

Abstract
Mitochondrial bioenergetic dysfunction is involved in neurodegeneration in Huntington’s disease (HD). Iron is critical for normal mitochondrial bioenergetics but can also contribute to pathogenic oxidation. The accumulation of iron in the brain occurs in mouse models and in human HD. Yet the role of mitochondria-related iron dysregulation as a contributor to bioenergetic pathophysiology in HD is unclear. We demonstrate here that human HD and mouse model HD (12-week R6/2 and 12-month YAC128) brains accumulated mitochondrial iron and showed increased expression of iron uptake protein mitoferrin 2 and decreased iron-sulfur cluster synthesis protein frataxin. Mitochondria-enriched fractions from mouse HD brains had deficits in membrane potential and oxygen uptake and increased lipid peroxidation. In addition, the membrane-permeable iron-selective chelator deferiprone (1 µM) rescued these effects ex-vivo, whereas hydrophilic iron and copper chelators did not. A 10-day oral deferiprone treatment in 9-week R6/2 HD mice indicated that deferiprone removed mitochondrial iron, restored mitochondrial potentials, decreased lipid peroxidation, and improved motor endurance. Neonatal iron supplementation potentiates neurodegeneration in mouse models of HD by unknown mechanisms. We found that neonatal iron supplementation increased brain mitochondrial iron accumulation and potentiated markers of mitochondrial dysfunction in HD mice. Therefore, bi-directional manipulation of mitochondrial iron can potentiate and protect against markers of mouse HD. Our findings thus demonstrate the significance of iron as a mediator of mitochondrial dysfunction and injury in mouse models of human HD and suggest that targeting the iron-mitochondrial pathway may be protective.
Keywords: neurodegeneration, mitochondria, iron, Huntington’s disease
Graphical abstract
Introduction
Iron-containing proteins are required for a large number of functions including cellular respiration, DNA replication and repair, and regulation of cell survival [1, 2]. Iron exists in ferrous (II) and ferric (III) oxidation states in biological systems and binds oxygen, properties that are harnessed by many proteins. However, when inappropriately coordinated, redox properties of labile iron are uncontrolled and can result in the catalytic generation of damaging oxygen radicals [1]. Iron homeostatic pathways regulate iron transport and its utilization from the organellar to the whole body level. These pathways have the important property of limiting the availability of labile iron.
Diverse diseases are linked with peripheral iron accumulation and iron-mediated oxidative injury [3, 4]. In addition, the accumulation of iron within the central nervous system is associated with neurodegenerative diseases, including Alzheimer disease, Parkinson disease, Huntington disease (HD), amyotrophic lateral sclerosis, and neurodegeneration with brain iron accumulation [5–7]. The precise role of iron in these disorders is a subject of debate; however, disruption of genes known to be directly involved in iron metabolism is sufficient to cause neurodegeneration [8, 9]. Efforts to understand and ameliorate the effects of iron dysregulation are therefore of broad interest, especially as there are no treatments that can slow the progression of these diseases.
HD is a progressive neurodegenerative disease caused by a trinucleotide CAG repeat expansion in exon 1 of the HTT gene [10]. HD is characterized by progressive motor, psychiatric, and cognitive deterioration, and weight loss [11]. Neurodegeneration, characterized by neuronal death and glial activation, primarily occurs in the striatum and cerebral cortex. Mutant huntingtin protein (mhtt) is expressed in neurons and glia [12] and results in the disruption of a number of downstream pathways [13]. The brain has high energy demands and is therefore vulnerable to the effects of disruption of energy metabolism. There is considerable evidence for bioenergetics dysfunction in HD, although the disease process begins years before clinical onset and the point at which mitochondrial involvement begins is unclear [14, 15]. Bioenergetic defects are, however, present in early clinical HD. For example, early HD patients demonstrate a decreased bioenergetics response to non-invasive cortical activation as measured by magnetic resonance spectroscopy [16]. Brains from patients with advanced HD obtained post-mortem demonstrate disruption of mitochondrial energy metabolism, which includes decreased activities of mitochondrial respiratory complexes II-IV and aconitase [17, 18]. Although interpretation of these findings is complicated by early neuronal loss and gliosis [19], studies of human HD support the presence of progressive bioenergetics dysfunction as part of the disease phenotype.
Mitochondrial alterations in HD result from a combination of disease-promoting pathways. Huntingtin is involved in the regulation of nuclear gene expression. Studies in mouse and cell models of HD have demonstrated that mitochondrial dysfunction is, in part, explained by disruption of nuclear transcriptional pathways important for encoding mitochondrial proteins [20–26]. However, huntingtin also interacts with mitochondria [27, 28] such that it triggers mitochondrial fragmentation, stimulates mitophagy, and impairs mitochondrial protein import [29–32]. Furthermore, energetic dysfunction is a prominent feature of HD human cybrids, implicating persistent mitochondrial dysfunction despite the absence of mhtt expression [33]. Mitochondrial dysfunction in HD therefore results from altered nuclear gene expression and the direct effects of mhtt, as well as persistent effects of prior injury. Mitochondria from mouse models of HD (hereafter referred to as ‘HD mice’) also demonstrate compensatory changes, such as an increased resistance to calcium-induced opening of the permeability transition pore [34]. Therefore, mitochondrial alterations in HD are complex and there remain significant gaps in our understanding of the mechanisms underlying these functional changes.
Huntingtin, while having numerous functions, modulates iron homeostasis. Developing zebrafish with morpholino-induced htt deficiency have an iron deficiency phenotype that is rescued by iron supplementation [35]. In addition, knockdown of htt in adult mice changes the expression of brain iron homeostatic proteins [36]. Although the relationship between the state of htt deficiency and HD is different, HD brain iron levels are increased in autopsy samples from patients with advanced disease [7, 37]. Magnetic resonance imaging supports changes in brain iron metabolism during early HD [7, 38, 39]. In the HD mouse brain, iron accumulates in both neurons and glia, suggesting pleiotropic roles in the disease [40, 41]. Iron supplementation of infants and children is widely carried out in the general human population to prevent nutritional deficiency; however, excess supplementation has the potential to promote neurodegeneration in adult life [42]. Modeling iron supplementation in neonatal rodents provides evidence for adverse effects on the adult brain both in wild-type animals and animal models of several neurodegenerative diseases [42]. In particular, neonatal iron supplementation of HD mice promotes markers of disease in adult life [43, 44]. In addition, a brain iron chelator improves behavioral and pathology markers of mouse HD [40]. However, the associated cellular and subcellular targets, as well as the pathways involved, are poorly understood.
Mitochondria use large amounts of iron for the synthesis of heme and iron-sulfur cluster proteins [2]. Iron is taken up by mitochondria by mitoferrin 2, an inner mitochondrial transmembrane protein [45]. Mitochondria have a labile iron pool that provides iron for assimilation into iron proteins [46]. Although these pathways are complex, frataxin is one protein that is important for the delivery of iron into mitochondrial iron-sulfur synthesis pathways [47]. Notably, frataxin deficiency results in Friedreich ataxia (FA), a disorder characterized by mitochondrial iron accumulation, defective synthesis of iron-sulfur proteins, and neurodegeneration [9]. The mitochondrial transporter ATP binding cassette subfamily B member 8 (ABCB8) exports heme or iron-sulfur clusters to other cell compartments [48]. Therefore, mitochondrial iron homeostasis is fundamental for many cell functions, and its disruption has far-reaching effects. The role of mitochondrial iron dysregulation in other neurodegenerative diseases is further supported in studies of pantothenate kinase-associated neurodegeneration, Alzheimer disease, and 6-hydroxy-dopamine-induced nigral degeneration [49–52]. In human HD, a role for mitochondrial iron is suggested by the co-occurrence of iron dysregulation and mitochondrial dysfunction. In addition, transcriptomic analyses indicate increased transcript expression of mitoferrin 2 in human HD [53]. As therapies that ameliorate the effects of brain iron accumulation may have protective effects, it is important to understand the associated mechanisms.
Based on these considerations, we set out to test the hypothesis that mitochondria are a key site of iron dysregulation in HD. We studied brain mitochondria from HD mice and from human HD tissues. We found that labile iron accumulated in HD mitochondria with disease progression and that functional and biochemical markers of mitochondrial dysfunction were increased. The membrane-permeable, iron-selective chelator deferiprone rescued mitochondrial markers of HD ex vivo and in vivo. We further showed that nutritionally relevant levels of neonatal iron supplementation in HD mice potentiated HD-associated mitochondrial dysfunction. These studies show that the bi-directional effects of iron chelation and supplementation in HD models affect mitochondria. Our findings support a role for labile mitochondrial iron as a mediator of HD mitochondrial dysfunction and suggest new therapeutic approaches.
MATERIALS AND METHODS
Materials
All chemicals were purchased from Sigma-Aldrich, unless otherwise stated.
