

Abstract
By nature of the reversibility of the addition of glutathione to low pKa cysteine residues, the post-translational modification of S-glutathionylation sanctions a cycle that can create a conduit for cell signaling events linked with cellular exposure to oxidative or nitrosative stress. The modification can also avert proteolysis by protection from over-oxidation of those clusters of target proteins that are substrates. Altered functions are associated with S-glutathionylation of proteins within the mitochondria and endoplasmic reticulum compartments, and these impact energy production and protein folding pathways. The existence of human polymorphisms of enzymes involved in the cycle (particularly glutathione S-transferase P) create a scenario for inter-individual variance in response to oxidative stress and a number of human diseases with associated aberrant S-glutathionylation have now been identified.
Keywords: Endoplasmic reticulum, glutathione, glutathione S-transferase, S-glutathionylation, mitochondria, polymorphisms, protein disulfide isomerase, unfolded protein response
Graphical abstract
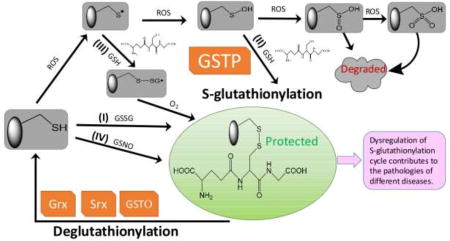
1. Concept of an S-Glutathionylation Cycle
1.1 S-glutathionylation - forward reaction
S-glutathionylation is the reversible post-translational modification of a protein cysteine thiol, with a resultant increase in molecular mass of ~305.6 (from glutathione, GSH) and a net increase in negative charge (from glutamic acid). Not only does this event protect the cysteine against further oxidative damage, it can create distinct structural and functional changes in the target protein. The pKa of the receptive cysteine thiol is critical for S-glutathionylation and the reaction is favored if the three-dimensional environment of the cysteine is basic (low pKa), for example, as may be provided by vicinal arginine, histidine or lysine residues [1]. The typical pKa of a cysteine thiol approximates 8.5, however, such values can range from 3.5 to >12, depending on the local microenvironment. Decreased cysteine pKa values may be attributed to stabilization of the thiolate anion by electron withdrawing groups, or an adjacent positive charge, whereas thiolate pKa may be higher when adjacent to negatively charged residues, or buried within protein folds [2, 3]. Protein S-glutathionylation proceeds either spontaneously or enzymatically (Figure 1) [4, 5]. The non-enzymatic reaction can proceed as follows:
-
(1)
thiol-disulfide exchange between protein thiol (PSH) and glutathione disulfide (GSSG). In this case, the ease of PSSG formation is a product of the equilibrium constant of the reaction (Kmix), expressed as . Therefore, for any protein, the extent of protein S-glutathionylation ( ) depends heavily on the local GSH:GSSG ratio [6, 7]. For most protein cysteines with Kmix ~1 [6], the intracellular GSH:GSSG ratio would have to decline dramatically (i.e. from 100:1 to 1:1) to achieve 50% conversion of PSH to PSSG. Such extreme conditions are rare in vivo, therefore for most proteins, spontaneous PSSG formation through PSH and GSSG exchange is uncommon [7]. However, there are some examples such as c-Jun that have a Kmix ~13 and therefore 50% of c-Jun may be S-glutathionylated when the GSH:GSSG ratio is ~13 [8].
-
(2)
PSH is oxidized by reactive oxygen species (ROS, e.g. H2O2) to a sulfenic acid (PSOH) which then rapidly reacts with GSH to form PSSG. Low µM H2O2 can rapidly oxidize PSH to -SOH, which is unstable and can undergo further oxidization to sulfinic (SO2H) and eventually sulfonic acid (SO3H) which generally irreversibly deactivates the protein. Thus, S-glutathionylation of an SOH group can prevent the target protein from over-oxidation [4, 5]. Under physiological conditions, intracellular H2O2 levels are expected to be in the sub-µM range (10−7–10−9 M) [9], thus spontaneous S-glutathionylation in vivo through this mechanism would occur rather slowly.
-
(3)
PSH is oxidized to a thiyl radical (PS•) which rapidly reacts with GSH to form a thiyl radical glutathionyl intermediate (PSSG•−) which can then react with O2 to form PSSG. Similarly, GS• can react with PSH to form PSSG•−. Thiyl radical mediated protein S-glutathionylation has been suggested to occur in vivo [5], perhaps involving glutaredoxin 1 and 2 (Grx1 and 2) [10, 11].
-
(4)
Nitric oxide (NO) induced S-glutathionylation. NO is itself a weak thiol oxidant, however, either S-nitrosylation (PSNO) or S-glutathionylation (PSSG) can be promoted through secondary generation of reactive nitrogen species (RNS). In addition, PSH may be modified by GSNO to form PSNO and/or PSSG. In vitro studies showed that proteins such as papain, creatine phosphokinase or glyceraldehyde-3-phosphate dehydrogenase are susceptible to both S-nitrosylation and S-glutathionylation by GSNO, whereas alcohol dehydrogenase, bovine serum albumin and actin appear merely to be S-nitrosylated [12]. Furthermore, our early reports showed that treatment of cells with PABA/NO (a glutathione S-transferase (GST) activated diazeniumdiolate prodrug (O2- [2,4-dinitro-5- (N methyl-N-4-carboxyphenylamino) phenyl] 1-N, N-dimethylamino) diazen-1-ium-1, 2-diolate)) resulted in a dose-dependent increase in intracellular NO levels with subsequent undetectable nitrosylation, but drug induced high levels of S-glutathionylation of such proteins as, β-lactate dehydrogenase, Rho GDP dissociation inhibitor β, ATP synthase, elongation factor 2, protein disulfide isomerase (PDI), nucleophosmin-1, chaperonin, actin, protein tyrosine phosphatase 1B, and glucosidase II [13, 14]. Intriguingly, it has been reported that the S-N bond of GSNO has some polarity, with the sulfur being more negatively charged than the nitrogen, indicating that nucleophilic attack of the protein thiolate anion (PS−) on nitrogen to form PSNO should be favored over sulfur to form PSSG [12, 15]. Taken together, such evidence suggests that two distinct pools of S-nitrosylated proteins might exist, one that is GSH stable and another that is GSH labile and subject to rapid conversion to S-glutathionylated products. However, those properties or conditions that favor the implementation of PSSG versus PSNO have yet to be determined.
Figure 1.

Protein S-glutathionylation and deglutathionylation cycle. Protein S-glutathionylation proceeds either spontaneously or enzymatically. The non-enzymatic reaction can proceed as follows: (1) thiol-disulfide exchange between protein thiol (PSH) and glutathione disulfide (GSSG); (2) PSH is oxidized by reactive oxygen species (ROS) to a sulfenic acid (PSOH) which then rapidly reacts with GSH to form PSSG, thus prevent the target protein from over-oxidation to sulfinic (SO2H) and eventually sulfonic acid (SO3H) which generally irreversibly deactivates the protein; (3) PSH is oxidized to a thiyl radical (PS•) which rapidly reacts with GSH to form a thiyl radical glutathionyl intermediate (PSSG•−) which can then react with O2 to form PSSG; (4) PSH may be modified by GSNO to form PSNO and/or PSSG. Protein S-glutathionylation can occur spontaneously, but the rates and magnitude are greatly enhanced by catalytic activity of GSTP. Following a forward reaction dependent on GSTP, deglutathionylation can be achieved by Grx, Srx or GSTO.
Thus, while in certain circumstances, protein S-glutathionylation can occur spontaneously, the rates and magnitude are greatly enhanced by catalytic activity of GST, primarily GSTP [16–22]. The first evidence of this came from reports on cells with depleted or deleted expression of GSTP, where total levels of cellular S-glutathionylation were markedly decreased, implicating GSTP as the causative factor in the forward reaction [18, 22, 23]. When GSTP knockout cells are exposed to ROS, S-glutathionylation of both global and identifiable individual proteins are decreased [18]. Particular examples include peroxiredoxin 6 (Prx6) [16, 17], aldose reductase [19], histone H3 [20], AMP-activated protein kinase (AMPK) [21], 78-kDa glucose-regulated protein/immunoglobulin heavy chain binding protein (GRP78/BiP), PDI, calnexin, calreticulin, endoplasmin, and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) [22]. Despite its primarily cytosolic location, GSTP has also been found in nuclear [24], mitochondrial [25] and endoplasmic reticulum (ER) [22] compartments of the cell. Catalytically, GSTP binds GSH and lowers the pKa of the cysteine residue of GSH from 9.2 [26] to 6.3 [27] through abstraction of the proton by Tyr7, resulting in a thiolate anion (GS−) at the active site. In addition to delivering this thiolate to small molecule substrates, receptive cysteines in proteins can also be targets for transfer. Accordingly, cells expressing a Tyr7 mutant of GSTP also have diminished levels of overall S-glutathionylation when exposed to ROS [18]. For example, reactivation of 1-cys Prx6, where oxidation of the catalytic Cys47 of Prx6 has been associated with its loss of peroxidase activity. However, heterodimerization of Prx6 with GSH-saturated GSTP mediates Cys47 S-glutathionylation in Prx6 and is followed by subsequent spontaneous reduction of the mixed disulfide, thereby restoring its peroxidase activity [16, 17]. A recent study provided evidence that even in the absence of strong oxidants (i.e. mild oxidation - physiological-like conditions), either GSTP or GSTM could interact with AMPK, catalyze S-glutathionylation at residues Cys299 and Cys304, and cause direct conformational changes that activate kinase activity[21].
1.2 Deglutathionylation – reverse reaction
It is meaningful to compare the cyclical nature of S-glutathionylation with the processes of protein phosphorylation and dephosphorylation. Where phosphorylation has forward and reverse reaction kinetics governed by kinases and phosphatases, the S-glutathionylation cycle depicted in Figure 1 has equivalent forward/reverse reactions. S-glutathionylation and deglutathionylation also regulate functional switches integral to homeostatic redox signal transduction [1, 28, 29]. Intriguingly, S-glutathionylation of certain kinases and phosphatases [28] can regulate their enzymatic activities providing an interesting convergence of cyclical post-translational modifications. As discussed above, following a forward reaction dependent on GSTP [16–22], deglutathionylation can be achieved by Grx [30–32], sulfiredoxin (Srx) or GSTP (omega; Figure 1). Mammalian Grx enzymes can be found in numerous sub-cellular locations. Grx1 primarily presents in cytosol (~1 µM) and the mitochondrial inter-membrane space (~0.1 µM); whereas Grx2 is localized predominantly to the mitochondrial matrix (~1 µM) [33, 34]. Grx2 shares ~34% sequence identity with Grx1, but like Grx1, it contains an active site CXXC motif (CSYC versus CPYC on Grx1), the so-called thioredoxin fold that is four mixed β-sheets surrounded by α-helices [35] and the glutathionyl stabilization site [31, 32]. Catalytic mechanisms of Grx1 and Grx2 in the reduction of S-glutathionylated substrates have been reviewed elsewhere [34]. Grx catalyzes the deglutathionylation of target proteins through two successive thiol-disulfide exchange reactions:
-
(1)
The Grx N-terminal active-site cysteine thiolate (Grx-S−) attacks the glutathionyl sulfur of PSSG, forming a Grx-SSG intermediate, releasing reduced PSH. This step is fast relative to the overall rate of the catalyzed reaction and Grx is highly selective and specific for glutathionyl mixed disulfides (PSSG) versus other mixed disulfides (e.g. PSS-Cys which is not a substrate) [11, 36]. The orientation of the γ-glutamyl moiety of GSH seems essential for the Grx selectivity [37]. The non-glutathionyl component of the disulfide substrate appears to be unrestricted and therefore Grx is thought to operate as a general deglutathionylating enzyme. However, the rate constants for deglutathionylation vary more than two orders of magnitude among PSSG substrates and good substrates are distinguished by a combination of high accessibility of the glutathionylated site and low pKa of the cysteine residue [38].
