

Abstract
T cell factor 1 (TCF-1) is expressed in both developing and mature T cells, and has been shown to restrain mature T cell-mediated Th17 responses by inhibiting IL-17 expression. However, it is not clear when is TCF-1 required in vivo to restrain the magnitude of peripheral Th17 responses and what are the molecular mechanisms responsible for TCF-1-regulated IL-17 gene expression. Here we showed that conditional deletion of TCF-1 at early but not later at CD4+CD8+ double positive (DP) stage in mice, enhanced Th17 differentiation and aggravated experimental autoimmune encephalomyelitis (EAE), which correlates with abnormally high IL-17 expression. Expression of TCF-1 in TCF-1 deficient thymocytes, but not TCF-1 deficient Th17 cells, inhibited IL-17 expression. TCF-1 binds to IL-17 promoter regions, and deletion of two TCF-1-binding sites relief TCF-1-mediated inhibition of IL-17 promoter activity. Lastly, WT TCF-1 but not a TCF-1 mutant that has no intrinsic histone deacetylase activity (HDAC) was able to inhibit IL-17 expression in TCF-1 deficient mouse thymocytes. Thus, our study first demonstrates requirement of TCF-1 in vivo at stages earlier than DP cells to restrain peripheral Th17 immunity by directly binding and inhibiting IL-17 promoter in its intrinsic HDAC-dependent manner.
Introduction
T cell development in thymus is a process to arm T cells with the capacity to mediate appropriate immune responses in peripheral tissues. Lymphoid progenitors which developed from hematopoietic stem cells in the bone marrow migrate into thymus to complete sequential maturation stages, including CD4-CD8- double negative (DN), CD4+CD8+ double positive (DP), and CD4+ or CD8+ single positive (SP) stages (1, 2). Mature single positive T cells then migrate to the peripheral lymphoid organs to participate adaptive immune responses against pathogens.
When the single CD4+ positive T cells just migrate out of thymus they are naïve and are not competent to mediate immune responses. To become effector T cells, they must undergo an activation and differentiation process. This process is initiated upon encountering antigens and eventually differentiates naïve T cells into T helpers that include Th1, Th2, Th17 and regulatory T (Treg) cells. Th17 cells secrete IL-17 and participate in protective immunity against pathogens (3, 4). Whereas inappropriately exaggerated Th17 responses contribute to pathological immune responses involved in the autoimmunity such as psoriasis and multiple sclerosis (5-9).
Besides shaping T cell repertoire that reacts to foreign but not self-antigens, thymocyte developmental process also controls the magnitude of T cell responses in the periphery. For example, T cell factor 1 (TCF-1), a transcription factor enriched in hematopoietic cell compartments, regulates T cell development in thymus (10-12). Our previous studies have shown that germline deletion of TCF-1 resulted in increased IL-17 expression both in thymus and peripheral T cells and hence led to enhanced Th17 differentiation and more severe EAE (13, 14), indicating the negative role of TCF-1 in the regulation of Th17 immunity. We have some in vitro evidence that TCF-1 loses the ability to regulate IL-17 gene expression in mature T cells. However, because it was a germline deletion, it is not clear when TCF-1 is required in vivo to limit IL-17 gene expression and thus control the scale of Th17 responses in the periphery. By using conditional deletion of TCF-1 at different developmental stages, we demonstrated that CD4-Cre-mediated deletion of TCF-1 at CD4+CD8+ DP stage did not significantly affect thymic T cell development, peripheral Th17 differentiation and EAE. Whereas, Vav1-Cre-mediated deletion of TCF-1 at earlier hematopoietic stages disrupts thymic T cell development and potentiates Th17 differentiation and development of EAE. Moreover, expression of TCF-1 in TCF-1-/- thymocytes but not TCF-1-/- Th17 cells was able to down-regulate IL-17 expression. We also found that TCF-1-mediated inhibition of IL-17 expression depends on its intrinsic histone deacetylase activity (15). We mapped the TCF-1-binding regions on IL-17 promoter, and deletion of the DNA fragments containing the TCF-1 binding sites prevented TCF-1 to inhibit IL-17 promoter. Therefore, we first demonstrated the state-specific requirement of TCF-1 in vivo during early development to control the scale of the peripheral Th17 immune responses via inhibiting IL-17 expression through the intrinsic HDAC activity of TCF-1.
Materials and Methods
Mice
TCF-1fl/fl mice were described previously (16) and obtained from Dr. Hai-Hui Xue (University of Iowa, Iowa City, IA). Rag1-/-, transgenic CD4-Cre and Vav1-Cre mice were purchased from The Jackson Laboratory. TCF-1f/fl/CD4-Cre and TCF-1f/f/Vav1-Cre were generated by crossing TCF-1f/f to CD4-Cre and Vav1-Cre, respectively. For all experiments, mice were 6-10 week-old. All mice were bred and maintained in the specific pathogen-free conditions and experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee at the Beckman Research Institute of City of Hope (IACUC#07023).
T cell isolation and In vitro differentiation
Mouse naïve CD4+ T cells were isolated from spleens of 6- to 10-week-old mice by negative selection using a CD4+ T cell isolation kit (Miltenyi Biotec, Bergisch-Gladbach, Germany). T cells were cultured and differentiated in Iscove's DMEM (Corning, VA, USA) containing 10% FBS, 50 mM β-mercaptoethanol, 100 U/ml penicillin-streptomycin at 37°C with 5% CO2. In brief, 4×105/ well naïve CD4+ T cells were first activated with 0.25 μg/ml anti-CD3 (145-2C11; eBioscience, CA, USA) and 1μg/ml anti-CD28 (37.51; eBioscience, CA, USA) in goat-anti-hamster (G-α-H) IgG (0.2 mg/ml, MP Biomedicals, Santa Ana, CA) pre-coated 24-well plates. Cells were then differentiated in the presence of polarizing cytokine and antibody cocktails for 72 h. For Th1: 10 ng/ml recombinant murine (rm) IL-2 (Biolegend), 20 ng/ml rmIL-12 (Biolegend) and 20μg/ml anti-IL4 (11B11; Biolegend). For Th17: 2 ng/ml rmTGF-β1 (eBioscience) and 25 ng/ml rmIL-6 (eBioscience). For Treg: 5 ng/ml rmTGF-β, 10 μg/ml anti-IFN-γ (XMG1.2; Biolegend) and 10 μg/ml anti-IL-4 (11B11; Biolegend).
