

Abstract
12/15 lipoxygenase (LOX) directs inflammation and lipid remodeling. However, the role of 12/15LOX in post-myocardial infarction (MI) left ventricular remodeling is unclear. To determine the role of 12/15LOX, 8-12 week-old C57BL/6J wild-type (WT; n=93) and 12/15LOX−/− (n=97) mice were subjected to permanent coronary artery ligation and monitored at day (d)1 and d5 post-operatively. Post-MI d28 survival was measured in male and female mice. No-MI surgery mice were maintained as d0 naïve controls. 12/15LOX-/- mice exhibited higher survival rates with lower cardiac rupture and improved LV function as compared with WT post-MI. Compared to WT, neutrophils and macrophages in 12/15LOX-/- mice were polarized towards N2 and M2 phenotypes, respectively, with increased of expression mrc-1, ym-1, and arg-1 post-MI. 12/15LOX-/- mice exhibited lower levels of pro-inflammatory 12-(S)-hydroperoxyeicosatetraenoic acid (12(S)-HETE) and higher CYP2J-derived epoxyeicosatrienoic acids (EETs) levels. CYP2J-derived 5,6-, 8,9-, 11,12-, and 14,15-EETs activated macrophage-specific hemeoxygenase (HO)-1 marked with increases in F4/80+/Ly6Clow and F4/80+/CD206high cells at d5 post-MI in 12/15LOX-/- mice. In contrast, inhibition of HO-1 led to total mortality in 12/15LOX-/- mice by post-MI d5. 12/15LOX-/- mice exhibited reduced collagen density and lower α-smooth muscle actin (SMA) expression at d5 post-MI, indicating delayed or limited fibroblast-to-myofibroblast differentiation. In conclusion, genetic deletion of 12/15LOX reduces 12(S)-HETE and activates CYP2J-derived EETs to promote effective resolution of inflammation post-MI leading to reduced cardiac rupture, improved LV function, and better survival.
Keywords: Heart failure, lipid mediators, macrophages, myocardial infarction, neutrophils
Introduction
Adverse cardiac remodeling is pathological process that develops after myocardial infarction (MI) and alters shape, size, and function of the left ventricle (LV) [1]. Functional and structural changes of the LV are widely associated with defective resolution of inflammation and extracellular matrix (ECM) responses, leading to increased mortality in heart failure patients [2, 3]. The post-MI inflammatory response initiates mobilization of immune cells (neutrophils and macrophages) studied in chemokine and cytokine inhibitory approaches. Due to the failure of non-aspirin containing anti-inflammatory products, the FDA recommends limiting the use of inflammation inhibiting agents in the heart failure setting [4]. Thus, it becomes an unmet medical need to resolve inflammation after MI. Resolution in response to MI-induced healing is coordinated by macrophages, lipid mediators and inflammation resolving enzymes such as lipoxygenases (LOXs) cyclooxygenases (COX) and cytochrome P450 epoxygenases (CYP) [5].
LOXs and CYPs are a series of iron-containing enzymes that metabolize arachidonic acid to form biologically active products, such as epoxyeicosatrienoic acids (EETs) hydroperoxyeicosatetraenoic acids (HETEs), prostaglandins, leukotrienes and thromboxanes [6]. Among these mediators, 12/15LOX has gained particulate attention because of its increased expression in inflammatory diseases, such as atherosclerosis, diabetes and diabetic cardiomyopathy [6-8]. 12/15LOX forms unstable 12(S)-HETE, which can activate a signaling cascade leading to cytokine-induced cell damage [9, 10]. Transgenic expression of 12/15LOX specifically in cardiomyocytes versus macrophage results in divergent effects on atheroprogressive inflammation and atherogenic cardiac pathology [11]. Disruption of LOXs, COX, and CYPs alters the metabolic transformation of arachidonic acid or other essential fatty acids to lipid mediators that have differential effects on resolving and non-resolving inflammatory responses [12-14].
