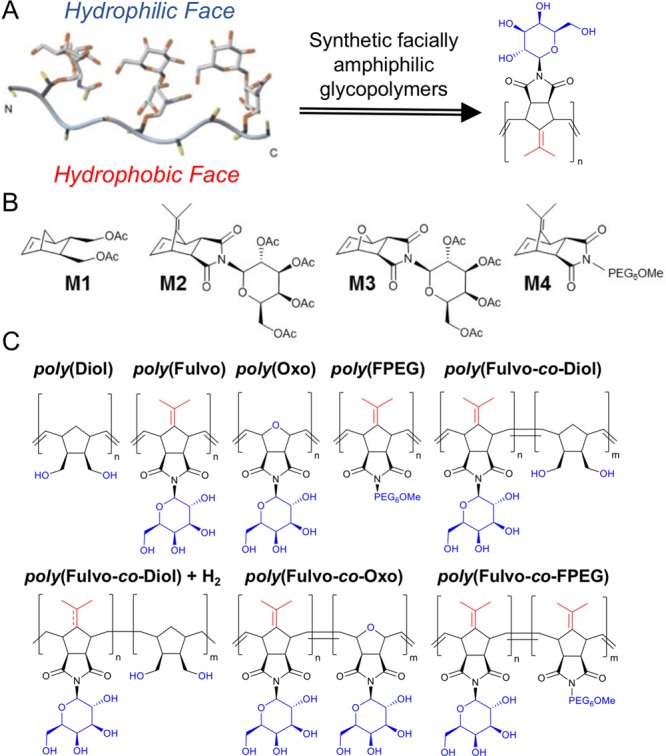
(A) Concept of facially amphipathic ROMP polymers to mimic AFGP. Adapted from ref (7). (B,C) Monomers and polymers synthesized here; hydrophilic groups are indicated in blue, and hydrophobic in red.
Table 1. Polymer Characterization.
|
Mn (g mol–1) |
|||||
|---|---|---|---|---|---|
| theor | exptla | Đ (−)a | DP (−)a | conv (%)b | |
| poly(Diol)c | 10 000 | 2 200 | 1.01 | 14 | 100 |
| 5 300 | 1.01 | 34 | |||
| 8 400 | 1.02 | 54 | |||
| poly(Fulvo) | 25 000 | 10 300 | 1.21 | 28 | 100 |
| poly(Oxo) | 10 000 | 7 300 | 1.18 | 22 | 100 |
| poly(FPEG) | 10 000 | 35 900 | 1.38 | 133 | 100 |
| poly(Fulvo-co-Diol)-11 | 5 000 | 10 700 | 1.12 | 14, 35 | 94/97 |
| poly(Fulvo-co-Diol)-17 | 10 000 | 16 800 | 1.10 | 22, 54 | 100 |
| poly(Fulvo-co-Diol)-35 | 25 000 | 34 600 | 1.26 | 47, 112 | 100 |
| poly(Fulvo-co-Oxo) | 10 000 | 7 700 | 1.35 | 11, 11 | 100/96 |
| poly(Fulvo-co-FPEG) | 10 000 | 55 600 | 1.47 | 76, 58 | 71 |
Mn = molar mass, Đ = dispersity, and DP = degree of polymerization, determined by SEC.
Conversion, determined by 1H NMR.
Single species.
The polymer library was assessed for IRI activity using a “splat” assay, where ice crystals are nucleated and their growth after 30 min at −8 °C was recorded. Activity is expressed as the mean grain area (MGA) relative to a phosphate-buffered saline (PBS) control, with smaller values representing more activity. Polymers derived from M2 containing the “fulvo” motif were significantly less soluble than those derived from M3 containing the “oxo”-ether units. Their solution concentrations were therefore determined by UV–vis absorption spectroscopy (see SI for Beer–Lambert plots) at saturation. In the case of poly(Fulvo), 1% v/v dimethylsulfoxide was required, and controls were adjusted to account for this.
