

Abstract
Silicosis is a lung inflammatory disease caused by chronic exposure to crystalline silica (CS). Leukotriene B4 (LTB4) plays an important role in neutrophilic inflammation that drives silicosis and promotes lung cancer. Here, we examined the mechanisms involved in CS-induced inflammatory pathways. Phagocytosis of CS particles is essential for the production of LTB4 and IL-1β in mouse macrophages, mast cells and neutrophils. Phagosomes enclosing CS particles trigger the assembly of “lipidosome” in the cytoplasm that is likely the primary source of CS-induced LTB4 production. Activation of JNK pathway is essential for both CS-induced LTB4 and IL-1β production. Studies with bafilomycin-A1 and NLRP3 deficient mice revealed that LTB4 synthesis in the lipidosome is independent of inflammasome activation. siRNA knockdown and confocal microscopy studies showed that GTPases Rab5c, Rab40c along with JNK1 are essential for lipidosome formation and LTB4 production. BI-78D3, a JNK inhibitor abrogated CS-induced neutrophilic inflammation in-vivo in an air pouch model. These results highlight an inflammasome independent and JNK activation dependent lipidosome pathway as a regulator of LTB4 synthesis and CS-induced sterile inflammation.
Introduction
Occupational exposure to Crystalline Silica (CS) is a major public health concern with millions of workers in the mining, drilling, quarrying industries and construction sites continuously exposed to CS (1–3). Around two million US workers and several million worldwide are occupationally exposed to CS every year (1). Fine CS particles can escape the mucociliary defense mechanism and enter the lung interstitium, causing an irreversible inflammatory disease called silicosis. Currently, there is no effective therapy to reverse or halt the disease progression and the only viable treatment option available is lung transplantation (3). Silicosis may eventually lead to several other complications including lung infections, autoimmunity and lung cancer (2). Despite confounding factors such as cigarette smoking, poor nutrition, and genetic susceptibility, epidemiological data suggests that CS exposure increases the risk of lung cancer, especially in cigarette smokers (2, 4, 5).
Although silicosis is extensively studied, little is known about the mechanisms that initiate and perpetuate the process. Resident macrophages, mast cells and lung epithelial cells engulf CS particles deposited in the lungs and in turn are activated. Once internalized, CS particles cause phagosomal destabilization and activation of the NLRP3 inflammasome complex that is accompanied by caspase-1 activation and apoptosis (6). Release of CS particles from the apoptotic cells triggers a continuous cycle of phagocytosis resulting in sustained inflammation characterized by secretion of mediators such as ROS/RNS, TNF-α, IL-1β, TGF-β, leukotriene B4 (LTB4) and CXCL1/2 (4, 7–12). This eventually leads to the formation of granulomatous nodules and tissue damage.
LTB4 is a potent neutrophil chemoattractant that acts via G-protein coupled receptor BLT1 (13). Our recent studies showed that LTB4 is an early and important mediator of CS-induced neutrophilic inflammation (14). Mast cell-produced LTB4 initiates an amplification loop of neutrophil recruitment. In macrophages, the CS-induced cell death and phagocytosis loop also contributes to the production of LTB4, thereby perpetuating a chronic inflammatory condition. In the absence of LTB4-BLT1 pathway in K-rasLA1 mice, there was significant abrogation of the CS-promoted lung tumor progression (14). Although LTB4 is a major player in CS-induced sterile inflammation, the key events leading to its production are not well defined. Here, we studied how pathways leading to LTB4 and IL-1β production are interconnected in different cell types to mediate an overall inflammatory response.
In this study, we identified that phagocytic uptake of CS induces rapid formation of “lipidosome” complex, which is the likely site of LTB4 synthesis. LTB4 production in the lipidosome is dependent on JNK activation but independent of the inflammasome pathway. Studies with inhibitors, siRNAs and confocal microscopy demonstrated that Rab GTPases Rab5c, Rab40c along with JNK1 activation are essential for the formation of lipidosome. Lastly, we demonstrated that a JNK inhibitor, BI-78D3, is effective in reducing CS induced sterile-inflammation in an air-pouch model in mice. These studies on the CS-induced inflammatory pathways may provide novel targets for therapeutic intervention of silicosis and associated diseases.
Materials and Methods
Reagents
Crystalline Silica (MIN-U-SIL-5; average particle diameter 1.7 mm) was obtained from US Silica Co., WV, and was baked at 200 °C overnight to make it endotoxin free. Zinc Oxide (ZnO) nanoparticles 10–30nm in size (US Research Nanomaterials, Inc., Houston, TX). Monosodium urate (MSU) crystals (tlrl-msu; Invivogen, San Diego, CA), mCherry expressing E. Coli was described earlier in (16) and fluorescent red latex beads (L3280; Sigma-Aldrich, St. Louis, Missouri) were used as controls in phagocytosis experiments. Calcium ionophone A23187 was purchased from Sigma-Aldrich. The following pharmacological inhibitors were used in the study: Zileuton, Cytocholasin D, SB202190, and BI-78D3 (all from Sigma-Aldrich), Bafilomycin-A1 (Santa Cruz, Dallas, TX) and AG-126 (Cayman Chemicals, Ann Arbor, MI).
Mice, cell lines and primary cells
C57BL/6, 5-LO-deficient Alox5−/− and NALP3-deficient Nlrp3−/− mice were purchased from Jackson laboratories and bred in our facility at the University of Louisville. 6–8 weeks age old mice were used in ex-vivo and in-vivo experiments. All mice were cared for in accordance with the institutional and National Institutes of Health (NIH) guidelines. The University of Louisville Institutional Animal Care and Use Committee (IACUC) approved all the procedures.
Bone marrow derived macrophages
Mice (6–8-week-old) of indicated genotypes were euthanized by cervical dislocation. The hind legs were dissected and the bone marrow cells were flushed out. The bone marrow cells were cultured in Dulbeco’s Modified Eagle Medium (DMEM) containing 10% FBS, 100 units/ml penicillin, 100 mg/ml streptomycin, 2 mM L-glutamine and 50 mM β-mercaptoethanol supplemented with 50 ng/ml recombinant mouse macrophage colony-stimulating factor (BioLegend; San Diego, CA). The cells were plated at a density of 1 million cells per 100-mm tissue culture dishes containing 10 ml of medium. After 3 days, the medium was replaced by 10 ml of fresh growth medium. The cultures were maintained for another 3 days before the experiments. The purity of the cells (>99%) was confirmed using flow cytometry by surface staining for F4/80 and CD11b.
Bone marrow derived mast cells (BMMC) were isolated from 6–8 weeks old mice and cultured as described previously (14)
RAW 264.7 cells (ATCC, TIB-71) were maintained in DMEM containing 10% FBS, 100 units/ml Penicillin, 100 μg/ml Streptomycin, 2mM L-Glutamine and 50 μM β-mercaptoethanol.
Human neutrophils were isolated from healthy donors using the plasma-Percoll gradient as previously described (15). The recruitment of healthy donors and all the procedures involved in blood draws were in accordance with the guidelines approved by the University of Louisville Institutional Review Board. Microscopy evaluation of isolated cells determined to be 90–95 % neutrophils and >97% of cells were viable by Trypan blue exclusion. Following Percoll gradient, a second purification step was performed to obtain 99.9% purity using EasySep Neutrophil Enrichment Kit (StemCell Technologies; Vancouver, Canada).
