

Abstract
The past decade has witnessed the burst of ultrasmall (<3nm) luminescent gold nanoparticles (AuNPs). Unlike semiconductor quantum dots, ultrasmall gold nanoparticles have very diverse emission mechanims, which are often involved with many structural factors such as size, valence state, surface ligands and crystallinity. In this frontier, we summarized our latest advancement in the fundamental understandings of emission mechanisms of ultrasmall gold nanoparticles, which are expected to help us more precisely control their emissions and broaden their applications from energy technologies to disease detection.
Introduction
Ultrasmall luminescent noble metal nanoparticles draw intensive attention because they serve as a unique platform to fundamentally understand the quantum confinement of free electrons on a size scale (De Broglie wavelength, ~1nm), 1, 2 which is much smaller than the well-known characteristic length scales for semiconductor quantum dots. As a result, luminescence of noble metal nanoparticles is much more sensitive to the particle size,3 valence state of metals,4 surface ligands5 as well as crystallinity,6 making it very difficult to fully understand luminescence of noble metals on the nano scale. Although more efforts are still needed to further elucidate luminescence mechanisms of noble metal nanoparticles, a brief summary of what we have achieved in the past two decades will be beneficial to the future advancement in this field. In this review, we will focus on size-dependent and size-independent luminescence from ultrasmall gold nanoparticles at the fundamental level and illustrate these seemingly contradictory optical behaviors of gold metal on an extremely small size scale.
The first study on luminescence mechanism of gold metal can be dated back to 1969, when Mooradia et al. observed emission from gold film at 567nm under the excitation of a 488nm laser.7 Considering the continuous conduction (sp) band of gold metal, the emission was not attributed to intraband energy transitions within sp bands but interband transition from sp-band to d-band. The recombination of electrons at the Fermi energy level with the holes in d-band with higher energy level was believed to give the emission of gold film. The proposed luminescence mechanism by Mooradia et al. remained mainstream for about 30 years until the big breakthroughs in atomically precise synthesis of ultrasmall gold nanoparticles in the early 21st century, making it possible to quantitatively correlate the emission with the size and surface chemistry of gold nanoparticles.
Size-dependency in the luminescence of few atom gold nanoclusters (AuNCs)
Size dependencies in the optical properties have been widely observed from a variety of inorganic engineered nanoparticles. For metal nanoparticles, the surface plasmon absorption is the most well-known optical response that can be tuned by modulating particles size.8 The origin of surface plasmon is due to the collective oscillation of a large number of free electrons shared by all the gold atoms, which can be quantitatively described with the Mie theory.9 However, with the decrease of the particle size to just few nanometers, the number of free electrons in the metal nanoparticles is dramatically reduced. As a result, the classical continuous band structure of metal nanoparticles breaks into the quantized discrete energy states and free electrons in the particles can no longer collectively oscillate but are confined on those discrete energy states. Under the light irradiation, gold nanoparticles can emit fluorescence, similar to the optical transitions in the small organic dyes, where only single-electron excitation and emission become allowable. Thus, a fundamental question is whether the emission from those ultrasmall gold nanoparticles is still size-dependent.
While fundamental understanding of the luminescence of metal nanoparticles was mainly limited by their low quantum yields (QY) and wide distribution in size at the early stage, in 2003, we first reported the synthesis of water-soluble poly(amidoamine) (PAMAM)- protected gold nanoclusters with high QY (41 ± 5%) and reaction yield and the AuNCs was determined to be mono-dispersed Au8 by electrospray ionization mass spectrometry (ESI-MS).10 This breakthrough in the synthesis of highly luminescent atomically precise AuNCs lays down a foundation to further explore the size-dependency in the luminescence of the few-atom AuNCs. In 2004, we further synthesized a series of dendrimer (PAMAM) -encapsulated highly luminescent AuNCs from Au5, Au8, Au13, Au23 to Au31 with emissions at 385nm, 456nm, 510nm, 751nm and 879nm, respectively, with high quantum yields (Figure 1a).3 The emissions of these AuNCs exhibited a red shift with the increase of number of gold atoms and can be quantitatively fit with a scaling relation of EFermi/N1/3 (Figure 1b) for smaller AuNCs from Au5 to Au13, where EFermi is the Fermi energy of bulk gold (5.53 eV) and N is the number of gold atoms in the AuNCs, which presents a size-dependent relationship between the luminescence property and the number of gold atoms and follows a free-electron model in a spherical harmonic potential (Figure 1b).3 For larger AuNCs, the anharmonicity increases due to the electron screening effect and the potential well that confines free electrons is gradually distorted and becomes flatten slightly, leading to a slightly deviated relationship between emission energy and number of gold atoms with an anharmonicity of U=0.033, following a modified free-electron model in a Woods – Saxon potential model (Figure 2b). Fundamentally, change in particles size on the size scale of electron Fermi wavelength leads to the change in the spacing of discrete energy level (Figure 1b, inserted).11 Using thermal energy as a criterion, when particles size is small, the energy level spacing of the sp bands is much higher than the thermal energy kT and thus can emit under this circumstance. However, with the increase in particle size, the energy level spacing becomes commensurate to the thermal energy and the luminescence would disappear. This newly discovered size-dependency also shed light on the origin of luminescence observed from the few-atom gold nanoclusters. In these dendrimer encapsulated gold nanoclusters, the free-electron model clearly indicates that emission is due to sp-sp intraband transition instead of sp-d interband transition. While the size-dependency in the emission was mainly observed from dendrimer encapsulated gold nanoclusters, many other gold clusters coated with different ligands also exhibit the similar emissions.12–14 In addition, these dendrimer-encapsulated AuNCs exhibited nanosecond-scale luminescence lifetime and small stokes shift, indicating that ligand to metal charge transfer (LMCT) was not involved and the luminescence solely originated from the core of AuNCs and was assigned to fluorescence, further supporting the size-dependent emission theory. Same size-dependent emission has also been found in pepsin-coated AuNCs later in 2011.12
Figure 1.