-
(2)
Free GSH attacks the glutathionyl sulfur of the Grx-SSG intermediate releasing reduced Grx and GSSG. GSSG is then reduced to 2GSH by GSSG reductase and NADPH. This is the rate-limiting step, and the rate enhancement by Grx over non-catalyzed rates has been attributed to the unusually low pKa of the active-site cysteine (serves as the leaving group in this step) on Grx 1 (~3.5) or Grx 2 (~4.6) [11], in comparison to the typical pKa ~ 8.5. For classic thiol-disulfide exchange reactions, each 1 pH unit decrease in the pKa of the leaving group thiol predicts a ~4-fold increase in the rate constant, which explains the majority of the rate enhancement of Grx1-mediated deglutathionylation [4ΔpKa = 45 = 1024-fold] [11]. Besides the leaving group effect, an additional enhancement exists for GSH as the second substrate, which suggests an enzyme-induced increase in nucleophilicity of the GS− for the Grx-SSG intermediate [34]. It is worth noting that Grx activities are also affected by the chemical environment, e.g. pH and ROS sources. The different pH values in cytosol (~7.4), mitochondrial inter-membrane space (~7.0), and mitochondrial matrix (~8.0), lead to ranging Grx activities among sub-cellular compartments. Accordingly, compared to cytosol, Grx activity would be ~66% lower in the mitochondrial inter-membrane space and 2.5 times higher in the mitochondrial matrix [34]. Local production of ROS may also favor catalysis of thiyl radical scavenging or protein S-glutathionylation by Grx.
Even though the deglutathionylation reaction has been largely attributed to the actions of Grx, both GSTO1-1 [39] and/or Srx [40, 41] can also serve similar functions. GSTO1-1 has structural similarities to Grx including the thioredoxin-like domain and the glutathionyl stabilization site where glutathione can form a disulfide bond with a conserved active site cysteine residue [39]. However, other GST isozyme families, including, GSTA, GSTM, GSTP, GSTT, GSTS and GSTZ, have catalytic tyrosine or serine residues. In addition, GSTO1-1 has a relatively open active-side pocket that could potentially accommodate a protein or a peptide as a substrate [42]. Therefore, GSTO1-1 catalyzes Grx-like protein deglutathionylation in two similar steps: First, Cys32 on GSTO1-1 attacks PSSG to form Cys32-SSG, releasing reduced PSH. Second, Cys32-SSG is recycled by free GSH to form GSSG and GSTO1-1 is free to catalyze the deglutathionylation of the next protein substrate [43]. The role (if any) that GSTO1-1 plays in S-glutathionylation of proteins is less clear [39, 44].
In vitro studies showed that human Prx1 can be S-glutathionylated at three of the four cysteine residues, including Cys52, Cys173 and Cys83, and the deglutathionylation of Cys83 and Cys173 is preferentially catalyzed by Srx, while deglutathionylation of Cys52 is preferentially catalyzed by Grx [41]. The deglutathionylation carried out by Srx seems have broader substrate specificity. In this regard, work from our laboratory has shown that HEK293 cells transfected with Srx exhibit a decrease in global protein S-glutathionylation following exposure to ROS treatments [40].
In each of the circumstances described above, quantitative amounts of each of these enzymes will be critical determinants of their relative contribution to the cycle. This feature takes on added significance when considering redox metabolism in cancers of various types. Dysregulated redox homeostasis and aberrant signaling are each hallmarks of the disease, but the variability of expression of, for example, GSTP, Srx and Grx in different tumor types can be quite profound. Many tumor cells exist at the biological boundaries of elevated oxidative stress, and yet have adapted their cellular milieu to maintain viability. There is even an indication that nutrient stress may be linked with redox signaling through glycosylation of KEAP1, impacting its interaction with Nrf2 [45]. In this regard, the use of glucose and the existence of dysregulation of redox homeostasis in tumors may be linked. Constructive lines of thought converge on the consideration that redox homeostasis may be the Achilles heel of many types of cancer. Telintra is an example of a cancer drug designed to target GSTP, and while its therapeutic value will be discussed in a later section, its pharmaceutical actions include inhibiting S-glutathionylation, providing more evidence for the role of GSTP in the S-glutathionylation cycle.
2. Examples of how S-glutathionylation alters protein structure/function
2.1 Protein Disulfide Isomerase
PDI is the most abundant chaperone in the lumen of the ER [46], participating in isomerase/chaperone activities in protein folding and in instigation of unfolded protein response (UPR) signaling pathways [47]. The 57 KDa protein has four thioredoxin domains, two of which have CGHC motifs critical to catalytic activities, with two other domains forming a central hydrophobic core pocket for substrate binding [48]. The C terminal KDEL sequence retains it within the ER and two conserved cysteines in the catalytic domains, cycle between disulfide and dithiol forms. From our own studies, drug induced nitrosative stress leads to PDI S-glutathionylation, resulting in diminished activity of the enzyme and activation of UPR [18]. Specific targeted cysteines were identified [13] and the modification was demonstrated to blunt its isomerase activity [46] and interfere with its chaperone activity, abolishing its capacity to form a complex with ERα [49]. S-glutathionylation of PDI alters the secondary and tertiary structure of the protein, measured by a number of distinct criteria. Circular dichroism identified a consistent small decrease in the α-helix content of the S-glutathionylated form of the enzyme. Using tandem mass spectrometry (increase in molecular mass of 305.6) the specific S-glutathionylated cysteine residues were identified. These were localized in the active sites of the a and a′ domains, each containing a CXXC motif. S-glutathionylation of PDI caused a substantial decrease in tryptophanyl fluorescence [14]. In the crystal structure, Cys61 and Cys64 are proximal to a Trp at position 60. Possible disulfides that form at positions Cys90 and Cys97 are too distal from this Trp to have an effect. However, a hypochromic shift of emission maximum of ~4nm after S-glutathionylation indicates some shielding of Trp60 from a polar environment through Cys61 and/or Cys64 S-glutathionylation [14]. Each of these observations indicate that S-glutathionylation alters the tertiary structure of PDI. The involvement of PDI in physiological processes such as thrombus formation, tissue factor regulation, platelet aggregation, cell adhesion and virus internalization [14], suggest that it may be an important factor as an emerging redox sensor and/or promising therapeutic target.
2.2 Peroxiredoxin 6
1-Cysteine Prx6 is an anti-oxidant enzyme that catalyzes the reduction of both H2O2 and organic hydroperoxides using GSH [50]. The strictly conserved cysteine (Cys47) located at its N terminus regulates peroxidase activity, where oxidation to a sulfenate inactivates the Prx6 homodimer [51]. The cysteine residues in the two monomers are buried in the hydrophobic globular core region [52], and restoration of activity is achieved initially through heterodimerization with GSH loaded GSTP where S-glutathionylation occurs [29, 53]. From the crystal structure of Prx6, the catalytic triad of S32, H26, D140 residues provides a basic environment (low pKa) around the single conserved cysteine (Cys47) to make it an appropriate target for S-glutathionylation [1]. The crystal structure depicts the peroxide mediated oxidation of the conserved Cys47 to sulfenic acid [54]. Because of its reactive nature, this oxidized sulfenate monomer is short-lived, although it acquires stability by being inaccessible inside the globular protein. The two oxidized Cys47 in the homodimer are separated by 32Å, not close enough to form a disulfide. Ralat et al.[51] characterized the complex of GSTP/Prx6 by determining: molecular weight of the complex; catalytic activity of the GSTP monomer; interaction sites between GSTP and Prx6. Heterodimer formation with GSTP is accompanied by GSH-dependent reduction, within the complex facilitating access of GSH to the oxidized Cys47 of Prx6, enabling its S-glutathionylation, and eventual regeneration [16, 17]. Active Prx6 is also known to be a major antioxidant in the neuronal tissue of dementia patients with Lewy bodies, and a critical enzyme in neurodegenerative diseases [55] and as discussed in section 6 (below), is an indicator of traumatic brain injury (TBI) [55].
3. S-glutathionylation in sub-cellular compartments
Regulation through S-glutathionylation has been ascribed to a large number of proteins that can be aligned into different functional clusters including, cytoskeletal, glycolysis/energy metabolism, kinase and signaling pathways, calcium homeostasis, antioxidant enzymes and protein folding. Detailed information on these can be obtained from some previous reviews [1, 56]. While it appears that proteins localized in a variety of sub-cellular compartments are represented in these clusters, the prominent role of ROS in both mitochondria and endoplasmic reticulum warrant further attention.
3.1 S-glutathionylation - mitochondrial proteins
Because of the unique physical properties of the matrix, mitochondrial energy metabolism is quite susceptible to adjustment by S-glutathionylation and the importance of S-glutathionylation in mitochondrial function or disease has been reviewed elsewhere [4]. The slight alkalinity of the mitochondrial matrix (pH~8.0) [34] favors protein thiolate formation and thereby enhances its reactivity toward GSH. Mitochondria also contain high concentrations of protein thiols (60–90 mM) and GSH (~1–5 mM) and are a major source of ROS, each of which render S-glutathionylation more likely [57]. Reversal of S-glutathionylation by Grx2 would also be favored by the basic environment and ROS. In principle, S-glutathionylation will provide a rapid, reversible response to redox fluctuations ensuing from the vicissitudes of energy metabolism resulting from, for example, excess mitochondrial superoxide production. There are three operative concerns where S-glutathionylation of target proteins might influence mitochondrial functions (Figure 2):
-
(I)
with enzymes involved in the supply of reducing equivalents to the electron transport system (ETS). Pyruvate dehydrogenase (vicinal lipoate thiols on the E2 subunit) [58], isocitrate dehydrogenase (Cys269) [59], α-ketoglutarate dehydrogenase (vicinal lipoate thiols on the E2 subunit) [60], and malate dehydrogenase [61] are each subject to reversible inhibition by S-glutathionylation. Each generates NADH in the Krebs cycle, thus their S-glutathionylation directly shrinks pools of NADH. Other proteins involved in Krebs cycle, such as aconitase (Acn) and succinyl-CoA transferase are also reversibly inhibited by S-glutathionylation [62]. Cysteine residues on Acn, specifically Cys126 and Cys385 in close proximity to the citrate binding site, are susceptible to S-glutathionylation, which lowers Acn activity, probably by blocking the active site [63]. In this case, S-glutathionylation protects the enzymes from irreversible oxidative deactivation, and ensures the enzymes can be reactivated by Grx2.