Mouse husbandry
Animal procedures were approved by the Institutional Animal Care and Use Committee and also followed NIH guidelines. Mice were maintained under standard environmental and husbandry conditions. They were fed ad libitum with a cereal-based rodent chow (LabDiet 5K67). R6/2 and YAC128 mice were obtained by breeding HD males with wild-type females. R6/2 males were crossed with B6/CBA F1 females, and YAC128 males were crossed with FVB females. Tail tips collected at 2.5 weeks of age were used for genotyping as described [54, 55]. Mice were grouped by genotype under standard conditions with a 12-hour dark/light cycle. Mice used for neonatal iron supplementation and deferiprone (DFP) treatment had a CAG repeat size in the range 150–160. Mice used in other studies had a CAG repeat size in the range of 180–190. We did not identify mitochondrial differences between the two cohorts. In the R6/2 study, female mice were used for all experiments, whereas in the YAC128 study, both male and female mice were used.
Human brains
Human HD and control cerebro-cortical brain samples were obtained from the NIH NeuroBioBank. Each group included four males and one female with an age range of 50–60 years. Post-mortem intervals were 14–32 hours. All the HD brains were characterized as Vonsattel grade 3 [19]. CAG repeat sizes were determined in cerebral cortex BA17 by Laragen, Inc (Culver City, CA) and were 44, 47, 48, 49, and 51.
Brain regional fractionation and purity validation
Mice were anesthetized with 90 mg/kg ketamine and 10 mg/kg xylazine and then were perfused intra-cardially with cold heparinized 0.9% (w/v) saline for 2 minutes. Cerebro-cortices and striata were dissected and extractions were completed on the same day essentially as described [56]. Briefly, tissues from mouse and human brains were homogenized (30 seconds) in isolation buffer containing 215 mM mannitol, 75 mM sucrose, 20 mM HEPES (pH 7.2), 1 mM EGTA, 1 mM EDTA, 1 mM PMSF, and a protease inhibitor cocktail (Roche). Homogenates were centrifuged at 1000×g for 5 minutes at 4°C, pellets were stored as the nuclear fraction for western blotting analysis, and the supernatants were centrifuged at 13000×g for 10 minutes at 4°C. The resulting supernatants were then collected as the cytosolic fraction, and the mitochondrial pellets were re-suspended in isolation buffer and centrifuged at 13000×g for 10 minutes at 4°C. The cytosolic fractions were stored at −70°C, whereas the mitochondrial fractions were either used immediately or stored at −70°C. Freshly isolated mitochondria were used for mitochondrial functional studies such as membrane potential, oxygen consumption rate, ATP, and lipid peroxidation analyses. The purity of the fractions was assessed using enzymatic and also western blot analyses (see below). Succinate and lactate dehydrogenase assays were performed as described [57].
Inductively coupled plasma mass spectrometry (ICP-MS) iron measurement
The analysis was conducted essentially as described [58, 59], with some modifications. Briefly, 300 µl of mitochondrial and cytosolic fractions containing 500 µg of protein were digested with 14% (v/v) of trace metal grade 70% HNO3 and a 4-hour incubation at 90°C in a block heater; the fractions were then cooled to room temperature for 10 minutes. To remove lipids, 6% (v/v) of 30% hydrogen peroxide was added and incubated for 10 minutes at room temperature before the fractions were heated to 90°C for 2 hours. The fractions were allowed to cool for 10 minutes and then 18 MΩ purified water was added to a final volume of 3 ml. Each sample was assayed for iron and also Cu, Zn, Co, and Mn concentrations by ICP-MS (7700× ICP-MS Agilent) with the following parameters: helium collision mode, three points per peak, 100 scans per replicate, and five replicates per sample. Plasma gas flow was 15 l/minute, auxiliary flow was 0.9 l/minute, and carrier gas was flow 0.70 l/minute. The radio-frequency power was 1.5 kW. The instrument was calibrated using Plasma CAL multielement calibration standards (SCP Science); 2000 ppb of scandium was used as an internal standard.
Mitochondrial labile iron analyses
A bleomycin-based assay was used as a sensitive and reliable method of measuring free iron in tissue samples as described [60, 61]. This method is based on the direct complexation of free iron with the antibiotic bleomycin. The iron-bleomycin complex causes degradation of DNA, and the degradation products were measured by the thiobarbituric acid test for the detection of free iron. All reagents, except bleomycin, were treated with Chelex 100 resin (0.3 g/10 ml) to remove contaminating iron. Mitochondrial samples (100 µg of protein in a 50 µl volume) were each added to 1 ml of 100 mM Tris-HCl (pH 7.4) containing 1 mg/ml calf thymus DNA, 1.5 U/ml bleomycin, 5 mM MgCl2, and 0.8 mM ascorbate, and the samples were then incubated for 1 hour at 37°C. The reaction was stopped by addition of 100 µl of 100 mM EDTA, followed by 500 µl thiobarbituric acid (1% [w/v] in 50 mM NaOH) and 500 µl of 25% HCl. The reaction tubes were incubated for 20 minutes in a water bath at 80°C to develop the chromogen. The chromogen was then extracted after cooling to room temperature by vortexing the sample with 3 ml chloroform followed by centrifugation at 1000×g for 20 minutes. The absorption of the organic layer was determined at 532 nm. Each sample was also assayed in the absence of bleomycin. Iron concentrations were determined using a series of standard iron concentrations (1–50 µM).
Immunoblotting
Brain proteins were extracted from mouse nuclear, cytosolic, and mitochondrial fractions as described [56, 62]. Proteins from human cortical regions BA4, BA9, and BA17 of HD and control brains were extracted with ice-cold lysis buffer containing 120 mM NaCl, 1 mM EDTA, 1 mM sodium fluoride, 50 mM Tris-HCl (pH 7.5), 1% (v/v) NP-40, 1 mM PMSF, and a protease inhibitor cocktail (Roche). Protein concentrations for mouse and human studies were determined using the Bradford method. Proteins from mouse and human samples (20 and 30 µg, respectively) were separated by SDS-PAGE and transferred to PVDF membranes. Blots were incubated in the following reagents in order (with standard wash steps): 5% fat-free milk as a blocking reagent, primary antibodies HDAC1 (Santa Cruz Biotechnology (sc)-81598), GAPDH (sc-365062), frataxin (sc-293431), mitoferrin 2 (sc-138430), ABCB8 (sc-68246), TOM20 (sc-17764), and β-actin (Sigma AC-4700) at dilutions of 1:500–1:1000; and horseradish peroxidase-conjugated anti-mouse/anti-goat/anti-rabbit secondary antibodies at dilutions of 1:1000–1:2000. The protein signal was visualized by chemiluminescence reagents in conjunction with X-ray film. Densitometry measurements of each protein were quantified using Image-J software (NIH).
Mitochondrial aconitase activity
Mitochondrial aconitase activity was determined using a commercially available kit (Sigma). This assay uses a coupled enzyme reaction in which citrate is converted to isocitrate by aconitase. Isolated mitochondrial fractions were sonicated three times for 30 seconds each on ice at 100% amplitude and centrifuged at 20,000× g for 15 minutes, and then the protein content of the supernatant fraction was determined. Mitochondrial aconitase activity was determined by measuring the absorbance at 450 nm. Results were expressed as nanomoles per minute per milligram mitochondrial protein.
Mitochondrial membrane potential
Mitochondrial membrane potentials (MMP) were measured using JC-1 dye as described [63]. In brief, isolated mitochondrial pellets were directly suspended in isotonic buffer comprising 10 mM phosphate buffer (pH 7.4) containing 145 mM KCl, 50 mM sucrose, 1 mM EGTA, and 1 mM MgCl2, and protein concentrations were then determined. The mitochondria samples containing 200 µg protein were then incubated in isotonic buffer containing 10 mM pyruvate, 10 mM succinate, and 1 mM ADP with 1 µM JC-1 for 20 minutes at 37°C in a 96-well plate. The samples were washed twice in isotonic buffer before being resuspended in 200 µl of isotonic buffer. The fluorescence emission ratio (596 nm/534 nm) was then determined using a fluorescence microplate reader (TECAN infinite M200). For the measurement of the effect of chelators. chemicals were added 10 minutes before the JC1 incubation. Five separate fluorescence ratio measurements were made at 1-minute intervals [64].
Mitochondrial oxygen consumption
Mitochondrial oxygen consumption was measured at 30°C using a Clark-type electrode (World Precision Instruments) with minor modifications of published methods [65]. Two-point calibration of the sensor was performed in air-saturated water at 37°C in an argon atmosphere on each day that measurements were made. Freshly prepared mitochondria pellets were suspended in 10 mM Tris-HCl (pH 7.4) buffer containing 0.25 mM sucrose, and protein concentrations were measured. Mitochondria containing 250 µg protein were added in 50 mM Tris/HCl buffer (pH 7.4) containing 0.2 M sucrose, 3 mM MgCl2, 5 mM sodium succinate, and 5 mM sodium pyruvate to a final volume of 1 ml. After equilibriation for 1 minute, basal respiration was measured for 3 minutes. DFP or vehicle was then added and respiration measured for a further 3 minutes. The following mitochondrial respiration control compounds were then used: carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) at 1 µM and antimycin A at 10 µM.