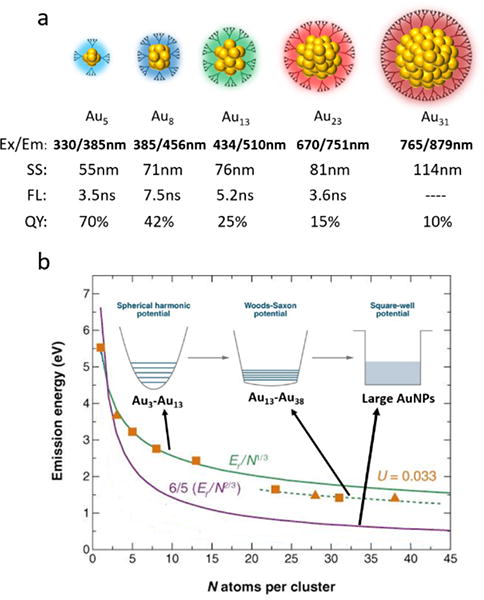
a) Dendrimer-coated AuNCs with size-dependent emissions and corresponding stokes shift (SS), luminescence lifetime (FL) and QY, b) quantitative correlation between emission energy and particle size and the evolution of potential energy model with particle size.
(Green solid line represents the scaling relation for Au5, Au8 and Au13; green dash line represents a modified scaling relation for Au23 and Au31; purple line represents the scaling relation in square-well potential model. (Reprinted from ref. 11 with permission from Annual Reviews)
Figure 2.
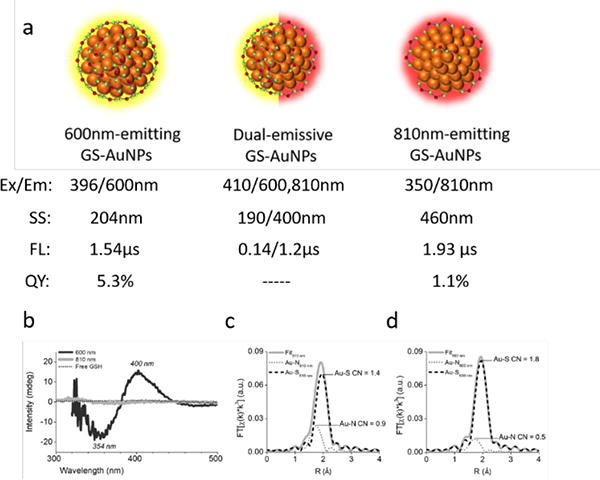
a) Size-independent emission in thiolated AuNPs, b) CD spectra for 600nm-emitting and 810nm-emitting AuNPs, c) EXAFS spectra of 810nm-emitting AuNPs, d) EXAFS spectra of 600nm-emitting AuNPs. (Reprinted from ref. 18. Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim)
Size-independent emissions in the thiolated AuNPs of 2nm
The size-dependent emission in observed from the dendrimer encapsulated gold nanoclusters suggested that the ligands have little interference on the optical transitions of gold clusters. However, gold atom is known for its high affinity to thiolated ligands. Whether strong ligand-metal charge interaction will impact the emissions of ultrasmall gold nanoparticles is worth extensive investigating. In 2004, we synthesized the first waters-soluble highly luminescent glutathione(GSH) coated gold nanoparticles of 2nm by fine controlling the polymerization and depolymerization of gold-glutathione oligomers in aqueous solution. Different from the previous synthesized glutathione gold clusters with very weak emissions,16, 17 highly luminescent gold nanoparticles were synthesized without using strong reducing agents such as NaBH4. We took advantage of reducing property of glutathione to allow the slow growth of gold nanoparticles at room temperature in aqueous solution and the obtained the luminescent gold nanoparticles of 2nm have QYS three orders higher than those glutathione coated gold nanoparticles obtained with NaBH4 reduction. The highly luminescent gold nanoparticles of 2nm, however, challenged the size-dependencies we observed from the dendrimer encapsulated gold nanoclusters. According to free-electron model, the gold nanoparticles of 2nm are not expected to be luminescent because the number of gold atoms and free electrons in the particles should be high enough to diminish any energy spacing.