Flow cytometry and cell sorting
Cultured T cells were stimulated with 50 ng/ml PMA (Sigma-Aldrich) and 1 μg/ml ionomycin (Sigma-Aldrich) in the presence of brefeldin A for 4 h before staining. Both cultured T cells and freshly prepared thymocytes were first stained with fluorescence-conjugated antibodies (Abs) to cell surface markers followed by fixed/permeabilized either with the BD Cytofix/Cytoperm kit (BD Biosciences) or Transcription Factor Staining Buffer Set (BD Pharmingen). Cells were then intracellularly strained with fluorescence-conjugated Abs to cytokines or transcription factors. Abs include FITC-CD4 (GK1.5, Biolegend), PE-CD8 (53-6.7, Biolegend), PE-Cy7-CD8 (53-6.7, eBioscience), APC-CD8 (53-6.7, BD Biosciences), PE-Thy1.2 (30-H12, BD Biosciences), PE-TCRβ (H57-597, BD Biosciences), APC-IL-17A (eBio17B7, eBioscience), APC-IFN-γ (XMG1.2, eBioscience), APC-FOXP3 (FJK-16s, eBioscience) and PE-RORγt (Q31-378, BD Biosciences). Flow cytometry were performed by using a FACSCantoII (BD Biosciences) and analyzed with FlowJo software (Tree Star). Double-negative (DN) thymocytes (Thy1.2+CD4-CD8-) were electronically sorted by using FACSAriaII (BD Biosciences).
Quantitative real-time PCR (qRT-PCR)
Total RNA was prepared using the RNeasy Isolation Kit according to the manufacturer's instructions (Qiagen). cDNA was synthesized using the Tetro cDNA Synthesis Kit (BIO-65043, Bioline). qRT-PCR was performed using SsoFast EvaGreen Supermic (Bio-Rad) in a CFX96 Real-Time PCR Detection System (Bio-Rad). Primers are as follows: IL17A-forward: 5′- tttaactcccttggcgcaaaa-3′, IL17A-reverse: 5′-ctttccctccgcattgacac-3′; IL17F-forward: 5′- tgctactgttgatgttgggac-3′, IL17F-reverse: 5′- aatgccctggttttggttgaa-3′; CCL20-forward: 5′-gcctctcgtacatacagacgc-3′, CCL20-reverse: 5′-ccagttctgctttggatcagc-3′; CCR6-forward: 5′- cctgggcaacattatggtggt-3′, CCR6-reverse: 5′-cagaacggtagggtgaggaca-3′.
Retroviral packaging and transduction
MSCV-IRES-EGFP based retroviral expression vectors encoding TCF-1 or empty vectors were transfected into Plat-E packaging cells by Lipofectamine 2000 (Invitrogen Life Technologies, CA, USA) mediated transfection. After 48 h, viral supernatants were collected and stored at −80°C until use. For transduction, naïve CD4+ T cells were first activated with 0.25 μg/ml anti-CD3 (145-2C11; eBioscience, CA, USA), 1μg/ml anti-CD28 (37.51; eBioscience, CA, USA) in G-α-H IgG pre-coated 24-well plate for 24 h followed by spin-infection with viral supernatants (2500 rpm, 30°C for 2 h) in the presence of 8 μg/ml polybrene (Sigma-Aldrich, MO, USA). After spin-infection, indicated cytokines described above were added to the culture media to induce Th17 differentiation.
Coculture and transduction of thymocytes on OP9-DL1 stromal cells
Thymocytes were allowed to differentiate on OP9-DL1 cells (gift from Dr. Timothy P. Bender, University of Virginia Health System, Charlottesville, VA). Briefly, 5×105 / well electronically sorted Thy1.2+CD4-CD8- thymocytes were cultured overnight on an 80% confluent OP9-DL1 monolayer in the flat-bottom 24-well plates in αMEM (STEMCELL Technologies) supplemented with 20% FBS, 100 U/ml penicillin-streptomycin, 2 mM L-glutamine (Invitrogen Life Technologies), and 5 ng/ml rmIL-7 (PeproTech). Co-cultures were then spin-infected (2500 rpm, 30°C for 2 h) with retroviral supernatants in the presence of 5 μg/ml polybrene. 72 h posttransduction, cocultures were harvested for flow cytometry.
Induction and assessment of EAE
Active EAE was induced according to the manufacturer's instructions (Hooke Laboratories, Lawrence, MA). Briefly, mice were immunized with 200 μ1 myelin oligodendrocyte glycoprotein 35–55 (MOG35–55) peptide emulsion subcutaneouly. On days 0 and 1 after immunization, mice were injected intraperitoneally with 200 ng Bordetella pertussis toxin (Hooke Laboratories, Lawrence, MA). For Th17- or Th1-induced passive EAE, donor mice were immunized with MOG35–55 subcutaneouly. 10 days later, cells were isolated from spleen and lymph node and cultured with 20ug/ml MOG35–55 for 3 days under either Th17-polarizing conditions (20ng/ml rmIL23, R&D) or Th1-polarizing conditions (20ng/ml rmIL-12, R&D; 2μg/ml α-IL23p19, eBioscience) (17). Rag1-/- recipient mice were then transferred intraperitoneally with 3.0×107 MOG35–55-specific Th17 or Th1 cells. The severity of EAE was monitored and evaluated on a scale from 0 to 5 according to Hooke Laboratories ' guideline. In brief: 0 = no disease; 1 = paralyzed tail; 2= hind limb weakness; 3= hind limb paralysis; 4 = hind and fore limb paralysis; 5= moribund and death. When a mouse was euthanized because of severe paralysis, a score of 5 was entered for that mouse for the rest of the experiment.
Isolation and analysis of CNS infiltrating cells
Brain and spinal cords were homogenized and single cell suspensions were prepared using 70-μm cell nylon cell strainers (Fisher Scientific). Cells were collected by centrifugation at 400 rcf for 10 min. Cells were then resuspended in 4 ml 30% Percoll (GE Healthcare) and centrifuged onto 5 ml 70% cushion for 20 min at 2000 rpm. Lymphocytes at 30-70% interface were collected and subjected to cell surface staining with fluorescence-conjugated Abs: FITC-CD45 (30-F11, eBioscience), APC-CD3 (145-2C11, BD Pharmingen), APC-Cy7-Ly6C (HK1.4, eBioscience), PE-Cy7-Ly6G (1A8, BioLegend), and PE-CD11b (M1/70, eBioscience). For intracellular cytokine staining, the CNS infiltrating cells were stimulated with 50 ng/ml PMA (Sigma-Aldrich) and 1μg/ml ionomycin (Sigma-Aldrich) in the presence of brefeldin A for 4 h, and then stained with PE-Cy7-CD4 followed by intracellular staining of FITC-IFN-γ (XMG1.2, BioLegend), PE-GM-CSF (MP1-22E9, BioLegend), and APC-IL-17A (eBio17B7, eBioscience).