ATP determination
A commercially available ATP assay kit (Abcam) was used to measure ATP concentrations in cytoplasmic and mitochondrial brain fractions. Freshly isolated mitochondrial fractions were sonicated three times for 20 seconds each at 100% amplitude and centrifuged at 20,000×g for 10 minutes, and then protein concentrations were determined. Fractions consisting of 500 µg of protein were de-proteinized in 4 M cold perchloric acid and 2 M potassium hydroxide. ATP levels were measured fluorometrically according to the manufacturer’s instructions.
Mitochondrial glutathione
Soluble extracts of mitochondria were prepared as described for the ATP assay. Mitochondrial extracts (500 µg protein per sample) were de-proteinized with 6% perchloric acid, and glutathione (GSH) and oxidized glutathione (GSSG) levels were determined as described [66]. In brief, total glutathione was measured by the reduction of GSSG to GSH using NADPH and glutathione reductase; GSH was then determined using the chromagen 5,5′-dithiobis-(2-nitrobenzoic acid) using a 3-minute kinetic assay with absorbance measured at 405 nm. GSSG was determined as above but with prior derivatization of GSH with the alkylating agent 2-vinylpyridine. True GSH was calculated by subtraction. GSSG standards were used.
Mitochondrial membrane lipid peroxidation
We used a previously described method to quantify aldehyde end products of lipid peroxidation [67]. Briefly, 100 µg of each mitochondria-enriched fraction in a 200-µl volume was added to 50 µl of 10% SDS and 450 µl of a mixture of 46.4 mM thiobarbituric acid and 50% glacial acetic acid. The mixture was incubated for 45 minutes in a 95°C water bath. The reaction tubes were cooled, and then 500 µl of chloroform was added. The sample was vortexed, centrifuged at 1000×g for 10 minutes, and allowed to cool for 10 minutes at room temperature. The upper aqueous layer was collected and absorbance determined at 532 nm. Malondialdehyde and buffer blanks were used for quantification. To measure the malondialdehyde production rate, fresh mitochondria were incubated in 10 mM HEPES buffer (pH 7.2) containing 125 mM sucrose, 65 mM KCl, 2 mM succinate, 2 mM pyruvate, and 2 mM citrate with or without 1 µM DFP for 30 minutes at 30°C as described [68]. Baseline control samples were stored at 4°C and assayed in parallel. The assays were completed as described above. Baseline lipid peroxidation values were subtracted, and the lipid peroxidation rate was determined by normalizing to protein and incubation time.
Neonatal iron supplementation study
We used a previously used dose of iron supplementation that is considered to model human infant iron supplementation levels [43]. Iron or saline treatments were randomly assigned to litters. Pups were administered saline or 120 mg/kg/day carbonyl iron via oral gavage from day 10 to 17 once daily. Pups were weighed daily before each dose.
Sub-acute deferiprone mouse study
Mice were randomized and the study was blinded for the behavioral analysis. Treatments were started after 9.5 weeks of age. Mice were treated with 150 mg/kg/dose DFP or saline vehicle twice a day for 10 days with 10 to 12-hour interval between doses. Motor endurance testing (Rota-rod) was completed at 5 and 9 weeks to obtain baseline values for each genotype; the final measurement was at 11 weeks (last 3 days of dosing) of age. The rod accelerated from 5 to 25 rpm over 15 minutes. At each of the times tested (5, 9, and 11 weeks), animals were trained on day 1 and tested on three consecutive days. On the training day, mice were placed on the rota-rod, and each time they fell off were replaced on the rod [during a 5-minute period]. On the testing day, mice were replaced on the apparatus if a fall happened within the first 30 seconds; otherwise the time of falling was recorded as the latency to fall [43].
Measurement of deferiprone levels in brain mitochondria
Mitochondrial pellets were mixed with 20 µl of 50% methanol and 80 µl of acetonitrile containing 100 ng/ml of theobromine as an internal standard (IS), and the samples were centrifuged at 1500×g for 10 minutes to precipitate the proteins. The supernatant was used for liquid chromatography–tandem mass spectrometry (LC-MS/MS). Five non-zero standard solutions were prepared with DFP concentrations ranging from 1.25 to 20 ng/ml. The linearity of the relationship between the peak area ratio and concentration was confirmed by the correlation coefficients obtained for the linear regression of DFP (r=0.9948). Concentrations of DFP were determined using an ABI-5500 Qtrap (Sciex, Ontario, Canada) mass spectrometer with electrospray ionization source interfaced with a Shimadzu high-performance liquid chromatography (HPLC) system. The Analyst software version 1.6 package from Applied Biosystems MDS SCIEX was used to control the LC-MS/MS system, as well as for data acquisition and processing. Separation was performed on an XBridge C18 (Waters) column (4.6 × 150 mm i.d., particle size 3.5-µm. The mobile phase consisted of A, 0.1% formic acid in purified deionized water, and B, 0.1% formic acid in acetonitrile. The gradient program was as follows: 0.01 to 0.5 minutes, 40% B; 0.5 to 0.8 minutes, 40% B to 95% B; 0.8 to 2 minutes, 95% B; 2 to 2.1 minutes, 95% B to 40% B; and 2.1 to 4.2 minutes, 40% B. The flow rate was 0.8 ml/minute, and the injection volume was 5 µl. MS/MS experiments were carried out with the Turbo-Ionspray™ Interface and positive ion-mode multiple-reaction monitoring (MRM-transitions: DFP m/z 140.1–96.1, DP 98 V, EP 10 V, CE 48.27 V, CXP 13 V; IS m/z 181.2–96.2, DP 100 V, EP 8 V, CE 30 V, CXP 16 V). Additional parameters were as follows: curtain gas, 30; collision gas, medium; ion spray voltage, 5000 V; temperature, 500°C; ion source gas 1, 50; ion source gas 2, 50.
Statistical analyses
All data were analyzed using SAS software (version 9.4, Cary, NC) using the GLM or MIXED procedures. MIXED procedures were used for repeat measures analyses and when the brain region was included in the analysis. Significant differences were determined by pair-wise comparisons if there was significant genotype or treatment effects, or interactions in the initial analysis. Otherwise, significant pair-wise comparisons are described as unprotected. All analyses used two-tailed tests. Results are expressed as the means ± SEM.
RESULTS
Mitochondrial iron accumulates in mouse and human HD brains
R6/2 and YAC128 mice are widely used transgenic models that express a fragment of and full-length human htt, respectively, containing a CAG triplet expansion [69]. These mice undergo progressive development of HD with rapid progression to death at 13–14 weeks (R6/2) or a normal lifespan (YAC128). To investigate the possible role of mitochondrial iron in HD, we carried out studies in which we obtained brain regional mitochondrial and cytoplasmic fractions and then determined the iron content by ICP-MS. We first demonstrated the purity of the fractions by enzymatic and Western blot analyses (Fig. S1). R6/2 mice showed a progressive increase in brain mitochondrial, but not cytoplasmic, iron content in the striatum and cerebral cortex (Fig. 1A–D), the main HD-affected areas.
Figure 1. Mitochondrial iron increases in mouse and human HD brains.

A–G. Iron distribution in mouse HD brain. Mitochondrial (A–B) but not cytoplasmic (C–D) total iron concentrations are increased from middle-stage HD in the striata (A, C) and cerebral cortices (B, D) of R6/2 HD mice as determined by ICP-MS. n = 8 or 9 at 4 and 8 weeks; n = 10–16 at 12 weeks. Striatal and cortical mitochondrial labile iron concentrations (E) are increased in R6/2 HD mice as determined using a bleomycin-based assay. n = 10. Mitochondrial (F) but not cytosolic (G) total iron concentrations are increased in the striata of 12-month-old YAC128 HD mice. n = 10. H–I. Iron distribution in human HD cortex. Mitochondrial (H) iron levels are increased across the three cortical brain areas. Cytosolic (I) iron levels are increased across the same three brain regions. n = 5. *p < 0.05, **p < 0.01, ***p < 0.001, BA4=Brodmann area 4; BA9=Brodmann area 9; BA17=Brodmann area 17.
Mitochondria are iron-rich because of their expression of many iron-sulfur and heme proteins. Therefore, increased mitochondrial iron content could result from increased levels of iron proteins. In contrast, iron-mediated neurodegeneration is linked with labile iron, which is able to generate reactive oxygen species. Therefore, we used an assay specifically for labile mitochondrial iron and found significant increases in R6/2 HD mouse brains (Fig. 1E). Twelvemonth-old YAC128 mice, which are approximately in middle-stage HD, also had elevated striatal mitochondrial, but not cytoplasmic, iron content (Fig. 1F–G). To determine if accumulation of mitochondrial iron is a feature of human HD, we studied autopsy-obtained primary motor cortex (BA4), dorso-lateral prefrontal cortex (BA9), and primary visual cortex (BA17). Despite the post-mortem intervals we found that there was a significant effect of HD on mitochondrial iron (Fig. 1H); cytosolic iron was also increased (Fig. 1I). Mouse and human HD data together suggest that brain iron accumulation occurs in HD mitochondria.