To unravel the origin of luminescence in the large glutathione coated gold nanoparticles, we adjusted the synthetic ratio between GSH surface ligand and HAuCl4, and luminescent AuNPs with identical core size (ca. 2.5nm) but different emission wavelength ranging from visible to NIR region (600nm-emitting GS-AuNPs and 810nm-emitting GS-AuNPs) can be obtained.18(Figure 2a) However, unlike the emission shift in dendrimer-encapsulated AuNCs, adjusting the ratio in between leads to an integration of emissions within one particle system, forming dual-emissive AuNPs (emission at both 600nm and 810nm) still with the same size scale. The size-independent emission observed in few-nanometer AuNPs can be easily controlled and tuned, providing a set of powerful tools to fundamentally investigate the size-independent property.
To account for the size-independent emission in few-nanometer AuNPs, elemental analysis, circular dichroism (CD) and extended X-ray absorption fine structure (EXAFS) spectroscopy were used to demonstrate the difference between them. Results of elemental analysis indicated that the surface ligand density of 600nm-emitting GS-AuNPs is almost two times higher than that of 810nm-emitting GS-AuNPs while the CD signal observed only from 600nm-emitting GS-AuNPs suggested a highly ordered and chiral conformation of GSH on particle surface.18 (Figure 2b) The EXAFS spectroscopy revealed different Au-S coordination numbers of these two types of GS-AuNPs (1.4 in 600-emitting GS-AuNPs vs 1.8 in 810nm-emitting GS-AuNPs), suggesting two distinct local Au bonding environment and two emission centers in these two types of GS-AuNPs.18 (Figure 2c and 2d) Luminescence of nonthiolated AuNCs exhibited small stokes shift with nanosecond lifetime (Figure 2a) and was assigned to fluorescence, which clearly indicated that ligand is not involved in luminescence. On the contrary, luminescence of thiolated AuNPs exhibited large stokes shift and microsecond lifetime (Figure 4) and was assigned to phosphorescence, consistent with the luminescence properties of Au(I) thiolate complexes. The luminescence mechanisms of Au(I) thiolate complexes have been intensively investigated due to relatively easier access to crystal structures and the emission is involved with ligand-to-metal charge transfer (LMCT) and ligand-to-metal-metal charge transfer (LMMCT) based on S – Au(I) interaction and Au(I) – Au(I) interaction due to aurophilicity.19–22 While a short Au(I) – Au(I) bonding distance is essential to the luminescence of Au(I) thiolate complexes, the Au(0) core in thiolated AuNCs can help to maintain such a short bonding distance by fixing and stabilizing the Au(I)- S staple units on the surface, as evidenced by the X-ray spectroscopy study conducted by Zhang et al.23, 24 Therefore, the size-independent emission in thiolated AuNPs of 2nm can be explained due to the involvement of LMCT and LMMCT in the luminescence mechanism, which are more sensitive to ligand type and Au(I)- Au(I) local bonding than size.
Figure 4.

a) Kohn-Sham orbital energy level diagram for based on time-dependent density functional theory calculation; b) the theoretical absorption spectrums of ; c) experimental UV-vis absorption spectrum of Au25 clusters. All the HOMO and LUMO orbitals also have a significant degree of the S(3p) character and the calculated energy transitions matched well with both the theoretical and experimentally observed absorption spectrums of Au25 clusters.
(Reprinted from ref. 31 with permission from American Chemical Society)
Size-independent emission in thiolated luminescent AuNCs
Strong ligand-metal charge transfer not only enables large 2-3nm AuNPs highly luminescent but also dramtically change fluorescence responses from the few-atom thiolated gold nanoclusters. For example, in glutathione coated AuNCs – Aun(SG)m, the Au18(SG)14, Au22(SG)18 and Au25(SG)18 have emissions at 745nm, 665nm and 700nm with QYs of 5.3%, 8% and 0.19%, respectively, and microseconds luminescence lifetimes (Figure 3).14, 25, 26 No quantitative correlation can be found between size (number of gold atoms) and emission energy of these thiolated GS-AuNCs. Besides, Wang et al. reported a series of AuNCs ranging from Au11, Au38, Au140 to Au201 coated with different surface ligands with comparable emissions in NIR range,27 which are also against the size-dependent emission theory.
