

Abstract
Background
Circadian genes have been considered as a possible biological mechanism for the observed relationship between circadian rhythm disruptions and increased risk of hormone-related cancers. In the current study, we investigated the relationship between circadian gene variants and prostate cancer risk and whether reducing bioavailable testosterone modifies the circadian genes-prostate cancer relationship.
Methods
We conducted a nested case-control study among Caucasian men in the Prostate Cancer Prevention Trial (PCPT), a randomized placebo-controlled clinical trial to assess if finasteride (an androgen bioactivation inhibitor) could prevent prostate cancer. We evaluated the associations between 240 circadian gene variations and prostate cancer risk among 1,092 biopsy-confirmed prostate cancer cases and 1,089 biopsy-negative controls in the study (642 cases and 667 controls from the placebo group; 450 cases and 422 controls from the finasteride group), stratified by treatment group.
Results
Among men in the finasteride group, there were suggestive associations between NPAS2 variants and total prostate cancer risk, with one SNP remaining statistically significant after Bonferroni correction (rs746924, odds ratio [OR] = 1.5, p = 9.6x10−5). However, we found little evidence of increased prostate cancer risk (overall or by low/high grade) associated with circadian gene variations in men of the placebo group, suggesting potential modification of genetic effects by treatment.
Conclusions
We did not find strong evidence that circadian gene variants influenced prostate cancer risk in men who were not on finasteride treatment. There were suggestive associations between NPAS2 variants and prostate cancer risk among men using finasteride, which warrants further investigations.
Keywords: circadian genes, genetic polymorphisms, prostate cancer, finasteride, Prostate Cancer Prevention Trial
INTRODUCTION
Disruption in circadian rhythms has been classified as a probable carcinogen to humans for several hormone-related cancers [1]. For men, observational studies on sleep duration [2], light at night [3], rotating shift workers [4,5], and male airline pilots [6–8] have all linked circadian rhythm disruptions to increased prostate cancer risk. Although no underlying molecular mechanism has been identified, hypotheses involving genes that regulate the endogenous circadian rhythm have been proposed [9]. The endogenous circadian rhythm has been shown to be controlled by 9 known circadian genes, including aryl hydrocarbon receptor nuclear translocator-like (ARNTL), clock homolog (mouse) (CLOCK), cryptochrome 1 (CRY1), CRY2, casein kinase 1, epsilon (CSNK1E), neuronal PAS domain protein 2 (NPAS2), period 1 (PER1), PER2, and PER3 [10].
Previously, only a few epidemiologic studies examined associations between circadian gene variants and prostate cancer risk, and some suggestive associations were reported [11–13]. In genome-wide association studies (GWAS), no genetic variants of circadian genes were significantly associated with prostate cancer risk. One possible reason for the inconclusive findings may be that the association between circadian genes and prostate cancer risk needs to be evaluated under an appropriate environmental exposure. One such exposure is androgens. It is well known that dihydrotestosterone (DHT), the most potent biologically active androgen in the prostate [14] that activates the androgen receptor (AR) and subsequent signaling pathways, contributes to prostate carcinogenesis [15]. Animal models show that androgens also have a profound effect on circadian rhythms. For instance, gonadectomy in male mice (a method for androgen ablation) lengthens the period of circadian rhythms independent of light, and these effects were reversible with androgen replacement [16]. It is unknown whether reduction in androgen exposure also influences the relationship between circadian genes, which control the endogenous circadian rhythm, and prostate cancer risk.
The Prostate Cancer Prevention Trial (PCPT) is a previously conducted, placebo-controlled randomized clinical trial that found daily treatment with 5 mg finasteride reduced the risk of prostate cancer by 25% in men without prostate cancer [17,18]. Finasteride is an inhibitor of 5-alpha reductase, the primary enzyme that converts testosterone to DHT. Thus, the trial provided a unique opportunity to investigate whether the associations between circadian genes and prostate cancer risk differ by exposure to DHT. One other unique aspect of the PCPT study is the existing biopsy, which confirms that all the controls in the study have negative biopsy results for prostate cancer.
Herein, we report the associations between variants in circadian genes and prostate cancer stratified by the use of finasteride in the PCPT study.