Mitochondrial iron homeostatic proteins are disrupted in mouse and human HD
Precise regulation of mitochondrial iron uptake, utilization, and export is essential for mitochondrial health. Mitoferrin 2 and ABCB8 are mitochondrial iron uptake and export proteins, respectively. Mitochondrial mitoferrin 2 levels were significantly increased in R6/2 HD striatum and cortex (Fig. 2A); however, there were no changes in ABCB8 in these brain areas (Fig. 2B). Frataxin is a mitochondrial protein important for iron assimilation into iron-sulfur proteins. Mitochondrial frataxin was significantly decreased in R6/2 HD striatum and cortex (Fig. 2C). We also found that activity of the iron-sulfur protein mitochondrial aconitase was decreased in R6/2 HD mice (Fig. S2). To determine if the findings in R6/2 mice extend to humans, we quantified mitoferrin 2 and frataxin levels in human HD and control brains. HD brains showed increased mitoferrin 2 in BA9 and BA17 (Fig. 2D). Frataxin was decreased in BA4 and BA9 (Fig. 2E) (the p-value for the genotype was 0.0503, so the pair-wise comparisons are unprotected). Together, the findings suggest that there is increased mitochondrial iron uptake with decreased utilization in HD brain which may lead to the accumulation of labile iron.
Figure 2. Human and mouse HD brains demonstrate disruption of mitochondrial iron homeostatic proteins.

A–C. Mitochondrial protein analyses in 12-week-old R6/2 HD mice. n = 6. Iron uptake mitoferrin 2 is increased in striatal and cortical HD mitochondria (A). Iron export protein ABCB8 is unaltered in striatal and cortical HD mitochondria (B). Iron-sulfur synthesis protein frataxin is decreased in striatal and cortical HD mitochondria of R6/2 HD mice (C). D–E. Mitochondrial protein analyses in human cerebral cortex. n = 5. Mitoferrin 2 levels are increased in BA9 and BA17 (D). Frataxin levels are decreased in BA4 and BA9 (E). Control values are set at 1. p < 0.05, **p < 0.01, ***p < 0.001. BA4, Brodmann area 4; BA9, Brodmann area 9; BA17, Brodmann area 17.
Iron chelation rescues HD mitochondrial function ex vivo
We hypothesized that increased mitochondrial labile iron would disrupt normal mitochondrial function. MMP was decreased from the middle stages of HD in the R6/2 and YAC128 mouse models (Fig. 3A–B, Fig. S3). To address a role for iron we used the lipophilic iron-selective and FDA-approved chelator DFP in purified mitochondria and showed significant rescue of membrane potential in HD mice (Fig. 3C Fig. S4); however, there was no effect of the hydrophilic iron-selective chelator deferoxamine (Fig. 3D) suggesting that the chelator must enter the mitochondrial matrix to mediate its effect. The mixed chelator clioquinol (Fig. 3E) did not rescue at ≤ 10 µM. Furthermore, the copper and calcium-selective chelators trientine and EGTA did not rescue the membrane potential deficit (Fig. 3F–G). The protective effect of DFP was significantly blocked by adding iron, but not by calcium, to HD mitochondria (Fig. 3H, Fig. S5). MMP deficits in HD mice correlated with decreased mitochondrial ATP and increased levels of the oxidative stress marker oxidized glutathione (Fig. S6A–C). Mitochondrial respiration is fundamental for the maintenance of the MMP. Mitochondrial oxygen uptake was decreased in R6/2 HD mice, and DFP rescued this effect (Fig. 3I). Taken together, our results indicate that labile iron promotes mitochondrial bioenergetic dysfunction in HD.
Figure 3. Iron-selective chelation rescues mitochondrial membrane potential and oxygen uptake deficits in HD mice.

Studies were completed in mitochondria isolated from brains of R6/2 HD or littermate mice. Unless stated otherwise, mice were 12 weeks old. A–B. HD disease stage-dependent decreases in striatal (A) and cortical (B) MMP. n = 9 or 10. C–G. Effect of metal chelators on MMP in mitochondria purified from cerebral cortices of R6/2 HD and wild-type mice. Log doses: 1 = 10 nM; 2 = 100 nM; 3 = 1 µM, etc. n= 10. C–D. Effect of iron-selective chelators. The lipophilic iron chelator DFP rescues MMP (C), but hydrophilic deferoxamine does not (D). E–G. Effect of non-iron-selective chelators on MMP. The mixed low-affinity chelator clioquinol (E), the copper-selective chelator trientine (F), and the calcium-selective chelator EGTA (G) do not rescue MMP in HD mice. n = 10. H. Rescue of MMP by DFP is iron dependent. The protective effect of DFP is blocked by low concentrations of added iron, but not calcium. n = 10. I. Decreased oxygen consumption rate (OCR) of cortical mitochondria from 12 week old R6/2 HD mice is rescued by DFP. n = 8. *p < 0.05, **p < 0.01, ***p < 0.001. Line graphs: black triangles, wild-type mice; white circles, R6/2 HD mice.
Deferiprone decreases lipid peroxidation rates in HD mitochondria
We further showed that R6/2 HD mitochondria had significantly increased levels of end-products of lipid peroxidation (Fig. 4A). To determine if rates of lipid peroxide production are increased in HD mitochondria and if this increase is iron dependent we measured lipid peroxide production rates in brain mitochondria in the presence and absence of DFP. Striatal and cortical mitochondrial lipid peroxide production rates were increased in HD mice, and the effect was significantly inhibited in striatum by DFP (Fig. 4B–C).
Figure 4. Iron-selective chelation decreases HD-associated mitochondrial lipid peroxidation ex vivo.

Studies were completed on mitochondria isolated from 12 week-old R6/2 HD mice and wild-type controls. A. Steady-state lipid peroxidation product levels are increased in mitochondria purified from striata and cerebral cortices. B–C. In vitro mitochondrial lipid peroxide production rates are increased in HD mitochondria purified from striata (B) and cerebral cortices (C). Lipid peroxidation in striatal mitochondria is decreased by lipophilic iron chelation with [1 µM] DFP. P-values: *p < 0.05, **p < 0.01, ***p < 0.001.
Short-term treatment of R6/2 mice with deferiprone rescues HD mitochondrial markers and improves motor endurance
DFP is an orally available drug that is approved by the FDA for blood transfusion-induced iron overload. Prior to testing deferiprone in HD mice, we first demonstrated that it penetrates brain brain mitochondria (Fig. 5A). We then conducted studies in which mice were treated for 10 days (with two treatments per day) with 150 mg/kg DFP by gavage starting at 9.5 weeks of age. Control and DFP groups of HD mice had performed similarly at 9 weeks of age, just before treatment initiation; however, 10 days later DFP-treated mice performed significantly better than controls (p = 0.0093). There was no effect of the drug on wild-type mice when pre-treatment baseline differences were taken into account (Fig. 5B). In addition, DFP normalized the mitochondrial iron content in the striata and cortices of HD mice (Fig. 5C–D). Interestingly, treatment significantly increased cytoplasmic iron in the HD striatum only (Fig. S7A–B) suggesting a partial transfer of iron from the mitochondria to the cytosol. Further, the treatment fully restored MMP deficits, resulted in a trend toward increased ATP (p = 0.0737), and decreased levels of lipid peroxide end products (Fig. 5E–G).
Figure 5. Oral deferiprone restores mitochondrial iron levels, rescues mitochondrial pathophysiology, and promotes motor endurance in HD mice.

A. Acute brain mitochondrial pharmacokinetics of DFP. Mice were dosed orally with 150 mg / kg DFP, and brains were harvested 2 hours later. DFP is present in a cortical mitochondrial fraction in HD and wild-type mice. n = 8 female mice. B–G. R6/2 and wild-type mice were dosed orally twice daily for 10 days starting at 9.5 weeks and brains were harvested 2–3 hours after the last dose. n = 9 or 10. Rota-rod activity in R6/2 HD and wild-type mice was measured at 5, 9, and 11 weeks of age (B). DFP treatment began after the 9 week measurement and increased motor endurance at 11 weeks in HD but not wild-type mice. C–D. DFP normalizes striatal (C) and cortical (D) mitochondrial iron levels in HD mice, but has no effect on wild-type littermates. E. Decreased cortical MMP in HD mice is rescued by DFP. F. Cortical mitochondrial ATP (mATP) levels are significantly decreased in HD mice. There is a trend toward increased mitochondrial ATP in DFP-treated HD mice. G. Deferiprone rescues increased mitochondrial lipid peroxidation product levels in striata of HD mice but has no effect in wild-type controls. *p < 0.05, **p < 0.01, ***p < 0.001.