Figure 3.
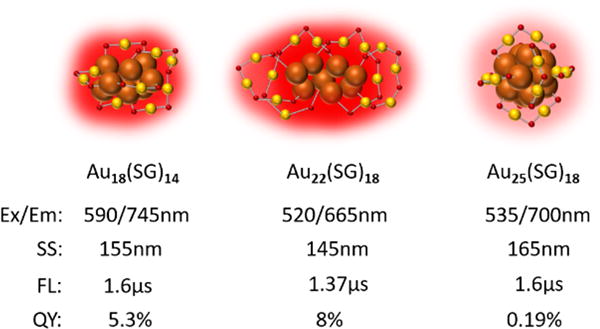
Size-independent emission in GS-AuNCs with corresponding stokes shift (SS), luminescence lifetime (FL) and QY. No quantitative relationship between emission energy and number of gold atoms is observed in Au18(SG)14, Au22(SG)18 and Au25(SG)18.
The breakthrough in unraveling the crystal structures of thiolated gold nanoclusters further advance our understandings of structure-property relationships in the luminescent gold nanoclusters and provide us a good opportunity to differentiate thiolated AuNCs from nonthiolated AuNCs from a structural point of view. Unlike nonthiolated AuNCs, thiolated AuNCs adopt a core-shell structure with an inside Au (0) core and an exterior shell made of Au (I) -S units.28–30 Taking Au25NCs as an example, single-crystal X-ray crystallographic study on phenylethanethiol-coated Au25 revealed a core-shell structure with a centered icosahedral Au13 core and a surface shell constituted of 6 S-Au-S-Au-S staple units.31 Besides, the time-dependent density functional theory study indicated that in the electronic structure of thiolated , the highest HOMO and the lowest three LUMOs are mainly composed of 6sp atomic orbitals from gold, constituting the sp band of Au25(SH)18, while the lower HOMOs are composed of 5d atomic orbitals of gold, constituting the d band of Au25(SH)18 (Figure 4a).31 All the HOMO and LUMO orbitals are composed almost exclusively of atomic orbital contributions from the Au13 core. However, all the HOMO and LUMO orbitals also have a significant degree of the S(3p) character, indicating hybridization of 3p orbitals of S with 5d and 6Sp orbitals of gold is involved in the electronic states of thiolated AuNCs and the calculated energy transitions matched well with both the theoretical and experimentally observed absorption spectrums of Au25 clusters (Figure 4b, 4c).31
A fundamental question in thiolated AuNCs is the role that the ultrasmall Au (0) core may play in luminescence in addition to stabilize and fix surface staple units. The Au (0) core in thiolated AuNCs should still have discrete energy states and may affect luminescence in a different manner. As we mentioned above, the Au (0) core is directly related to the electronic states of thiolated AuNCs based on TDDFT calculations,31, 32 therefore, the Au (0) core may also significantly affect the luminescence and play a critical role in it. Some recent reports regarding the influence of doping other metal into the core of AuNCs can shed some new light on this issue. Negishi et al. reported that doping silver (Ag) into the core of Au25(SR)18 can continuously modulate the electronic state of original Au25(SR)18 based on the doping ratio of Ag as proved by the shift in absorption spectrum.33 A blue shift in emission with the increase of this ratio clearly indicated that the Au (0) core is involved in luminescence. Besides, Jin et al. reported a 200-fold increase in QY of rod-shaped Au25NCs upon doping of 13 Ag atoms into the Au core.34 Such a dramatic increase is believed to be caused by the perturbation on electronic states of AuNCs by Ag atoms, which can also be observed from absorption spectrums. All of these studies can partly and indirectly reflect the influence of Au (0) core on the luminescence. However, despite of the progress at this stage, further studies are highly demanded in the future.
The next question is what causes the difference in the size-independent emission in thiolated AuNCs and AuNPs. The integration of two different emission centers in one particle system can partially prove that the Au (0) core is not involved in the luminescence of thiolated AuNPs. The Au (0) core in thiolated AuNPs is too big to exhibit discrete energy levels compared to the small Au (0) core in thiolated AuNCs. The role that the Au (0) core plays in the luminescence of thiolated AuNPs is more likely to be simply stabilizing and fixing the Au-S motif on the surface, and this also explains the high QYs of some thiolated AuNPs in aqueous solution compared with Au-S complexes.35–37 Certainly, more detailed study into the role of Au (0) core in the luminescence of thiolated few-nanometer AuNPs are in great need in our future research.