METHODS
Study Population and Subject Selection
The PCPT trial is registered in ClinicalTrials.gov (NCT00288106) and details of the trial have been described previously [17,18]. Briefly, 18,882 prostate cancer-free men 55 years of age or older were randomly assigned to treatment with finasteride (5 mg per day) or placebo over a 7-year period. The current study includes participants who were selected for a nested case-control study designed to examine multiple hypotheses about prostate cancer risk factors in the PCPT [19,20]. Cancer cases and controls in this report included participants from both the placebo- and finasteride-treated study arms. Cases were men with biopsy-determined prostate cancer identified either by a for-cause or end-of-study biopsy, and who had sufficient DNA from white blood cells available for genotyping. Controls were men who completed the end-of study biopsy procedure with no evidence of prostate cancer, had sufficient DNA samples from white blood cells available for genotyping, and were frequency matched to cases on distributions of treatment arm, age (in 5-year age groups) and positive family history for first degree relative with prostate cancer. Controls were oversampled on race to include all eligible non-whites. Of the participants included in the original nested case-control study, 1,169 cases and 1,365 controls satisfied all selection criteria. Participants who were excluded due to lack of adequate DNA were comparable to participants with adequate DNA in terms of demographic characteristics such as age, BMI, race, and family history (data not shown).
DNA from cases and controls were genotyped for single nucleotide polymorphism (SNP) in 9 known circadian core genes (gene and SNP selection is discussed below) at the genotyping facility at The University of Texas, San Antonio Genomics Research Core. We further excluded non-whites from the analysis because there was insufficient power to reliably detect associations in non-whites (43 cases and 118 controls from the placebo group and 34 cases and 158 controls from the finasteride group). In total, we included 1,092 prostate cancer cases and 1,089 controls in the study, including 642 cases and 667 controls in the placebo group, and 450 cases and 422 controls in the finasteride group.
Tumors were graded centrally and categorized as low (Gleason < 7) and high (Gleason ≥ 7) grade; this definition is consistent with that in the original trial report [17]. Of the prostate cancer included in the placebo group, there were 499 low-grade and 122 high-grade cancers. Among cases in the finasteride group, there were 283 low-grade and 151 high-grade cancers.
Circadian genes and SNP selection
We selected 9 genes that are known to control the endogenous circadian rhythm, including ARNTL, CLOCK, CSNK1E, CRY1, CRY2, NPAS2, PER1, PER2, and PER3 [10]. We included both putatively functional and tag SNPs. Two types of putatively functional SNPs were included: non-synonymous polymorphisms and polymorphisms in non-coding regions that may have functional consequences due to their effects on mRNA processing or splicing. Non-synonymous SNPs were identified by the programs SIFT [21], PolyPhen [22] and ESEfinder [23,24] as well as a search of the NCBI SNP database (https://www.ncbi.nlm.nih.gov/snp/; accessed January 9, 2009). Selection of Tag SNP was done using the software application TagZilla, developed at the National Cancer Institute (v1.1; http://tagzilla.nci.nih.gov/). Tagzilla employs the pairwise binning algorithm of Carlson et al. [25] For each gene, SNPs within the region 10 kb upstream (5′) of the ATG-translation initiation codon and 10 kb downstream (3′) of the end of the last exon (gene boundaries) were binned using a binning threshold of r2=0.80. SNP locations are based on Genome Reference Consortium Human genome build 37 (GRCh37). We included SNPs with predicted minor allele frequencies (MAF) of 5% or greater (n=282). After genotyping, we excluded 20 SNPs from subsequent statistical analysis because their data were missing in more than 25% of the study subjects, 9 SNPs because the MAF of the SNPs was less than 5% in the participants of the study, and 13 SNPs due to deviation from Hardy–Weinberg equilibrium. Thus, a total of 240 SNPs was included for analysis in the study.
Statistical Analysis
To evaluate the relationship between circadian gene variations and prostate cancer risk (total, low-grade and high-grade cancers), and potential modification of the relationship by treatment, we used covariate-adjusted logistic regression models stratified by treatment group. Covariates included in the statistical models included matching factors of age (continuous) and family history, as well as potential risk factor for prostate cancer including diabetes status (dichotomous) [26,27], and BMI (continuous) [28]. Analyses were conducted using SAS software version 9.4 (SAS Institute, Cary, NC) and the R SNPAssoc package (https://cran.r-project.org/web/packages/SNPassoc/index.html). We consider a Bonferroni-corrected p-value of 0.05/240 = 0.0002 as significant; the Bonferroni-correction does not account for LD between SNPs.