In-vitro CS stimulation assays
BMDMs were plated at a density of 0.3 million cells per well in a 12 well culture dish in 1 ml or 0.1 million cells per well in a 96 well culture dish in 200 μl of DMEM containing 10% FBS and allowed to attach overnight. BMDMs were primed with 10 ng/ml of LPS (LPS-EK; InvivoGen, San Diego, CA) for 3 hours. The media was changed to 1% FBS containing media to achieve a final volume of 400 μl in 12 well plate or 200 μl in a 96 well plate. The cells were then pre-treated with the pharmacological inhibitors at the indicated concentrations for 1 hour prior to stimulation with 100 μg/cm2 of CS for 3–6 hours.
RAW 264.7 cells were plated at a density of 0.1 million cells in 100 μl in a 96 well plate in 10% FBS containing media. They were primed for 3 hours with 10 ng/ml LPS. The media was changed to 1% FBS containing media. The cells were pre-treated with the compounds for an hour before stimulation with 100 μg/cm2 CS for 3 hours.
Mast cells were plated at a density of 0.1 million cells per well in a 12 well culture dish in 400 μl of serum-free media. Mast cells required no LPS priming. The cells were pre-treated with the inhibitors for an hour before stimulation with 100 μg/cm2 CS for 3 hours.
Neutrophils plated at 0.1 million cells per well in a 96 well plate with 100 μl of serum free media, were primed with 50 ng/ml LPS and stimulated with 100 μg/cm2 CS for 3 hours after pre-treatment with the inhibitors for an hour.
ELISA
Levels of LTB4 and IL-1β in the supernatants of cell culture and air pouch lavage fluid were measured using LTB4 EIA Kit (Cayman Chemicals) and Mouse IL-1β ELISA MAX Standard Kit (Biolegend) respectively, using manufacturer’s instructions. Absorbance was measured using a BioTek (Winooski, VT) microplate reader at 405nm and 450nm for LTB4 and IL-1β respectively.
Confocal microscopy
1 × 106 BMDMs or RAW 264.7 cells were plated in a 35 mm cover-glass bottom dish (World precision Instruments, Sarasota, FL) in DMEM with 10% FBS for attachment overnight. The media was changed to DMEM with 1% FBS and the cells were primed with LPS as described above. The cells were stimulated with 35 μg/cm2 CS and processed for different staining protocols for microscopy as described below. The confocal images were captured using Nikon A1R confocal microscope at 60 x magnification with appropriate lasers as indicated. A minimum of five fields were captured for each sample. Confocal reflection microscopy combined with florescence microscopy was used to visualize internalized CS particles. Reflection was captured by allowing the laser light to directly pass to the detector channel.
BODIPY staining
After 3 hours of treatment with 35 μg/cm2 CS, cells were washed with Hanks-buffered salt solution without calcium chloride and magnesium chloride (HBSS−/−) and incubated with 1 μM BODIPY: 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-sindacene (ThermoFisher Scientific, Waltham, MA) for 1 hour at 37 °C to stain the lipid bodies. Cells were washed again with HBSS−/− and fixed with 4% paraformaldehyde for 15 min. Subsequently, cells were permeabilized with 0.1% saponin and stained with nuclear stain DAPI (4′,6-Diamidino-2-Phenylindole, Dihydrochloride; ThermoFisher Scientific) (17).
Acridine orange staining
BMDMs were loaded with 5 μg/ml acridine orange (Sigma-Aldrich) for 30 min post CS treatment and washed with HBSS−/− before imaging.
Phagosome staining
BMDMs were loaded with 40 μg/ml pHrodo Red Dextran (ThermoFisher Scientific) together with 1 μM BODIPY and DAPI for 1 hour at 37°C, after 3 hours of CS treatment. The cells were washed thoroughly with HBSS−/− before analyzing by confocal microscopy.
Antibody staining
The cells were fixed with 4% paraformaldehyde for 15 min post CS treatment. The cells were permeabilized with 0.1% saponin and blocked with 5% BSA for 1 hour. Subsequently, the cells were incubated with either anti-Caspase-1, anti-Rab-40c, anti-FLAP (sc-56036, sc-514826, sc-28815; Santa Cruz), anti-5-LO (160402; Cayman Chemicals) or anti-LTA4H (ab196607) antibodies at 1:500 dilution overnight at 4°C. The cells were washed three times with 1xPBS and incubated with the appropriate secondary antibody (Alexa Fluor 594 goat anti-mouse IgG A-11032 or Alexa Fluor 594 goat anti-rabbit IgG A-11012; ThermoFisher Scientific) at 1:500 dilution for 1 hour. The cells were washed and stained with DAPI. Additionally, for phagocytosis experiments, Alexa Fluor 488 or 594-Cholera toxin Subunit B (ThermoFisher Scientific) was used at the last step along with DAPI. The cells were washed again before analyzing by confocal microscopy.
For time-lapse studies, LPS-primed BMDMs were loaded with 1 μM BODIPY or Active Caspase-1 stain (ab219935; abcam) along with nuclear stain Hoechst. After an hour, cells were stimulated with 35 μg/cm2 CS for different times, as indicated. The cells were washed with HBSS−/− before analyzing by confocal microscopy.
Air pouch experiment
5 mL of sterile air was injected subcutaneously into the back of 6–8 weeks old mice to generate an air pouch. An additional 3 ml of sterile air was injected into the pouch 3 days later to maintain the integrity of air pouch. 3 days later, the animals were injected with either BI-78D3 (25 mg/kg) or vehicle into the air pouch. After an hour, 1 mg of CS in 1 ml PBS was injected into the air pouch. The control animals received PBS. After 6 hours, the air pouch was rinsed by injecting 3 ml of cold PBS and the lavage fluid was collected.
Flow Cytometry
Cells from the air pouch lavage were stained with CD45-PE-Cy7, CD11b-APC, Ly6G-FITC, F4/80-PerCP, SiglecF-PE fluorochrome labelled antibodies purchased from BD Biosciences or Biolegend, following standard protocols. Cells labeled with isotype-matched antibodies were used as controls. Flow cytometry data were acquired on FACS Calibur or FACS Canto (BD Biosciences) and analyzed using Flowjo software (Tree Star).
Cytospin
The air pouch lavage fluid cells were spun down Shandon Cytospin centrifuge (Shandon Lipshaw) followed by staining with Hema-3 reagents (ThermoFisher Scientific) according to the manufacturer’s recommendations.
siRNA transfection
For siRNA transfections, 20 μl of 168 nM Silencer siRNA (Life Technologies) diluted in Opti-MEM (Life Technologies) was mixed with 10 μl of 0.03% (v/v) Lipofectamine RNAiMax/Opti-MEM (Life Technologies) as described by the manufacturer. siRNA-lipofectamine mixture (30 μl) was added to each well of a white flat-bottom 96-well plate (Greiner). After 10 min at room temperature, 1x104 RAW264.7 macrophages suspended in 80 μl of DMEM+10% FBS were added. Cells were incubated for 48 h at 37 °C with 5% CO2. 48h later, the cells were re-suspendend in DMEM+1% FBS medium, primed with 10ng/ml of LPS for 3 h and stimulated with 100 μg/cm2 CS. After 6h, the supernatant was analyzed for LTB4 production. A total of 110 genes were screened using a pool of three distinct siRNAs (final 10nM with respect to each siRNA) for each gene. 5-LO siRNA and scrambled siRNAs served as positive and negative controls, respectively.