Surface-ligand effect on luminescence of thiolated luminescent gold nanoparticles
Electron-donating capability and charge state effects
Since surface ligands play critical roles in the luminescence of thiolated ultrasmall AuNPs due to the LMCT/LMMCT mechanisms, tuning surface ligands is expected to impact the luminescence property of thiolated gold nanoparticles. In 2010, Wu et al. reported that the electron-donating capability of surface ligands and the charge states can affect the QYs of Au25NCs.5 By using surface ligands with different electron-donating capabilities (PhCH2CH2 > C12H25 > C6H13), they found that surface ligand with higher electron-donating capability led to higher QY of Au25 clusters ([Au25(SC2H4Ph)18]− > [Au25(SC12H25)18]− > [Au25(SC6H13)18]−) with a maximum increase of about 5 times (QY: 1 × 10−4 > 5 × 10−5 > 2 × 10−5). (Figure 5a) Similarly, changing the charge state of metal core via adding oxidants can also affect the QYs, increasing the charge state from −1 to +2 led to a 10-fold increase in QYs. (Figure 5b)
Figure 5.
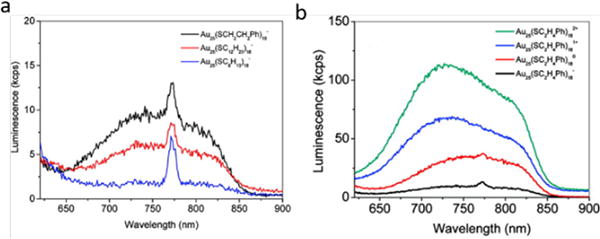
a) Effect of electron-donating capability of ligand, and b) charge state effect on the luminescence of Au25NCs.
(Reprinted from ref. 5 with permission from American Chemical Society)
Surface ligand density, valence state effect, aggregation-induced emission and rigidity
There have been a lot of reports regarding increased QYs of thiolated AuNPs/AuNCs during the past decade. To explain the strong luminescence and elimination of surface plasmon resonance absorption in 2nm GS-AuNPs (Emission: 565nm, QY: ~4%), we first proposed the valence state effect in few-nanometer AuNPs.4 The X-ray photoelectron spectroscopic (XPS) study indicated a high portion (~40-50%) of Au (I) state, which are responsible for the unique optical properties in 2nm luminescent GS-AuNPs. Such a high portion of Au (I) state assures strong Au(I)- Au(I) interaction, which is also in agreement with the previous luminescence study on Au(I) thiolate complexes. Later in 2012, Luo et al. synthesized a set of GS-AuNCs with 610nm emission and extremely high QYs (~15%).35 The XPS studies showed that the Au (I) portion in these GS-AuNCs is ~75%. Meanwhile, noticing the luminescence of Au (I) -S complexes dramatically increased upon aggregation in organic/water mixture solvent, they therefore attributed the high QYs of these GS-AuNCs to the aggregation of Au (I) – S units on the surface which induced strong emission. (Figure 6a) The aggregation-induced emission of AuNCs is consistent with the same phenomenon observed in organic molecules by Tang et. al38 and the enhanced emission can be explained due to the restricted motion of Au-S units on the surface of ultrasamll AuNCs. However, in 2015, Pyo et al. reported that the QY of Au22(SG)18 can be dramatically increased to ~60% upon rigidifying by tetraoctylammonium with slight blue shift of about 35nm.39 (Figure 6b) Noting that the fundamental idea of aggregation-induced emission (AIE) is the restriction of intramolecular motion,38 the effect of rigidity on luminescence is also due to the restraint of molecular motion, which reduces energy loss.40 It is very hard to evaluate which one plays a more important role in accounting for the increased QY in thiolated AuNPs. But it is clear that the Au(I)- Au(I) interactions are strengthened in both AIE and rigidity effects. The surface ligand density has also been reported to affect the QY of AuNPs. The QY of 11-mercaptoundecanoic acid-capped luminescent AuNPs was reported to increase with the increase of surface ligand density without any shift in emission wavelength by adding extra surface ligands to as-synthesized AuNPs.41 A same idea has also been reported very recently in lipoic acid-based sulfobetaine capped AuNCs, however, in this case, adding extra surface ligands not only led to increased QY, but also a blue shift in emission due to the formation of an extended ligand shell via electrostatic interactions between ligands, which means the additional surface ligands don’t bond to the surface gold of the AuNCs. Apparently, surface ligand density is proportional to the Au (I) amount in AuNPs. Therefore, the increase in QY due to surface ligand density may also be interpreted with the same idea in valence state effect. In a word, there is no doubt that the Au (I) ratio plays a critical role in the luminescence of ultrasmall thiolated AuNPs, but fundamentally, it is how this ratio affects the QYs and emission wavelength that matters.