-
(II)
with proteins involved in the electron flux through ETS. Complex I is the entry point for electrons into the ETS from NADH to ubiquinone, coupled to the translocation of four protons across the inner mitochondrial membrane, and is a major source of ETS derived superoxide [64]. Several subunits of complex I have been identified as key sites for regulation by S-glutathionylation, including Ndufv1 (51-kDa) [65], Ndufs1 (75-kDa) [66], Ndufs7 [67], Ndufa11 [67], and ND3 [4]. Specifically, Cys531 and Cys704 on Ndufs1 are proximal to the NADH binding site and thus it is probable that S-glutathionylation induces a conformational change which lessens NADH oxidation [66]. Reversibility of S-glutathionylation of Ndufs1 occurs through Grx2 [4, 68]. Mitochondrial complex I can cycle between active (A) and de-activated (D) forms. When the A-form of complex I is left idle in the absence of substrates (NADH and ubiquinone), it converts to the D-form, which can be reactivated by adding substrates to induce catalytic turnover [69]. Of Note, upon de-activation, Cys39 of the ND3 subunit (located in the vicinity of the ubiquinone binding site) becomes exposed and modification of Cys39 by oxidation, nitrosylation, S-glutathionylation or any kind of SH-reagent like N-ethylmaleimide or iodoacetamide prevents reactivation [4, 70, 71]. It would be important to know whether all subunits of complex I (Ndufv1, Ndufs1, Ndufs7, Ndufa11and ND3) can be S-glutathionylated simultaneously or whether these subunits might be differentially S-glutathionylated under physiological or pathological conditions. S-glutathionylation of complex II, specifically Cys90 on the 70-kDa FAD-binding subunit (succinate dehydrogenase A, SdhA), is required to maintain activity. SdhA is actually deglutathionylated during myocardial ischemia/reperfusion [72]. In addition, S-glutathionylation of ATP synthase (Cys294 on the α-subunit of the F1 complex) is associated with low respiratory flux and reversibly inhibited ATPase activity [62, 73].
-
(III)
with transporters involved in the membrane potential of mitochondria. Uncoupling proteins 2 and 3 (UCP 2 and −3, specifically Cys25 and Cys259 on UCP3) appear to be deglutathionylated by Grx1 under high oxidative stress conditions, leading to an increase of the proton leak across the inner mitochondrial membrane and dissipating the membrane potential to reduce mitochondrial ROS production [74, 75]. Other proton leak pathways influenced by the adenine nucleotide transporter (ANT) may be regulated by reversible S-glutathionylation, where the forward reaction prevents mitochondrial membrane permeabilization and protects astrocytes from apoptosis [76, 77].
Figure 2.

S-glutathionylation targets in mitochondria. There are three concerns where S-glutathionylation of target proteins might influence mitochondrial functions: (1) with enzymes involved in the supply of reducing equivalents to the electron transport system (ETS), e.g. pyruvate dehydrogenase, aconitase, isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, succinyl-CoA transferase, and malate dehydrogenase in the Krebs cycle; (2) with proteins involved in the electron flux through ETS, e.g. complex I, complex II and ATP synthase; (3) with transporter involved in the membrane potential of mitochondria, e.g. uncoupling proteins 2 and 3 (UCP2 and −3), and adenine nucleotide transporter (ANT). The specific S-glutathionylation sites of each individual protein and the effect of S-glutathionylation on the protein function are discussed in the text.
Also, relevant to mitochondrial energy metabolism, our own studies with bone-marrow derived dendritic cells (BMDDC), have shown that GSTP mediated S-glutathionylation of estrogen receptor α (ERα) is implicated in switching energy production from oxidative phosphorylation to glycolysis, a metabolic shift universally important in regulating bone marrow proliferation and differentiation pathways [78].
3.2 S-glutathionylation - endoplasmic reticulum proteins
As the organelle responsible for folding of newly synthesized, secreted and membrane-bound proteins, the ER maintains a redox homeostasis that is shifted toward a more oxidized state when compared to the remainder of the cell; the GSH/GSSG ratio ranges from 1:1 to 3:1, compared to the overall cellular ratio of 30:1 to 100:1 [79]. The protein folding machinery of the ER is highly sensitive to ER luminal Ca2+ fluctuations and it has evolved a sophisticated system of quality control, in addition to an UPR pathway to deal with mis-folded proteins [80]. Our work and a review of the literature confirms that several ER proteins enmeshed in a diverse array of ER-processes are subject to S-glutathionylation (Figure 3).
Figure 3.

S-glutathionylation targets in sarco/endoplasmic reticulum (SR/ER). The maintenance of the high Ca2+ concentrations in the SR/ER is primarily achieved by crosstalk between (1) molecular chaperones that bind and buffer Ca2+; (2) Ca2+ pumps that use ATP to work against electrochemical gradients; (3) channel pores that leak or release from SR/ER to the cytosol. Alteration in this balance have the potential to perturb ER proteostasis, resulting in ER stress and the induction of UPR. The effects of S-glutathionylation on Ca2+ release from SR ryanodine receptors (RyRs) and ER inositol 1,4,5-triphosphate receptors (IP3Rs), and on Ca2+ reuptake by sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), have been well reported. The activities of the molecular chaperones in the ER, e.g. calnexin, calreticulin, GRP78/BiP, and PDI, that bind and buffer Ca2+ are also affected by S-glutathionylation. The specific S-glutathionylation sites of each individual protein and the effect of S-glutathionylation on the protein function are discussed in the text. A proportion of the cellular content of GSTP is found in the ER compartment, substantiating the concept that S-glutathionylation may be a crucial regulatory process in the ER.
Intracellular Ca2+ as an important second messenger for signal transduction, is generally stored within the sarcoplasmic reticulum (SR) in muscle cells and the ER in other cell types. When extracellular signaling causes the release of intracellular Ca2+ into the cytosol, the result can be the engagement of various pathways that might promote processes as divergent as gene regulation, proliferation or cell death. The maintenance of the high Ca2+ concentrations in the SR/ER is primarily achieved by crosstalk between (1) molecular chaperones that bind and buffer Ca2+; (2) Ca2+ pumps that use ATP to work against electrochemical gradients; (3) channel pores that leak or release from SR/ER to the cytosol [81]. The effects of S-glutathionylation on Ca2+ release from SR ryanodine receptors (RyRs), ER inositol 1,4,5-triphosphate receptors (IP3Rs) and on Ca2+ reuptake by SERCA have been studied previously. RyRs exist in three isoforms (RyR 1–3) that can exist as homo-tetramers. 12 of the 100 cysteine residues in the RyR1 monomer have been identified as subject to redox modifications. 10 of these (Cys36, 253, 315, 811, 906, 1591, 2326, 2363, 3193 and 3635) are susceptible to S-glutathionylation, and while Cys1040 and 1303 are exclusively S-nitrosylated, whereas Cys1591 and 3193 are exclusively S-glutathionylated. Cys253, Cys315, Cys811 and Cys906 can be S-glutathionylated and S-nitrosylated, Cys36, Cys2326 and Cys2363 can be S-glutathionylated and oxidized to form a disulfide. Cys3635 can be S-glutathionylated, S-nitrosylated, and oxidized to form a disulfide [82]. The combination of S-glutathionylation and S-nitrosylation of RyR1 induced by GSNO seems to decrease the binding of FKBP12 and/or calmodulin binding to RyR1 (both inhibit RyR1), resulting in activation of Ca2+-induced Ca2+ release kinetics [83]. The physiological relevance of each individual cysteine residue identified on RyR1 has yet to be defined. RyR2 is the major intracellular Ca2+ release channel that drives heart contraction. RyR2 contains as many as 90 cysteines per monomer, and the oxidative PTM of these residues, particularly S-glutathionylation and S-nitrosylation, have been suggested to be responsible in regulating RyR2 activity [84]. For example, tachycardia-stimulated NADPH oxidase-dependent RyR2 S-glutathionylation, contributes to sustained faster calcium release rates during conditions of increased cardiac activity. Tachycardia-induced preconditioning was proven to reduce infarct size after ischemia [84, 85]. In addition, in non-excitable cells, Ca2+-induced Ca2+ release by IP3R is enhanced by S-glutathionylation. IP3R S-glutathionylation may represent a fundamental mechanism for regulating IP3R activity during physiological redox signaling during pathological oxidative stress [86].
The SERCA pump is encoded by a family of three genes, SERCA1, 2, and 3, each of which are highly conserved. SERCA1-3 have multiple cysteine residues (24, 29, and 27 respectively) and many of these are susceptible to S-glutathionylation, e.g. Cys344, Cys349, Cys364, Cys498, Cys525, and Cys614 for SERCA1 [87], and Cys344, Cys349, Cys364, Cys498, Cys524, Cys613 and Cys674 for SERCA2 [88]. SERCA (especially SERCA2) S-glutathionylation represents a physiological, cGMP-independent mechanism that stimulates vessel relaxation. Low concentrations of peroxynitrite, generated physiologically from nitric oxide, directly increases the SERCA activity through S-glutathionylation to stimulate the uptake of cytosolic Ca2+ and relax cardiac, skeletal and vascular smooth muscle. This modification is blocked by irreversible cysteine sulfonation during atherosclerosis which may be a contributory factor in the impaired vasodilation of atherosclerotic smooth muscle. Site-directed mutagenesis analysis suggested that S-glutathionylation of Cys674, located in the cytosolic-facing hinge domain, was accountable for SERCA2 activation [88].
The activities of the molecular chaperones in the ER that bind and buffer Ca2+ are also affected by the oxidation state of cysteine residues [89]. Numerous ER-localized chaperones have the capacity to buffer Ca2+ since either the number of binding sites are high, or the Ca2+ association/dissociation ratios are low. These chaperones include calnexin, calreticulin, GRP78/BiP, GRP94 and PDI, each of which contribute to protein folding and quality control [81]. We recently showed that a proportion of the cellular content of GSTP is found in the ER compartment, substantiating the concept that S-glutathionylation may be a crucial regulatory process in the ER [22]. Moreover, there are examples where S-glutathionylation may elicit different outcomes. For example, S-glutathionylation of PDI inhibits both its isomerase and chaperone activities and can be linked with the cytotoxicity of PABA/NO [14, 46]. However, BiP S-glutathionylation increases its holdase activities, but enhances cell proliferation during and after oxidative stress [90]. GSTP promotes S-glutathionylation of ER proteins, and protects the cells from UPR induced cell death caused by drugs that target the ER, such as, thapsigargin (ThG) or tunicamycin (TuM) [22]. ThG is a specific inhibitor of SERCA, altering ER Ca2+ homeostasis and interfering with the functional activities of calcium-dependent chaperones. TuM inhibits N-linked glycosylation leading to the accumulation of misfolded, non-glycosylated proteins in the ER. In either case, saturation of the ER folding capacity can, in turn, induce cell death through influencing the UPR [22] and it appears that reversible S-glutathionylation of key proteins can provide the rheostat for their control.
4. Evidence for possible individual differences in S-glutathionylation?
Evidence for polymorphic SNP variants of GST isozymes has perfused the scientific literature for some time. Correlative associations of disease susceptibility and response to drug therapies or environmental conditions are numerous (over 450 publications in PubMed), but have frequently provided conflicting information about definitive associations or relationships. Since GSTP has a role in the S-glutathionylation cycle, we will focus attention on this isozyme. Human polymorphic variants of the GSTP gene (and their distribution in Caucasian populations) include: wild type GSTP1-1A (Ile105, Ala114; 57%) and three variants, GSTP1-1B (Ile105Val, Ala114; 30%), GSTP1-1C (Ile105Val, Ala114Val; 12%) and GSTP1-1D (Ile105, Ala114Val; 1% [91]). In all instances, mutation of the tyrosine at position 7 precludes the capacity to carry out any of the catalytic functions ascribed to GSTP. In terms of disease associations, the GSTP1-1B allele has been associated with bladder and testicular cancer and decreased GSTP1-1A has been linked with prostate cancer, but associations with colorectal or lung cancer have not been reported [92, 93]. In the context of oxidative stress response, present and ongoing studies consider whether there are population or ethnic differences in the capacity to respond to ROS or RNS. Our group has carried out a series of studies to investigate whether such GSTP variants might influence response to drugs [18], hydrogen peroxide [94] or the capacity to promote reactivation of Prx6 through its S-glutathionylation [53]. In a HEK293 transfection setting, GSTP1-1A possessed the greatest S-glutathionylation activity, with GSTP1-1B having the least of the three polymorphic variants (approximately 50% of 1-1A). It does not seem unreasonable to predict that susceptibility to the impacts of oxidative stress will vary amongst those individuals who express these GSTP polymorphic variants, perhaps in combination with other critical enzymes that maintain redox homeostasis.