We have tested the hypothesis that reduced levels of 12 (S)-HETE, achieved using genetic deletion of 12/15LOX, would improve post-MI LV function and survival by accelerating the resolution of inflammation. Post-MI, 12/15LOX null mice augment CYP-mediated mediated EETs through the splenocardiac axis in coordination with resolving neutrophils, macrophages and reduced levels of 12(S)-HETE. Our results indicate that 12/15LOX null mice orchestrate effective resolution of inflammation leading to improved LV function, limited fibroblast to myofibroblast transition, and increased survival post-MI.
Methods
A brief description of methods used is given below.
Animal care compliance
All animal procedures were conducted according to the “Guide for the Care and Use of Laboratory Animals” (8th Edition. 2011), AVMA Guidelines for the Euthanasia of Animals: (2013 Edition) and were approved by the Institutional Animal Care and Use Committees at the University of Alabama, Birmingham, USA.
Mice and coronary ligation surgery
MI was induced in WT and 12/15LOX-/- by surgical ligation of the left anterior descending coronary artery, as described previously [15]. The study design and output measurements for the complete study are defined in supplementary figure 1 and supplementary table 1.
Autopsy and post-MI survival analysis
The mice were checked daily for 28 days post-MI for survival analysis. At autopsy, cardiac rupture was confirmed by the presence of clotted blood in the thoracic cavity and direct identification of the LV rupture site. To determine sex comparison between male and female mice on post-MI rupture and non-rupture congestive heart failure, chi-square analysis was done.
Echocardiography
LV function was evaluated using the Vevo 770 imaging system (VisualSonics Inc.), as described previously [15].
Necropsy and infarct area analysis
Tissue and blood samples were collected as detailed in our previous reports [16]. The infarct area was calculated as the percentage of infarct area to total LV area [15].
Mass spectrometric analysis of plasma and spleen lipid mediators
Lipid mediators in the plasma and spleen were analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS) (AB/Sciex API-6500QTRAP mass spectrometer) as per the protocol described [17, 18].
Isolation of mononuclear LV cells for FACs
Single mononuclear cells were isolated from no-MI controls day (d0) and at post-MI-d5 from LV following methods of Ishmahil et al. with slight modification [19, 20]. The viability was measured by using the trypan blue exclusion method.
Spleen cell preparation for FACs
The splenocytes for flow cytometry were isolated as described previously with slight modification [19, 20].
Surface markers strategy for FACs
LV mononuclear cell or splenocytes were defined as neutrophils, classical or alternative macrophages based on the surface markers as detailed in supplementary data in supplemental figure 2.
Isolation of neutrophils and macrophages from LV infarct
Neutrophils and macrophages were isolated from LV infarct at post-MI d1 and d5 using Ly6G+ and CD11b+ -specific magnetic beads [21].
LV histology, Immunohistochemistry, and confocal microscopy
LV Immunohistochemistry for neutrophils, macrophages, COX-2, HO-1, SMA and DDR2 samples were performed as detailed in the protocol in supplementary methods.
Collagen measurements using picrosirius red staining
For collagen deposition measurement picrosirus red (PSR) was done as detailed in supplementary methods.
Image analysis for IHC and PSR staining
The image analysis was done with a microscope (BX43) using the cellSens Dimension program (Olympus 1.9), then analyzed for percent area stained using Image-Pro Premier 64-bit software.
RT2profiler inflammatory and ECM PCR array
Real-Time RT2-PCR gene array for inflammatory cytokines and receptors (Qiagen, PAMM-011E) and ECM and adhesion molecules (Qiagen, PAMM-013E) were performed to quantify gene expression levels as per manufacturer's protocol, on ABI7900HT. Gene levels were normalized to hypoxanthine phosphoribosyltransferase (Hprt-1) as the housekeeping gene control. The results were reported as 2-ΔCt (ΔΔCt) values.
LV and spleen protein extraction and immunoblotting
Protein extraction and immunoblotting performed as described previously [15] and the total protein was used as internal loading control. Image J was used for densitometry analysis.