Neonatal iron supplementation (NIS) promotes mitochondrial dysfunction in adult HD mice
Previous work from our laboratory has demonstrated that supplementation of iron in neonates potentiates the HD phenotype in R6/2 and YAC128 HD mice [43, 44]. NIS also potentiates disease in models of Parkinson and Alzheimer diseases [42], although the mechanisms for these effects are poorly understood. Here we analyzed whether NIS affects brain mitochondrial function. The dose of iron supplementation used was designed to model human infant supplementation and did not result in short-term weight effects; however, with advancing disease, that same level of iron supplementation resulted in potentiation of HD-associated weight loss in R6/2 mice (Fig. 6A). NIS also potentiated the increases in brain mitochondrial iron in HD mice, but not in wild-type littermates (Fig. 6B–C). There were, however, no effects of iron in the cytoplasmic fraction (Fig. S8A–B). To investigate if NIS affected the levels of mitochondrial iron homeostatic proteins, we analyzed mitoferrin 2, ABCB8, and frataxin protein levels but found no effect in HD and wild-type mice (Fig. S9A–F). NIS potentiated declines in MMP (Fig. 6D–E), and this effect was partly rescued ex vivo by DFP (Fig. 6F). NIS did not significantly alter mitochondrial ATP levels in the cortex but did potentiate increases in brain mitochondrial lipid peroxidation (Fig. 6G–I).
Figure 6. Neonatal iron supplementation (NIS) potentiates mitochondrial dysfunction in HD but not wild-type mice.

A. NIS decreases body weight in HD, but not wild-type mice from 7 to 12 weeks. n = 15–20. B–C. Effect of NIS on mitochondrial iron levels. Iron supplementation increases striatal (B) and cortical (C) mitochondrial iron levels in HD, but not wild-type mice. n = 11 or 12. D–E. Effect of NIS on MMP. The HD-associated decrease in MMP is potentiated by NIS in striatal (D) and cortical (E) mitochondria, but there is no effect in wild-type mice. n = 11 or 12. F. In vitro DFP partially rescues the loss of MMP in iron-supplemented HD mice. n = 11 or 12. G. Cortical mitochondrial ATP (mATP) levels are decreased in HD mice. n = 11 or 12. DFP does not alleviate this decrease. H–I. Effect of NIS on striatal (H) and cortical (I) mitochondrial lipid peroxide levels. Lipid peroxide products are increased by HD. Iron supplementation potentiates lipid peroxidation in HD, but not wild-type mice. n = 11 or 12. *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
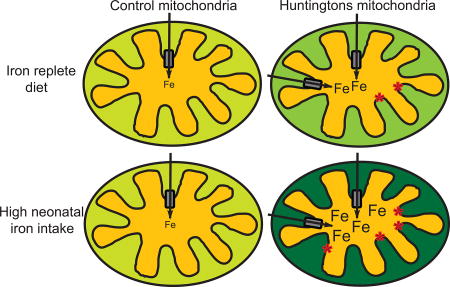
Cellular iron homeostasis is regulated by iron-regulatory proteins 1 and 2 (IRP1/2), which mediate post-transcriptional control of expression of iron homeostatic proteins [70]. Increased and decreased cellular labile iron results in decreased and increased IRP1/2, respectively. Consistent with a state of iron stress in HD brain we have previously reported that IRP1/2 are decreased in the R6/2 mouse model, while the iron-bound ferritin pool is increased [40]. However, the specific mechanisms by which iron stress may potentiate HD have remained unclear. The present study exposes elevated mitochondrial labile iron as a mediator of energetic and oxidative dysfunction in HD. In addition to demonstrating that brain mitochondrial iron levels were increased in two HD models, and in human HD brain, we provided evidence that mitochondrial labile iron altered key markers of bioenergetic status. We further showed that NIS in HD mice, a model for iron nutrition promotion of neurodegeneration [42], was linked with further potentiation of HD mitochondrial disease markers.
Mitochondrial homeostasis is regulated by a number of proteins including iron uptake mitoferrin 2, which was increased in the mouse model and human HD brain samples studied. Interestingly, NIS of HD mice had no effect on the expression of the mitoferrin 2 and frataxin, even though there was promotion of iron accumulation. It is possible that mitochondrial iron accumulation in HD is initially a compensatory response to limit cytosolic iron stress, as supported by decreased IRP1/2 [40]. Alternatively, mitochondrial iron accumulation may be disease promoting, not serving any adaptive role. Taken together however, findings show that HD mitochondria are more prone to iron accumulation in the presence of an iron challenge, and identify mitoferrin 2 as the putative protein involved.
Mitochondrial iron accumulation also occurs in other disorders including red cell precursors in sideroblastic anemias as a result of defective iron assimilation into heme [71]. Mitochondrial iron accumulation is also a feature of FA, where it results from a recessive deficiency of frataxin [9]. We found decreased frataxin in mouse and human HD brains. As transcriptional dysregulation has an important role in HD we searched transcriptional databases but found no evidence to support decreased frataxin transcripts in human and mouse HD [53, 72]. Frataxin is involved in iron-sulfur protein synthesis [73]. Activity of the iron-sulfur enzyme aconitases are deficient in human HD striatum and cortex [74]. Consistent with this, we also demonstrated here a deficit of mitochondrial aconitase in mouse HD. While decreased aconitase activity could be the result of impaired iron-sulfur protein synthesis resulting from frataxin deficiency, other explanations are possible. For example, aconitases are vulnerable to oxidative inactivation by superoxide [75, 76]. More studies are needed to address the extent to which decreased frataxin and oxidative stress contribute to loss of aconitase activity in HD. Consistent with our finding of increased mitochondrial iron-uptake mitoferrin 2 in mouse models and human HD, decreased frataxin expression results in a secondary increase in mitoferrin expression in a Drosophila model [77]. Therefore, whereas HD and FA are different disorders our findings suggest that they share a common mitochondrial-iron disease pathway.
The mitochondrial membrane potential (MMP) is the electrical gradient across the inner mitochondrial membrane and is formed by gradients of protons, potassium, and other ions. The magnitude of the MMP varies according to a number of factors which include the activity of the respiratory chain and proton pumping by complexes I, III, and IV; permeability of the inner mitochondrial membrane to protons and other ions; and also proton usage for ATP generation and proton-driven membrane pumps. R6/2 HD mice have a rapidly progressive phenotype and show advanced HD by 12 weeks of age. In these mice, we found decreased MMP, as determined by using the JC-1 dye, at 12 weeks in cortex and striatum, and also in the cortex at 8 weeks. These findings are consistent with findings from another study that used brain slices from 12-week-old R6/2 mice and CMXRos dye to estimate MMP [78]. To elucidate a potential mechanism for iron in mouse HD that results in these MMP changes, we used the membrane-permeable and -impermeable iron chelators DFP and deferoxamine, respectively. DFP, but not deferoxamine, rescued MMP in HD mice; there were no effects in control mitochondria. Modification of MMP by DFP specifically in HD mice strongly implicates the involvement of labile iron, as iron coordinated in proteins and co-enzymes is unavailable to interact with DFP. Supporting this interpretation, we measured directly and found increases in labile iron in brain mitochondrial fractions. Rescue of MMP by DFP took minutes in our ex vivo studies, indicating a rapid effect of labile iron on mitochondrial function. The mechanisms by which DFP restores MMP and oxygen uptake are currently unclear. One possibility is that there is reactivation of bioenergetics pathways inhibited by oxidative processes. For example, labile iron may promote protein thiol oxidative damage in specific tricarboxylic acid cycle or respiratory chain proteins; this damage could be repaired by mitochondrial enzymes once labile iron is removed [79]. Interestingly, in a HEK-293 cell culture model of FA (frataxin-deficient cells), DFP also restored the impaired MMP, an effect attributed to chelation of mitochondrial labile iron [80].
We found decreased mitochondrial oxygen consumption in brain mitochondrial fractions from 12-week-old R6/2 mice, as compared with controls. The brain mitochondrial OCR was not, however, altered in 6–8-week-old R6/2 mice [81] indicating a disease stage-dependent effect. Mejia et al. [82] reported a 60% reduction in OCR in lymphoblasts isolated from the blood of human HD patient, as compared with controls. In our studies, DFP increased the OCR in HD mitochondria but had no effect on control mitochondria. Although the mechanism of this increase in OCR in HD mitochondria by DFP is unclear, the finding that DFP also increases the MMP is not consistent with an uncoupling effect. It is important to note that the basal respiration of the HD mitochondria was lower than that of control mitochondria. Consistent with this, the resting membrane potential of HD mitochondria was lower than that of control mitochondria, and DFP reversed it. The proton ionophore and uncoupler FCCP increased the OCR in both HD and control mitochondria. However, the increase was smaller with HD mitochondria relative to the control mitochondria, indicating decreased maximum energygenerating capacity. This finding is similar to findings from a magnetic resonance spectroscopy study that measured OCR in vivo in R6/2 brains at 9 weeks of age [83]. The authors found no difference in striatal oxygen uptake in the resting state but a decreased OCR response in the presence of the uncoupler dinitrophenol. Therefore, we interpret our findings as a restorative effect of DFP on HD mitochondrial OCR.