Increased expression of Prx6 has also been associated with cancers such as breast [95], and Prx6 also sustains the growth, invasiveness and metastasis of breast cancer cells [96]. Activation of Prx6 as a peroxidase is delimited by heterodimerization with GSTP1-1 [17, 97, 98] and S-glutathionylation enhances enzyme activity [99]. Differences in molecular volume (Ala − 69Å3; Val − 120Å3 and Ile − 204Å3) and side-chain hydrophobicity impact affinity of GSTP1-1 for Prx6 and our previous results showed the affinities of GSTP1-1A or 1-1C for Prx6 to be higher (KD~ 61–68 nM) than those of GSTP1-1B or 1-1D (KD~86–78 nM). Such affinity depends on proximity between the catalytic Cys47-sulfenate of Prx6 and activated GSH (thiolate) bound to the GSTP1-1 allelic variant, regulating the activation of the sulfenate through S-glutathionylation [17]. Peroxidase activities of cells transfected with GSTP1-1A or 1-1C were substantially higher than those with either GSTP1-1B or 1-1D [53]. So in this setting, GSTPB1-1 also has a lower capacity to mediate a specific antioxidant response. Those individuals constitute 30% of the Caucasian population, implying that a greater understanding of polymorphic penetrance would likely facilitate better scrutiny of epidemiological data pertinent to antioxidant response.
5. Inhibiting S-glutathionylation with small molecules
In the 1990’s, a collaborative academic/private sector endeavor led to the synthesis and early testing of an isozyme specific inhibitor of GSTP [100]. Initially named TLK199, preclinical testing has advanced to human clinical trials and in parallel advanced the name to Telintra or Ezatiostat [101, 102]. The phase 2 study evaluated two extended dose schedules of oral Ezatiostat in 89 heavily pretreated patients with low to Intermediate 1 risk myelodysplastic syndromes (MDS). The most common Ezatiostat-related adverse events were grade 1 and 2 gastrointestinal, including nausea, diarrhea and vomiting. Overall, 29% of the RBC transfusion-dependent patients had transfusion reduction, with 11% achieving transfusion independence. The median duration of response was 34 weeks. Multilineage responses were observed and higher responses to Ezatiostat were observed in subsets of patients previously treated with lenalidomide, suggested a potential role for combining the two drugs [101]. Additionally, there are two case reports of MDS patients who responded particularly well to Ezatiostat [103, 104]. The first was a 77-year-old male who relapsed and discontinued lenalidomide, and was randomized to receive Ezatiostat tablets at 3000 mg/day for 14 days of a 21-day cycle, resulting in improvement in all three blood counts that continued to remain high a year post-therapy [103]. The other was a 64-year-old female who suffered from long-standing idiopathic chronic neutropenia (ICN) with frequent episodes of sepsis and had an inadequate response to G-CSF. She responded by the end of the first cycle of treatment with stabilization of her absolute neutrophil count (ANC), clearing of fever and healing of areas of infection. Following 8 cycles of treatment, she continued to show remarkable improvement of ANC [104]. Overall, the available clinical data have shown favorable tolerability and hematopoietic promoting activity profiles for Ezatiostat in MDS patients and indicated that the drug was worthy of further evaluation in randomized phase 2 and phase 3 trials.
The specificity of the drug for GSTP is accommodated by the fact that its binding affinity to the G-site of GSTP1-1 is greater than that of GSH and its selectivity for GSTP1-1 is at least 50-fold greater (Ki ~ 400 nM) than GSTA or GSTM families of isozymes [105, 106]. As discussed, the number of S-glutathionylated proteins in the proteome is not proportionally large but their functional importance can in some cases, make them attractive drug targets in cancer. These might include enzymes with catalytically important cysteines especially those involved with protein folding/stability, nitric oxide regulation and redox homeostasis; cytoskeletal; signaling -particularly kinases and phosphatases; transcription factors; ras proteins; heat shock proteins; ion channels, calcium homeostasis; energy metabolism and glycolysis. Under stress conditions, the half-life of S-glutathionylation approximates 4 h [18]. However, relevant to the use of Telintra, there are instances where interference with S-glutathionylation can have a plausible therapeutic effect. In this regard, the enhanced myeloproliferative phenotype of the GSTP knockout mouse [107], together with the other indications where pharmacological inhibition of GSTP influences bone marrow proliferation and migration [23] dictate that S-glutathionylation of certain key proteins is an important in regulating myeloproliferation in the bone marrow. Most recently, our own studies have suggested that estrogen receptor alpha (ERα) is one such protein, where Cys221, 245, 417, 447 alters the binding affinity for estradiol and regulates bone marrow proliferation and differentiation pathways [78]. Clinical trials with Telintra have been enacted and there appears to be a niche for the drug in myelodysplastic syndrome patients [101, 102]. In addition, certain preclinical studies with TLK117, the activated form of Telintra, have suggested opportunities for its use in lung diseases such as idiopathic pulmonary fibrosis [108].
6. Aberrant redox homeostasis and human disease
Oxidative or nitrosative stress result from exposure to a number of chemical entities that can include, free radical superoxide anion (O2·−) and hydroxyl radicals (OH·), nitric oxide (NO) and non-radical molecules such as hydrogen peroxide (H2O2) and peroxynitrite (ONOO2). Radical groups constituted by reactive sulfur species include hydrogen sulfide, hydrogen polysulfide, thiols and various S-modified cysteine adducts [109] of which S-glutathionylation is one. Based on the emerging fact that the redox network is a mélange of these species, a recent review [110] has introduced reactive species interactome as an integrative biology concept in redox regulation. While sustained, elevated levels of these species can cause imbalance, cell damage and impact the etiology of a number of human diseases, at lower concentrations they can perform as second messengers in physiological situations [111]. Conditions such as hypertension, cardiovascular disease, aging, obesity, asthma, diabetes, neurodegenerative diseases and certain cancers are associated with aberrant redox homeostasis [112, 113].
These result from reversible or irreversible modifications of biomolecules; lipid peroxidation, post-translational modification of proteins: S-glutathionylation, disulfide formation, S-nitrosylation, thiol formation, carbonylation, tyrosine nitration, and mutations in DNA. Specific dysregulation of S-glutathionylation may contribute to the pathogenesis of disorders including obesity, diabetes, inflammation, cancer, cardiovascular, lung, and neurodegenerative diseases covered in recent reviews [1, 4]. Perhaps less well known, are a number of other connections. For example, increased S-glutathionylation of α-tubulin (as well as decreased tyrosination of α-tubulin) has been associated with Friedrich’s ataxia, a neuropathological condition characterized by degeneration of spinal cord pathways [114]. By protein-protein interaction analysis, GSTP1 was identified as a binding partner of the human papilloma virus (HPV-16) E7 oncoprotein. Following mapping of the region in the HPV-16 E7 sequence involved in the interaction, a mutant molecule of HPV-16 E7 with strongly reduced affinity for GSTP1-1 was created. When expressed in human keratinocytes, HPV-16 E7 modified the equilibrium between the oxidized and reduced forms of GSTP1-1 (and in this way presumably altering S-glutathionylation), inhibiting JNK phosphorylation and its ability to induce apoptosis. From the data, a pivotal role for GSTP1-1 in the pro-survival program elicited by its binding with HPV-16 E7 was uncovered [115].
There are also linkages between S-glutathionylation and immune modulatory pathways. For example, cysteine containing peptides are an important class of T cell epitopes and naturally processed MHC-bound peptides are presented on cell surfaces in an MHC allomorph-dependent manner. A significant proportion of these peptides are modified through S-glutathionylation. Alterations in the cellular redox state induced by viral infection are communicated to the immune system through the presentation of S-glutathionylated viral peptides, resulting in altered T cell recognition. Collectively, oxidative stress mediated S-glutathionylation represents a mechanism for modulating the virus-specific T cell response [116]. STAT1 and STAT3 are transcription factors with roles in regulating immune response. Oxidative stress induces S-glutathionylation of both STAT1 and STAT3, but has opposing role in regulation of their signaling, particularly in microglia cells and this impacts immune response [117]. S-glutathionylation also plays a pivotal role in allergic inflammation and airway hyper-responsiveness, suggesting a role for the S-glutathionylation cycle in controlling the nature of the adaptive immune response by promoting Type-2-driven inflammation and restricting IL-17A [118]. From our own recent report, bone marrow derived dendritic cells isolated from GSTP knockout animals had increased cell division, differentiation and decreased protein S-glutathionylation [23]. These same cells also showed that ablation of GSTP was coupled with enhanced expression of estrogen receptor alpha [78]. ERα signaling is directly invested in pathways of development and maturation of dendritic cells and the general sustainability of an immune response [119]. Moreover, ERα broadly impacts glucose metabolism and metabolic gene expression patterns that stimulate resting T cells to proliferate and differentiate into mature T effector cells [120]. GSTP forms a complex with ERα, stimulating S-glutathionylation and this produces an altered ERα binding affinity for estradiol and 3-fold reduced overall binding potential (receptor density and affinity). BMDDC differentiated by granulocyte-macrophage colony-stimulating factor elevate their ERα levels, and this is particularly exaggerated in cells from GSTP knockout mice. When stimulated with lipopolysaccharide, these same BMDDC exhibit: (i) augmented endocytosis, maturation rate, cytokine secretion and T-cell activation; (ii) heightened glucose uptake and glycolysis; (iii) increased Akt signaling (in the mTOR pathway); and (iv) decreased AMPK-mediated phosphorylation of proteins. Taken together, these findings suggest that quantitative changes in levels of GSTP-mediated S-glutathionylation of ERα control BMDDC differentiation and alter metabolic function in dendritic cells.
Traumatic brain injury patients have tissue destruction consistent with consequences of acute oxidative stress. Cerebrospinal fluid (CSF) from patients with severe TBI showed substantial levels of Prx6 and its associated binding partner GSTP. Prx6 was fully active in CSF of control patients but was significantly inactivated (oxidized) in TBI with increased oxidation correlating with severity of trauma. Recovery of Prx6 activity 24h following diagnosis was associated with a more favorable patient outcome. In this regard, Prx6 redox status has the potential to be a biomarker for TBI outcome and perhaps an indicator of therapeutic response [55].
The organism that causes plague, Yersinia pestis, secrets a protein (LcrV) that caps the type III secretion machine. This protein is S-glutathionylated at Cys273 and this modification promotes association with host ribosomal protein S3 (RPS3) and moderates Y pestis type III effector transport and macrophage killing, thereby enhancing bubonic plague pathogenesis in rodents. This activity is abolished by codon substitution C273A in LcrV, and this is accompanied by enhanced animal survival in models of bubonic plague. The mechanism responsible for these virulence attributes is linked to RPS3 as a ligand of LcrV, an association that is perturbed by the lack of S-glutathionylation of Cys273. Macrophages infected with the LcrVC273A variant displayed accelerated apoptotic death and diminished pro-inflammatory cytokine release. LcrV was S-glutathionylated after its transport to the type III needle via disulfide bond formation with extracellular (host organism) oxidized glutathione. Such results show that Y. pestis exploits glutathione in host tissues to activate a virulence strategy, thereby accelerating plague pathogenesis [121].