ChIP on qRT-PCR
ChIP on qRT-PCR was performed with the ChIP-IT High Sensitivity kit (Active Motif, 53040). Briefly, 1.5×107 cells per ChIP were obtained from thymus or differentiated Th17 cells. Cells were fixed and sheared as described in the ChIP-IT High Sensitivity manual. ChIP reactions were then performed on 20 μg of the prepared chromatin using 4 μg anti-TCF-1 antibody (C63D9, Cell Signaling) or antiacetyl-histone H3 (06-599, Millipore). 4 μg of IgG was used as negative control. Input DNA and DNA pull-downed after immunoprecipitation were analyzed by qRT-PCR as described above. Primers which cover ∼2000 base pair of IL17A promoter region were designed as follows: P1-forward: 5′-aaaccctatgcagttggtaca-3′, P1-reverse: 5′-catctctccagctccatgga-3′; P2-forward: 5′-agcccataaagaagccaatgt-3′; P2-reverse: 5′-tcctggctttctacgtgtca-3′; P3-forward: 5′-gggtgaaagaggacattgcc-3′, P3-reverse: 5′-tgccaggtcttttcccattc-3′; P4-forward: 5′-tgacacgtagaaagccagga-3′, P4-reverse: 5′-gattgccaggtcttttccca-3′; P5-forward: 5′-ggcaatcagaggtgtgtgtg-3′, P5-reverse: 5′-ctcagaagttgcagcacctc-3′; P6-forward: 5′-ctatcggtccacctcatgct-3′, P6-reverse: 5′-ggagatgagggatgagaaggg-3′; P7-forward: 5′-gttagtagtctccacccggc-3′, P7-reverse: 5′-tgaggttcggtatcaagcct-3′; P8-forward: 5′-gagtgggtttctttgggcaa-3′, P8-reverse: 5′-agcatgacttcttgggagct-3′; P9-forward: 5′-agctcccaagaagtcatgct-3′, P9-reverse: 5′-tacgtcaagagtgggttggg-3′.
Results
Vav1-Cre but not CD4-Cre-mediated deletion of TCF-1 potentiates Th17 differentiation and EAE
Previous studies of TCF-1 function mostly depend on utilizing a strain of germline knockout mice that cannot determine tissue and time specific function of TCF-1 (10, 18). To determine the critical stages that TCF-1 is required to restrain peripheral Th17 responses, we crossed conditional TCF-1 (TCF-1fl/fl) mice (16) to CD4-Cre and Vav1-Cre mice, respectively. CD4-Cre induces gene deletion at CD4+CD8+ DP stage (19), whereas Vav1-Cre induces deletion at earlier hematopoietic stages including CD4-CD8- DN stage (20). We performed PCR analysis of TCF-1 locus to detect the deletion of TCF-1 gene (Fig. 1A). As expected, there was no deletion at all in the absence of Cre as shown in TCF-1fl/fl mice. However, TCF-1 deletion was detected in CD4+CD8+ thymocytes and peripheral T cells but not in CD4-CD8- thymocytes of the TCF-1fl/fl/CD4-Cre mice. In contrast, TCF-1fl/fl /Vav1-Cre mice deleted TCF-1 in CD4−CD8−, CD4+CD8+ and peripheral T cells, confirming that Vav1-Cre induced TCF-1 deletion at the stage earlier than CD4-CD8- DN cells. To determine the effects of deletion of TCF-1 at different developmental stages on peripheral Th17 responses, EAE was induced (Fig. 1B). Interestingly, no obvious differences of EAE development in terms of disease onset and severity were observed between TCF-1fl/fl and TCF-1fl/fl/CD4-Cre mice, with a comparable mean peak disease score about 3. However, the clinical score of TCF-1fl/fl/Vav1-Cre mice reached over 3.5, indicating more severe EAE than that observed in TCF-1fl/fl/CD4-Cre mice. These results demonstrated that deletion of TCF-1 at earlier stages, but not later DP stage, potentiated Th17 immunity responsible for EAE. Moreover, TCF-1fl/fl/Vav1-Cre mice had significantly reduced number of peripheral T cells compared to TCF-1fl/fl and TCF-1fl/fl/CD4- Cre mice (Fig. 1C), suggesting less T cells but with higher potency in the induction of EAE when TCF-1 was deleted by Vav1-Cre.
Figure 1.
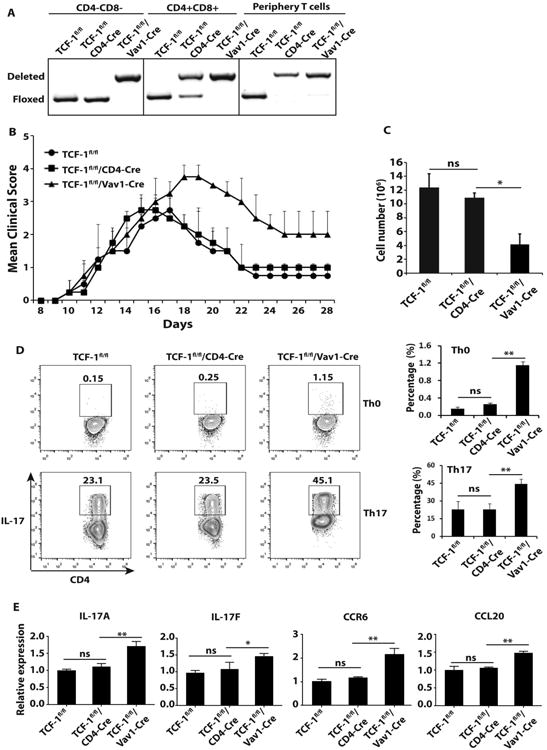
Vav1-Cre but not CD4-Cre induced deletion of TCF-1 potentiates EAE and Th17 differentiation. A, Floxed and deleted TCF-1 loci in indicated population of thymocytes and peripheral T cells from indicated genotypes of mice, as determined by PCR analysis. Data shown is the representative of three independent experiments. B, Mean clinical score of EAE in indicated genotypes of mice different days after MOG35–55 immunization. (TCF-1Fl/Fl n=6, TCF-1Fl/Fl/CD4-Cre n=6, and TCF-1Fl/Fl/Vav1-Cre n=6). C, Total number of CD4+ T cells in the spleens of indicated genotypes of mice (n=3). D, Percentage of IL-17+ cells among indicated genotypes of naïve CD4+ T cells stimulated under Th0 or Th17 priming conditions, as determined by flow cytometric analysis. Right panels are the quantification of three biological replica. E, Expression levels of indicated critical Th17 genes in differentiated Th17 cells of indicated genotypes, as determined by qPCR. ns, no significant difference; * < 0.05 and **<0.01.