DFP is an orally available iron chelator licensed for the treatment of blood transfusion-induced iron overload in thalassemia major patients. The drug is brain permeable and has also been tested in early-phase clinical trials for Parkinson disease, FA, and amyotrophic lateral sclerosis [84–86]. We proceeded to test HD mice in a short-term dosing study to determine if effects found ex vivo were recapitulated in vivo. Ten days of dosing was sufficient to decrease mitochondrial iron and rescue the MMP.
Lipid peroxidation is increased in mouse and human HD brains [78]. In addition, inhibition of lipid peroxidation in R6/2 HD mice increases the MMP [78]. DFP also decreased mitochondrial membrane lipid peroxidation in our ex vivo and in vivo experiments. Consistent with these findings, there was a significant improvement in motor endurance in DFP-treated HD mice. Our own findings and those of others [87] support a role for mitochondrial oxidative stress in HD. Interestingly, lipid peroxidation and labile iron are also required for ferroptotic cell death [88]. Consistent with this, one study has demonstrated that the ferroptosis inhibitor ferrostatin-1 inhibits cell death in a brain slice model of HD [89].
We and others have used NIS to study its neurodegeneration-promoting effects in mouse models of HD and other neurodegenerative diseases [43, 44, 90, 91]. This dietary model is relevant to human brain aging and neurodegeneration as a result of the high level of iron supplementation in many populations. Whereas numerous studies have demonstrated adverse effects of high iron intake in neonatal rodents on brain outcomes in adults, the mechanisms involved are poorly understood [42]. We showed here that NIS increased iron in mitochondrial fractions and potentiated other mitochondrial markers of HD in the R6/2 model. Iron supplementation in rats causes functional defects in hepatocyte mitochondria [92]. However, we are unaware of prior reports on the effects of NIS on brain mitochondria.
In conclusion, our findings demonstrate that positive and negative regulation of iron has opposite effects on the phenotype of mitochondria from HD brain. These results support a role for the identified mitochondrial-iron pathway as one mediator of HD progression. Importantly, at least in the R6/2 HD model, there is increased mitochondrial susceptibility to the effects of increased iron intake during the neonatal period. Illumination of mechanisms relating to iron and mitochondria in HD may lead to the identification of novel therapeutic targets.
Supplementary Material
Highlights.
Human and mouse Huntington disease (HD) mitochondria dysregulate iron
Mitochondrial labile iron promotes lipid oxidation in mouse HD
Deferiprone normalizes mitochondrial iron and associated HD markers in mice
Neonatal iron supplementation in HD mice potentiates mitochondrial injury
Acknowledgments
We thank Justin Hildenbrand for proving oversight of the ICP-MS analyses; Dr. Feng Li, Pharmakokinetics core facility, University of Michigan, for the HPLC-MS/MS analysis of DFP; David Donley for assisting with mouse experiments; and Dr. Gino Cortopassi, at the University of California, Davis, for providing advice.
Funding:
This work was supported primarily by NIH NINDS R01 NS079450 [to JF], and in part by thematic project support from NIGMS (2P20GM103432) and AHA award number 15BGIA23250030 (to BT). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
List of Abbreviations
- DFP
deferiprone
- HD
Huntington’s disease
- PD
Parkinson’s disease
- AD
Alzheimer’s disease
- TBARS
thiobarbituric acid-reactive substances
- ABCB8
ATP binding cassette subfamily B member 8
- ISC
iron-sulfur cluster
- SDH
succinate dehydrogenase
- LDH
lactate dehydrogenase
- DFO
deferoxamine
- TET
trientine
- EGTA
ethylene glycol-bis-(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid
- FCCP
trifluorocarbonylcyanide phenylhydrazone
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting galley proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gozzelino R, Arosio P. Iron Homeostasis in Health and Disease. International journal ofmolecular sciences. 2016;17 doi: 10.3390/ijms17010130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gille G, Reichmann H. Iron-dependent functions of mitochondria--relation to neurodegeneration. Journal of neural transmission. 2011;118:349–359. doi: 10.1007/s00702-010-0503-7. [DOI] [PubMed] [Google Scholar]
- 3.Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016;388:706–716. doi: 10.1016/S0140-6736(15)01315-X. [DOI] [PubMed] [Google Scholar]
- 4.Allali S, de Montalembert M, Brousse V, Chalumeau M, Karim Z. Management of iron overload in hemoglobinopathies. Transfus Clin Biol. 2017 doi: 10.1016/j.tracli.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Hadzhieva M, Kirches E, Mawrin C. Review: iron metabolism and the role of iron in neurodegenerative disorders. Neuropathology and applied neurobiology. 2014;40:240–257. doi: 10.1111/nan.12096. [DOI] [PubMed] [Google Scholar]
- 6.Schneider SA. Neurodegenerations with Brain Iron Accumulation. Parkinsonism Relat Disord. 2016;22(Suppl 1):S21–25. doi: 10.1016/j.parkreldis.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Rosas HD, Chen YI, Doros G, Salat DH, Chen NK, Kwong KK, Bush A, Fox J, Hersch SM. Alterations in brain transition metals in Huntington disease: an evolving and intricate story. Archives of neurology. 2012;69:887–893. doi: 10.1001/archneurol.2011.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyajima H. Aceruloplasminemia, an iron metabolic disorder. Neuropathology. 2003;23:345–350. doi: 10.1046/j.1440-1789.2003.00521.x. [DOI] [PubMed] [Google Scholar]
- 9.Koeppen AH, Mazurkiewicz JE. Friedreich ataxia: neuropathology revised. Journal of neuropathology and experimental neurology. 2013;72:78–90. doi: 10.1097/NEN.0b013e31827e5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.A novel gene containing a trinucleotide repeat that is expanded unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 11.Vonsattel JP, DiFiglia M. Huntington disease. Journal of neuropathology and experimentalneurology. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Jansen AH, van Hal M, Op den Kelder IC, Meier RT, de Ruiter AA, Schut MH, Smith DL, Grit C, Brouwer N, Kamphuis W, Boddeke HW, den Dunnen WF, van Roon WM, Bates GP, Hol EM, Reits EA. Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia. 2017;65:50–61. doi: 10.1002/glia.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labbadia J, Morimoto RI. Huntington's disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci. 2013;38:378–385. doi: 10.1016/j.tibs.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polyzos AA, McMurray CT. The chicken or the egg: mitochondrial dysfunction as a cause or consequence of toxicity in Huntington's disease. Mech Ageing Dev. 2017;161:181–197. doi: 10.1016/j.mad.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubinsky JM. Towards an Understanding of Energy Impairment in Huntington's Disease Brain. Journal of Huntington's disease. 2017;6:267–302. doi: 10.3233/JHD-170264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mochel F, N'Guyen TM, Deelchand D, Rinaldi D, Valabregue R, Wary C, Carlier PG, Durr A, Henry PG. Abnormal response to cortical activation in early stages of Huntington disease. Mov Disord. 2012;27:907–910. doi: 10.1002/mds.25009. [DOI] [PubMed] [Google Scholar]
- 17.Brennan WA, Jr, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington's disease brain. Journal of neurochemistry. 1985;44:1948–1950. doi: 10.1111/j.1471-4159.1985.tb07192.x. [DOI] [PubMed] [Google Scholar]
- 18.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington's disease caudate nucleus. Annals of neurology. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 19.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. Journal of neuropathology and experimental neurology. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 21.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 22.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 23.Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RL, Olson JM, Jones L, Luthi-Carter R. Regional and cellular gene expression changes in human Huntington's disease brain. Human molecular genetics. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 24.Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Human molecular genetics. 2002;11:1953–1965. doi: 10.1093/hmg/11.17.1953. [DOI] [PubMed] [Google Scholar]