Poly(Oxo) was found to inhibit ice crystal growth by ∼50% MGA at concentrations above 5 mg·mL–1 (Figure 2A), which makes it more active than many previously reported IRI-active polymers.28,29 The poly(Fulvo) derivative featuring the hydrophobic face, however, was considerably more active, inhibiting by ∼50% MGA at just 0.5 mg·mL–1 (solubility limit), supporting the facially amphipathic hypothesis for IRI. Molecular models corroborate this (Figure 2B) and illustrate the relative increase in hydrophobicity across the poly(Oxo) and poly(Fulvo) homopolymers. To improve the solubility, a 1:1 statistical copolymer of M2/M3, poly(Fulvo-co-Oxo), was prepared. This co-polymer had significantly improved solubility and comparable overall IRI activity to poly(Fulvo), showing that some co-monomer incorporation is tolerated, unlike PVA,30 and example ice wafers are shown in Figure 2C. However, the non-ideal copolymerization kinetics of the oxo (M2) and fulvo (M3) co-monomers led to a blocky rather than statistical copolymerization.31,32 Infrared analysis confirmed incomplete acetate removal (in contrast to the homopolymers), suggesting an internalized domain structure and/or aggregation, with some (hydrophobic) surfaces being solvent inaccessible and hence limiting the total activity of poly(Fulvo-co-Oxo). The monomers alone also had no activity (SI), confirming that a macromolecular architecture is essential.
Figure 2.
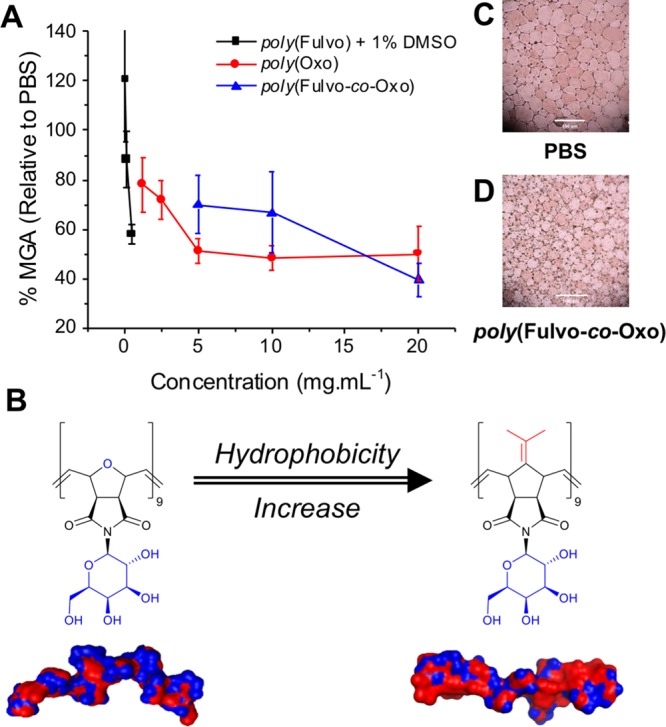
(A) IRI activities of the poly(Fulvo), poly(Oxo), and copolymer series. (B) Hydrophobic surface map of poly(Fulvo) and poly(Oxo). (C,D) Optical microscopy of ice crystal wafers of PBS and poly(Fulvo-co-Oxo).
To improve solubility, a norbornenediol monomer, M1, with a non-hydrophilic bridgehead was investigated. De-acetylated homopolymers of M1, poly(Diol), were found to have surprisingly low solubility and no activity at their solubility limit of 0.5 mg·mL−1. However, when M1 was incorporated as a co-monomer with the (IRI-active) “fulvo” monomer M2 to give poly(Fulvo-co-Diol), an overall increase in solubility was achieved. Poly(Fulvo-co-Diol)-17 had remarkable IRI activity: 40% MGA at just 1.3 mg·mL–1 (Figure 3). This polymer showed some molecular weight dependence on activity, with 17 kDa being more active than 11 kDa (and far more than the monomer, indicating the need for a macromolecular architecture). Increasing the molecular weight further to 35 kDa lowered the solubility of the copolymer, and hence the activity, highlighting a “sweet spot”. Work undertaken by Inada et al. described the molecular weight dependence on IRI of PVA.33 Similarly, a previous study by Deswal et al. reported on the IRI activity of proteins extracted from the leaves of the freeze-tolerant plant Seabuckthorn, of which superior antifreeze activity was observed only for polypeptides of elevated molecular weights.34 Replacing the glycan with a short oligo(ethylene glycol) PEG chain, to give both poly(Fulvo-co-FPEG) and poly(FPEG), decreased activity, as the (flexible) PEG can access numerous conformations, reducing the overall amphipathy (see SI). Hydrogenation of the alkene backbone to increase flexibility resulted in a wholly insoluble polymer (see SI). These observations demonstrate that precise macromolecular engineering is essential to achieve a potent IRI mimetic.