Statistical Analysis
All in-vitro experiments were repeated at least three times and the representative data is shown as mean ± S.E of triplicate cultures. All data were analyzed with Graph Pad Prism4 Software (San Diego, CA). Statistical significance was determined using unpaired Student’s t-test.
Results
Phagocytosis is required for CS-induced LTB4 and IL-1β production
Phagocytosis is essential for the production of various CS-induced pro-inflammatory mediators including IL-1β (6, 18, 19). To examine the requirement of phagocytosis for CS-induced LTB4 production, LPS-primed BMDMs were treated with phagocytosis inhibitor cytocholasin D (CytD). CytD, an F-actin polymerase inhibitor, blocked the production of CS-induced IL-1β and LTB4 (Fig. 1a–b). To determine whether uptake was necessary in other cell types that produce LTB4 in response to CS, murine mast cells, neutrophils and human neutrophils were stimulated with CS in the presence of CytD. CytD completely blocked CS-induced LTB4 production in all these cell types (Fig. 1c). Mast cells and neutrophils did not make any detectable levels of IL-1β. Reflective microscopy in combination with fluorescence staining techniques were used to visualize CS particles inside the cell. In the presence of CytD, the cells did not take-up CS particles (Fig. 1d). Together, these results indicate that phagocytosis of CS particles is necessary for LTB4 production.
Figure 1. Phagocytosis is required for CS-induced LTB4 and IL-1β production.

BMDMs from WT mice (3 × 106 cells) were cultured O/N in 12 well dishes, LPS-primed and stimulated with 100 μg/cm2 CS for 6 h in the presence or absence of Cytocholasin D (CytD, 10 μM) as described in methods. ELISA was performed to assess levels of (a) LTB4 and (b) IL-1β in the culture supernatants. (c) Mouse bone marrow-derived mast cells, mouse bone marrow-derived neutrophils and human neutrophils were stimulated with 100 μg/cm2 CS in the presence or absence of CytD as described in methods. Levels of LTB4 production was measured using ELISA. Data is representative of one of the three experiments. Data are expressed as mean ± SEM. *p< 0.05, **p< 0.01, ***p< 0.001 non-parametric t-test. (d) CS uptake in the presence and absence of CytD was analyzed 2h post stimulation with 100 μg/cm2 CS in LPS-primed BMDM. Cell membrane was stained with Alexa fluor 594 Cholera toxin Subunit B (red), nucleus with DAPI (blue) and CS particles were observed using reflective microscopy. Representative images shown are from one of the three independent experiments.
Inhibition of phagolysosome formation enhances LTB4 production
The production of several CS-induced pro-inflammatory mediators including IL-1β, bFGF, and HMGB1 are directly linked to the activation of inflammasome pathway (20). Bafilomycin-A1 (Baf-A1), an ATPase inhibitor prevents fusion of phagosome and lysosome by inhibiting vacuolar acidification. To investigate whether phagolysosome formation is necessary for LTB4 production, LPS-primed BMDMs were treated with varying doses of Baf-A1 prior to CS stimulation. As previously reported, Baf-A1 completely blocked CS-induced IL-1β production (6) (Fig. 2a). Interestingly, Baf-A1 increased LTB4 production in a dose dependent manner (Fig. 2b). A similar trend was also observed in RAW 264.7 cells, mast cells and neutrophils (Fig. 2c). Approximately three-fold increase in LTB4 production over CS control was observed at 5 μM, 0.5 μM and 1 μM in BMDM, RAW 264.7 cells and human neutrophils, respectively. It is important to note that Baf-A1 alone did not cause LTB4 production (Fig. 2a–c). The effect of Baf-A1 on vacuolar acidification was verified using acridine orange (AO) staining. AO emits green fluorescence in nuclear and cytoplasmic compartments, whereas acidic lysosomes or phagolysosomes are stained orange-red. CS-treated cells showed enlarged lysosomal compartments whereas; in Baf-A1 treated CS exposed cells no lysosomal staining was observed (Fig. 2d). These results suggest that LTB4 production could be upstream of inflammasome activation.
Figure 2. Inhibition of phagolysosome formation enhances LTB4 production.

BMDMs from WT mice (0.1x 106 cells in 200 μl media) were LPS-primed and stimulated with 100 μg/cm2 CS for 6 h in the presence or absence of varying doses of Bafilomycin-A1. ELISA was performed to assess (a) IL-1β and (b) LTB4 levels in the cell culture supernatants. LTB4 production by 0.1x 106 (c) RAW264.7 cells treated with 100μg/cm2 CS for 6h in the presence or absence of varying doses of Bafilomycin-A1. LTB4 production by 0.1x 106 mouse and human neutrophils upon stimulation with 100 μg/cm2 CS in the presence or absence of Baf-A1 (1 μM). Data presented is from one of the three representative experiments. Data are expressed as mean ± SEM. **p< 0.01, ***p< 0.001 non-parametric t test. (d) LPS-primed BMDMs were stimulated with 35 μg/cm2 CS for 3 h in the presence or absence of Baf-A1. Cells were stained with acridine orange (red lysosomes and green DNA/RNA) for 30 min after CS treatment and visualized by confocal microscopy. Images are representative of one of the three experiments. (e) LPS-primed BMDMs from WT, 5-LO−/− and NLRP3−/− were stimulated with 100 μg/cm2 CS for 6 h. ELISA was performed to assess LTB4 levels and IL-1β levels in the cell culture supernatants. Data are expressed as mean ± SEM.
To determine whether LTB4 production is independent of inflammasome activation, BMDMs from 5LO−/− and NLRP3−/− mice were LPS-primed and stimulated with CS for 6 hours. BMDMs from 5LO−/− mice were deficient in CS-induced LTB4 production but produced IL-1β. Similarly, BMDMs derived from NLRP3−/− mice produced LTB4 in response to CS but did not produce IL-1β. Collectively, these results show that CS-induced LTB4 production is an early event that does not require inflammasome activation or IL-1β production (Fig. 2e).
Activation of JNK pathway is essential for CS-induced LTB4 and IL-1β production
Studies have found JNK signaling to play an important role in COPD associated mucus overproduction and tobacco smoke-induced lung tumor promotion (21, 22). To investigate the involvement of MAP Kinases in CS-induced LTB4 production, LPS-primed BMDMs were pre-treated with specific MAPK inhibitors prior to CS exposure. MEK inhibitor, AG-126 had no effect on production of CS-induced LTB4 or IL-1β. p38 inhibitor, SB202190, moderately inhibited LTB4 production but not IL-1β production. JNK inhibitor, BI-78D3, significantly inhibited production of both LTB4 and IL-1β in BMDM (Fig. 3a–b). Treatment with BI-78D3 also completely inhibited CS-induced LTB4 production in mouse mast cells and neutrophils as well as in human neutrophils (Fig. 3c). Real-time PCR analysis revealed that BI-78D3 significantly decreased the expression of neutrophil active chemokines CXCL1, 2, 3 and 5 (Supplementary Fig. 1a). BI-78D3 also blocked Baf-A1-induced increase in LTB4 production in BMDM (data not shown). MTT assay showed BI-78D3 treatment did not significantly increase toxicity of cells over CS vehicle control (Supplementary Fig. 1b). These results demonstrate that JNK activation is required for both CS-induced LTB4 and IL-1β production.