Since Th17 cells contribute to EAE development (21), we wondered how stage specific deletion of TCF-1 affects Th17 differentiation. Even under Th0 conditions, we have already observed increased percentage of IL-17+ cells in naïve CD4+ T cells from TCF-1fl/fl/Vav1-Cre mice, while IL-17 expression was hardly detectable in TCF-1fl/fl and TCF-1fl/fl/CD4-Cre T cells. Under Th17 polarization conditions, T cells from TCF-1fl/fl and TCF-1fl/fl/CD4-Cre mice were equivalent in the generation of IL-17+ cells, whereas about 100% more IL-17+ cells were developed from TCF-1fl/fl/Vav1-Cre T cells (Fig. 1D). Increased IL-17+ TCF-1fl/fl/Vav1-Cre T cells correlated with the significant increased expression of Th17 signature genes, including IL-17A, IL-17F, CCR6 and CCL20 (Fig. 1E). In contrast, naïve CD4+ T from TCF-1fl/fl/Vav1-Cre mice had no defects in Th1 and Treg differentiation (Fig.S1), suggesting the selective role of TCF-1 in Th17 differentiation. These results demonstrate that TCF-1 is required at early but not later CD4+CD8+ stage during T cell development to restrain peripheral Th17 immunity by selectively controlling the potential of Th17 differentiation.
Deletion of TCF-1 by Vav1-Cre potentiates Th17 but not Th1-mediated EAE
TCF-1 was reported to modulate the differentiation of Th1 cells (22). Besides Th17 cells, Th1 cells can also contribute to EAE induction, but these two forms of EAE display distinct lesion sites and immune profiles in central nervous system (CNS) (23, 24), indicating different mechanisms are involved in the development of Th17 and Th1-mediated EAE. Although our in vitro experiment did not show significant difference in Th1 differentiation between TCF-1fl/fl/Vav1-Cre mice and TCF-1fl/fl and TCF-1fl/fl/CD4-Cre control (Fig.S1), it is not clear whether deletion of TCF-1 affects Th1 function in term of induction of EAE in vivo. We thus compared the function of differentiated Th1 and Th17 cells by means of induction of passive EAE. Interestingly, Rag1-/- recipient mice (which have a congenital deficiency in mature B and T cells) reconstituted with cells from spleen and lymph node of TCF-1fl/fl, TCF-1fl/fl/CD4-Cre or TCF-1fl/fl/Vav1-Cre mice stimulated under Th1 priming conditions developed equivalent EAE (Fig. 2A, B). However, cells from TCF-1fl/fl/Vav1-Cre mice stimulated under Th17 priming conditions induced more severe EAE in Rag1-/- recipient mice (Fig. 2C), which was also indicated by significantly increased absolute number of central nervous system (CNS)-infiltrating lymphocytes, including T cells, neutrophils and monocytes (Fig. 2D, E, gate strategy for different cells shown in Fig.S2). In addition, intracellular staining of CNS-infiltrating cells showed greatly increased percentage of CD4+IL-17+IFN-γ+ and CD4+IL-17+GM-CSF+ cells, which are believed to be pathogenic for EAE (5-7), in the CNS of Rag1-/- mice adoptively transferred with Th17-priming conditions treated cells from TCF-1fl/fl/Vav1-Cre mice (Fig. 2F, G). Taken together, these findings supported that the exaggerated EAE resulted from deletion of TCF-1 at early T cell developmental stage was due to enhanced Th17 but not Th1 responses.
Figure 2.
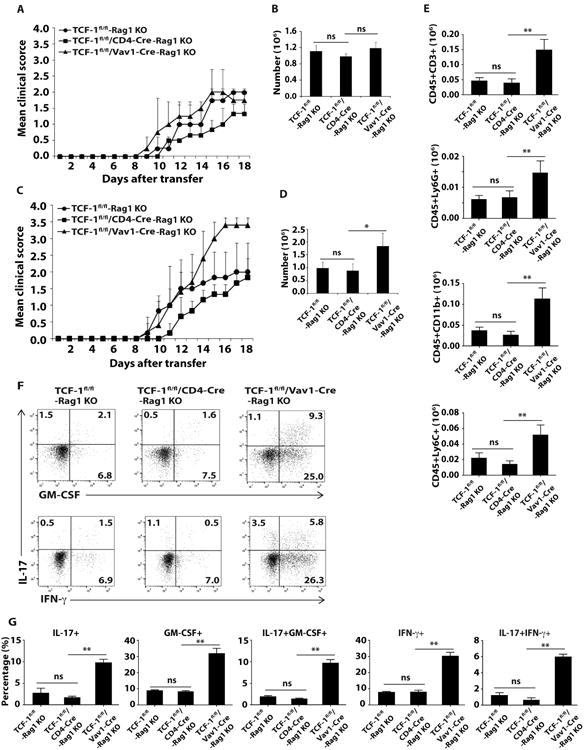
Deletion of TCF-1 by Vav1-Cre potentiates Th17 but not Th1-mediated EAE. A, C, Mean clinical scores of EAE in Rag1-/- mice different days after adoptively transferred with indicated genotypes of MOG33–35-expanded Th1 (A) or Th17 (C) cells. B, D, Number of lymphocytes infiltrated into the CNS of Rag1-/- mice adoptively transferred with indicated Th1 (B) or Th17 (D) cells at the peak of disease as shown in A or C. E, Quantification of cells expressing surface markers characteristic of various types of lymphocytes as determined by flow cytometric analysis at the peak of disease from Rag1-/- mice receiving Th17 cells. F, Flow cytometric analysis of intracellular IL-17, GM-CSF and IFN-γ in CD4+ T cells that infiltrated CNS of Rag1-/- mice receiving Th17 cells at the peak of disease. G, Percentage of cells positive for indicated cytokines among CD4+ T cells infiltrated into CNS of Rag1-/- mice. Data are pooled from three experiments. ns, no significant difference; * < 0.05 and **<0.01.