Figure 3.
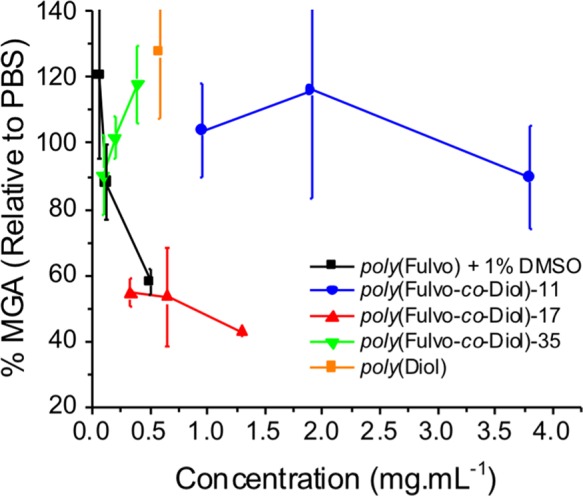
IRI activities of the poly(Fulvo-co-Diol) molecular weight series.
AF(G)Ps bind to specific ice crystal faces,35,36 leading to dynamic ice shaping (unwanted in cryopreservation3). Control ice crystals (Figure 4A) showed no dynamic ice shaping, but addition of AFGPs (Figure 4B) produced distinctive spicular (needle-like) crystals. Poly(Fulvo-co-Diol) (Figure 4C) did not lead to ice shaping, ruling out strong and specific ice face recognition and showing that these effects can be separated by macromolecular design.
Figure 4.

Optical microscopy ice morphology analysis: (A) water, −6 °C; (B) AFGP-8, −5 °C; (C) poly(Fulvo-co-Diol)-17 (0.72 mg·mL–1), −8 °C.
SANS was employed to evaluate the solution conformation and rigidity of the poly(Fulvo-co-Diol) series (Figure 5 and SI). The persistence lengths, bt, were estimated from the position of the characteristic crossover between the scattering profile typical for fractal aggregates (q–3.5) and that of rigid rods (q–1) (see SI).37,38 The estimated bt values for poly(Fulvo-co-Diol)-11 and poly(Fulvo-co-Diol)-17 are 38.9 and 44.4 Å, respectively. It should be noted that the overlap may actually occur at a lower q region, but is masked by aggregate scattering. Thus, these values should be taken as the minimum persistence length for each polymer. Nevertheless, each bt is much larger than the monomer length (∼10 Å), which suggests that the chain backbones are locally stiff.38 Furthermore, given the approximate contour length, L, of both polymer chains (490 and 760 Å for poly(Fulvo-co-Diol)-11 and poly(Fulvo-co-Diol)-17, respectively), the large bt suggests rigid rather than highly flexible aggregates of potentially rod-like structures. This rigidity, coupled with the intrinsic amphipathy of the polymers, is aligned with the hypothesized semi-rigid (and generally amphipathic) ice binding faces of AFPs,14,39 and the flexible hydrophilic “glycan face” of AFGPs, providing evidence that facial amphipathy is a key motif for introducing IRI activity into a diverse range of polymers.
Figure 5.
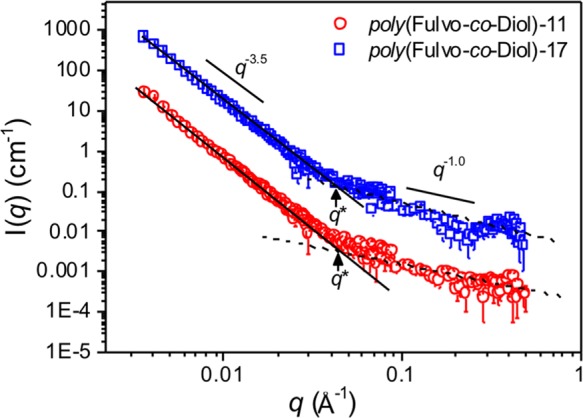
SANS data for poly(Fulvo-co-Diol)-11 (1 mg.mL−1, red) and poly(Fulvo-co-Diol)-17 (1 mg.mL−1, blue) in D2O at 25 °C. Straight lines show −3.5 and −1 decays for comparison.