Figure 3. Activation of JNK pathway is essential for CS-induced LTB4 and IL-1β production.
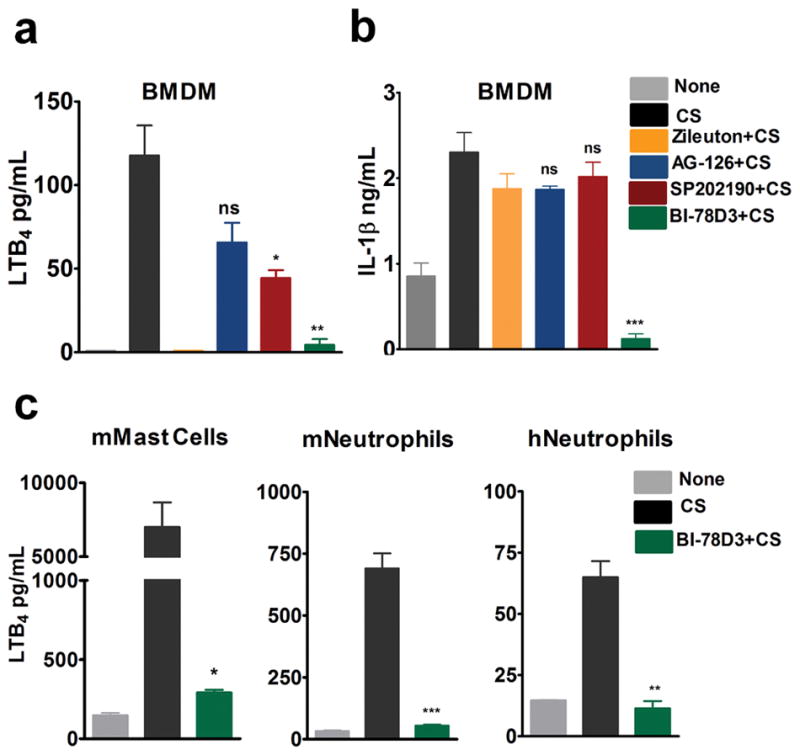
LPS-primed BMDMs (0.3 × 106 cells in 400 μl media) were stimulated with 100 μg/cm2 CS for 6 h in the presence or absence of MAPK inhibitors: AG-126 (10 μM), SB-202190 (10 μM) and BI-78D3 (10 μM). Zileuton was used as a positive control for inhibition of LTB4 synthesis. ELISA was performed to assess (a) LTB4 levels and (b) IL-1β levels in the BMDM cell culture supernatants. CS (100 μg/cm2) induced LTB4 levels were also measured in (c) mouse mast cells, mouse and human neutrophils in the presence of JNK inhibitor. Data representative of n=3. Data are expressed as mean ± SEM. **p< 0.01, ***p< 0.001 non-parametric t-test.
CS-induced lipidosome formation and LTB4 production is inflammasome independent
To visualize the divergence in CS-induced LTB4 and IL-1β pathways, we used BODIPY and caspase-1 staining. LPS-primed BMDMs with/without pre-treatment with pharmacological inhibitors were stimulated with CS for 3 hours, followed by staining with BODIPY, a yeast extract that stains lipid bodies in the cell. An increase in accumulation of lipid bodies, here after referred to as “lipidosome” was observed in the cytoplasm of CS- stimulated cells (Fig. 4a). In the presence of CytD, uptake of CS and lipidosome formation were blocked. There was a significant increase in lipidosome activation in cells that were pre-treated with Baf-A1 prior to CS stimulation. The JNK inhibitor, BI-78D3 completely abrogated the activation of lipidosome (Fig. 4a, upper panel). Confocal microscopy images are quantified in terms of fluorescence intensity of lipidosome and the number of lipid bodies per cell. Increase in lipidosome formation in a cell correlated with the increase in LTB4 produced under these conditions allowing us to speculate that lipidosome may be a source of CS-induced LTB4 production. Inflammasome activation under similar conditions was measured by staining for active caspase-1 as described in Methods. CS-exposed cells showed caspase-1 activation, whereas the cells that were pre-treated with CytD, Baf-A1 or BI-78D3 prior to CS-stimulation showed no caspase 1 staining (Fig. 4b). Both lipidosome and inflammasome pathways are regulated independent of each other as observed in BODIPY and caspase 1 staining of CS-stimulated BMDMs from NLRP3−/− and 5-LO−/− mice. (Fig. 4c). Together, these data provided the evidence that inhibition of phagocytosis or JNK inhibits both lipidosome (LTB4 producing) and inflammasome (IL-1β producing) pathways, whereas Baf-A1 enhances lipidosome activation while blocking the inflammasome pathway.
Figure 4. CS-induced lipidosome formation and LTB4 production is inflammasome independent.

LPS-primed BMDMs were stimulated with 35μg/cm2 CS for 3h in the presence or absence (control) of CytD, Baf-A1 or BI-78D3. (a) The cells were loaded with BODIPY (green) for 60 min. Subsequently, cells were fixed, permeabilized (saponin 0.01%), stained for nucleus using DAPI (blue), and analyzed by confocal microscopy to visualize lipidosome. Lipidosome activation was quantified based on the intensity of BODIPY (each point refers to a unit area in a cell, left panel) and number of lipidosome/cell (right panel). Images shown are representative from one of the five experiments. A minimum of five fields were captured for each sample in every experiment. Data are expressed as mean ± SEM. **p< 0.01, ***p< 0.001 non-parametric t-test. (b) The cells were fixed, permeabilized and stained for: Caspase-1 (red), cell membrane (Alexa fluor 488 Cholera toxin Subunit B, green) and nucleus (DAPI, blue) post CS-treatment to visualize inflammasome activation. (c) LPS-primed BMDMs from 5-LO−/− and NLRP3−/− were stimulated with 35 μg/cm2 CS for 3 h. They were stained with: BODIPY (green) and DAPI (blue) to visualize lipidosome (upper panels). The cells were also stained for Caspase-1 (red), cholera toxin (green) and DAPI (blue) to visualize inflammasome activation (lower panels). Images shown are representative from one of the three experiments.
Kinetics of lipidosome and inflammasome activation
To examine the kinetics, production of LTB4 and IL-1β in the supernatants were measured at time points ranging from 1 hour to 6 hours (Fig. 5a). Significant levels of LTB4 and IL-1β secretion was observed at 1 hour that reached peak levels by 2 hours post CS treatment. At 30 min time point no significant increase in LTB4 or IL-1β levels over untreated cells was observed (data not shown). The kinetics of lipidosome and inflammasome activation was followed in cells loaded with BODIPY or active caspase-1 stain, stimulated with CS and live cell images were captured at various time points (Fig. 5b). Although the accumulation of both LTB4 and IL1β in the supernatant appears to follows a similar time course, live staining data revealed that lipidosome formation occurs as early as 5 min, whereas the earliest caspase-1 activation was observed only at 30 min after CS exposure. Interestingly, stimulation with calcium ionophore A23187 resulted in significant amounts of LTB4 secretion within 5 min of exposure but did not show any lipidosome activation (Fig. 5c).