Vav1-Cre but not CD4-Cre induced deletion of TCF-1 leads to increased IL-17 expression in thymus
Since we observed increased IL-17 expression in mature CD4+ T cells from TCF-1fl/fl/Vav1-Cre mice (Fig. 1D), we next wondered whether these increased IL-17 producing cells originated from thymus. Vav1-Cre but not CD4-Cre-induced deletion of TCF-1 affected thymocyte development indicated by thymic weight (Fig. 3A, left panel) and cellularity (Fig. 3A, right panel) as well as developmental stages defined by CD4 and CD8 surface markers (Fig. 3B), suggesting that TCF-1 plays a major role at rather earlier developmental stages to ensure normal thymocyte development. Intracellular IL-17 staining indicated more mature CD4+ TCRhi thymocytes from TCF-1fl/fl/Vav1-Cre mice produce IL-17 (3.10%) than that from TCF-1fl/fl (0.03%) and TCF-1fl/fl/CD4-Cre (0.2%) mice (Fig. 3B). This was consistent with our observation that peripheral CD4+ cells from TCF-1fl/fl/Vav1-Cre mice generated more IL-17 producing cells even in Th0 neutral conditions (Fig. 1D). When gated on earlier CD4-CD8- TCR- stage, we still found more CD4-CD8- thymocytes from TCF-1fl/fl/Vav1-Cre mice produced IL-17 (4.51%) compared to that from TCF-1fl/fl (0.42%) and TCF-1fl/fl/CD4-Cre (0.48%) mice (Fig. 3B). Therefore, Vav1-Cre-induced deletion of TCF-1 resulted in heightened IL-17 expression at as early as CD4-CD8- DN stage, and such heightened IL-17 expression was maintained at mature CD4+ T cell stage even after migrating out of thymus to the periphery. Since RORγt is required to stimulate IL-17 expression (25), RORγt expression was assessed at different thymocyte developmental stages in all three groups (Fig. 3C). Consistent with previous observation (26), RORγt was up-regulated at CD4+CD8+ DP stage but down-regulated at mature CD4+ cells in control TCF-1fl/fl mice. RORγt expression levels overlapped well between TCF-1fl/fl and TCF-1fl/fl/CD4-Cre thymocytes at CD4-CD8-, CD4+CD8+ and CD4+ stages. In contrast, a portion of CD4-CD8- DN and CD4+ thymocytes expressed higher levels of RORγt in TCF-1fl/fl/Vav1-Cre mice (Fig. 3C, arrows), which correlated with the observed increased IL-17 expression. Taken together, these data indicate that TCF-1 is required at early but not late developmental stages to inhibit IL-17 expression during T cell development.
Figure 3.
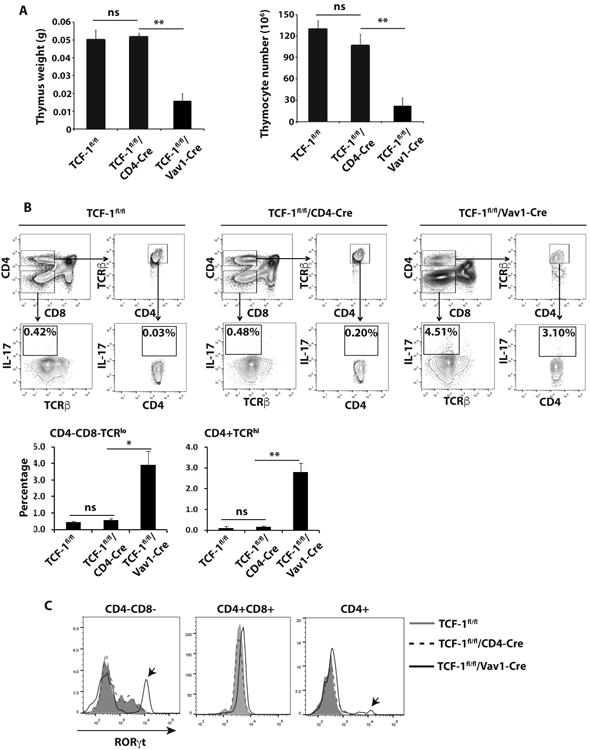
Vav1-Cre induced deletion of TCF-1 leads to abnormal IL-17 expression in thymus. A, Weight (left panel) and number of total thymocytes (right panel) of the thymus (n=3) from indicated genotypes of mice. B, Thymocytes of different genotypes of mice were either gated on CD4-CD8- or CD4+ and TCRβhi (top panels). Percentage of IL-17+ cells in CD4-CD8- TCRβlo and CD4+ TCRβhi population of indicated genotype of mice was determined (middle panels). Bottom panels are the quantification of percentage of IL-17+ cells shown on middle panels from three mice of each genotype. C, Expression of RORγt in indicated population of cells from indicated genotypes of mice, detected by intracellular staining. Arrow indicates the very high levels of RORγt population in CD4-CD8- and CD4+ thymocytes of TCF-1f/fl /Vav1-Cre mice.