RESULTS
Table 1 shows selected characteristics of the PCPT participants who were included in the current study. In the placebo group, cases and controls were similar on the original matching factors of age at baseline and family history of prostate cancer. Cases and controls were also similar on BMI, alcohol consumption, and smoking status. Controls were more likely to have diabetes than cases (6.6% vs. 3.3%). Characteristics of individuals treated with finasteride were in general similar to those of the placebo group. Among cases included in the study, there were more high-grade cancers in the finasteride group than in the placebo group, consistent with results of the PCPT [17], although distribution of stage of cancer between the two treatment groups were similar.
Table 1.
Selected characteristics of study participants in the Prostate Cancer Prevention Trial (PCPT)
| Characteristics | Placebo Group | Finasteride Group | ||
|---|---|---|---|---|
|
|
|
|||
| Controls | Cases | Controls | Cases | |
| Number of Participants | 667 | 642 | 422 | 450 |
| Age at Baseline, Mean (SD) | 63.8 (5.7) | 63.5 (5.6) | 64.3 (5.8) | 63.6 (5.6) |
| Family History of Prostate Cancer, N (%) | 142 (21.3) | 135 (21.0) | 106 (25.1) | 101 (22.4) |
| BMI, Mean (SD) | 27.6 (4.0) | 27.1 (4.0) | 27.6 (3.9) | 27.6 (3.8) |
| Diabetes, N (%) | 44 (6.6) | 21 (3.3) | 17 (4.0) | 21 (4.7) |
| Alcohol Categories, N (%) | ||||
| Non-drinker | 150 (22.5) | 132 (20.6) | 99 (23.5) | 90 (20.0) |
| >0 to <30 g/day Alcohol | 458 (68.7) | 455 (70.9) | 281 (66.6) | 308 (68.4) |
| >=30 g/day Alcohol | 59 (8.8) | 55 (8.6) | 42 (10.0) | 52 (11.6) |
| Smoking Status, N (%) | ||||
| Never Smoker | 230 (34.5) | 229 (35.7) | 149 (35.3) | 159 (35.3) |
| Current Smoker | 45 (6.7) | 34 (5.3) | 25 (5.9) | 26 (5.8) |
| Former Smoker | 392 (58.8) | 379 (59.0) | 248 (58.8) | 265 (58.9) |
| Incident Cases (Diagnosed <2 yrs into Trial), N (%) | 19 (13.0) | 14 (3.1) | ||
| Gleason Grade | ||||
| Low Grade (Gleason Grade <7) | 499 (80.3) | 283 (65.2) | ||
| High Grade (Gleason Grade ≥7) | 122 (19.6) | 151 (34.8) | ||
| Tumor TNM Stage | ||||
| T-stage | ||||
| T1 a/b/c | 506 (80.8) | 341 (78.0) | ||
| T2 a/b/c | 113 (18.1) | 90 (20.6) | ||
| T ¾ | 7 (1.1) | 6 (1.4) | ||
| N-stage | ||||
| N X/0 | 607 (99.3) | 428 (99.5) | ||
| N ½ | 4 (0.7) | 2 (0.5) | ||
| M-stage | ||||
| M X/0 | 609 (99.7) | 429 (99.8) | ||
| M1 | 2 (0.3) | 1 (0.2) | ||
Figure 1 shows the significance of association between SNPs in circadian genes and prostate cancer risk by treatment arm. For men who were treated with finasteride, a cluster of SNPs in NPAS2 had suggestive associations with prostate cancer risk, with SNP (rs746924) remaining statistically significant after Bonferroni correction (OR per minor allele, 1.5; 95% CI, 1.2–1.8; p = 9.6*10−5). For men in the placebo arm, there were no associations between circadian genes variants and prostate cancer risk with p-values below 0.001; results were similar when analyses were stratified by low and high Gleason grade (data not shown). P values for interaction between treatment and individual SPs were performed; of the 240 SNPs tested, only 7 had significant interactive p values for total, high-grade, and low-grade prostate cancer. It is noteworthy that for SNP rs746924, all three interactive p values were statistically significant, although only the one for total prostate cancer achieved a p value less than 0.001.