7. Methods to measure S-glutathionylation
One of the primary challenges in detecting and measuring S-glutathionylated proteins is their generally low levels in cells. Recent technical advances, especially in proteomics, have facilitated enhanced sensitivity of detection and a number of informative reviews have appeared [77, 122, 123]. To detect the protein-SSG bonds two principle approaches (indirect and direct) have been employed. Given the general lack of direct methods, indirect techniques are still a popular choice and can be achieved using the following sequence of steps: (i) The free thiols are initially blocked by thiol-reactive agents such as N-acetylmaleimide (NEM) or iodoacetamide (IAM) during tissue homogenization or cell lysis. NEM reacts with the deprotonated thiolate anions via Michael addition, whereas IAM reacts with the thiolate via nucleophilic substitution as the nucleophile, and iodine as the leaving group. Compared to IAM, NEM is faster by 1–2 orders of magnitude, more specific and less pH-dependent [77]. (ii) Protein-SSG is selectively reduced to free thiols by use of a Grx reduction cocktail (GSH/Glutathione reductase/NADPH). It should be noted that the selective reduction strategy is not perfect in terms of specificity. For example, even with the mutated form of Grx to reduce the risk of disulfide reduction, non-specific reduction of the Grx enzyme cocktail in the absence of Grx is still observed [124]. Therefore, when applying indirect methods, inclusion of proper negative and positive controls is important to enhance confidence in the identification of the redox modification. (iii) The newly reduced thiols are subjected to different enrichment and labeling strategies using thiol-reactive affinity resins [124, 125], biotin tags [126, 127], clickable tags [128], radiolabels, fluorophores [129, 130], isotopic or isobaric mass tags, e.g. heavy/light ICAT (isotope-coded affinity tags) containing biotin linker [82] and 6-plex iodoTMT (tandem mass tags) [131]. Compared to the biotin avidin-based approaches, the resin-assisted capture (RAC) procedure provides a simpler workflow, higher enrichment specificity (>95%), and better sensitivity, as demonstrated by a side-by-side comparison [132]. The RAC also provides the flexibility for enabling multiplex quantification across 4–10 biological samples by allowing on-resin digestion and isobaric labeling with the amine-reactive 4-plex or 8-plex iTRAQ (isobaric tags for relative and absolute quantification), 6-plex or 10-plex TMT [124, 125]. The iodoTMT approach offers the flexibility of multiplexed quantification as well; however, the RAC should provide higher enrichment specificity due to the covalent capture process when compared with the non-covalent anti-TMT immunoaffinity enrichment. (iv) S-glutathionylated proteins can then be separated, visualized, detected or identified by fluorescent-, immunoblotting- or proteomic-based approaches (Table 1, Figure 4).
Table 1.
Comparative methods to measure S-glutathionylation.
| Direct methods | Indirect methods | ||||
|---|---|---|---|---|---|
| Methods | Advantage | Disadvantage | Methods | Advantage | Disadvantage |
| In vivo relevance. | Limited in multiplex capability to compare S-glutathionylation levels of an individual protein. | Most of the indirect methods have multiplex capability to compare S-glutathionylation levels of individual proteins. | Selective reduction strategy is not perfect in terms of specificity. | ||
| Anti-GSH antibodies | Easy to perform and compare global S-glutathionylation levels between different samples. When coupled with immunoprecipitation, the site of the modification can be identified. | Low sensitivities of these antibodies impart a limited detection potential and many of the proteins identified are abundant. | Radiolabel, fluorescent or biotin tag | Easy to perform and compare the global S-glutathionylation levels between different samples. When coupled with two dimensional gel and mass spectrometry, the site of the modification can be identified. | Does not offer the flexibility of quantification of S-glutathionylation levels of individual proteins in different samples. |
| [35S] GSH | Radiolabeled S-glutathionylated proteins can be detected by radiography. When coupled with two dimensional gel and mass spectrometry, the site of the modification can be identified. | This method does not permit discrimination between proteins modified by [35S]GSH and those ligated to [35S]cysteines.. In addition, it requires preincubation with cycloheximide to block protein synthesis, likely perturbing cell physiology. | Isotopically labeled azido-biotin and ICAT | Allows for enrichment of S-glutathionylated proteins on avidin and offers the flexibility of quantification of S-glutathionylation levels of individual proteins in 2 samples. | Time consuming, labor intensive and expensive. Requires mass spectrometry for identification. |
| Biotinlated GSSG or bintinlated GSH ethyl esters | Allows for enrichment of S-glutathionylated proteins on streptavidin. | The bulky nature of biotin is likely to interfere with enzyme-mediated (de)glutathionylation and the artificial introduction of biotinylated GSH or GSSG may alter the endogenous redox status. | IodoTMT | Allows for enrichment of S-glutathionylated proteins with anti-TMT antibodies and offers the flexibility of multiplexed quantification of S-glutathionylation levels of individual proteins (6 samples). | Time consuming, labor intensive and expensive. Requires mass spectrometry for identification. |
| Clickable GSH | The small versatile azide handle seems not to interfere with enzyme-mediated (de) glutathionylation and can couple with a variety of different applications. No exogenous thiol is introduced. | The production of the azido-GSH is dependent on the transfection efficiencies of mutant glutathione synthetase. | Resin-assisted capture | The covalent capture by resin provides simpler workflow, higher enrichment specificity (>95%), and better sensitivity compared to the other indirect methods (ICAT and iodoTMT). It also offers the flexibility of multiplexed quantification of S-glutathionylation levels of individual proteins (4–10 samples). | Time consuming, labor intensive and expensive. Requires mass spectrometry for identification. |
Figure 4.

Indirect methods for identification and quantification of protein S-glutathionylation can be achieved using the following sequence of steps: (1) The free thiols are initially blocked by thiol-reactive agents such as N-acetylmaleimide (NEM) or iodoacetamide (IAM) during tissue homogenization or cell lysis; (2) Protein-SSG is selectively reduced to free thiols by use of a Grx reduction cocktail; (3) The newly reduced thiols are subjected to different enrichment and labeling strategies using thiol-reactive radiolabels, fluorescent groups, affinity resins, biotin tags, and isotopic or isobaric mass tags; (4) S-glutathionylated proteins can then be identified and quantified by radiation-, fluorescent-, immunoblotting- or proteomic-based approaches.
The direct method is based on the use of (i) anti-GSH antibodies to pull down the target S-glutathionylated proteins. However, low sensitivities of these antibodies impart a limited detection potential and many of the proteins identified are abundant [1]. (ii) [35S]cysteine to form radiolabeled GSH. The subsequent radiolabeled S-glutathionylated proteins can be detected by radiography [133]. However, this method does not permit discrimination between proteins modified by [35S]GSH and those ligated to [35S]cysteines. In addition, it requires pre-incubation with cycloheximide and/or chloramphenicol to block protein synthesis, likely perturbing cell physiology. (iii) biotinylated GSSG [134] or the cell permeable biotinylated GSH ethyl esters [135] which allows for enrichment of S-glutathionylated proteins on streptavidin. However, the bulky nature of biotin is likely to interfere with enzyme-mediated (de)glutathionylation and the artificial introduction of biotinylated GSH or GSSG may alter the endogenous redox status. (iv) a mutant of glutathione synthetase (GS), capable of coupling γGlu-Cys to azido-Ala in place of Gly. Transfection of this GS mutant and incubation of azido-Ala in cells efficiently produces the azido-GSH which is glutathionylated on proteins, and the subsequent click reaction allows for enrichment and detection [136, 137]. Compared to [35S]GSH and biotinylated GSH, a major advantage of “clickable” GSH is the small versatile azide handle, which seems not to interfere with enzyme-mediated (de)glutathionylation and can couple with a variety of different applications. Moreover, no exogenous thiol is introduced. After enrichment, the S-glutathionylated proteins can then be detected or identified by fluorescent-, immunoblotting- or proteomic-based approaches (Table 1).
8. Perspectives/conclusions
Over the decades, many published studies have implied that cellular GSH levels perform primarily as buffers against ROS/RNS or that GSH:GSSG ratios directly correlate with cell survival. Arguably, the relative proportion of “activated” cysteines in a proteome may be the most critical regulator of cellular redox (thiol) homeostasis. The facile cycling of phosphate groups (serines, threonines or tyrosines) controls a surfeit of biological functions. In this regard, similarly profound affects can surely be associated with a sulfur-based S-glutathionylation cycle. Ironically, many of these same kinases and phosphatases are themselves subject to regulation by S-glutathionylation. GSTP has always been an enigmatic member of the GST gene superfamily, with properties not always commensurate with detoxification. With a role in catalyzing the forward reaction of the S-glutathionylation cycle, the high levels of expression of this GSTP isozyme in highly proliferating tissues or tumors can perhaps be more readily appreciated. Either genetic (knockout or over-expression) or pharmaceutical (Telintra) manipulation of GSTP leads to unassailable evidence for the importance of GSTP in S-glutathionylation. Independent of exogenous ROS, endogenous oxidative signaling (primarily attributable to hydrogen peroxide) is securely regulated and the S-glutathionylation cycle would appear to provide a tunable rheostat that might easily have evolved through selective pressures afforded by the use of oxidative phosphorylation as a means of energy generation. As the field advances, databases of proteins, the structure and function of which are impacted by S-glutathionylation will help to shed light on the critical nature of this cycle in regulating cell functions.
Highlights FRBM.
Addition of glutathione to cysteines alters the structure/function of a protein
The process, S-glutathionylation, is influenced by oxidative stress
The cyclical nature of the process allows for redox mediated signaling events
Abnormal S-glutathionylation is linked with human diseases.
Acknowledgments
This work was supported by grants from the National Institutes of Health (CA08660, CA117259, NCRR P20RR024485 - COBRE in Oxidants, Redox Balance and Stress Signaling) and support from the South Carolina Centers of Excellence program and was conducted in a facility constructed with the support from the National Institutes of Health, Grant Number C10 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources.