Forced expression of TCF-1 in CD4CD8- thymocytes of TCF-1fl/fl/Vav1-Cre mice inhibits IL-17 expression
We next determined whether forced expression of TCF-1 could inhibit IL-17 expression in developing and mature T cells from TCF-1fl/fl/Vav1-Cre mice. Just like in mature T cells with germline deletion of TCF-1 (13), expression of TCF-1 by retrovirus-mediated gene transduction in mature T cells from TCF-1fl/fl/Vav1-Cre mice was unable to reduce IL-17 expression to WT levels under Th17 priming conditions (Fig. 4A). Next, we determined the effects of TCF-1 on thymocytes co-cultured with OP9-DL1 stroma cells (27). TCF-1fl/fl CD4-CD8- DN thymocytes could develop to CD4+CD8+ DP thymocytes with or without expression of exogenous TCF-1 from retrovirus (Fig. 4B). TCF-1fl/fl/CD4-Cre CD4-CD8- DN thymocytes were able to develop to CD4+CD8+ DP thymocytes, however, with reduced efficiency indicated by reduced percentage of CD4+CD8+ cells. Exogenous TCF-1 could restore the percentage of CD4+CD8+ cells in TCF-1fl/fl/CD4-Cre thymocytes to WT level (Fig. 4B), indicating the intact function of exogenous TCF-1. In contrast to TCF-1fl/f and TCF-1fl/fl/CD4-Cre thymocytes, majority of TCF-1fl/fl/Vav1-Cre CD4-CD8- DN thymocytes stayed at CD4-CD8- DN stage even in the presence of exogenous TCF-1 (Fig. 4B). We next determined the effects of exogenous TCF-1 on IL-17 expression. IL-17 was hardly detectable in thymocytes (including DN and developed DP) from TCF-1fl/fl and TCF-1fl/fl/CD4-Cre mice with or without exogenous TCF-1 (Fig. 4C). Whereas, CD4-CD8- DN thymocytes from TCF-1fl/fl/Vav1-Cre mice had abundant IL-17 expression (Fig. 4C), consistent with what we observed in vivo (Fig. 3B). Indeed, exogenous TCF-1 significantly suppressed IL-17 expression in TCF-1fl/fl/Vav1-Cre thymocytes (Fig. 4C). Taken together, these data further demonstrated TCF-1 is required to suppress IL-17 expression at early T cell developmental stage.
Figure 4.
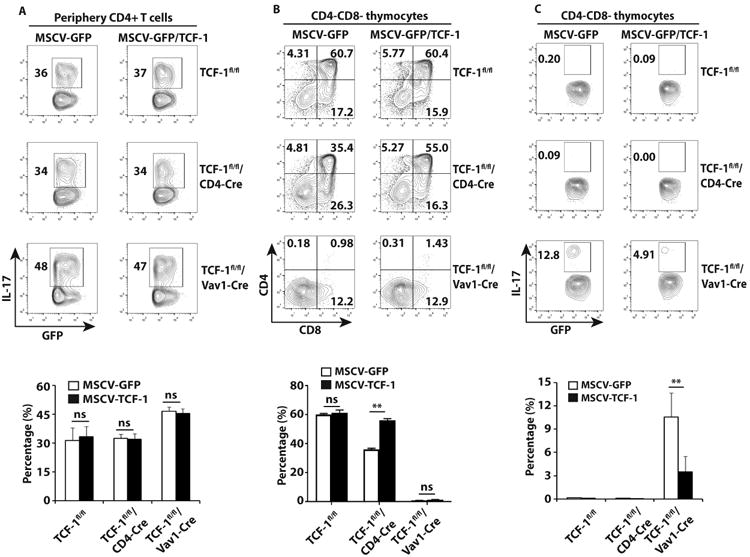
Forced expression of TCF-1 in TCF-1-deficient CD4-CD8- thymocytes but not mature CD4+ T cells from TCF-1fl/fl/Vav1-Cre mice inhibits IL-17 expression. A, Percentage of IL-17+/GFP+ cells among indicated genotypes of CD4+ T cells transduced with retrovirus expressing GFP along or together with TCF-1 and differentiated under Th17-priming conditions for 3 days, as determined by flow cytometry analysis. Bottom panel is the quantification of three biological replica. B, Flow cytometric analysis of CD4 and CD8 among thymocytes ex vivo differentiated from CD4−CD8− thymocytes sorted from indicated genotypes of mice, transduced with retrovirus expressing GFP along or together with TCF-1, and co-cultured for 3 days with OP9-DL1 stroma cells and IL-7. The number in each quadrant indicated the percentage of cells in gated area among GFP+ cells. Bottom panel is the quantification the percentage of CD4+CD8+ cells shown on top panels. C, Percentage of IL-17+/GFP+ cells among ex vivo differentiated thymocytes described in B. Bottom panel is the quantification of the results.
TCF-1 binds to IL-17 promoter and inhibits IL-17 expression through its intrinsic HDAC activity
Histone deacetylation is an evolutionary conserved epigenetic modification mechanism responsible for gene repression (28). Recent studies have shown that TCF-1 had intrinsic HDAC activity (15). Considering the dual capacity of DNA binding and intrinsic HDAC activity in TCF-1, we wondered whether TCF-1 binds to IL-17 locus and silences IL-17 expression via its intrinsic HDAC activity. To answer this question, we first determined whether TCF-1 binds to IL-17 promoter by chromatin immunoprecipitation (ChIP) assays with nine primer pairs covering the IL-17A promoter regions (P1 to p9) about 2000 base-pair (bp) upstream of transcriptional starting site (TSS) (Fig. 5A). The enrichment of TCF-1 binding to IL-17 promoter was observed in the region from P4 to P8 in TCF-1fl/fl thymus versus differentiated Th17 cells (Fig. 5B). To determine which TCF-1-binding regions are functional, each individual region from P4 to P8 was deleted to assess the effects on IL-17 reporter activity. Consistent with previous results (29, 30), RORγt stimulated IL-17 reporter. Whereas TCF-1 inhibited RORγt-dependent IL-17 reporter, deletion of P4 or P7 but not P5, P6 or P8 region of IL-17 promoter significantly relieved TCF-1-mediated inhibition of IL-17 reporter (Fig. 5C), suggesting that P4 and P7 are two functional TCF-1-binding sites responsible for the inhibition of IL-17 promoter activity. To determine whether TCF-1 is required for epigenetic modification critical for IL-17 expression, we compared histone H3 acetylation levels in the regions from P4 to P8. Due to the deletion of TCF-1 in TCF-1fl/fl/Vav1-Cre thymus, P5 and P8 had higher H3 acetylation levels (Fig. 5D), indicating these two regions underwent TCF-1-dependent histone deacetylation in the WT thymus. These data indicate the possibility that TCF-1 mediates histone deacetylation in the regions around these binding sites. We further observed that in contrast to WT TCF-1, TCF-1-5aa mutant which diminished HDAC activity (15) was unable to inhibit IL-17 expression in DN thymocytes obtained from TCF-1fl/fl/Vav1-Cre mice, comparable to virus expressing GFP alone (Fig. 5E). Taken together, these results demonstrated that TCF-1 binds to IL-17 promoter and silences IL-17 expression through its intrinsic HDAC activity.
Figure 5.