Figure 6.
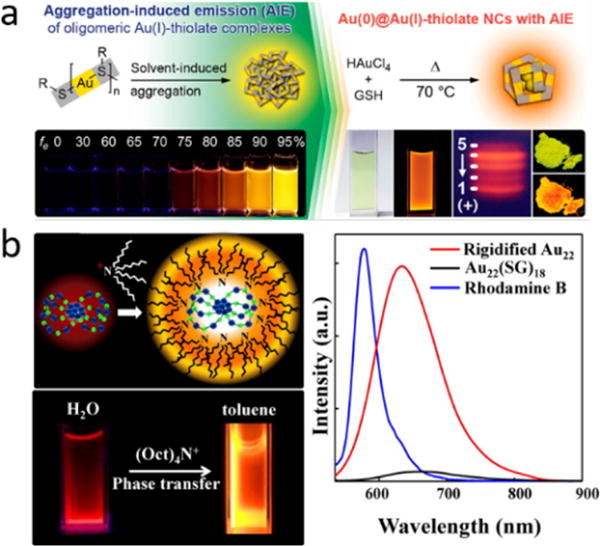
a) Highly luminescent AuNCs with AIE, b) effect of rigidity on the luminescence of Au22(SG)18.
(Reprinted from ref. 35 and 39, with permission from American Chemical Society)
Outlook and perspective
While the size-dependent emission in nonthiolated ultrasmall AuNPs has been well demonstrated over the past two decades, size-independent emission in thiolated ultrasmall AuNPs is discussed herein as well as other aspects that may have impact on luminescence of thiolated ultrasmall AuNPs. Admittedly, fundamental understandings on luminescent ultrasmall AuNPs have been greatly advanced in the past decade. These deeper understandings help us better design luminescent AuNPs and find new applications for luminescent AuNPs. However, there are also fundamental questions that need to be addressed: 1) What is the role that Au (0) core plays in the luminescence of thiolated AuNCs? 2) How to correlate the luminescence of Au (I) – S complexes with luminescent thiolated AuNPs? 3) How the bonding conformation of surface Au (I) – S shell affect the luminescence of thiolated ultrasmall AuNPs? For the future directions of this field, apart from fundamental studies, we also need to aim at developing new applications for ultrasmall luminescent AuNPs. A typical example is the recent study using atomically precise GS-AuNCs with different numbers of gold atoms to investigate the renal clearance efficiency at sub-nanometer regime.42 One limitation of ultrasmall luminescent AuNPs in bioimaging now is the low QYs in NIR range, which requires a relatively high injection dosage in in-vivo imaging. Also, the short excitation wavelength of NIR-emitting AuNPs has low light penetration depth. Therefore, future efforts should be dedicated into exploring new synthetic methods to obtain luminescent AuNPs with high QYs and excitation and emission in NIR range. Another future direction of ultrasmall luminescent AuNPs is the surface modification. Surface modification endows AuNPs with new functions and favorable optical properties, which can further broaden their applications such as in drug delivery and cancer imaging and therapy, etc.43–51 Taking GSH-AuNPs as an example, it has been reported that by introducing cysteamine as a second surface ligand of GS-AuNPs, the QY of GS-AuNPs increased by two times.43 Besides, Jin et. al reported that silver ions can enhance the fluorescence of Au25(SG)18 with nanomolar concentration.52 Surface modification can potentially change the LMCT in luminescent AuNPs and the rigidity of AuNPs, which will consequently change their optical properties and may serve as an efficient method to tune the luminescence of AuNPs. Therefore, fundamental understanding on how surface modification affects the luminescence as well as other properties of ultrasmall AuNPs will be of great significance in the future research.