Figure 5. Kinetics of Lipidosome and inflammasome activation.

LPS-primed BMDMs (0.3 × 106 cells in 400μl media) were stimulated with 100 μg/cm2 CS. (a) LTB4 and IL-1β levels were measured in the supernatants collected at the indicated time points from 1 hour through 6 hours. Data are expressed as mean ± SEM. **p< 0.01, ***p< 0.001 non-parametric t-test. (b) LPS-primed BMDMs were loaded with BODIPY or active caspase-1 stain along with nuclear Hoechst stain, stimulated with 35 μg/cm2 CS and live cell images were captured at indicated time points to observe lipidosome and inflammasome activation. Images shown are representative of one of the three experiments. (c) LPS-primed BMDMs were stimulated with A23187 (10 μM), LTB4 was measured in the supernatants. Representative experiment of n=3. Data are expressed as mean ± SEM. **p< 0.01, ***p< 0.001 non-parametric t-test. In parallel cultures, the cells were stained with BODIPY at 5, 10 15 and 30 min during A23187 (10 μM) treatment and confocal images were obtained. Representative Images from one of the three independent experiments are shown.
CS-induced LTB4 synthesis occurs in phagosome associated lipidosome
To investigate whether the observed lipidosome is in fact the source of CS-induced LTB4 production, LPS-primed BMDMs were stained for 5-LO, FLAP and LTA4H along with BODIPY 3h after CS stimulation. All of the enzymes required for LTB4 synthesis appear to colocalize with the lipidosome, suggesting that lipidosome may not merely be a storage vesicle but likely the site of CS-induced LTB4 production (Fig. 6a). Given that CS-induced lipidosome formation occurs after phagocytosis and before phagolysosome fusion, the cells were loaded with phagosome stain pHrodo dextran along with BODIPY, to spatially localize lipidosome. A z-section and quantification using the intensity plot shows the close association of phagosome and lipidosome (Fig. 6b). To further explore the correlation between phagocytosis and lipidosome formation, we used an array of particles; Monosodium Urate (MSU) crystals, Zinc oxide (ZnO) nanoparticles, inert latex beads and mCherry expressing E. Coli in similar experiments. LPS-primed BMDMs stimulated with CS, MSU, E. Coli particles induced LTB4 production and lipidosome formation (Supplementary Fig. 2). Whereas, phagocytosis of ZnO nanoparticles or inert latex beads did not induce LTB4 production or lipidosome formation. In agreement with previous studies, CS, MSU and latex beads were also capable of activating the inflammasome pathway and IL-1β production (Supplementary Fig. 2) (23, 24).
Figure 6. CS-induced LTB4 synthesis occurs in phagosome associated lipidosome.

(a) LPS-primed BMDMs were stimulated with 35μg/cm2 CS for 3h. The cells were stained for either 5-LO, FLAP or LTA4H (red) and BODIPY (green) as described in Materials and Methods. The intensity plot shows the colocalization of green and red signals. Images shown are representative from one of three independent experiments. (b) LPS-primed BMDMs were loaded with pHrodo red dextran (red) and BODIPY (green) post CS stimulation. A slice of a z-stack showing appearance of lipidosome and phagosomes. The intensity plot shows the proximity of non-overlapping green and red signals. The intensity plot shown is representative of ten separate measurements.
Molecular mediators of CS-induced lipidosome formation
Since LTB4 production in response to CS occurs after phagocytosis and before the fusion of phagosome with the lysosome, we sought to identify specific components in the phagosome maturation pathway by siRNA knockdown experiments to identify the key molecules necessary for lipidosome formation. In previous studies, screening 18,000 murine genes for interference with Yersenia pestis persistence in macrophages led to identification of ~300 genes involved in the phagocytosis and autophagy pathway to be important for Y. pestis survival in macrophages (16). Of those genes, 110 were selected to investigate their roles in CS-induced LTB4 production in RAW264.7 cells. RAW264.7 cells were transfected with siRNA for 48 hours followed by CS stimulation as described in the Materials and Methods. Inhibition of many of these proteins led to more than 50% inhibition of LTB4 levels compared to the scrambled siRNA control. These included genes involved in phagocytosis such as clathrin, dynamin-2; GTPases like Arf6, Rab1, Rab14, Rab40c; signaling kinases like PI3K, JNK1/2, PKA, PKC. In a second screen, we specifically examined proteins involved in lipid metabolism and adapters/scaffold proteins and found that 5-LO, cPLA2-α, Pld2 as well as Plin3, Hip1r, Akap1 were also critical in CS-induced LTB4 production (Supplementary Fig. 3). Almost sixty other genes in our screen such as Caveolin-1, Rab7, Rab8a, Rab20 and Seipin reduced the LTB4 levels by less than 50% relative to scrambled siRNA control.
To further explore the contribution of some of these molecules in lipidosome formation, 7 genes (Table1) were selected. RAW264.7 cells plated in confocal dishes were transfected with siRNA for 48 hours. The cells were then LPS-primed, stimulated with CS for 3 hours and stained with BODIPY and nucleus stain DAPI to analyze lipidosome formation. LTB4 levels were measured in supernatants collected from cells plated in parallel after 6 hours of CS exposure. Knockdown of early endosome marker Rab5c, components of JNK pathway (JNK1, JIP1) and Rab40c inhibited CS-induced LTB4 production whereas knockdown of ASC and Rab18 did not (Fig. 7a). Complete abrogation of lipidosome formation was observed in the presence of Rab5c, JNK1 and Rab40c siRNAs. In concurrence with previous results, knockdown of inflammasome activation with LAMP1 and ASC enhanced lipidosome formation (Fig. 7b). To further validate whether Rab40c is an integral part of the lipidosome structure, LPS-primed BMDMs were stained with Rab40c along with BODIPY 3h after CS stimulation. Fig. 7c shows co-localization of Rab40c with lipidosome.
Table 1.
Select siRNAs tested for their effects on lipidosome formation and LTB4 production
| Sl. No | Gene Symbol | Full Gene Name | Gene ID |
|---|---|---|---|
| 1 | Alox5 | Arachidonate 5-lipoxygenase | 11689 |
| 2 | Rab5c | RAB5C, member RAS oncogene family | 19345 |
| 3 | Pycard | ASC, inflammasome | 66824 |
| 4 | Mapk8 | mitogen activated protein kinase 8 | 26419 |
| 5 | Mapk8ip | mitogen activated protein kinase 8 interacting protein 1 | 19099 |
| 6 | Rab18 | RAB18, member RAS oncogene family | 19330 |
| 7 | Rab40c | Rab40c, member RAS oncogene family | 224624 |
Figure 7. Molecular mediators of CS-induced lipidosome formation.