To conclude, we have designed and synthesized facially amphipathic glycopolymers to mimic the solution confirmation and selective functions of antifreeze glycoproteins. It was found that the addition of hydrophobic faces, opposing the glycan units, introduced potent IRI activity, but that substitution with a more hydrophilic ether unit removed activity. These results support a mechanism for IRI activity which is dependent upon local water ordering rather than an essential ice binding unit, and there was no evidence of dynamic ice shaping. Small-angle neutron scattering supports a locally rigid confirmation, as seen for AF(G)Ps, supporting the hypothesis of amphipathy as the driver for activity.
Acknowledgments
M.I.G. holds an ERC Starter Grant (CRYOMAT 638661). Thanks to A. L. DeVries for supplying the antifreeze glycoprotein AFGP-8. The SANS portion of this work is based upon experiments performed at the KWS-2 instrument operated at the Heinz Maier-Leibnitz Zentrum(MLZ), Garching, Germany.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b02066.
Full experimental details, including synthesis/characterization, additional IRI data, and SANS analysis (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Harding M. M.; Anderberg P. I.; Haymet A. D. J. Eur. J. Biochem. 2003, 270 (7), 1381. 10.1046/j.1432-1033.2003.03488.x. [DOI] [PubMed] [Google Scholar]
- Congdon T.; Dean B. T.; Kasperczak-Wright J.; Biggs C. I.; Notman R.; Gibson M. I. Biomacromolecules 2015, 16 (9), 2820. 10.1021/acs.biomac.5b00774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao H.; Davies P. L.; Carpenter J. F. J. Exp. Biol. 1996, 199 (Pt 9), 2071. [DOI] [PubMed] [Google Scholar]
- Graham B.; Bailey T. L.; Healey J. R. J.; Marcellini M.; Deville S.; Gibson M. I. Angew. Chem., Int. Ed. 2017, 56, 15941. 10.1002/anie.201706703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies P. L. Trends Biochem. Sci. 2014, 39, 548–555. 10.1016/j.tibs.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Wilkinson B. L.; Stone R. S.; Capicciotti C. J.; Thaysen-Andersen M.; Matthews J. M.; Packer N. H.; Ben R. N.; Payne R. J. Angew. Chem., Int. Ed. 2012, 51 (15), 3606. 10.1002/anie.201108682. [DOI] [PubMed] [Google Scholar]
- Tachibana Y.; Fletcher G. L.; Fujitani N.; Tsuda S.; Monde K.; Nishimura S. I. Angew. Chem., Int. Ed. 2004, 43 (7), 856. 10.1002/anie.200353110. [DOI] [PubMed] [Google Scholar]
- Biggs C. I.; Bailey T. L.; Ben Graham; Stubbs C.; Fayter A.; Gibson M. I. Nat. Commun. 2017, 8 (1), 1546. 10.1038/s41467-017-01421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson M. I. Polym. Chem. 2010, 1 (8), 1141. 10.1039/c0py00089b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deller R. C.; Vatish M.; Mitchell D. A.; Gibson M. I. Nat. Commun. 2014, 5, 3244. 10.1038/ncomms4244. [DOI] [PubMed] [Google Scholar]
- Mitchell D. E.; Cameron N. R.; Gibson M. I. Chem. Commun. 2015, 51 (65), 12977. 10.1039/C5CC04647E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. E.; Lovett J. R.; Armes S. P.; Gibson M. I. Angew. Chem., Int. Ed. 2016, 55 (8), 2801. 10.1002/anie.201511454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deller R. C.; Pessin J. E.; Vatish M.; Mitchell D. A.; Gibson M. I. Biomater. Sci. 2016, 4, 1079. 10.1039/C6BM00129G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom C. S.; Liu X. Y.; Jia Z. J. Biol. Chem. 2004, 279 (31), 32407. 10.1074/jbc.M401712200. [DOI] [PubMed] [Google Scholar]
- Nguyen D. H.; Colvin M. E.; Yeh Y.; Feeney R. E.; Fink W. H. Biophys. J. 2002, 82 (6), 2892. 10.1016/S0006-3495(02)75630-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capicciotti C. J.; Leclere M.; Perras F. A.; Bryce D. L.; Paulin H.; Harden J.; Liu Y.; Ben R. N. Chem. Sci. 2012, 3 (5), 1408. 10.1039/c2sc00885h. [DOI] [Google Scholar]