- 25.Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Human molecular genetics. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 26.Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, Deverasetty S, Tebbe A, Schaab C, Lavery DJ, Howland D, Kwak S, Botas J, Aaronson JS, Rosinski J, Coppola G, Horvath S, Yang XW. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci. 2016;19:623–633. doi: 10.1038/nn.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 28.Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Human molecular genetics. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 29.Shirendeb UP, Calkins MJ, Manczak M, Anekonda V, Dufour B, McBride JL, Mao P, Reddy PH. Mutant huntingtin's interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington's disease. Human molecular genetics. 2012;21:406–420. doi: 10.1093/hmg/ddr475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo X, Sun X, Hu D, Wang YJ, Fujioka H, Vyas R, Chakrapani S, Joshi AU, Luo Y, Mochly-Rosen D, Qi X. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington's disease. Nat Commun. 2016;7:12646. doi: 10.1038/ncomms12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yano H, Baranov SV, Baranova OV, Kim J, Pan Y, Yablonska S, Carlisle DL, Ferrante RJ, Kim AH, Friedlander RM. Inhibition of mitochondrial protein import by mutant huntingtin. Nat Neurosci. 2014;17:822–831. doi: 10.1038/nn.3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA, Hayden MR, Masliah E, Ellisman M, Rouiller I, Schwarzenbacher R, Bossy B, Perkins G, Bossy-Wetzel E. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nature medicine. 2011;17:377–382. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferreira IL, Cunha-Oliveira T, Nascimento MV, Ribeiro M, Proenca MT, Januario C, Oliveira CR, Rego AC. Bioenergetic dysfunction in Huntington's disease human cybrids. Exp Neurol. 2011;231:127–134. doi: 10.1016/j.expneurol.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 34.Brustovetsky N, LaFrance R, Purl KJ, Brustovetsky T, Keene CD, Low WC, Dubinsky JM. Age-dependent changes in the calcium sensitivity of striatal mitochondria in mouse models of Huntington's Disease. Journal of neurochemistry. 2005;93:1361–1370. doi: 10.1111/j.1471-4159.2005.03036.x. [DOI] [PubMed] [Google Scholar]
- 35.Lumsden AL, Henshall TL, Dayan S, Lardelli MT, Richards RI. Huntingtin-deficient zebrafish exhibit defects in iron utilization and development. Human molecular genetics. 2007;16:1905–1920. doi: 10.1093/hmg/ddm138. [DOI] [PubMed] [Google Scholar]
- 36.Dietrich P, Johnson IM, Alli S, Dragatsis I. Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet. 2017;13:e1006846. doi: 10.1371/journal.pgen.1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD. Alterations in the levels of iron, ferritin and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia. Brain. 1991;114(Pt 4):1953–1975. doi: 10.1093/brain/114.4.1953. [DOI] [PubMed] [Google Scholar]
- 38.van Bergen JM, Hua J, Unschuld PG, Lim IA, Jones CK, Margolis RL, Ross CA, van Zijl PC, Li X. Quantitative Susceptibility Mapping Suggests Altered Brain Iron in Premanifest Huntington Disease AJNR. American journal of neuroradiology. 2016;37:789–796. doi: 10.3174/ajnr.A4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dominguez JF, Ng AC, Poudel G, Stout JC, Churchyard A, Chua P, Egan GF, Georgiou-Karistianis N. Iron accumulation in the basal ganglia in Huntington's disease: cross-sectional data from the IMAGE-HD study. J Neurol Neurosurg Psychiatry. 2016;87:545–549. doi: 10.1136/jnnp-2014-310183. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Marks E, Lai B, Zhang Z, Duce JA, Lam LQ, Volitakis I, Bush AI, Hersch S, Fox JH. Iron accumulates in Huntington's disease neurons: protection by deferoxamine. PloS one. 2013;8:e77023. doi: 10.1371/journal.pone.0077023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington's disease. Glia. 2007;55:1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- 42.Agrawal S, Berggren KL, Marks E, Fox JH. Impact of high iron intake on cognition and neurodegeneration in humans and in animal models: a systematic review. Nutrition reviews. 2017 doi: 10.1093/nutrit/nux015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berggren KL, Chen J, Fox J, Miller J, Dodds L, Dugas B, Vargas L, Lothian A, McAllum E, Volitakis I, Roberts B, Bush AI, Fox JH. Neonatal iron supplementation potentiates oxidative stress, energetic dysfunction and neurodegeneration in the R6/2 mouse model of Huntington's disease. Redox biology. 2015;4:363–374. doi: 10.1016/j.redox.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berggren KL, Lu Z, Fox JA, Dudenhoeffer M, Agrawal S, Fox JH. Neonatal Iron Supplementation Induces Striatal Atrophy in Female YAC128 Huntington's Disease Mice. Journal of Huntington's disease. 2016;5:53–63. doi: 10.3233/JHD-150182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 46.Petrat F, Weisheit D, Lensen M, de Groot H, Sustmann R, Rauen U. Selective determination of mitochondrial chelatable iron in viable cells with a new fluorescent sensor. The Biochemical journal. 2002;362:137–147. doi: 10.1042/0264-6021:3620137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muhlenhoff U, Richhardt N, Ristow M, Kispal G, Lill R. The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Human molecular genetics. 2002;11:2025–2036. doi: 10.1093/hmg/11.17.2025. [DOI] [PubMed] [Google Scholar]
- 48.Ichikawa Y, Bayeva M, Ghanefar M, Potini V, Sun L, Mutharasan RK, Wu R, Khechaduri A, Jairaj Naik T, Ardehali H. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:4152–4157. doi: 10.1073/pnas.1119338109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santambrogio P, Dusi S, Guaraldo M, Rotundo LI, Broccoli V, Garavaglia B, Tiranti V, Levi S. Mitochondrial iron and energetic dysfunction distinguish fibroblasts and induced neurons from pantothenate kinase-associated neurodegeneration patients. Neurobiology of disease. 2015;81:144–153. doi: 10.1016/j.nbd.2015.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi ZH, Nie G, Duan XL, Rouault T, Wu WS, Ning B, Zhang N, Chang YZ, Zhao B. L Neuroprotective mechanism of mitochondrial ferritin on 6-hydroxydopamine-induced dopaminergic cell damage: implication for neuroprotection in Parkinson's disease. Antioxidants & redox signaling. 2010;13:783–796. doi: 10.1089/ars.2009.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, Yang H, Zhao S, Sato H, Konishi Y, Beach TG, Abdelalim EM, Bisem NJ, Tooyama I. Expression and localization of mitochondrial ferritin mRNA in Alzheimer's disease cerebral cortex. PloS one. 2011;6:e22325. doi: 10.1371/journal.pone.0022325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu WS, Zhao YS, Shi ZH, Chang SY, Nie GJ, Duan XL, Zhao SM, Wu Q, Yang ZL, Zhao BL, Chang YZ. Mitochondrial ferritin attenuates beta-amyloid-induced neurotoxicity: reduction in oxidative damage through the Erk/P38 mitogen-activated protein kinase pathways. Antioxidants & redox signaling. 2013;18:158–169. doi: 10.1089/ars.2011.4285. [DOI] [PubMed] [Google Scholar]
- 53.Narayanan M, Huynh JL, Wang K, Yang X, Yoo S, McElwee J, Zhang B, Zhang C, Lamb JR, Xie T, Suver C, Molony C, Melquist S, Johnson AD, Fan G, Stone DJ, Schadt EE, Casaccia P, Emilsson V, Zhu J. Common dysregulation network in the human prefrontal cortex underlies two neurodegenerative diseases. Mol Syst Biol. 2014;10:743. doi: 10.15252/msb.20145304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fox JH, Connor T, Chopra V, Dorsey K, Kama JA, Bleckmann D, Betschart C, Hoyer D, Frentzel S, Difiglia M, Paganetti P, Hersch SM. The mTOR kinase inhibitor Everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington's disease. Molecular neurodegeneration. 2010;5:26. doi: 10.1186/1750-1326-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Human molecular genetics. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 56.Agrawal S, Singh A, Tripathi P, Mishra M, Singh PK, Singh MP. Cypermethrin-induced nigrostriatal dopaminergic neurodegeneration alters the mitochondrial function: a proteomics study. Molecular neurobiology. 2015;51:448–465. doi: 10.1007/s12035-014-8696-7. [DOI] [PubMed] [Google Scholar]
- 57.Burte F, De Girolamo LA, Hargreaves AJ, Billett EE. Alterations in the mitochondrial proteome of neuroblastoma cells in response to complex 1 inhibition. Journal of proteome research. 2011;10:1974–1986. doi: 10.1021/pr101211k. [DOI] [PubMed] [Google Scholar]
- 58.Cavadini P, Gellera C, Patel PI, Isaya G. Human frataxin maintains mitochondrial iron homeostasis in Saccharomyces cerevisiae. Human molecular genetics. 2000;9:2523–2530. doi: 10.1093/hmg/9.17.2523. [DOI] [PubMed] [Google Scholar]