RAW 264.7 cells (0.1 × 106 cells in 100 μl media) were incubated with the indicated siRNAs for 48 h. Subsequently, the cells were LPS-primed and stimulated with 100 μg/cm2 CS. (a) LTB4 production after 6 h of CS-stimulation was measured using ELISA. Error bars denote SEM. (b) The cells were stained for lipidosome with BODIPY (green) and nucleus with DAPI (blue) 3 h after CS stimulation. (c) After CS treatment, LPS-primed RAW 264.7 cells were stained for lipidosome (BODIPY, green). The cells were then fixed, permeabilized (saponin 0.01%) and stained for Rab40c (red) and DAPI (blue). Representative images from one of the three experiments are shown. A minimum of five fields were captured for each sample in every experiment.
JNK signaling is required for CS-induced neutrophilic inflammation
Neutrophil mediated inflammation is the driving factor for progression of silicosis and CS-promoted lung cancer. Given that BI-78D3 completely blocked the production of LTB4, IL-1β and CXC chemokines in macrophages, mast cells and neutrophils in-vitro, we sought to test the efficacy of the compound in controlling CS-induced inflammation in-vivo by using the air pouch model. Analysis of cellular infiltrates in air pouch upon CS administration using cytospin and flow cytometry revealed that CS induced recruitment of leukocytes, mainly neutrophils and macrophages into the air pouch (Fig. 8a, b). Pre-treatment with BI-78D3 resulted in significant reduction in leukocytes especially neutrophils into the air pouch. BI-78D3 was effective in reducing CS-induced sterile inflammation in-vivo.
Figure 8. JNK signaling is required for CS-induced neutrophilic inflammation.

Sterile air was injected subcutaneously on the back of the WT mice to form an air pouch. CS was injected into air-pouch in the presence/absence of BI-78D3 and 6 hours later inflammation was assessed. (a) Leukocytes on cytospin slides and (b) total leukocytes, neutrophils and macrophages as identified by flow cytometry in the air pouch lavage fluid. Error bars denote SEM. Data are from at least nine mice per group from two separate experiments.
Discussion
LTB4 is an established mediator of diverse inflammatory condtions. In this study, we demonstrate that uptake of CS-particles trigger the formation of a cytoplasmic complex, termed lipidosome, as the site of LTB4 synthesis. Furthermore, CS-induced LTB4 and IL-1β pathways are independently regulated (Fig. 9).
Figure 9. Inflammasome independent lipidosome activation by CS.

Cells take up silica by phagocytosis. Early during phagosome maturation process lipidosome is activated in the cytosol as the primary source of CS-induced LTB4 production. Enzymes required for LTB4 production as well as Rab40c are essential structural components of the CS-induced lipidosome. Inflammasome activation and IL-1β production requires the phagosome fusion with lysosomes. JNK activation is required for both LTB4 and IL-1β production.
The uptake mechanisms of CS largely depend on the size, shape and physiochemical properties of the CS particles. Several studies have suggested that scavenger receptor (SR-A) and MARCO play an important role in CS uptake (8, 25, 26). SR-A is known to go through both clathrin and caveolin mediated endocytosis (27). Other reports suggest that CS can also be taken up through actin-dependent endocytosis (6). In this study, blocking uptake of CS using CytD significantly abrogated IL-1β and LTB4 production (Fig 1). Inflammatory pathways triggered by phagocytosis of CS particles appears closely linked to the phagosome maturation pathway with different mediators such as LTB4, IL-1β and CXC chemokines produced at different stages. Treatment of cells with Baf-A1 prevents phagolysosome formation (28). Although this is a key step in IL-1β production (6), based on the results presented here, it appears to be downstream of LTB4 synthesis. However, a completely independent mechanism for an increase in LTB4 production with Baf-A1 cannot be ruled out. The complete independence of LTB4 and IL-1β pathways is confirmed by the use of primary cells from NLRP3−/− and 5-LO−/− mice (Fig 2).
CS is known to activate several kinases such as ERK1/2, JNK, and p38 MAPKs (25, 29–31). Treatment with MAPK inhibitors revealed that pERK and p38 had little effect on CS-induced LTB4 or IL-1β production at 10 μM. Further studies are needed to illustrate the role, if any, of the ERK kinase family members in CS-induced LTB4 production and inflammation. However, treatment with the JNK inhibitor, BI-78D3 (10 μM) completely blocked both LTB4, IL-1β and CXC chemokine production. These results are in agreement with other reports that suggest JNK activation to be crucial in the development of silicosis (32). Hara et al. have shown the importance of Syk or JNK kinases in activation of NLRP3 and AIM2 inflammasome pathway induced by pathogens. They showed that, both the use of Syk and Jnk kinase inhibitors and knockdown using siRNA resulted in complete reduction of IL-18 as well as caspase-1. The importance of JNK pathway in tobacco smoke induced lung cancer promotion has also been demonstrated (21) suggesting a critical function for JNK in mediating inflammatory pathways in lungs.
Leukotriene biosynthesis was suggested to occur exclusively on the nuclear membrane after the translocation of 5-lipoxygenase (5-LO) into the nucleus (33–35). However, recent studies have demonstrated that leukotriene synthesis can also take place in cytosolic lipid bodies (36–38). Diverse physiological functions including lipid homeostasis, testosterone production and eicosanoid generation were attributed to lipid bodies that are variably named as lipid droplets, adiposomes, lipid bodies etc. (39–41). Here, we used the term “lipidosome” to identify not only the physical form but also a functional complex that allows the coupling of phagocytosis to LTB4 production. The lipidosome can compartmentalize the enzymatic machinery necessary for leukotriene production. An increase in lipid bodies have been observed in various disease conditions like inflammatory arthritis, Crohn’s disease, sepsis and allergy (42). Localization of eicosanoid synthesis enzymes within lipid bodies have confirmed that they could be sites of leukotriene synthesis depending on the cell type and the state of activation (42–46). In this study, we observed that phagocytosis of CS particles triggers the formation of lipidosome in the cytoplasm that is completely blocked by CytD and BI-78D3 treatment. The dissociation of inflammasome activation that requires phagosme fusion with lysosome clearly places the lipidosome activation upstream of this phagosome maturation event (Fig. 4). Kinetics study showing lipidosome activation as early as 5 min after CS exposure as opposed to 30 min for caspase-1 activation (Fig. 5) also supports the hypothesis that lipidosome activation might precede inflammasome activation.
The appearance of the lipidosome is correlated with the LTB4 production under similar conditions. Additionally, antibody staining with 5-LO, FLAP and LTA4H showed that all enzymes involved in LTB4 biosynthesis are localized within the lipidosomes (Fig. 6), indicating that they were the primary sites of CS-induced LTB4 production. Whereas the calcium ionophone A23187 induced LTB4 production did not require the lipidosome formation (Fig. 5c). Simultaneous staining with pHrodo dextran and BODIPY showed non–overlaping but close association of phagosome and lipidosome, providing evidence that it is an early event initiated by phagocytosis. Interestingly, phagocytosis of Urate crystals and E. Coli, which induced LTB4 production in a similar fashion, also triggered lipidosome activation. Whereas, phagocytosis of other particles such as ZnO nano particles or non-toxic latex beads did not induce LTB4 production or the appearance of lipidosome. Thus, the differences in the kinetics of appearance, composition and functional significance of lipidosome activation during the uptake different materials needs further investigation.