- Capicciotti C. J.; Kurach J. D. R.; Turner T. R.; Mancini R. S.; Acker J. P.; Ben R. N. Sci. Rep. 2015, 5, 9692. 10.1038/srep09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K.; Molinero V. J. Am. Chem. Soc. 2018, 140, 4803. 10.1021/jacs.7b13630. [DOI] [PubMed] [Google Scholar]
- Mitchell D. E.; Clarkson G.; Fox D. J.; Vipond R. A.; Scott P.; Gibson M. I. J. Am. Chem. Soc. 2017, 139 (29), 9835. 10.1021/jacs.7b05822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng H.; Liu X.; Shi G.; Bai G.; Ma J.; Chen J.; Wu Z.; Song Y.; Fang H.; Wang J. Angew. Chem., Int. Ed. 2017, 56 (4), 997. 10.1002/anie.201609230. [DOI] [PubMed] [Google Scholar]
- Liu K.; Wang C.; Ma J.; Shi G.; Yao X.; Fang H.; Song Y.; Wang J. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (51), 14739. 10.1073/pnas.1614379114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclère M.; Kwok B. K.; Wu L. K.; Allan D. S.; Ben R. N. Bioconjugate Chem. 2011, 22 (9), 1804. 10.1021/bc2001837. [DOI] [PubMed] [Google Scholar]
- Mai Y.; Eisenberg A. Chem. Soc. Rev. 2012, 41 (18), 5969. 10.1039/c2cs35115c. [DOI] [PubMed] [Google Scholar]
- Ilker M. F.; Nüsslein K.; Tew G. N.; Coughlin E. B. J. Am. Chem. Soc. 2004, 126 (48), 15870. 10.1021/ja045664d. [DOI] [PubMed] [Google Scholar]
- Arnt L.; Tew G. N. J. Am. Chem. Soc. 2002, 124 (26), 7664. 10.1021/ja026607s. [DOI] [PubMed] [Google Scholar]
- Lienkamp K.; Madkour A. E.; Musante A.; Nelson C. F.; Nüsslein K.; Tew G. N. J. Am. Chem. Soc. 2008, 130 (30), 9836. 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak S.; Nelson C. F.; Nusslein K.; Tew G. N. Biomacromolecules 2009, 10 (2), 353. 10.1021/bm801129y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs C.; Lipecki J.; Gibson M. I. Biomacromolecules 2017, 18 (1), 295. 10.1021/acs.biomac.6b01691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capicciotti C. J.; Trant J. F.; Leclère M.; Ben R. N. Bioconjugate Chem. 2011, 22 (4), 605. 10.1021/bc100394k. [DOI] [PubMed] [Google Scholar]
- Congdon T.; Notman R.; Gibson M. I. Biomacromolecules 2013, 14 (5), 1578. 10.1021/bm400217j. [DOI] [PubMed] [Google Scholar]
- Kiessling L. L.; Strong L. E. In Alkene Metathesis in Organic Synthesis; Fuerstner A., Ed.; Springer-Verlag: Berlin/eidelberg, 2013; pp 219–220. [Google Scholar]
- Tallon M. A. In Handbook of Maleic Anhydride Based Materials; Musa O. M., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp 387–388. [Google Scholar]
- Inada T.; Lu S. S. Cryst. Growth Des. 2003, 3 (5), 747. 10.1021/cg0340300. [DOI] [Google Scholar]
- Deswal R.; Gupta R.; Sharma B. J. Proteins Proteomics 2016, 7 (3), 199. [Google Scholar]
- Raymond J. A.; Wilson P.; DeVries A. L. Proc. Natl. Acad. Sci. U. S. A. 1989, 86 (3), 881. 10.1073/pnas.86.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight C. A.; Driggers E.; DeVries A. L. Biophys. J. 1993, 64 (1), 252. 10.1016/S0006-3495(93)81361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.; Paradossi G.; Burchard W. Makromol. Chem., Rapid Commun. 1985, 6 (11), 767. 10.1002/marc.1985.030061111. [DOI] [Google Scholar]
- Buhler E.; Boué F. Macromolecules 2004, 37 (4), 1600. 10.1021/ma0215520. [DOI] [Google Scholar]
- Howard E. I.; Blakeley M. P.; Haertlein M.; Haertlein I. P.; Mitschler A.; Fisher S. J.; Siah A. C.; Salvay A. G.; Popov A.; Dieckmann C. M.; Petrova T.; Podjarny A. J. Mol. Recognit. 2011, 24 (4), 724. 10.1002/jmr.1130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.