Figure 1.
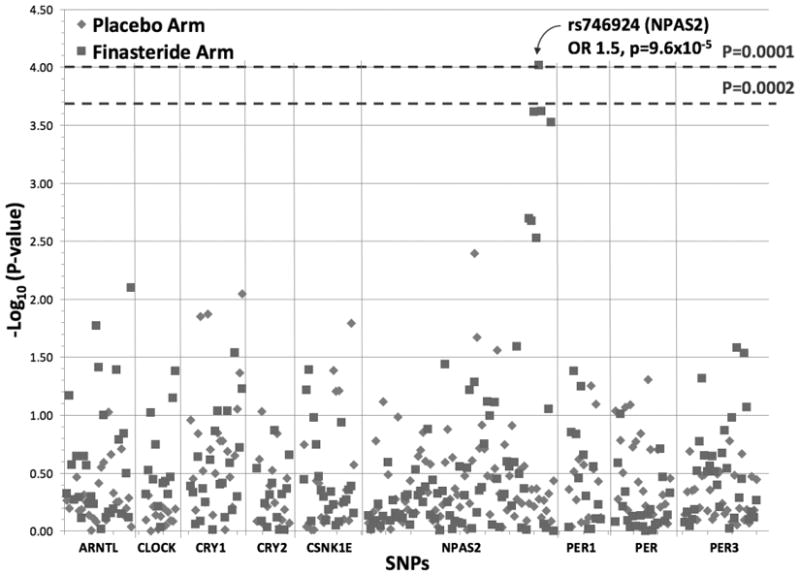
DISCUSSION
In this study, of the 9 core circadian genes and 240 SNPs, we observed suggestive associations between NPAS2 gene variations and prostate cancer risk among men taking finasteride in a randomized clinical trial; however, only one SNP remained statistically significant after Bonferroni correction. In addition, no association was observed among men on the placebo group of the trial. Of these SNPs, 7 had significant interaction with treatment for total, high, and low-grade tumors, including the SNP on the NPAS2. Overall, our results do not provide strong support that circadian gene polymorphisms serve as an underlying mechanism for the link between circadian rhythm disruptions and increased risk of prostate cancer.
Only a few prior epidemiologic studies have assessed the associations between variations in the circadian genes and prostate cancer. A small case-control study in China (187 cases and 242 controls) examined 5 SNPs and reported that men with the CRY2 rs1401417 variant had an increased risk of prostate cancer (p = 0.03) [13]. A case-control study among Caucasian men (1,308 cases and 1,266 controls) reported that of 41 SNPs in the nine core circadian genes, at least one SNP in each of the nine genes was associated with prostate cancer risk (lowest p value = 0.02) [11]. In a study of fatal prostate cancer using data from three cohorts with a total of 169 Caucasian cases, eight SNPs (out of 96 examined) across the core circadian genes were nominally associated with fatal prostate cancer in at least one cohort (lowest p value = 0.003) [12]. However, given the p values observed, it is likely that most of the positive findings will become null after accounting for multiple comparison, which was not performed in any of the studies discussed above.
Overall, our study does not provide strong support for an association between circadian genes and prostate cancer risk, which is generally consistent with previous studies. Nevertheless, we observed a positive association between one NPAS2 gene variation (rs746924) and prostate cancer risk, but only among men taking finasteride (thus, with reduced DHT in the blood and the prostate gland). rs746924 is a missense variant but its function and subsequent effect on NPAS2 is unknown. To date, neither rs746924 nor any SNPs in strong LD (r2 ≥ 0.8; as determined by SNP Annotation and Proxy Search version 2.2 accessed on October 4, 2017 [29]) has been associated with prostate cancer. Although the molecular mechanisms underlying the observed association is unclear, experimental studies have suggested that NPAS2 may interact with DHT to influence the AR-dependent signaling pathway [30], a pathway that is thought to contribute to prostate carcinogenesis [15]. Alternatively, it is possible that our findings are due to chance, and replication of our results using a larger sample size is needed. It should also be noted that some of the tag SNPs may also be associated with genes that reside in close proximity to the circadian genes. The genomic boundaries used to define the gene are to enable the capture of genetic variation of enhancer elements that may lie outside of coding sequences. This approach in selecting tag SNPs is used in many genetic association studies. In the current study, SNPs identified in NPAS2 also lie in the region of two other genes (TBC1D8 and RPL31) based on proximity to those genes. It is not possible to determine whether the effects of the NPAS2 SNPs, if any, are related to NPAS2, other nearby genes, or both. Thus, our results should be interpreted with caution.