- 59.Maynard CJ, Cappai R, Volitakis I, Cherny RA, Masters CL, Li QX, Bush AI. Gender and genetic background effects on brain metal levels in APP transgenic and normal mice: implications for Alzheimer beta-amyloid pathology. Journal of inorganic biochemistry. 2006;100:952–962. doi: 10.1016/j.jinorgbio.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 60.Burkitt MJ, Milne L, Raafat A. A simple, highly sensitive and improved method for the measurement of bleomycin-detectable iron: the ‘catalytic iron index' and its value in the assessment of iron status in haemochromatosis. Clinical science. 2001;100:239–247. [PubMed] [Google Scholar]
- 61.Evans PJ, Halliwell B. Measurement of iron and copper in biological systems: bleomycin and copper-phenanthroline assays. Methods in enzymology. 1994;233:82–92. doi: 10.1016/s0076-6879(94)33010-7. [DOI] [PubMed] [Google Scholar]
- 62.Agrawal S, Dixit A, Singh A, Tripathi P, Singh D, Patel DK, Singh MP. Cyclosporine A and MnTMPyP Alleviate alpha-Synuclein Expression and Aggregation in Cypermethrin-Induced Parkinsonism. Molecular neurobiology. 2015;52:1619–1628. doi: 10.1007/s12035-014-8954-8. [DOI] [PubMed] [Google Scholar]
- 63.Banerjee K, Sinha M, Pham Cle L, Jana S, Chanda D, Cappai R, Chakrabarti S. Alpha-synuclein induced membrane depolarization and loss of phosphorylation capacity of isolated rat brain mitochondria: implications in Parkinson's disease. FEBS letters. 2010;584:1571–1576. doi: 10.1016/j.febslet.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 64.Gall J, Skrha J, Jr, Buchal R, Sedlackova E, Verebova K, Platenik J. Induction of the mitochondrial permeability transition (MPT) by micromolar iron: liberation of calcium is more important than NAD(P)H oxidation. Biochimica et biophysica acta. 2012;1817:1537–1549. doi: 10.1016/j.bbabio.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 65.Kollau A, Hofer A, Russwurm M, Koesling D, Keung WM, Schmidt K, Brunner F, Mayer B. Contribution of aldehyde dehydrogenase to mitochondrial bioactivation of nitroglycerin: evidence for the activation of purified soluble guanylate cyclase through direct formation of nitric oxide. The Biochemical journal. 2005;385:769–777. doi: 10.1042/BJ20041354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fox JH, Barber DS, Singh B, Zucker B, Swindell MK, Norflus F, Buzescu R, Chopra R, Ferrante RJ, Kazantsev A, Hersch SM. Cystamine increases L-cysteine levels in Huntington's disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. Journal of neurochemistry. 2004;91:413–422. doi: 10.1111/j.1471-4159.2004.02726.x. [DOI] [PubMed] [Google Scholar]
- 67.Ogura R, Sakanashi T, Nagata O, Sugiyama M, Kajiyama K, Nakagawa T, Shin G, Hidaka T. Assay for lipid peroxide content in mitochondria by the thiobarbituric acid reaction. The Kurume medical journal. 1987;34:53–58. doi: 10.2739/kurumemedj.34.53. [DOI] [PubMed] [Google Scholar]
- 68.Santos NC, Castilho RF, Meinicke AR, Hermes-Lima M. The iron chelator pyridoxal isonicotinoyl hydrazone inhibits mitochondrial lipid peroxidation induced by Fe(II)-citrate. European journal of pharmacology. 2001;428:37–44. doi: 10.1016/s0014-2999(01)01291-2. [DOI] [PubMed] [Google Scholar]
- 69.Ferrante RJ. Mouse models of Huntington's disease and methodological considerations for therapeutic trials. Biochimica et biophysica acta. 2009;1792:506–520. doi: 10.1016/j.bbadis.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klausner RD, Rouault TA, Harford JB. Regulating the fate of mRNA: the control of cellular iron metabolism. Cell. 1993;72:19–28. doi: 10.1016/0092-8674(93)90046-s. [DOI] [PubMed] [Google Scholar]
- 71.Cazzola M, Invernizzi R. Ring sideroblasts and sideroblastic anemias. Haematologica. 2011;96:789–792. doi: 10.3324/haematol.2011.044628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, Geschwind DH, Gottesfeld JM. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chiang S, Kovacevic Z, Sahni S, Lane DJ, Merlot AM, Kalinowski DS, Huang ML, Richardson DR. Frataxin and the molecular mechanism of mitochondrial iron-loading in Friedreich's ataxia. Clinical science. 2016;130:853–870. doi: 10.1042/CS20160072. [DOI] [PubMed] [Google Scholar]
- 74.Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH. Biochemical abnormalities and excitotoxicity in Huntington's disease brain. Annals of neurology. 1999;45:25–32. doi: 10.1002/1531-8249(199901)45:1<25::aid-art6>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 75.Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–355. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- 76.Liang LP, Patel M. Iron-sulfur enzyme mediated mitochondrial superoxide toxicity in experimental Parkinson's disease. Journal of neurochemistry. 2004;90:1076–1084. doi: 10.1111/j.1471-4159.2004.02567.x. [DOI] [PubMed] [Google Scholar]
- 77.Navarro JA, Botella JA, Metzendorf C, Lind MI, Schneuwly S. Mitoferrin modulates iron toxicity in a Drosophila model of Friedreich's ataxia. Free radical biology & medicine. 2015;85:71–82. doi: 10.1016/j.freeradbiomed.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 78.Lee J, Kosaras B, Del Signore SJ, Cormier K, McKee A, Ratan RR, Kowall NW, Ryu H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington's disease mice. Acta neuropathologica. 2011;121:487–498. doi: 10.1007/s00401-010-0788-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bak DW, Weerapana E. Cysteine-mediated redox signalling in the mitochondria. Mol Biosyst. 2015;11:678–697. doi: 10.1039/c4mb00571f. [DOI] [PubMed] [Google Scholar]
- 80.Kakhlon O, Manning H, Breuer W, Melamed-Book N, Lu C, Cortopassi G, Munnich A, Cabantchik ZI. Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood. 2008;112:5219–5227. doi: 10.1182/blood-2008-06-161919. [DOI] [PubMed] [Google Scholar]
- 81.Hamilton J, Pellman JJ, Brustovetsky T, Harris RA, Brustovetsky N. Oxidative metabolism and Ca2+ handling in isolated brain mitochondria and striatal neurons from R6/2 mice, a model of Huntington's disease. Human molecular genetics. 2016;25:2762–2775. doi: 10.1093/hmg/ddw133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mejia EM, Chau S, Sparagna GC, Sipione S, Hatch GM. Reduced Mitochondrial Function in Human Huntington Disease Lymphoblasts is Not Due to Alterations in Cardiolipin Metabolism or Mitochondrial Supercomplex Assembly. Lipids. 2016;51:561–569. doi: 10.1007/s11745-015-4110-0. [DOI] [PubMed] [Google Scholar]
- 83.Lou S, Lepak VC, Eberly LE, Roth B, Cui W, Zhu XH, Oz G, Dubinsky JM. Oxygen consumption deficit in Huntington disease mouse brain under metabolic stress. Human molecular genetics. 2016;25:2813–2826. doi: 10.1093/hmg/ddw138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martin-Bastida A, Ward RJ, Newbould R, Piccini P, Sharp D, Kabba C, Patel MC, Spino M, Connelly J, Tricta F, Crichton RR, Dexter DT. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson's disease. Sci Rep. 2017;7:1398. doi: 10.1038/s41598-017-01402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Elincx-Benizri S, Glik A, Merkel D, Arad M, Freimark D, Kozlova E, Cabantchik I, Hassin-Baer S. Clinical Experience With Deferiprone Treatment for Friedreich Ataxia. J Child Neurol. 2016;31:1036–1040. doi: 10.1177/0883073816636087. [DOI] [PubMed] [Google Scholar]
- 86.Moreau C, Danel V, Devedjian JC, Grolez G, Timmerman K, Laloux C, Petrault M, Gouel F, Jonneaux A, Dutheil M, Lachaud C, Lopes R, Kuchinski G, Auger F, Kyheng M, Duhamel A, Perez T, Pradat PF, Blasco H, Veyrat-Durebex C, Corcia P, Oeckl P, Otto M, Dupuis L, Garcon G, Defebvre L, Cabantchik IZ, Duce J, Bordet R, Devos D. Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis? Antioxidants & redox signaling. 2017 doi: 10.1089/ars.2017.7493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Polyzos A, Holt A, Brown C, Cosme C, Wipf P, Gomez-Marin A, Castro MR, Ayala-Pena S, McMurray CT. Mitochondrial targeting of XJB-5-131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Human molecular genetics. 2016;25:1792–1802. doi: 10.1093/hmg/ddw051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, Stockwell BR. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–4556. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaur D, Peng J, Chinta SJ, Rajagopalan S, Di Monte DA, Cherny RA, Andersen JK. Increased murine neonatal iron intake results in Parkinson-like neurodegeneration with age. Neurobiol Aging. 2007;28:907–913. doi: 10.1016/j.neurobiolaging.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 91.Fernandez LL, Carmona M, Portero-Otin M, Naudi A, Pamplona R, Schroder N, Ferrer I. Effects of increased iron intake during the neonatal period on the brain of adult AbetaPP/PS1 transgenic mice. J Alzheimers Dis. 2010;19:1069–1080. doi: 10.3233/JAD-2010-1304. [DOI] [PubMed] [Google Scholar]
- 92.Walter PB, Knutson MD, Paler-Martinez A, Lee S, Xu Y, Viteri FE, Ames BN. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2264–2269. doi: 10.1073/pnas.261708798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.