Our SiRNA knockdown studies allowed further examination of the phagosome maturation pathway to lipidosome activation. Clearly knock down of large number of genes involved in phagocytosis pathways reduced the levels of CS-induced LTB4 synthesis. Consistent with the results using Baf-A1 and inflammasome KO, knockdown of Rab7a, LAMP1 (data not shown) and ASC did not affect LTB4 production or lipidosome formation. In agreement with our inhibitor studies and known roles of certain Rab proteins (47) knock down of Rab5c and JNK pathway completely abrogated LTB4 production and lipidosome formation. Proteomic studies of lipid bodies in adipocytes have shown high concentration of Rab18 and Rab40c along with ADRP (48, 49). Rab18 which is mainly associated with lipid body biogenesis from ER (48, 50) surprisingly was not found to be essential for the formation CS-induced lipidosome or LTB4 synthesis. However, Rab40c was clearly required for lipidosome formation. Antibody staining with Rab40c also revealed that it is co-localised with BODIPY, suggesting that it is an important structural component of the lipidosome. Further studies are are needed to determine the biochemical composition and activation mechanisms of the CS-induced lipidosome.
Activation of inflammasome pathway and IL-1β production is a hallmark of sterile inflammation (6, 18, 19). Studies with ASC and Nalp3 KO mice showed decreased pulmonary inflammation and fibrosis after CS instillation (3, 51). Therefore, IL-1R antagonists and inflammasome inhibitors have been tested in various mouse models and in clinical trials for silicosis (19). IL-1R antagonists showed moderate reduction in fibrosis and inflammation in some trials, however, its overall therapeutic benefit remains to be established. Similarly, although LTB4 has been implicated in various inflammatory diseases, LTB4 receptor antagonists and LTA4H inhibitors have not seen much success due to the complexities involved in the pathway. Therefore, targetting both LTB4 and IL-1β production through JNK pathway could be an effective way of reducing CS-induced sterile inflammation.
Supplementary Material
Acknowledgments
We thank Becca VonBaby for assistance with mice colony maintenance.
This work was supported by the NIH grant AI-130756 and by the research grants from the Kentucky Lung Cancer Research Board as well as Institutional support from The James Graham Brown Cancer Center.
References
- 1.Leung CC, I, Yu T, Chen W. Silicosis. Lancet. 2012;379:2008–2018. doi: 10.1016/S0140-6736(12)60235-9. [DOI] [PubMed] [Google Scholar]
- 2.Steenland K, Ward E. Silica: a lung carcinogen. CA Cancer J Clin. 2014;64:63–69. doi: 10.3322/caac.21214. [DOI] [PubMed] [Google Scholar]
- 3.Lopes-Pacheco M, Bandeira E, Morales MM. Cell-Based Therapy for Silicosis. Stem Cells Int. 2016;2016:5091838. doi: 10.1155/2016/5091838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox LA., Jr An exposure-response threshold for lung diseases and lung cancer caused by crystalline silica. Risk Anal. 2011;31:1543–1560. doi: 10.1111/j.1539-6924.2011.01610.x. [DOI] [PubMed] [Google Scholar]
- 5.Tse LA, I, Yu T, Qiu H, Leung CC. Joint effects of smoking and silicosis on diseases to the lungs. PLoS One. 2014;9:e104494. doi: 10.1371/journal.pone.0104494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huaux F. New developments in the understanding of immunology in silicosis. Curr Opin Allergy Clin Immunol. 2007;7:168–173. doi: 10.1097/ACI.0b013e32802bf8a5. [DOI] [PubMed] [Google Scholar]
- 8.Hamilton RF, Jr, Thakur SA, Holian A. Silica binding and toxicity in alveolar macrophages. Free Radic Biol Med. 2008;44:1246–1258. doi: 10.1016/j.freeradbiomed.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimbori C, Shiota N, Okunishi H. Involvement of leukotrienes in the pathogenesis of silica-induced pulmonary fibrosis in mice. Exp Lung Res. 2010;36:292–301. doi: 10.3109/01902140903585517. [DOI] [PubMed] [Google Scholar]
- 10.Fubini B, Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med. 2003;34:1507–1516. doi: 10.1016/s0891-5849(03)00149-7. [DOI] [PubMed] [Google Scholar]
- 11.Davis GS, Pfeiffer LM, Hemenway DR. Persistent overexpression of interleukin-1beta and tumor necrosis factor-alpha in murine silicosis. J Environ Pathol Toxicol Oncol. 1998;17:99–114. [PubMed] [Google Scholar]
- 12.Tomaru M, Matsuoka M. The role of mitogen-activated protein kinases in crystalline silica-induced cyclooxygenase-2 expression in A549 human lung epithelial cells. Toxicol Mech Methods. 2011;21:513–519. doi: 10.3109/15376516.2011.568982. [DOI] [PubMed] [Google Scholar]
- 13.Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387:620–624. doi: 10.1038/42506. [DOI] [PubMed] [Google Scholar]
- 14.Satpathy SR, V, Jala R, Bodduluri SR, Krishnan E, Hegde B, Hoyle GW, Fraig M, Luster AD, Haribabu B. Crystalline silica-induced leukotriene B4-dependent inflammation promotes lung tumour growth. Nat Commun. 2015;6:7064. doi: 10.1038/ncomms8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uriarte SM, Rane MJ, Luerman GC, Barati MT, Ward RA, Nauseef WM, McLeish KR. Granule exocytosis contributes to priming and activation of the human neutrophil respiratory burst. J Immunol. 2011;187:391–400. doi: 10.4049/jimmunol.1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connor MG, Pulsifer AR, Price CT, Abu Kwaik Y, Lawrenz MB. Yersinia pestis Requires Host Rab1b for Survival in Macrophages. PLoS Pathog. 2015;11:e1005241. doi: 10.1371/journal.ppat.1005241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melo RC, D’Avila H, Bozza PT, Weller PF. Imaging lipid bodies within leukocytes with different light microscopy techniques. Methods Mol Biol. 2011;689:149–161. doi: 10.1007/978-1-60761-950-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA, Sutterwala FS. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105:9035–9040. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jessop F, Hamilton RF, Rhoderick JF, Shaw PK, Holian A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol Appl Pharmacol. 2016;309:101–110. doi: 10.1016/j.taap.2016.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell. 2010;17:89–97. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mercer BA, D’Armiento JM. Emerging role of MAP kinase pathways as therapeutic targets in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1:137–150. doi: 10.2147/copd.2006.1.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262–2271. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Zhang Y, Xia M, Gulbins E, Boini KM, Li PL. Activation of Nlrp3 inflammasomes enhances macrophage lipid-deposition and migration: implication of a novel role of inflammasome in atherogenesis. PLoS One. 2014;9:e87552. doi: 10.1371/journal.pone.0087552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holian A, Kelley K, Hamilton RF., Jr Mechanisms associated with human alveolar macrophage stimulation by particulates. Environ Health Perspect. 