There are several strengths of this study. We have a more extensive coverage of SNPs (240 SNPs) than any prior studies, allowing for a more comprehensive evaluation of circadian genes and prostate cancer risk. Case-control status was determined using biopsy, which reduced the likelihood of outcome misclassification. We are also the only epidemiologic study that examined the potential interaction between circadian genes and DHT. A major limitation of this study is the lack of replication of our positive findings in a larger population, preferably with concurrent use of finasteride. Also, this study consisted of only U.S. Caucasian men, which limited the generalizability of our results to men in other racial/ethnic groups.
In conclusion, our study did not find strong evidence that circadian gene variants influence prostate cancer risk, except for one variant in the NPAS2 gene. This finding should be replicated in future large studies.
Supplementary Material
Acknowledgments
Grant support: This study was supported by a grant from the Department of Defense Prostate Cancer Research Program (Grant number PC0731134) awarded to AWH. Research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under Award Numbers U10CA37429 (Charles D. Blanke) and UM1CA182883 (CMT).
The authors would like to express their gratitude to the participants of the PCPT. This study was supported by a grant from the Department of Defense Prostate Cancer Research Program (Grant number PC0731134) awarded to AWH. Research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under Award Numbers U10CA37429 (Charles D. Blanke) and UM1CA182883 (CMT). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- AR
androgen receptor
- BMI
body mass index
- CI
confidence interval
- DHT
dihydrotestosterone
- MAF
minor allele frequencies
- OR
odds ratio
- PCPT
Prostate Cancer Prevention Trial
- SNP
single nucleotide polymorphism
References
- 1.Straif K, Baan R, Grosse Y, et al. Carcinogenicity of shift-work, painting, and fire-fighting. The Lancet Oncology. 2007;8(12):1065–1066. doi: 10.1016/S1470-2045(07)70373-X. [DOI] [PubMed] [Google Scholar]
- 2.Kakizaki M, Inoue K, Kuriyama S, et al. Sleep duration and the risk of prostate cancer: the Ohsaki Cohort Study. Br J Cancer. 2008;99(1):176–178. doi: 10.1038/sj.bjc.6604425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kloog I, Haim A, Stevens RG, Portnov BA. Global Co-Distribution of Light at Night (LAN) and Cancers of Prostate, Colon, and Lung in Men. Chronobiol Int. 2009;26(1):108– 125. doi: 10.1080/07420520802694020. [DOI] [PubMed] [Google Scholar]
- 4.Kubo T, Ozasa K, Mikami K, et al. Prospective cohort study of the risk of prostate cancer among rotating-shift workers: findings from the Japan Collaborative Cohort Study. Am J Epidemiol. 2006;164(6):549–555. doi: 10.1093/aje/kwj232. [DOI] [PubMed] [Google Scholar]
- 5.Conlon M, Lightfoot N, Kreiger N. Rotating shift work and risk of prostate cancer. Epidemiology. 2007;18(1):182–183. doi: 10.1097/01.ede.0000249519.33978.31. [DOI] [PubMed] [Google Scholar]
- 6.Band PR, Le ND, Fang R, et al. Cohort study of Air Canada pilots: mortality, cancer incidence, and leukemia risk. Am J Epidemiol. 1996;143(2):137–143. doi: 10.1093/oxfordjournals.aje.a008722. [DOI] [PubMed] [Google Scholar]
- 7.Irvine D, Davies DM. British Airways flightdeck mortality study, 1950–1992. Aviat Space Environ Med. 1999;70(6):548–555. [PubMed] [Google Scholar]
- 8.Pukkala E, Aspholm R, Auvinen A, et al. Cancer incidence among 10,211 airline pilots: a Nordic study. Aviat Space Environ Med. 2003;74(7):699–706. [PubMed] [Google Scholar]