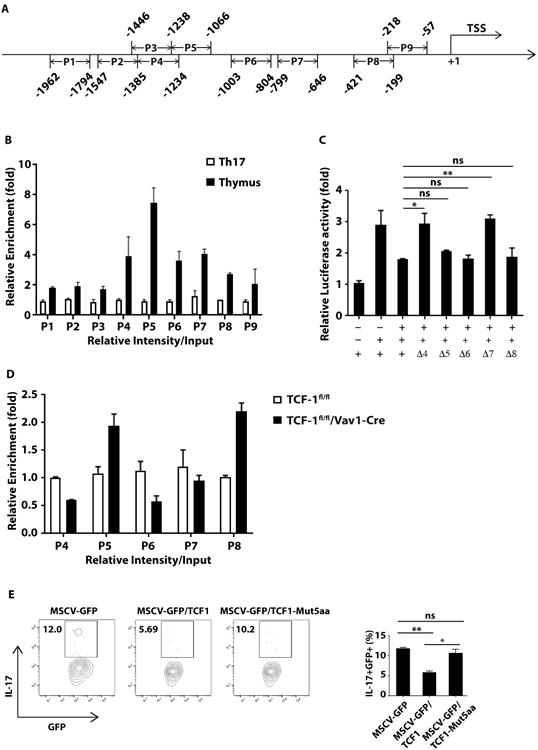
TCF-1 binds to IL-17 gene locus and inhibits IL-17 expression through its intrinsic HDAC activity. A, Schematic representation of the location of 9 regions, P1-P9, on IL-17 promoter. B, Enrichment of TCF-1 binding to 9 regions on IL-17 promoter, detected by ChIP assay using anti-TCF-1 antibody or IgG control in WT thymus or differentiated Th17 cells. C, IL-17 promoter-luciferase reporter activity in HEK293T cells transfected with the indicated expression plasmids. Δ4, deletion of P4; Δ5, deletion of P5; Δ6, deletion of P6; Δ7, deletion P7; Δ8, deletion of P8 region shown in A. Luciferase data are normalized to Renilla luciferase activity. D, DNA was precipitated with anti-acetylhistone H3 antibodies. qRT-PCR-based ChIP analysis was used to measure histone H3 acetylation levels in indicated regions. E, Percentage of IL-17+/GFP+ cells among thymocytes ex vivo differentiated from CD4-CD8- cells sorted from TCF-1fl/fl/Vav1-Cre mice, transduced with retrovirus expressing GFP along or together with TCF-1 or TCF1-Mut5aa, and co-cultured with OP9-DL1 stromal cells for 3 days, as determined by flow cytometry. Right panel is the quantification of three biological replica.
Discussion
Previous studies have shown TCF-1 was able to restrain peripheral Th17 responses and in vitro experiments show that TCF-1 negatively regulates peripheral Th17 responses at T cell developmental stage in thymus rather than mature T cells in periphery (13, 14). However, there was no in vivo evidence to support stage specific requirement of TCF-1 for limiting peripheral Th17 immunity. Furthermore, it remains unknown about the mechanisms responsible for TCF-1-mediated inhibition of Th17 responses. In this study, we first answered the critical question when is TCF-1 required to control the peripheral Th17 responses, given that T cell development is a sequential multi-step process. We found that deletion of TCF-1 early at hematopoietic stage, but not later DP stage, resulted in exaggerated Th17 differentiation and EAE development. In addition, since deletion of TCF-1 at early but not later DP stage leads to significant disruption of thymocyte development, demonstrating that TCF-1 plays a major role during early thymocyte development. Mechanically, we show that TCF-1 binds to and inhibit IL-17 promoter to prevent IL-17 expression via its intrinsic HDAC activity.
IL-17 gene locus must be kept inactive by TCF-1 prior to developing thymocytes entering DP stage. This is indicated by that deletion of TCF-1 at DP stage does not cause enhanced IL-17 expression in thymus, which means suppressive function of TCF-1 for IL-17 gene is not required at DP stage or later. Once TCF-1-mediated inhibition is imprinted on IL-17 gene locus, which we believe occurred no later than DN stage, it can last even after T cell migrating out of the thymus, resulting in prevention of exaggerated Th17 responses in periphery. Furthermore, our results highlight the specific requirement of HDAC activity of TCF-1 for suppression of IL-17 gene activity during T cell development. The HDAC family consists of a large family members with many of which play indispensable roles in regulating T cell development, including DN to DP transition, cell survival and TCR signaling in DP thymocytes (31-34). Although several HDAC proteins are expressed in thymus, it seems they are unable to compensate for the HDAC activity of TCF-1, as indicated by abnormal high levels of IL-17 expression (or natural Th17 cells) in thymus of TCF-1fl/fl/Vav1-Cre mice. This phenomenon highlights the specific requirement of HDAC activity of TCF-1 for keeping IL-17 gene inactive early during T cell development.
Interestingly, if IL-17 gene locus does not undergo TCF-1-mediated inhibition like TCF-1 deficiency at early development stage, it will lead to an elevated Th17 differentiation and Th17 immunity. This consequence is irreversible as reconstitution of TCF-1 in mature T cells was unable to reduce IL-17 expression to normal levels. The mechanism of why TCF-1 close IL-17 gene locus in early developing thymocytes rather than in mature T cells remains largely unknown. Our results suggest that IL-17 gene locus in mature T cells was relatively inaccessible for TCF-1 compared to that in early T cell developmental stage. This may result from chromatin remolding during later developmental stage and/or lack of cofactors that present in early thymocytes but not in mature T cells, which could be an interesting topic for future investigation.
Exaggerated Th17 responses are associated with many types of autoimmune diseases (8, 35). To develop the effective treatment for autoimmune diseases, many studies have focused on understanding the mechanisms of regulating Th17 differentiation and Th17-Tregs balance in peripheral immune system. Our study suggests that peripheral Th17 responses can be greatly influenced by specific developmental events in central immune system, and thus provide additional layer of control to prevent autoimmune diseases.
Supplementary Material
Acknowledgments
We thank Dr. Hai-Hui Xue for generously sharing TCF-1fl/fl mice. We appreciate the help by city of hope supported cores including animal, genomic and flow cytometer cores. In addition, we thank Chris Gandhi and Keely Walker for their careful proof/editing work on the manuscript.