Measurements of macrophage and neutrophil phenotype using real-time quantitative PCR
M1(F4/80+/Ly6chigh, IL-6 and TNF-α) and M2 macrophage (F4/80+/Ly6clow, CD206, Arg-1 and YM-1) or N1(F4/80-/Ly6G+/Ly6chigh, IL-6 and TNF-α) and N2(F4/80-/Ly6G+, CD206, Arg-1 and YM-1) neutrophil phenotype were measured using qPCR; results were reported as 2-ΔCt (ΔΔCt) values.
Venn diagram, hierarchical clustering of ECM and inflammatory array
Venn diagram, hierarchical cluster, and heat maps were generated for RT2profiler inflammatory and ECM PCR array data using cluster 3.0 and java tree view software. The values of genes were normalized by taking geometric mean of genes for statistical significance by student-t-test (unpaired). p<0.05 was considered as statistically significant.
Statistical analysis
Data are expressed as mean per group and SEM. Statistical analyses were performed using GraphPad Prism 5. Analysis of variance (ANOVA), followed by Newman–Keuls posthoc test, was for multiple comparisons of post-MI-d1, d5 compared to d0 naïve control. Kaplan-Meier test and the log-rank test was followed for survival analysis. For 2 groups comparison, student-t-test (unpaired) was applied and p<0.05 was considered as statistically significant.
Results
12/15LOX deletion increased survival post-MI
12/15LOX deletion was confirmed in 12/15LOX-/- mice by evaluating mRNA levels and immunohistochemistry (supplementary Fig 3A and B). Then, to determine the role of 12/15LOX, post-MI survival was monitored in WT and 12/15LOX-/- mice. Post-MI survival (to d28) was increased in 12/15LOX-/- mice compared to WT controls (58% in WT vs 85% in 12/15LOX-/- mice; p<0.01). Sex-specific analysis showed that WT males (58%) had lower survival compared to 12/15LOX-/- female (68%) mice (Fig 1A). Autopsy of WT male mice during the first week revealed that 37% (7/19) had LV rupture and 5% had congestive heart failure (CHF) compared with 12/15LOX-/- male mice having 7% rupture and 7% CHF. The 12/15LOX-/- female mice showed lower rates of rupture (7%) and heart failure compared to WT females. Thus, deletion of 12/15LOX improved survival and reduced rupture rates in both male and female mice (Fig 1B). To define the role of 12/15LOX in inflammation and the resolution of inflammation, further studies were done at two time points in male mice; (1) initiation of the acute inflammatory response (post-MI d1) and (2) the resolving phase of inflammation (post-MI d5). Since activated 12/15LOX uses arachidonic acid as a substrate to biosynthesize multiple bioactive lipid mediators (Fig 1C), we used an LC-MS/MS-based quantitative lipidomic approach. WT mice exhibited elevated plasma levels of arachidonic acid-derived 12(S)-HETE at d1 post-MI as compared with no-MI controls (p<0.05). Plasma 12(S)-HETE levels were lower at both d1 and d5 post-MI in 12/15LOX-/- mice compared to WT (p<0.05; Fig 1D-E). Loss of 12/15LOX decreased the levels of pro-inflammatory 15(S)-HETE (Fig 1F) but increased the pro-resolving metabolite 5(S)-HETE (Fig 1G).
Figure 1. 12/15LOX-/- deletion increased survival and reduced ruptures post-MI.