- 9.Zhu Y, Zheng T, Stevens RG, Zhang Y, Boyle P. Does “clock” matter in prostate cancer? Cancer Epidemiol Biomarkers Prev. 2006;15(1):3–5. doi: 10.1158/1055-9965.EPI-05-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu L, Lee CC. The circadian clock: pacemaker and tumour suppressor. Nat Rev Cancer. 2003;3(5):350–361. doi: 10.1038/nrc1072. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Stevens RG, Hoffman AE, et al. Testing the circadian gene hypothesis in prostate cancer: a population-based case-control study. Cancer Res. 2009;69(24):9315–9322. doi: 10.1158/0008-5472.CAN-09-0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Markt SC, Valdimarsdottir UA, Shui IM, et al. Circadian clock genes and risk of fatal prostate cancer. Cancer Causes Control. 2015;26(1):25–33. doi: 10.1007/s10552-014-0478-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu LW, Zhu Y, Yu K, et al. Variants in circadian genes and prostate cancer risk: a population-based study in China. Prostate Cancer Prostatic Dis. 2008;11(4):342–348. doi: 10.1038/sj.pcan.4501024. [DOI] [PubMed] [Google Scholar]
- 14.Walsh PC, Hutchins GM, Ewing LL. Tissue content of dihydrotestosterone in human prostatic hyperplasis is not supranormal. J Clin Invest. 1983;72(5):1772–1777. doi: 10.1172/JCI111137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jenster G. The role of the androgen receptor in the development and progression of prostate cancer. Semin Oncol. 1999;26(4):407–421. [PubMed] [Google Scholar]
- 16.Butler MP, Karatsoreos IN, LeSauter J, Silver R. Dose-dependent effects of androgens on the circadian timing system and its response to light. Endocrinology. 2012;153(5):2344–2352. doi: 10.1210/en.2011-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349(3):215–224. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 18.Feigl P, Blumenstein B, Thompson I, et al. Design of the Prostate Cancer Prevention Trial (PCPT) Control Clin Trials. 1995;16(3):150–163. doi: 10.1016/0197-2456(94)00xxx-m. [DOI] [PubMed] [Google Scholar]
- 19.Goodman PJ, Tangen CM, Kristal AR, et al. Transition of a clinical trial into translational research: the prostate cancer prevention trial experience. Cancer Prev Res (Phila) 2010;3(12):1523–1533. doi: 10.1158/1940-6207.CAPR-09-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chau CH, Price DK, Till C, et al. Finasteride concentrations and prostate cancer risk: results from the Prostate Cancer Prevention Trial. PLoS One. 2015;10(5):e0126672. doi: 10.1371/journal.pone.0126672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu HX, Chew SL, Cartegni L, Zhang MQ, Krainer AR. Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Mol Cell Biol. 2000;20(3):1063–1071. doi: 10.1128/mcb.20.3.1063-1071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu HX, Zhang M, Krainer AR. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998;12(13):1998–2012. doi: 10.1101/gad.12.13.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74(1):106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leitzmann MF, Ahn J, Albanes D, et al. Diabetes mellitus and prostate cancer risk in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Cancer Causes Control. 2008;19(10):1267–1276. doi: 10.1007/s10552-008-9198-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsilidis KK, Allen NE, Appleby PN, et al. Diabetes mellitus and risk of prostate cancer in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer. 2015;136(2):372–381. doi: 10.1002/ijc.28989. [DOI] [PubMed] [Google Scholar]
- 28.Barrington WE, Schenk JM, Etzioni R, et al. Difference in Association of Obesity With Prostate Cancer Risk Between US African American and Non-Hispanic White Men in the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA Oncol. 2015;1(3):342–349. doi: 10.1001/jamaoncol.2015.0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O’Donnell CJ, de Bakker PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24(24):2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mukhopadhyay NK, Ferdinand AS, Mukhopadhyay L, et al. Unraveling androgen receptor interactomes by an array-based method: discovery of proto-oncoprotein c-Rel as a negative regulator of androgen receptor. Exp Cell Res. 2006;312(19):3782–3795. doi: 10.1016/j.yexcr.2006.07.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.