Abbreviations
- Acn
aconitase
- AMPK
AMP-activated protein kinase
- ANT
adenine nucleotide transporter
- BMDDC
bone-marrow derived dendritic cells
- ERα
estrogen receptor α
- CSF
cerebrospinal fluid
- ER
endoplasmic reticulum
- ETS
electron transport system
- GRP78/BiP
78-kDa glucose-regulated protein/immunoglobulin heavy chain binding protein
- Grx
glutaredoxin
- GSH
glutathione
- GSSG
glutathione disulfide
- GST
glutathione S-transferase
- HPV
human papilloma virus
- IAM
iodoacetamide
- IP3Rs
inositol 1,4,5-triphosphate receptors
- iTRAQ
isobaric tags for relative and absolute quantification
- MDS
myelodysplastic syndromes
- NEM
N-acetylmaleimide
- PDI
protein disulfide isomerase
- Prx6
peroxiredoxin 6
- RAC
resin-assisted capture
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RPS3
ribosomal protein S3
- RyRs
ryanodine receptors
- SdhA
succinate dehydrogenase A
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
- Srx
sulfiredoxin
- TBI
traumatic brain injury
- ThG
thapsigargin
- TuM
tunicamycin
- UCP
uncoupling proteins
- TMT
tandem mass tags
- UPR
unfolded protein response
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grek CL, Zhang J, Manevich Y, Townsend DM, Tew KD. Causes and consequences of cysteine S-glutathionylation. J Biol Chem. 2013;288(37):26497–504. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bechtel TJ, Weerapana E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics. 2017;17(6) doi: 10.1002/pmic.201600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11(11):2807–50. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 4.Mailloux RJ, Willmore WG. S-glutathionylation reactions in mitochondrial function and disease. Front Cell Dev Biol. 2014;2:68. doi: 10.3389/fcell.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007;7(4):381–91. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert HF. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 7.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 8.Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999;13(12):1481–90. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 9.Arbault S, Pantano P, Sojic N, Amatore C, Best-Belpomme M, Sarasin A, Vuillaume M. Activation of the NADPH oxidase in human fibroblasts by mechanical intrusion of a single cell with an ultramicroelectrode. Carcinogenesis. 1997;18(3):569–74. doi: 10.1093/carcin/18.3.569. [DOI] [PubMed] [Google Scholar]
- 10.Starke DW, Chock PB, Mieyal JJ. Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J Biol Chem. 2003;278(17):14607–13. doi: 10.1074/jbc.M210434200. [DOI] [PubMed] [Google Scholar]
- 11.Gallogly MM, Starke DW, Leonberg AK, Ospina SM, Mieyal JJ. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: implications for intracellular roles. Biochemistry. 2008;47(42):11144–57. doi: 10.1021/bi800966v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giustarini D, Milzani A, Aldini G, Carini M, Rossi R, Dalle-Donne I. S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione. Antioxid Redox Signal. 2005;7(7–8):930–9. doi: 10.1089/ars.2005.7.930. [DOI] [PubMed] [Google Scholar]
- 13.Townsend DM, Findlay VJ, Fazilev F, Ogle M, Fraser J, Saavedra JE, Ji X, Keefer LK, Tew KD. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol Pharmacol. 2006;69(2):501–8. doi: 10.1124/mol.105.018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Townsend DM, Manevich Y, L He, Xiong Y, Bowers RR, Jr, Hutchens S, Tew KD. Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 2009;69(19):7626–34. doi: 10.1158/0008-5472.CAN-09-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konorev EA, Kalyanaraman B, Hogg N. Modification of creatine kinase by S-nitrosothiols: S-nitrosation vs. S-thiolation. Free Radic Biol Med. 2000;28(11):1671–8. doi: 10.1016/s0891-5849(00)00281-1. [DOI] [PubMed] [Google Scholar]
- 16.Manevich Y, Feinstein SI, Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc Natl Acad Sci U S A. 2004;101(11):3780–5. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ralat LA, Manevich Y, Fisher AB, Colman RF. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry. 2006;45(2):360–72. doi: 10.1021/bi0520737. [DOI] [PubMed] [Google Scholar]
- 18.Townsend DM, Manevich Y, L He, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J Biol Chem. 2009;284(1):436–45. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wetzelberger K, Baba SP, Thirunavukkarasu M, Ho YS, Maulik N, Barski OA, Conklin DJ, Bhatnagar A. Postischemic deactivation of cardiac aldose reductase: role of glutathione S-transferase P and glutaredoxin in regeneration of reduced thiols from sulfenic acids. J Biol Chem. 2010;285(34):26135–48. doi: 10.1074/jbc.M110.146423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Luca A, Moroni N, Serafino A, Primavera A, Pastore A, Pedersen JZ, Petruzzelli R, Farrace MG, Pierimarchi P, Moroni G, Federici G, Sinibaldi Vallebona P, Lo Bello M. Treatment of doxorubicin-resistant MCF7/Dx cells with nitric oxide causes histone glutathionylation and reversal of drug resistance. Biochem J. 2011;440(2):175–83. doi: 10.1042/BJ20111333. [DOI] [PubMed] [Google Scholar]
- 21.Klaus A, Zorman S, Berthier A, Polge C, Ramirez S, Michelland S, Seve M, Vertommen D, Rider M, Lentze N, Auerbach D, Schlattner U. Glutathione S-transferases interact with AMP-activated protein kinase: evidence for S-glutathionylation and activation in vitro. PLoS One. 2013;8(5):e62497. doi: 10.1371/journal.pone.0062497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye ZW, Zhang J, Ancrum T, Manevich Y, Townsend DM, Tew KD. Glutathione S-Transferase P-Mediated Protein S-Glutathionylation of Resident Endoplasmic Reticulum Proteins Influences Sensitivity to Drug-Induced Unfolded Protein Response. Antioxid Redox Signal. 2017;26(6):247–261. doi: 10.1089/ars.2015.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Ye ZW, Gao P, Reyes L, Jones EE, Branham-O’Connor M, Blumer JB, Drake RR, Manevich Y, Townsend DM, Tew KD. Glutathione S-transferase P influences redox and migration pathways in bone marrow. PLoS One. 2014;9(9):e107478. doi: 10.1371/journal.pone.0107478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamada K, Goto S, Okunaga T, Ihara Y, Tsuji K, Kawai Y, Uchida K, Osawa T, Matsuo T, Nagata I, Kondo T. Nuclear glutathione S-transferase pi prevents apoptosis by reducing the oxidative stress-induced formation of exocyclic DNA products. Free Radic Biol Med. 2004;37(11):1875–84. doi: 10.1016/j.freeradbiomed.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Goto S, Kawakatsu M, Izumi S, Urata Y, Kageyama K, Ihara Y, Koji T, Kondo T. Glutathione S-transferase pi localizes in mitochondria and protects against oxidative stress. Free Radic Biol Med. 2009;46(10):1392–403. doi: 10.1016/j.freeradbiomed.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 26.Tajc SG, Tolbert BS, Basavappa R, Miller BL. Direct determination of thiol pKa by isothermal titration microcalorimetry. J Am Chem Soc. 2004;126(34):10508–9. doi: 10.1021/ja047929u. [DOI] [PubMed] [Google Scholar]
- 27.Dirr H, Reinemer P, Huber R. X-ray crystal structures of cytosolic glutathione S-transferases. Implications for protein architecture, substrate recognition and catalytic function. Eur J Biochem. 1994;220(3):645–61. doi: 10.1111/j.1432-1033.1994.tb18666.x. [DOI] [PubMed] [Google Scholar]
- 28.Tew KD, Manevich Y, Grek C, Xiong Y, Uys J, Townsend DM. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free Radic Biol Med. 2011;51(2):299–313. doi: 10.1016/j.freeradbiomed.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiong Y, Uys JD, Tew KD, Townsend DM. S-glutathionylation: from molecular mechanisms to health outcomes. Antioxid Redox Signal. 2011;15(1):233–70. doi: 10.1089/ars.2010.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mannervik B, Axelsson K. Role of cytoplasmic thioltransferase in cellular regulation by thiol-disulphide interchange. Biochem J. 1980;190(1):125–30. doi: 10.1042/bj1900125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gladyshev VN, Liu A, Novoselov SV, Krysan K, Sun QA, Kryukov VM, Kryukov GV, Lou MF. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J Biol Chem. 2001;276(32):30374–80. doi: 10.1074/jbc.M100020200. [DOI] [PubMed] [Google Scholar]
- 32.Lundberg M, Johansson C, Chandra J, Enoksson M, Jacobsson G, Ljung J, Johansson M, Holmgren A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J Biol Chem. 2001;276(28):26269–75. doi: 10.1074/jbc.M011605200. [DOI] [PubMed] [Google Scholar]
- 33.Pai HV, Starke DW, Lesnefsky EJ, Hoppel CL, Mieyal JJ. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid Redox Signal. 2007;9(11):2027–33. doi: 10.1089/ars.2007.1642. [DOI] [PubMed] [Google Scholar]
- 34.Gallogly MM, Starke DW, Mieyal JJ. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid Redox Signal. 2009;11(5):1059–81. doi: 10.1089/ars.2008.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin JL. Thioredoxin--a fold for all reasons. Structure. 1995;3(3):245–50. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 36.Gravina SA, Mieyal JJ. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry. 1993;32(13):3368–76. doi: 10.1021/bi00064a021. [DOI] [PubMed] [Google Scholar]
- 37.Peltoniemi MJ, Karala AR, Jurvansuu JK, Kinnula VL, Ruddock LW. Insights into deglutathionylation reactions. Different intermediates in the glutaredoxin and protein disulfide isomerase catalyzed reactions are defined by the gamma-linkage present in glutathione. J Biol Chem. 2006;281(44):33107–14. doi: 10.1074/jbc.M605602200. [DOI] [PubMed] [Google Scholar]
- 38.Jensen KS, Pedersen JT, Winther JR, Teilum K. The pKa value and accessibility of cysteine residues are key determinants for protein substrate discrimination by glutaredoxin. Biochemistry. 2014;53(15):2533–40. doi: 10.1021/bi4016633. [DOI] [PubMed] [Google Scholar]
- 39.Menon D, Board PG. A role for glutathione transferase Omega 1 (GSTO1-1) in the glutathionylation cycle. J Biol Chem. 2013;288(36):25769–79. doi: 10.1074/jbc.M113.487785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Findlay VJ, Townsend DM, Morris TE, Fraser JP, L He, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66(13):6800–6. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park JW, Mieyal JJ, Rhee SG, Chock PB. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J Biol Chem. 2009;284(35):23364–74. doi: 10.1074/jbc.M109.021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Board PG, Coggan M, Chelvanayagam G, Easteal S, Jermiin LS, Schulte GK, Danley DE, Hoth LR, Griffor MC, Kamath AV, Rosner MH, Chrunyk BA, Perregaux DE, Gabel CA, Geoghegan KF, Pandit J. Identification, characterization, and crystal structure of the Omega class glutathione transferases. J Biol Chem. 2000;275(32):24798–806. doi: 10.1074/jbc.M001706200. [DOI] [PubMed] [Google Scholar]
- 43.Board PG, Menon D. Structure, function and disease relevance of Omega-class glutathione transferases. Arch Toxicol. 2016;90(5):1049–67. doi: 10.1007/s00204-016-1691-1. [DOI] [PubMed] [Google Scholar]