1994;102(Suppl 10):69–74. doi: 10.1289/ehp.94102s1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishijima N, Hirai T, Misato K, Aoyama M, Kuroda E, Ishii KJ, Higashisaka K, Yoshioka Y, Tsutsumi Y. Human Scavenger Receptor A1-Mediated Inflammatory Response to Silica Particle Exposure Is Size Specific. Front Immunol. 2017;8:379. doi: 10.3389/fimmu.2017.00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu XD, Zhuang Y, Ben JJ, Qian LL, Huang HP, Bai H, Sha JH, He ZG, Chen Q. Caveolae-dependent endocytosis is required for class A macrophage scavenger receptor-mediated apoptosis in macrophages. J Biol Chem. 2011;286:8231–8239. doi: 10.1074/jbc.M110.145888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jessop F, Hamilton RF, Jr, Rhoderick JF, Fletcher P, Holian A. Phagolysosome acidification is required for silica and engineered nanoparticle-induced lysosome membrane permeabilization and resultant NLRP3 inflammasome activity. Toxicol Appl Pharmacol. 2017;318:58–68. doi: 10.1016/j.taap.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Hu Y, Jin Z, Jiang H, Wen J. Silica-induced TNF-alpha and TGF-beta1 expression in RAW264.7 cells are dependent on Src-ERK/AP-1 pathways. Toxicol Mech Methods. 2009;19:51–58. doi: 10.1080/15376510802354201. [DOI] [PubMed] [Google Scholar]
- 30.Shen F, Fan X, Liu B, Jia X, Gao A, Du H, Ye M, You B, Huang C, Shi X. Downregulation of cyclin D1-CDK4 protein in human embryonic lung fibroblasts (HELF) induced by silica is mediated through the ERK and JNK pathway. Cell Biol Int. 2008;32:1284–1292. doi: 10.1016/j.cellbi.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 31.Ding M, Shi X, Dong Z, Chen F, Lu Y, Castranova V, Vallyathan V. Freshly fractured crystalline silica induces activator protein-1 activation through ERKs and p38 MAPK. J Biol Chem. 1999;274:30611–30616. doi: 10.1074/jbc.274.43.30611. [DOI] [PubMed] [Google Scholar]
- 32.Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, Mizuguchi J, Schweighoffer E, Tybulewicz V, Mitsuyama M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol. 2013;14:1247–1255. doi: 10.1038/ni.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandal AK, Jones PB, Bair AM, Christmas P, Miller D, Yamin TT, Wisniewski D, Menke J, Evans JF, Hyman BT, Bacskai B, Chen M, Lee DM, Nikolic B, Soberman RJ. The nuclear membrane organization of leukotriene synthesis. Proc Natl Acad Sci U S A. 2008;105:20434–20439. doi: 10.1073/pnas.0808211106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulkarni S, Das S, Funk CD, Murray D, Cho W. Molecular basis of the specific subcellular localization of the C2-like domain of 5-lipoxygenase. J Biol Chem. 2002;277:13167–13174. doi: 10.1074/jbc.M112393200. [DOI] [PubMed] [Google Scholar]
- 35.Luo M, Jones SM, Peters-Golden M, Brock TG. Nuclear localization of 5-lipoxygenase as a determinant of leukotriene B4 synthetic capacity. Proc Natl Acad Sci U S A. 2003;100:12165–12170. doi: 10.1073/pnas.2133253100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bozza PT, Bakker-Abreu I, Navarro-Xavier RA, Bandeira-Melo C. Lipid body function in eicosanoid synthesis: an update. Prostaglandins Leukot Essent Fatty Acids. 2011;85:205–213. doi: 10.1016/j.plefa.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 37.Pacheco P, Vieira-de-Abreu A, Gomes RN, Barbosa-Lima G, Wermelinger LB, Maya-Monteiro CM, Silva AR, Bozza MT, Castro-Faria-Neto HC, Bandeira-Melo C, Bozza PT. Monocyte chemoattractant protein-1/CC chemokine ligand 2 controls microtubule-driven biogenesis and leukotriene B4-synthesizing function of macrophage lipid bodies elicited by innate immune response. J Immunol. 2007;179:8500–8508. doi: 10.4049/jimmunol.179.12.8500. [DOI] [PubMed] [Google Scholar]
- 38.Weller PF, Dvorak AM. Arachidonic acid incorporation by cytoplasmic lipid bodies of human eosinophils. Blood. 1985;65:1269–1274. [PubMed] [Google Scholar]
- 39.Arrese EL, Saudale FZ, Soulages JL. Lipid Droplets as Signaling Platforms Linking Metabolic and Cellular Functions. Lipid insights. 2014;7:7–16. doi: 10.4137/LPI.S11128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Manen HJ, Kraan YM, Roos D, Otto C. Single-cell Raman and fluorescence microscopy reveal the association of lipid bodies with phagosomes in leukocytes. Proc Natl Acad Sci U S A. 2005;102:10159–10164. doi: 10.1073/pnas.0502746102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melo RC, Dvorak AM. Lipid body-phagosome interaction in macrophages during infectious diseases: host defense or pathogen survival strategy? PLoS pathogens. 2012;8:e1002729. doi: 10.1371/journal.ppat.1002729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bozza PT, Magalhaes KG, Weller PF. Leukocyte lipid bodies - Biogenesis and functions in inflammation. Biochim Biophys Acta. 2009;1791:540–551. doi: 10.1016/j.bbalip.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bozza PT, Melo RC, Bandeira-Melo C. Leukocyte lipid bodies regulation and function: contribution to allergy and host defense. Pharmacol Ther. 2007;113:30–49. doi: 10.1016/j.pharmthera.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 44.Bozza PT, Viola JP. Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids. 2010;82:243–250. doi: 10.1016/j.plefa.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 45.Vieira-de-Abreu A, Assis EF, Gomes GS, Castro-Faria-Neto HC, Weller PF, Bandeira-Melo C, Bozza PT. Allergic challenge-elicited lipid bodies compartmentalize in vivo leukotriene C4 synthesis within eosinophils. Am J Respir Cell Mol Biol. 2005;33:254–261. doi: 10.1165/rcmb.2005-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weinstein J. Synovial fluid leukocytosis associated with intracellular lipid inclusions. Arch Intern Med. 1980;140:560–561. [PubMed] [Google Scholar]
- 47.Kiss RS, Nilsson T. Rab proteins implicated in lipid storage and mobilization. J Biomed Res. 2014;28:169–177. doi: 10.7555/JBR.28.20140029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ozeki S, Cheng J, Tauchi-Sato K, Hatano N, Taniguchi H, Fujimoto T. Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J Cell Sci. 2005;118:2601–2611. doi: 10.1242/jcs.02401. [DOI] [PubMed] [Google Scholar]
- 49.Martin S, Parton RG. Characterization of Rab18, a lipid droplet-associated small GTPase. Methods Enzymol. 2008;438:109–129. doi: 10.1016/S0076-6879(07)38008-7. [DOI] [PubMed] [Google Scholar]
- 50.Martin S, Driessen K, Nixon SJ, Zerial M, Parton RG. Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J Biol Chem. 2005;280:42325–42335. doi: 10.1074/jbc.M506651200. [DOI] [PubMed] [Google Scholar]
- 51.Cavalli G, Fallanca F, Dinarello CA, Dagna L. Treating pulmonary silicosis by blocking interleukin 1. Am J Respir Crit Care Med. 2015;191:596–598. doi: 10.1164/rccm.201412-2150LE. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.