Acknowledgments
We greatly acknowledge the financial support from NIH (RO1DK103363), CPRIT (RP120588, RP140544 and RP160866), and a start-up fund from The University of Texas at Dallas (J.Z.). Also, we want to thank Xingya Jiang in Zheng group for discussion during the preparation of the manuscript.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Kubo R. Journal of the Physical Society of Japan. 1962;17:975–986. [Google Scholar]
- 2.Schaaff TG, Knight G, Shafigullin MN, Borkman RF, Whetten RL. The Journal of Physical Chemistry B. 1998;102:10643–10646. [Google Scholar]
- 3.Zheng J, Zhang C, Dickson RM. Physical Review Letters. 2004;93:077402. doi: 10.1103/PhysRevLett.93.077402. [DOI] [PubMed] [Google Scholar]
- 4.Zhou C, Sun C, Yu M, Qin Y, Wang J, Kim M, Zheng J. The Journal of Physical Chemistry C. 2010;114:7727–7732. doi: 10.1021/jp9122584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Z, Jin R. Nano Letters. 2010;10:2568–2573. doi: 10.1021/nl101225f. [DOI] [PubMed] [Google Scholar]
- 6.Zhou C, Yu J, Qin Y, Zheng J. Nanoscale. 2012;4:4228–4233. doi: 10.1039/c2nr30212h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mooradian A. Physical Review Letters. 1969;22:185–187. [Google Scholar]
- 8.Kreibig U, Vollmer M. Optical properties of metal clusters, Springer Science & Business Media. 2013 [Google Scholar]
- 9.Physik in unserer Zeit. 1978;9:33–33. [Google Scholar]
- 10.Zheng J, Petty JT, Dickson RM. Journal of the American Chemical Society. 2003;125:7780–7781. doi: 10.1021/ja035473v. [DOI] [PubMed] [Google Scholar]
- 11.Zheng J, Nicovich PR, Dickson RM. Annual review of physical chemistry. 2007;58:409–431. doi: 10.1146/annurev.physchem.58.032806.104546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawasaki H, Hamaguchi K, Osaka I, Arakawa R. Advanced Functional Materials. 2011;21:3508–3515. [Google Scholar]
- 13.Duan H, Nie S. Journal of the American Chemical Society. 2007;129:2412–2413. doi: 10.1021/ja067727t. [DOI] [PubMed] [Google Scholar]
- 14.Habeeb Muhammed MA, Ramesh S, Sinha SS, Pal SK, Pradeep T. Nano Research. 2008;1:333–340. [Google Scholar]
- 15.Zheng J. PhD thesis. Georgia Institute of Technology; 2005. [Google Scholar]
- 16.Negishi Y, Nobusada K, Tsukuda T. Journal of the American Chemical Society. 2005;127:5261–5270. doi: 10.1021/ja042218h. [DOI] [PubMed] [Google Scholar]
- 17.Negishi Y, Takasugi Y, Sato S, Yao H, Kimura K, Tsukuda T. Journal of the American Chemical Society. 2004;126:6518–6519. doi: 10.1021/ja0483589. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Duchesne PN, Yu M, Jiang X, Ning X, Vinluan RD, Zhang P, Zheng J. Angewandte Chemie International Edition. 2016;55:8894–8898. doi: 10.1002/anie.201602795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forward JM, Bohmann D, Fackler JP, Staples RJ. Inorganic Chemistry. 1995;34:6330–6336. [Google Scholar]
- 20.Cha SH, Kim JU, Kim KH, Lee JC. Chemistry of Materials. 2007;19:6297–6303. [Google Scholar]
- 21.Schneider J, Lee YA, Pérez J, Brennessel WW, Flaschenriem C, Eisenberg R. Inorganic Chemistry. 2008;47:957–968. doi: 10.1021/ic701763x. [DOI] [PubMed] [Google Scholar]
- 22.Li CK, Lu XX, Wong KMC, Chan CL, Zhu N, Yam VWW. Inorganic Chemistry. 2004;43:7421–7430. doi: 10.1021/ic049094u. [DOI] [PubMed] [Google Scholar]
- 23.Zhang P. The Journal of Physical Chemistry C. 2014;118:25291–25299. [Google Scholar]
- 24.Chevrier Daniel M, Yang R, Chatt A, Zhang P. Journal. 2015;4:193. [Google Scholar]
- 25.Yu Y, Luo Z, Chevrier DM, Leong DT, Zhang P, Jiang D-E, Xie J. Journal of the American Chemical Society. 2014;136:1246–1249. doi: 10.1021/ja411643u. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh A, Udayabhaskararao T, Pradeep T. The Journal of Physical Chemistry Letters. 2012;3:1997–2002. doi: 10.1021/jz400332g. [DOI] [PubMed] [Google Scholar]
- 27.Wang G, Huang T, Murray RW, Menard L, Nuzzo RG. Journal of the American Chemical Society. 2005;127:812–813. doi: 10.1021/ja0452471. [DOI] [PubMed] [Google Scholar]