- 44.Kim K, Kim SH, Kim J, Kim H, Yim J. Glutathione s-transferase omega 1 activity is sufficient to suppress neurodegeneration in a Drosophila model of Parkinson disease. J Biol Chem. 2012;287(9):6628–41. doi: 10.1074/jbc.M111.291179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen PH, Smith TJ, Wu J, Siesser PF, Bisnett BJ, Khan F, Hogue M, Soderblom E, Tang F, Marks JR, Major MB, Swarts BM, Boyce M, Chi JT. Glycosylation of KEAP1 links nutrient sensing to redox stress signaling. EMBO J. 2017;36(15):2233–2250. doi: 10.15252/embj.201696113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uys JD, Xiong Y, Townsend DM. Nitrosative stress-induced S-glutathionylation of protein disulfide isomerase. Methods Enzymol. 2011;490:321–32. doi: 10.1016/B978-0-12-385114-7.00018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghezzi P. Regulation of protein function by glutathionylation. Free Radic Res. 2005;39(6):573–80. doi: 10.1080/10715760500072172. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe MM, Laurindo FR, Fernandes DC. Methods of measuring protein disulfide isomerase activity: a critical overview. Frontiers in chemistry. 2014;2:73. doi: 10.3389/fchem.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiong Y, Manevich Y, Tew KD, Townsend DM. S-Glutathionylation of Protein Disulfide Isomerase Regulates Estrogen Receptor alpha Stability and Function. Int J Cell Biol. 2012;2012:273549. doi: 10.1155/2012/273549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hofmann B, Hecht HJ, Flohe L. Peroxiredoxins. Biological chemistry. 2002;383(3–4):347–64. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 51.Ralat LA, Misquitta SA, Manevich Y, Fisher AB, Colman RF. Characterization of the complex of glutathione S-transferase pi and 1-cysteine peroxiredoxin. Archives of biochemistry and biophysics. 2008;474(1):109–18. doi: 10.1016/j.abb.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mieyal JJ, Starke DW, Gravina SA, Dothey C, Chung JS. Thioltransferase in human red blood cells: purification and properties. Biochemistry. 1991;30(25):6088–97. doi: 10.1021/bi00239a002. [DOI] [PubMed] [Google Scholar]
- 53.Manevich Y, Hutchens S, Tew KD, Townsend DM. Allelic variants of glutathione S-transferase P1-1 differentially mediate the peroxidase function of peroxiredoxin VI and alter membrane lipid peroxidation. Free Radic Biol Med. 2013;54:62–70. doi: 10.1016/j.freeradbiomed.2012.10.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi HJ, Kang SW, Yang CH, Rhee SG, Ryu SE. Crystal structure of a novel human peroxidase enzyme at 2.0 A resolution. Nature structural biology. 1998;5(5):400–6. doi: 10.1038/nsb0598-400. [DOI] [PubMed] [Google Scholar]
- 55.Manevich Y, Hutchens S, Halushka PV, Tew KD, Townsend DM, Jauch EC, Borg K. Peroxiredoxin VI oxidation in cerebrospinal fluid correlates with traumatic brain injury outcome. Free Radic Biol Med. 2014;72:210–21. doi: 10.1016/j.freeradbiomed.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J, Grek C, Ye ZW, Manevich Y, Tew KD, Townsend DM. Pleiotropic functions of glutathione S-transferase P. Adv Cancer Res. 2014;122:143–75. doi: 10.1016/B978-0-12-420117-0.00004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murphy MP. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal. 2012;16(6):476–95. doi: 10.1089/ars.2011.4289. [DOI] [PubMed] [Google Scholar]
- 58.O’Brien M, Chalker J, Slade L, Gardiner D, Mailloux RJ. Protein S-glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free Radic Biol Med. 2017;106:302–314. doi: 10.1016/j.freeradbiomed.2017.02.046. [DOI] [PubMed] [Google Scholar]
- 59.Kil IS, Park JW. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J Biol Chem. 2005;280(11):10846–54. doi: 10.1074/jbc.M411306200. [DOI] [PubMed] [Google Scholar]
- 60.Applegate MA, Humphries KM, Szweda LI. Reversible inhibition of alpha-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry. 2008;47(1):473–8. doi: 10.1021/bi7017464. [DOI] [PubMed] [Google Scholar]
- 61.Eaton P, Shattock MJ. Purification of proteins susceptible to oxidation at cysteine residues: identification of malate dehydrogenase as a target for S-glutathiolation. Ann N Y Acad Sci. 2002;973:529–32. doi: 10.1111/j.1749-6632.2002.tb04694.x. [DOI] [PubMed] [Google Scholar]
- 62.Garcia J, Han D, Sancheti H, Yap LP, Kaplowitz N, Cadenas E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J Biol Chem. 2010;285(51):39646–54. doi: 10.1074/jbc.M110.164160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han D, Canali R, Garcia J, Aguilera R, Gallaher TK, Cadenas E. Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry. 2005;44(36):11986–96. doi: 10.1021/bi0509393. [DOI] [PubMed] [Google Scholar]
- 64.Baradaran R, Berrisford JM, Minhas GS, Sazanov LA. Crystal structure of the entire respiratory complex I. Nature. 2013;494(7438):443–8. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Passarelli C, Tozzi G, Pastore A, Bertini E, Piemonte F. GSSG-mediated Complex I defect in isolated cardiac mitochondria. Int J Mol Med. 2010;26(1):95–9. doi: 10.3892/ijmm_00000439. [DOI] [PubMed] [Google Scholar]
- 66.Hurd TR, Requejo R, Filipovska A, Brown S, Prime TA, Robinson AJ, Fearnley IM, Murphy MP. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J Biol Chem. 2008;283(36):24801–15. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumar V, Kleffmann T, Hampton MB, Cannell MB, Winterbourn CC. Redox proteomics of thiol proteins in mouse heart during ischemia/reperfusion using ICAT reagents and mass spectrometry. Free Radic Biol Med. 2013;58:109–17. doi: 10.1016/j.freeradbiomed.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 68.Mailloux RJ, Xuan JY, McBride S, Maharsy W, Thorn S, Holterman CE, Kennedy CR, Rippstein P, deKemp R, da Silva J, Nemer M, Lou M, Harper ME. Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. J Biol Chem. 2014;289(21):14812–28. doi: 10.1074/jbc.M114.550574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grivennikova VG, Kapustin AN, Vinogradov AD. Catalytic activity of NADH-ubiquinone oxidoreductase (complex I) in intact mitochondria. evidence for the slow active/inactive transition. J Biol Chem. 2001;276(12):9038–44. doi: 10.1074/jbc.M009661200. [DOI] [PubMed] [Google Scholar]
- 70.Drose S, Brandt U, Wittig I. Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochim Biophys Acta. 2014;1844(8):1344–54. doi: 10.1016/j.bbapap.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 71.Galkin A, Meyer B, Wittig I, Karas M, Schagger H, Vinogradov A, Brandt U. Identification of the mitochondrial ND3 subunit as a structural component involved in the active/deactive enzyme transition of respiratory complex I. J Biol Chem. 2008;283(30):20907–13. doi: 10.1074/jbc.M803190200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen YR, Chen CL, Pfeiffer DR, Zweier JL. Mitochondrial complex II in the postishemic heart: oxidative injury and the role of protein S-glutathionylation. J Biol Chem. 2007;282(45):32640–54. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 73.Wang SB, Foster DB, Rucker J, O’Rourke B, Kass DA, Van Eyk JE. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res. 2011;109(7):750–7. doi: 10.1161/CIRCRESAHA.111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mailloux RJ, Harper ME. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med. 2011;51(6):1106–15. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 75.Mailloux RJ, Seifert EL, Bouillaud F, Aguer C, Collins S, Harper ME. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J Biol Chem. 2011;286(24):21865–75. doi: 10.1074/jbc.M111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Queiroga CS, Almeida AS, Martel C, Brenner C, Alves PM, Vieira HL. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J Biol Chem. 2010;285(22):17077–88. doi: 10.1074/jbc.M109.065052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kramer PA, Duan J, Qian WJ, Marcinek DJ. The Measurement of Reversible Redox Dependent Post-translational Modifications and Their Regulation of Mitochondrial and Skeletal Muscle Function. Front Physiol. 2015;6:347. doi: 10.3389/fphys.2015.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang J, Ye ZW, Chen W, Manevich Y, Mehrotra S, Ball LE, Janssen-Heininger YM, Tew KD, Townsend DM. S-Glutathionylation of estrogen receptor alpha affects dendritic cell function. J Biol Chem. 2018 doi: 10.1074/jbc.M117.814327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257(5076):1496–502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 80.Coe H, Michalak M. Calcium binding chaperones of the endoplasmic reticulum. Gen Physiol Biophys 28 Spec No Focus. 2009:F96–F103. [PubMed] [Google Scholar]
- 81.Carreras-Sureda A, Pihan P, Hetz C. Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses. Cell Calcium. 2017 doi: 10.1016/j.ceca.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 82.Aracena-Parks P, Goonasekera SA, Gilman CP, Dirksen RT, Hidalgo C, Hamilton SL. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. 2006;281(52):40354–68. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- 83.Aracena P, Tang W, Hamilton SL, Hidalgo C. Effects of S-glutathionylation and S-nitrosylation on calmodulin binding to triads and FKBP12 binding to type 1 calcium release channels. Antioxid Redox Signal. 2005;7(7–8):870–81. doi: 10.1089/ars.2005.7.870. [DOI] [PubMed] [Google Scholar]
- 84.Zima AV, Mazurek SR. Functional Impact of Ryanodine Receptor Oxidation on Intracellular Calcium Regulation in the Heart. Rev Physiol Biochem Pharmacol. 2016;171:39–62. doi: 10.1007/112_2016_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sanchez G, Pedrozo Z, Domenech RJ, Hidalgo C, Donoso P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J Mol Cell Cardiol. 2005;39(6):982–91. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 86.Lock JT, Sinkins WG, Schilling WP. Protein S-glutathionylation enhances Ca2+-induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. J Physiol. 2012;590(15):3431–47. doi: 10.1113/jphysiol.2012.230656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Viner RI, Huhmer AF, Bigelow DJ, Schoneich C. The oxidative inactivation of sarcoplasmic reticulum Ca(2+)-ATPase by peroxynitrite. Free Radic Res. 1996;24(4):243–59. doi: 10.3109/10715769609088022. [DOI] [PubMed] [Google Scholar]
- 88.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10(11):1200–7. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 89.Vangheluwe P, Raeymaekers L, Dode L, Wuytack F. Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: cell biological implications. Cell Calcium. 2005;38(3–4):291–302. doi: 10.1016/j.ceca.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 90.Wang J, Sevier CS. Formation and Reversibility of BiP Protein Cysteine Oxidation Facilitate Cell Survival during and post Oxidative Stress. J Biol Chem. 2016;291(14):7541–57. doi: 10.1074/jbc.M115.694810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ali-Osman F, Akande O, Antoun G, Mao JX, Buolamwini J. Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase Pi gene variants. Evidence for differential catalytic activity of the encoded proteins. J Biol Chem. 1997;272(15):10004–12. doi: 10.1074/jbc.272.15.10004. [DOI] [PubMed] [Google Scholar]
- 92.Harries LW, Stubbins MJ, Forman D, Howard GC, Wolf CR. Identification of genetic polymorphisms at the glutathione S-transferase Pi locus and association with susceptibility to bladder, testicular and prostate cancer. Carcinogenesis. 1997;18(4):641–4. doi: 10.1093/carcin/18.4.641. [DOI] [PubMed] [Google Scholar]
- 93.Harris MJ, Coggan M, Langton L, Wilson SR, Board PG. Polymorphism of the Pi class glutathione S-transferase in normal populations and cancer patients. Pharmacogenetics. 1998;8(1):27–31. doi: 10.1097/00008571-199802000-00004. [DOI] [PubMed] [Google Scholar]
- 94.Grek CL, Reyes L, Townsend DM, Tew KD. S-glutathionylation of buccal cell proteins as biomarkers of exposure to hydrogen peroxide. BBA Clin. 2014;2:31–9. doi: 10.1016/j.bbacli.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Karihtala P, Mantyniemi A, Kang SW, Kinnula VL, Soini Y. Peroxiredoxins in breast carcinoma. Clin Cancer Res. 2003;9(9):3418–24. [PubMed] [Google Scholar]