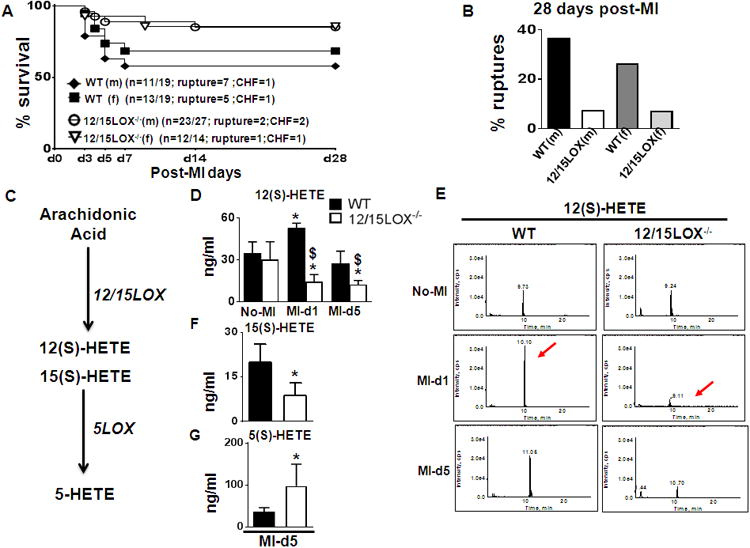
(A) Survival rates post-MI in WT and 12/15LOX-/- mice analyzed by log-lank test. *p<0.01 vs. WT, n=19 WT (male), n=19 WT (female), n=27 12/15LOX-/- (male) and n=14 12/15LOX (female). (B) Bar graph representing post-MI ruptures and congestive heart failure in WT and 12/15LOX-/- male and female mice. (C) Pathway scheme representing generation of lipid mediator from arachidonic acid. (D) Quantitative measurement of plasma 12(S)-HETE by mass spectrometry (LC-MS/MS). n=4; *p<0.05 vs no-MI, $p<0.05 vs WT post-MI. (E) Representative chromatograms of 12(S)-HETE at d1 and d5 post-MI. (F) Quantitative measurement of plasma 15(S)-HETE. n=4; *p<0.05 vs WT (G) Quantitative measurement of plasma 5(S)-HETE n=4; *p<0.05 vs WT.
12/15LOX-/- mice attenuated LV remodeling, dysfunction and edema index post-MI
Echocardiographic and gravimetric parameters were assessed to determine the impact of 12/15LOX deletion on LV function and remodeling in the post-MI setting. The LV mass-to-body weight ratio was reduced in 12/15LOX-/- mice compared to WT mice. Lung mass-to-body weight ratio was reduced in 12/15LOX-/- mice compared to WT mice at d5 post-MI. At d5 post-MI, right ventricle (RV) to the mass-to-body weight ratio was lower in 12/15LOX-/- mice compared to WT. Thus, necropsy data of LV and lung to body mass ratios suggested alleviation of post-MI LV remodeling and pulmonary edema in 12/15LOX-/- mice (Table 1). Echocardiography data (Table 1) revealed decreased ESD and EDD post-MI-d5 and increased fractional shortening in 12/15LOX-/- mice compared to WT, indicative of improved LV function post-MI.
Table 1.
Necropsy and echocardiography parameters indicating reduced LV remodeling, pulmonary edema and LV function in 12/15LOX-/- mice post-MI.
| Control d0 | MI day 1 | MI day 5 | MI day 28 | |||||
|---|---|---|---|---|---|---|---|---|
| Parameters | WT (n=6) | 12/15-LOX-/- (n=8) | WT (n=7) | 12/15-LOX-/- (n=10) | WT (n=6) | 12/15-LOX-/- (n=9) | WT (n=9) | 12/15-LOX-/- (n-18) |
| Necropsy parameters | ||||||||
| Body weight (g) | 23 ± 2 | 20 ±1 | 20 ± 1 | 19 ± 1 | 21 ± 1 | 19 ± 1 | 24 ± 1 | 20 ± 1 |
| LV (mg) | 74± 5 | 62 ± 2$ | 79 ± 4 * | 76 ± 2*$ | 108 ± 5* | 88 ± 3*$ | 96 ±7 | 78 ± 4*$ |
| LV/BW (mg/g) | 3.2± 0.04 | 3.2 ± 0.14 | 4.0 ± 0.24* | 3.9 ± 0.12* | 5.3 ± 0.31* | 4.8 ± 0.25* | 4.8 ± 0.18* | 3.9 ± 0.1 * |
| Right ventricle (mg) | 17 ± 2 | 17± 1 | 19 ± 1* | 17 ± 1 | 19 ± 2* | 16 ± 2$ | 21 ± 1* | 19 ± 1$ |
| Lung weight (mg) | 127± 8 | 113 ± 2$ | 184 ± 22* | 155 ± 15*$ | 211 ± 12* | 158 ± 17 $ | 127 ± 4 | 117 ± 5*$ |
| Lung wt (dry; mg) | 31 ± 1 | 27 ± 1 | 34 ± 2 | 31 ± 2 | 42 ± 2* | 34 ± 3$ | 30 ± 1 | 26 ±1* |
| Tibia (mm) | 16 ± 0.7 | 16 ± 0.3 | 16 ± 0.3 | 17. ± 0.2 | 17 ± 0.2 | 16 ± 0.1 | 17 ± 0.1 | 17 ± 0.2 |