This work was supported by grants from NIH R01-AI053147, NIH R01-AI109644 and institutional pilot funding. In addition, research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under award number P30CA33572, which includes work performed in the animal, genomic, flow cytometer and mass spectrometric cores supported by this grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Disclosures: The authors have no financial conflicts of interest
References
- 1.Jameson SC, Hogquist KA, Bevan MJ. Positive selection of thymocytes. Annual review of immunology. 1995;13:93–126. doi: 10.1146/annurev.iy.13.040195.000521. [DOI] [PubMed] [Google Scholar]
- 2.Ma J, Wang R, Fang X, Sun Z. beta-catenin/TCF-1 pathway in T cell development and differentiation. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2012;7:750–762. doi: 10.1007/s11481-012-9367-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see) Nature reviews Immunology. 2014;14:377–391. doi: 10.1038/nri3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annual review of immunology. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 5.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 7.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elloso MM, Gomez-Angelats M, Fourie AM. Targeting the Th17 pathway in psoriasis. Journal of leukocyte biology. 2012;92:1187–1197. doi: 10.1189/jlb.0212101. [DOI] [PubMed] [Google Scholar]
- 9.Skepner J, Ramesh R, Trocha M, Schmidt D, Baloglu E, Lobera M, Carlson T, Hill J, Orband-Miller LA, Barnes A, Boudjelal M, Sundrud M, Ghosh S, Yang J. Pharmacologic inhibition of RORgammat regulates Th17 signature gene expression and suppresses cutaneous inflammation in vivo. J Immunol. 2014;192:2564–2575. doi: 10.4049/jimmunol.1302190. [DOI] [PubMed] [Google Scholar]
- 10.Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, te Riele H, van de Wetering M, Oosterwegel M, Wilson A, MacDonald HR, Clevers H. An HMG-box-containing T cell factor required for thymocyte differentiation. Nature. 1995;374:70–74. doi: 10.1038/374070a0. [DOI] [PubMed] [Google Scholar]
- 11.Schilham MW, Wilson A, Moerer P, Benaissa-Trouw BJ, Cumano A, Clevers HC. Critical involvement of Tcf-1 in expansion of thymocytes. J Immunol. 1998;161:3984–3991. [PubMed] [Google Scholar]
- 12.Okamura RM, Sigvardsson M, Galceran J, Verbeek S, Clevers H, Grosschedl R. Redundant regulation of T cell differentiation and TCRalpha gene expression by the transcription factors LEF-1 and TCF-1. Immunity. 1998;8:11–20. doi: 10.1016/s1074-7613(00)80454-9. [DOI] [PubMed] [Google Scholar]
- 13.Ma J, Wang R, Fang X, Ding Y, Sun Z. Critical role of TCF-1 in repression of the IL-17 gene. PLoS One. 2011;6:e24768. doi: 10.1371/journal.pone.0024768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu Q, Sharma A, Ghosh A, Sen JM. T cell factor-1 negatively regulates expression of IL-17 family of cytokines and protects mice from experimental autoimmune encephalomyelitis. J Immunol. 2011;186:3946–3952. doi: 10.4049/jimmunol.1003497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, Li Y, Phillips FC, Maina PK, Qi HH, Liu C, Zhu J, Pope RM, Musselman CA, Zeng C, Peng W, Xue HH. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nature immunology. 2016;17:695–703. doi: 10.1038/ni.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinke FC, Yu S, Zhou X, He B, Yang W, Zhou B, Kawamoto H, Zhu J, Tan K, Xue HH. TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells. Nat Immunol. 2014;15:646–656. doi: 10.1038/ni.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, Liu L, Qian W, Ransohoff RM, Bergmann C, Stohlman S, Tuohy VK, Li X. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma J, Wang R, Fang X, Sun Z. beta-Catenin/TCF-1 Pathway in T Cell Development and Differentiation. J Neuroimmune Pharmacol. 2012 doi: 10.1007/s11481-012-9367-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 20.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- 21.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 22.Yu Q, Sharma A, Oh SY, Moon HG, Hossain MZ, Salay TM, Leeds KE, Du H, Wu B, Waterman ML, Zhu Z, Sen JM. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. 2009;10:992–999. doi: 10.1038/ni.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. The Journal of experimental medicine. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nature medicine. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 26.Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, Littman DR. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science. 2000;288:2369–2373. doi: 10.1126/science.288.5475.2369. [DOI] [PubMed] [Google Scholar]
- 27.Holmes R, Zuniga-Pflucker JC. The OP9-DL1 system: generation of Tlymphocytes from embryonic or hematopoietic stem cells in vitro. Cold Spring Harbor protocols. 2009;2009:pdb.prot5156. doi: 10.1101/pdb.prot5156. [DOI] [PubMed] [Google Scholar]
- 28.Rusche LN, Kirchmaier AL, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annual review of biochemistry. 2003;72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Zhang Y, Yang XO, Nurieva RI, Chang SH, Ojeda SS, Kang HS, Schluns KS, Gui J, Jetten AM, Dong C. Transcription of Il17 and Il17f is controlled by conserved noncoding sequence 2. Immunity. 2012;36:23–31. doi: 10.1016/j.immuni.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sen S, Wang F, Zhang J, He Z, Gwack Y, Xu J, Sun Z. SRC1 promotes Th17 differentiation by overriding Foxp3 suppression to stimulate RORgammat activity in a PKC-theta dependent manner. Proceedings of the National Academy of Sciences of the United States of America. 2017 doi: 10.1073/pnas.1717789115. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haery L, Thompson RC, Gilmore TD. Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes & cancer. 2015;6:184–213. doi: 10.18632/genesandcancer.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dovey OM, Foster CT, Conte N, Edwards SA, Edwards JM, Singh R, Vassiliou G, Bradley A, Cowley SM. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood. 2013;121:1335–1344. doi: 10.1182/blood-2012-07-441949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cabrero JR, Serrador JM, Barreiro O, Mittelbrunn M, Naranjo-Suarez S, Martin-Cofreces N, Vicente-Manzanares M, Mazitschek R, Bradner JE, Avila J, Valenzuela-Fernandez A, Sanchez-Madrid F. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Molecular biology of the cell. 2006;17:3435–3445. doi: 10.1091/mbc.E06-01-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kasler HG, Young BD, Mottet D, Lim HW, Collins AM, Olson EN, Verdin E. Histone deacetylase 7 regulates cell survival and TCR signaling in CD4/CD8 double-positive thymocytes. Journal of immunology. 2011;186:4782–4793. doi: 10.4049/jimmunol.1001179. [DOI] [PubMed] [Google Scholar]
- 35.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nature reviews Immunology. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.