- 96.Chang XZ, Li DQ, Hou YF, Wu J, Lu JS, Di GH, Jin W, Ou ZL, Shen ZZ, Shao ZM. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007;9(6):R76. doi: 10.1186/bcr1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Manevich Y, Feinstein SI, Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc Natl Acad Sci U S A. 2004;101(11):3780–5. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med. 2005;38(11):1422–32. doi: 10.1016/j.freeradbiomed.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 99.Townsend DM. S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Mol Interv. 2007;7(6):313–24. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Morgan AS, Ciaccio PJ, Tew KD, Kauvar LM. Isozyme-specific glutathione S-transferase inhibitors potentiate drug sensitivity in cultured human tumor cell lines. Cancer Chemother Pharmacol. 1996;37(4):363–70. doi: 10.1007/s002800050398. [DOI] [PubMed] [Google Scholar]
- 101.Raza A, Galili N, Smith SE, Godwin J, Boccia RV, Myint H, Mahadevan D, Mulford D, Rarick M, Brown GL, Schaar D, Faderl S, Komrokji RS, List AF, Sekeres M. A phase 2 randomized multicenter study of 2 extended dosing schedules of oral ezatiostat in low to intermediate-1 risk myelodysplastic syndrome. Cancer. 2012;118(8):2138–47. doi: 10.1002/cncr.26469. [DOI] [PubMed] [Google Scholar]
- 102.Mahadevan D, Sutton GR. Ezatiostat hydrochloride for the treatment of myelodysplastic syndromes. Expert Opin Investig Drugs. 2015;24(5):725–33. doi: 10.1517/13543784.2015.1021003. [DOI] [PubMed] [Google Scholar]
- 103.Quddus F, Clima J, Seedham H, Sajjad G, Galili N, Raza A. Oral Ezatiostat HCl (TLK199) and Myelodysplastic syndrome: a case report of sustained hematologic response following an abbreviated exposure. J Hematol Oncol. 2010;3:16. doi: 10.1186/1756-8722-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lyons RM, Wilks ST, Young S, Brown GL. Oral ezatiostat HCl (Telintra(R), TLK199) and idiopathic chronic neutropenia (ICN): a case report of complete response of a patient with G-CSF resistant ICN following treatment with ezatiostat, a glutathione S-transferase P1-1 (GSTP1-1) inhibitor. J Hematol Oncol. 2011;4:43. doi: 10.1186/1756-8722-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Flatgaard JE, Bauer KE, Kauvar LM. Isozyme specificity of novel glutathione-S-transferase inhibitors. Cancer chemotherapy and pharmacology. 1993;33(1):63–70. doi: 10.1007/BF00686025. [DOI] [PubMed] [Google Scholar]
- 106.Lyttle MH, Hocker MD, Hui HC, Caldwell CG, Aaron DT, Engqvist-Goldstein A, Flatgaard JE, Bauer KE. Isozyme-specific glutathione-S-transferase inhibitors: design and synthesis. Journal of medicinal chemistry. 1994;37(1):189–94. doi: 10.1021/jm00027a024. [DOI] [PubMed] [Google Scholar]
- 107.Gate L, Majumdar RS, Lunk A, Tew KD. Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. J Biol Chem. 2004;279(10):8608–16. doi: 10.1074/jbc.M308613200. [DOI] [PubMed] [Google Scholar]
- 108.McMillan DH, van der Velden LJ, Lahue KG, Qian X, Schneider RW, Iberg MS, Nolin JD, Abdalla S, Casey DT, Tew KD, Townsend DM, Henderson CJ, Wolf CR, Butnor KJ, Taatjes DJ, Budd RC, Irvin CG, van der Vliet A, Flemer S, Anathy V, Janssen-Heininger YM. Attenuation of lung fibrosis in mice with a clinically relevant inhibitor of glutathione-S-transferase pi. JCI Insight. 2016;1(8) doi: 10.1172/jci.insight.85717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang J, Hao X, Sang W, Yan Q. Hydrogen Polysulfide Biosignal-Responsive Polymersomes as a Nanoplatform for Distinguishing Intracellular Reactive Sulfur Species (RSS) Small. 2017 doi: 10.1002/smll.201701601. [DOI] [PubMed] [Google Scholar]
- 110.Cortese-Krott MM, Koning A, Kuhnle GGC, Nagy P, Bianco CL, Pasch A, Wink DA, Fukuto JM, Jackson AA, van Goor H, Olson KR, Feelisch M. The Reactive Species Interactome: Evolutionary Emergence, Biological Significance, and Opportunities for Redox Metabolomics and Personalized Medicine. Antioxid Redox Signal. 2017;27(10):684–712. doi: 10.1089/ars.2017.7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chemical reviews. 2013;113(7):4633–79. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Casas AI, Dao VT, Daiber A, Maghzal GJ, Di Lisa F, Kaludercic N, Leach S, Cuadrado A, Jaquet V, Seredenina T, Krause KH, Lopez MG, Stocker R, Ghezzi P, Schmidt HH. Reactive Oxygen-Related Diseases: Therapeutic Targets and Emerging Clinical Indications. Antioxid Redox Signal. 2015;23(14):1171–85. doi: 10.1089/ars.2015.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Watson JD. Type 2 diabetes as a redox disease. Lancet. 2014;383(9919):841–3. doi: 10.1016/S0140-6736(13)62365-X. [DOI] [PubMed] [Google Scholar]
- 114.Sparaco M, Gaeta LM, Santorelli FM, Passarelli C, Tozzi G, Bertini E, Simonati A, Scaravilli F, Taroni F, Duyckaerts C, Feleppa M, Piemonte F. Friedreich’s ataxia: oxidative stress and cytoskeletal abnormalities. Journal of the neurological sciences. 2009;287(1–2):111–8. doi: 10.1016/j.jns.2009.08.052. [DOI] [PubMed] [Google Scholar]
- 115.Mileo AM, Abbruzzese C, Mattarocci S, Bellacchio E, Pisano P, Federico A, Maresca V, Picardo M, Giorgi A, Maras B, Schinina ME, Paggi MG. Human papillomavirus-16 E7 interacts with glutathione S-transferase P1 and enhances its role in cell survival. PLoS One. 2009;4(10):e7254. doi: 10.1371/journal.pone.0007254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Trujillo JA, Croft NP, Dudek NL, Channappanavar R, Theodossis A, Webb AI, Dunstone MA, Illing PT, Butler NS, Fett C, Tscharke DC, Rossjohn J, Perlman S, Purcell AW. The cellular redox environment alters antigen presentation. J Biol Chem. 2014;289(40):27979–91. doi: 10.1074/jbc.M114.573402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Butturini E, Cozzolino F, Boriero D, Carcereri de Prati A, Monti M, Rossin M, Canetti D, Cellini B, Pucci P, Mariotto S. S-glutathionylation exerts opposing roles in the regulation of STAT1 and STAT3 signaling in reactive microglia. Free Radic Biol Med. 2018;117:191–201. doi: 10.1016/j.freeradbiomed.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 118.Hoffman SM, Nolin JD, Jones JT, Lahue KG, Chapman DG, Aliyeva M, Daphtary N, Lundblad LK, Abdalla S, Ather JL, Ho YS, Irvin CG, Anathy V, Wouters EF, Poynter ME, Janssen-Heininger YM. Ablation of the Thiol Transferase Glutaredoxin-1 Augments Protein S-Glutathionylation and Modulates Type 2 Inflammatory Responses and IL-17 in a House Dust Mite Model of Allergic Airway Disease in Mice. Ann Am Thorac Soc 13 Suppl. 2016;1:S97. doi: 10.1513/AnnalsATS.201510-656MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kovats S. Estrogen receptors regulate an inflammatory pathway of dendritic cell differentiation: mechanisms and implications for immunity. Horm Behav. 2012;62(3):254–62. doi: 10.1016/j.yhbeh.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang CY, Dwyer MA, Nelson ER, Pollizzi KN, Ilkayeva O, Giguere V, Zuercher WJ, Powell JD, Shinohara ML, McDonnell DP, Rathmell JC. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(45):18348–53. doi: 10.1073/pnas.1108856108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mitchell A, Tam C, Elli D, Charlton T, Osei-Owusu P, Fazlollahi F, Faull KF, Schneewind O. Glutathionylation of Yersinia pestis LcrV and Its Effects on Plague Pathogenesis. MBio. 2017;8(3) doi: 10.1128/mBio.00646-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.L Gu, Robinson RA. Proteomic approaches to quantify cysteine reversible modifications in aging and neurodegenerative diseases. Proteomics Clin Appl. 2016;10(12):1159–1177. doi: 10.1002/prca.201600015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang J, Carroll KS, Liebler DC. The Expanding Landscape of the Thiol Redox Proteome. Mol Cell Proteomics. 2016;15(1):1–11. doi: 10.1074/mcp.O115.056051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Su D, Gaffrey MJ, Guo J, Hatchell KE, Chu RK, Clauss TR, Aldrich JT, Wu S, Purvine S, Camp DG, Smith RD, Thrall BD, Qian WJ. Proteomic identification and quantification of S-glutathionylation in mouse macrophages using resin-assisted enrichment and isobaric labeling. Free Radic Biol Med. 2014;67:460–70. doi: 10.1016/j.freeradbiomed.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guo J, Gaffrey MJ, Su D, Liu T, Camp DG, 2nd, Smith RD, Qian WJ. Resin-assisted enrichment of thiols as a general strategy for proteomic profiling of cysteine-based reversible modifications. Nat Protoc. 2014;9(1):64–75. doi: 10.1038/nprot.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, von Lowenhielm HB, Holmgren A, Cotgreave IA. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Archives of biochemistry and biophysics. 2002;406(2):229–40. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 127.Reynaert NL, Ckless K, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. In situ detection of S-glutathionylated proteins following glutaredoxin-1 catalyzed cysteine derivatization. Biochim Biophys Acta. 2006;1760(3):380–7. doi: 10.1016/j.bbagen.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 128.Abo M, Weerapana E. A Caged Electrophilic Probe for Global Analysis of Cysteine Reactivity in Living Cells. J Am Chem Soc. 2015;137(22):7087–90. doi: 10.1021/jacs.5b04350. [DOI] [PubMed] [Google Scholar]
- 129.Hurd TR, Prime TA, Harbour ME, Lilley KS, Murphy MP. Detection of reactive oxygen species-sensitive thiol proteins by redox difference gel electrophoresis: implications for mitochondrial redox signaling. J Biol Chem. 2007;282(30):22040–51. doi: 10.1074/jbc.M703591200. [DOI] [PubMed] [Google Scholar]
- 130.Comini MA. Measurement and meaning of cellular thiol:disufhide redox status. Free Radic Res. 2016;50(2):246–71. doi: 10.3109/10715762.2015.1110241. [DOI] [PubMed] [Google Scholar]
- 131.Pan KT, Chen YY, Pu TH, Chao YS, Yang CY, Bomgarden RD, Rogers JC, Meng TC, Khoo KH. Mass spectrometry-based quantitative proteomics for dissecting multiplexed redox cysteine modifications in nitric oxide-protected cardiomyocyte under hypoxia. Antioxid Redox Signal. 2014;20(9):1365–81. doi: 10.1089/ars.2013.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27(6):557–9. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Michelet L, Zaffagnini M, Vanacker H, Le Marechal P, Marchand C, Schroda M, Lemaire SD, Decottignies P. In vivo targets of S-thiolation in Chlamydomonas reinhardtii. J Biol Chem. 2008;283(31):21571–8. doi: 10.1074/jbc.M802331200. [DOI] [PubMed] [Google Scholar]
- 134.Brennan JP, Miller JI, Fuller W, Wait R, Begum S, Dunn MJ, Eaton P. The utility of N,N-biotinyl glutathione disulfide in the study of protein S-glutathiolation. Mol Cell Proteomics. 2006;5(2):215–25. doi: 10.1074/mcp.M500212-MCP200. [DOI] [PubMed] [Google Scholar]
- 135.Yang Y, Shi W, Cui N, Wu Z, Jiang C. Oxidative stress inhibits vascular K(ATP) channels by S-glutathionylation. J Biol Chem. 2010;285(49):38641–8. doi: 10.1074/jbc.M110.162578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Samarasinghe KT, Munkanatta Godage DN, VanHecke GC, Ahn YH. Metabolic synthesis of clickable glutathione for chemoselective detection of glutathionylation. J Am Chem Soc. 2014;136(33):11566–9. doi: 10.1021/ja503946q. [DOI] [PubMed] [Google Scholar]
- 137.Samarasinghe KT, Munkanatta Godage DN, Zhou Y, Ndombera FT, Weerapana E, Ahn YH. A clickable glutathione approach for identification of protein glutathionylation in response to glucose metabolism. Mol Biosyst. 2016;12(8):2471–80. doi: 10.1039/c6mb00175k. [DOI] [PMC free article] [PubMed] [Google Scholar]