| Infarct area (%) | - | - | 47 ± 1 | 47 ± 1 | 49 ± 1 | 53 ± 1 | 51+3 | 51+2 |
| Echocardiography parameters | ||||||||
| Heart Rate (bpm) | 454 ± 16 | 435 ± 6 | 473 ± 18 | 439 ± 13 | 491 ± 16 | 427 ± 8 | 445 ± 13 | 400 ± 6 |
| EDD (mm) | 3.7 ± 0.12 | 3.7 ± 0.12 | 4.5 ± 0.12* | 4.4 ± 0.13*$ | 6.5 ± 0.02* | 5.2 ± 0.15 *$ | 5.1± 0.32 *$ | 4.9 ± 0.14 |
| ESD (mm) | 2.3 ± 0.14 | 2.4 ± 0.04 | 4.2 ± 0.13* | 3.9 ± 0.15*$ | 6.1 ± 0.18* | 4.8 ± 0.14*$ | 4.7± 0.31 | 4.4 ± 0.16 |
| Fractional Shortening % | 37 ± 2 | 34± 2 | 7 ± 2* | 11± 2*$ | 5 ± 1* | 7± 1*$ | 6± 2* | 9± 1$ |
| PWTs (mm) | 1.05± 0.02 | 1.00 ± 0.02 | 0.56 ± 0.06* | 0.57 ± 0.05* | 0.47 ± 0.05* | 0.51 ± 0.03*$ | 0.39 ± 0.05*$ | 0.50 ± 0.03 |
Values are means ± SEM. n indicates sample size; LV, Left Ventricle; BW. Body weight; RV, Right Ventricle; - = No infarct in d0 naïve controls; bpm, beats per minute; EDD, end-diastolic dimension; ESD, end-systolic dimension; PWT, posterior wall thickness, S, systole; mm, millimeter.
p<0.05 vs the d0 control,
p< 0.05 vs. day 0 wild-type (WT) control;
p< 0.05 vs. WT with respective time point.
12/15LOX deletion shifted arachidonic acid metabolism to the generation of EETs
12/15LOX uses arachidonic acid as a substrate to biosynthesize pro-inflammatory 12(S)-HETE. As the spleen is a major myeloid cell reservoir, we determined splenic levels of metabolites using LC-MS/MS for comprehensive lipidometabolomic analysis (Table 5). This revealed that levels of the pro-inflammatory metabolites 12(S)-HETE, 15(S)-HETE, LTB4, 8,10-DiHOME, 12(13)DiHOME, 9-HODE, 13-HODE and 20-Oxo-LTB4 were decreased in 12/15LOX-/- mice as compared with WT (Fig 2A). Interestingly, 12/15-LOX-/- mice displayed an increase in several bioactive EETs, including 5,6-, 8,9-, 11,12-, and 14,15-EpETrE, and 16(17)-EpDPE (Fig 2B). To validate increased levels of EETs, we measured CYP2J in the spleen and LV. Compared to WT, 12/15LOX deletion induced CYP2J cytochrome P450 in the LV and spleen (Fig 2C-D). Since we observed that EETs were high in 12/15LOX-/- mice, we further measured mRNA level of Ephx2 which encodes for soluble epoxide hydrolase (sEH), an enzyme that degrades EETs. mRNA expression of Ephx2 was lower in 12/15LOX-/- mice compared to WT at d5 post-MI in both the LV and spleen (Fig 2E). Moreover, EPA-derived pro-resolving-metabolites 5-, 9-, and 18-HEPE were increased in 12/15LOX-/- mice post-MI (Fig 2F). Thus, comprehensive bioactive lipid mediator analysis (supplementary table 2) indicated that genetic deletion of 12/15LOX decreased 12-(S)-HETE, a pro-inflammatory metabolite, and increased CYP2J expression in LV and spleen leading to increased CYP P450-derived endogenous anti-inflammatory lipid metabolites post-MI.