- 28.Jadzinsky PD, Calero G, Ackerson CJ, Bushnell DA, Kornberg RD. Science. 2007;318:430–433. doi: 10.1126/science.1148624. [DOI] [PubMed] [Google Scholar]
- 29.Zeng C, Chen Y, Kirschbaum K, Lambright KJ, Jin R. Science. 2016;354:1580–1584. doi: 10.1126/science.aak9750. [DOI] [PubMed] [Google Scholar]
- 30.Azubel M, Koivisto J, Malola S, Bushnell D, Hura GL, Koh AL, Tsunoyama H, Tsukuda T, Pettersson M, Häkkinen H, Kornberg RD. Science. 2014;345:909–912. doi: 10.1126/science.1251959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu M, Aikens CM, Hollander FJ, Schatz GC, Jin R. Journal of the American Chemical Society. 2008;130:5883–5885. doi: 10.1021/ja801173r. [DOI] [PubMed] [Google Scholar]
- 32.Yao H. The Journal of Physical Chemistry Letters. 2012;3:1701–1706. doi: 10.1021/jz300396u. [DOI] [PubMed] [Google Scholar]
- 33.Negishi Y, Iwai T, Ide M. Chemical Communications. 2010;46:4713–4715. doi: 10.1039/c0cc01021a. [DOI] [PubMed] [Google Scholar]
- 34.Wang S, Meng X, Das A, Li T, Song Y, Cao T, Zhu X, Zhu M, Jin R. Angewandte Chemie International Edition. 2014;53:2376–2380. doi: 10.1002/anie.201307480. [DOI] [PubMed] [Google Scholar]
- 35.Luo Z, Yuan X, Yu Y, Zhang Q, Leong DT, Lee JY, Xie J. Journal of the American Chemical Society. 2012;134:16662–16670. doi: 10.1021/ja306199p. [DOI] [PubMed] [Google Scholar]
- 36.Vickery JC, Olmstead MM, Fung EY, Balch AL. Angewandte Chemie International Edition in English. 1997;36:1179–1181. [Google Scholar]
- 37.Wing-Wah Yam V, Kam-Wing Lo K. Chemical Society Reviews. 1999;28:323–334. [Google Scholar]
- 38.Mei J, Leung NLC, Kwok RTK, Lam JWY, Tang BZ. Chemical Reviews. 2015;115:11718–11940. doi: 10.1021/acs.chemrev.5b00263. [DOI] [PubMed] [Google Scholar]
- 39.Pyo K, Thanthirige VD, Kwak K, Pandurangan P, Ramakrishna G, Lee D. Journal of the American Chemical Society. 2015;137:8244–8250. doi: 10.1021/jacs.5b04210. [DOI] [PubMed] [Google Scholar]
- 40.Valeur B, Berberan-Santos MN. Molecular fluorescence: principles and applications. John Wiley & Sons; 2012. [Google Scholar]
- 41.Chang HY, Chang HT, Hung YL, Hsiung TM, Lin YW, Huang CC. RSC Advances. 2013;3:4588–4597. [Google Scholar]
- 42.Du B, Jiang X, Das A, Zhou Q, Yu M, Jin R, Zheng J. Nature Nanotechnology. 2017;12:1096. doi: 10.1038/nnano.2017.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu M, Zhou C, Liu J, Hankins JD, Zheng J. Journal of the American Chemical Society. 2011;133:11014–11017. doi: 10.1021/ja201930p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu M, Zhou C, Liu L, Zhang S, Sun S, Hankins JD, Sun X, Zheng J. Angewandte Chemie International Edition. 2017;56:4314–4319. doi: 10.1002/anie.201612647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Xianyu Y, Jiang X. Accounts of Chemical Research. 2017 doi: 10.1021/acs.accounts.6b00506. [DOI] [PubMed] [Google Scholar]
- 46.Chompoosor A, Han G, Rotello VM. Bioconjugate Chemistry. 2008;19:1342–1345. doi: 10.1021/bc8000694. [DOI] [PubMed] [Google Scholar]
- 47.Ghosh P, Han G, De M, Kim CK, Rotello VM. Advanced Drug Delivery Reviews. 2008;60:1307–1315. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh PS, Kim CK, Han G, Forbes NS, Rotello VM. ACS Nano. 2008;2:2213–2218. doi: 10.1021/nn800507t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim B, Han G, Toley BJ, Kim C-K, Rotello VM, Forbes NS. Nat Nano. 2010;5:465–472. doi: 10.1038/nnano.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Y, Peng J, Li J, Huang L, Yang J, Huang K, Li H, Jiang N, Zheng S, Zhang X, Niu Y, Han G. Nano Letters. 2017;17:4096–4100. doi: 10.1021/acs.nanolett.7b00828. [DOI] [PubMed] [Google Scholar]
- 51.Mercado-Lubo R, Zhang Y, Zhao L, Rossi K, Wu X, Zou Y, Castillo A, Leonard J, Bortell R, Greiner DL, Shultz LD, Han G, McCormick BA. Nature Communications. 2016;7:12225. doi: 10.1038/ncomms12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu Z, Wang M, Yang J, Zheng X, Cai W, Meng G, Qian H, Wang H, Jin R. Small. 2012;8:2028–2035. doi: 10.1002/smll.201102590. [DOI] [PubMed] [Google Scholar]