Figure 2. 12/15LOX deletion upregulated cytochrome (450) subfamily CYP2J that promoted epoxyeicosatrienoic acids (EETs) biosynthesis in spleen.
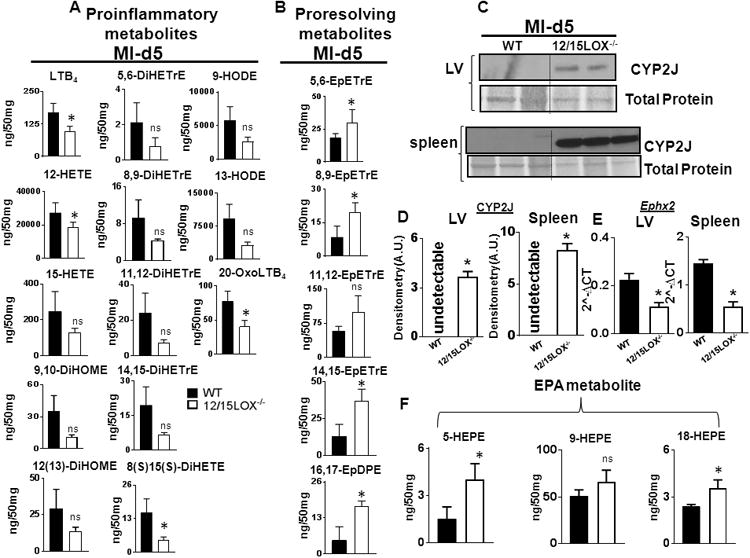
(A) Quantification of proinflammatory metabolites in spleens of WT and 12/15LOX-/- mice at d5 post-MI. (B) Quantification of proresolving metabolites: EETs and EpDPE in WT and 12/15LOX-/- mice at d5 post-MI. (C) Immunoblot representing CYP2J expression (upper panel) and in LV (lower panel) and spleen at d5 post-MI. n=2-3;*p<0.05 vs WT. (D) Densitometric analysis of CYP2J in LV and spleen normalized to total protein. n=2-3 mice; *p<0.05 vs MI-WT at d5 post-MI. (E) mRNA expression of Ephx2 in LV and spleen at d5 post-MI normalized to HPRT-1; n=4 mice; *p<0.05 vs MI-WT at d5 post-MI. (F) Quantification of HEPEs (5, 9 and 18) in spleens of WT and 12/15LOX-/- mice at d5 post-MI. n=4 mice; *p<0.05 vs MI-WT at d5 post-MI.
12/15LOX-/- mice shifted neutrophil polarization towards proresolving neutrophils
To determine the post-MI healing profile in 12/15LOX-/- mice, the biphasic inflammatory response was measured. Both WT and 12/15LOX-/- mice displayed similar neutrophil infiltration at d1 post-MI, but neutrophils were cleared faster in the 12/15-LOX-/- mice compared to WT at d5 post-MI (Fig 3A-B). The 84 inflammatory gene array data at d1 post-MI showed higher expression levels of cx3cl1, ccr55 and ccl5 in 12/15LOX-/- mice (Fig 3C, supplementary figure 8A, supplementary table 3 and 4). To confirm the precise quantity of LV mononuclear cells, we explored temporal dynamics on the basis of Ly6G+ and Ly6C+ expression using flow cytometry. Isolated cells showed a robust increase in CD11b+ cells with sub-populations having dual pattern of Ly6G+/Ly6Chigh LV mononuclear cells at d1 post-MI in both mice, indicating increased infiltration of classical macrophages with neutrophils (Fig 3D). Compared to WT (15.3±1.6%), 12/15LOX-/- (27.4±1.9%) mice showed high percentage of Ly6G+/Ly6Chigh cells (Fig. 3E). Isolation of the Ly6G+ neutrophils from infarcted LV displayed a distinct pattern of N1 (pro-inflammatory markers) and N2 proresolving markers. The neutrophils isolated from 12/15LOX-/- mice displayed higher levels of Tnf-α and IL-6 (Fig 3F) compared with WT indicating N1 phenotype at d1 post-MI with moderate increase in N2 markers, Mrc-1, Arg-1 and Ym-1, at d5 post-MI (Fig 3G). Thus, 12/15LOX deletion altered the biphasic response post-MI, first by priming the inflammatory response in the acute phase at d1 post-MI and then expediting resolution in the subsequent reparative phase at d5 post-MI.
Figure 3. 12/15LOX-/- deletion polarized neutrophils towards proresolving phenotype.
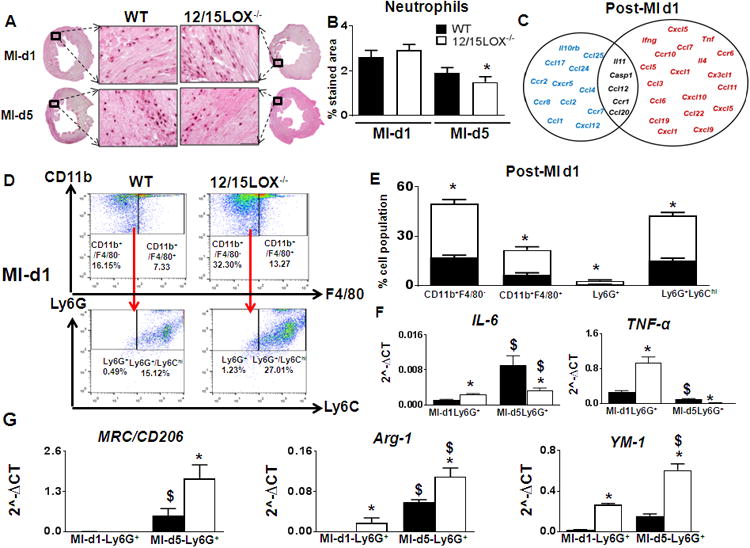
(A) Representative neutrophil-stained LV images at 40× with their accompanying 1.25× sections at d1, and d5 post-MI; scale bar = 50 μm n=4. (B) Quantification of the neutrophils stained area in the LV at post-MI d1 and d5. n=4; *p<0.05 vs WT. (C) Venn diagram representing post-MI inflammatory gene expression in LVI regions of 12/15LOX-/- mice at post-MI-d1. Post-MI-d1 gene expressions are normalized to respective no-MI. Red indicates ↑ed genes. Blue indicates ↓ed genes. Black indicates no change in genes. n=4; p<0.05 vs WT post-MI-d1. (D) Representative fluorescence-activated cell (FACS) sorting dot plots showing increased neutrophils by Ly6Ghigh strategy in LV mononuclear cells n=4-6 mice. (E) Quantification of CD11b+/F4/80-, CD11b+/F4/80+, Ly6G+ and Ly6G+/Ly6Chigh LV mononuclear cells post-MI-d1. n=4-6 mice; *p<0.05 vs WT. (F) mRNA expression of IL-6, tnf-α, from Ly6G+(N1) cells isolated from LV infarct. n=4mice; *p<0.05 vs WT $p<0.05 vs WT at d1 post-MI. (G) mRNA expression of Mrc-1/CD206, Arg-1 and Ym-1 from Ly6G+(N2) cells isolated from LV infarct. n=4-6 mice; *p<0.05 vs WT control, $p<0.05 vs WT at d1 post-MI.
12/15LOX-/- mice dominated macrophage polarization at LV
After MI, macrophages play important roles in cardiac wound healing. Immunohistochemistry based semi-quantitative analyses revealed higher density of macrophages in 12/15LOX-/- compared to WT mice at d5 (Fig. 4A-B). We further quantitated macrophage sub-populations by flow cytometry. Based on CD206 expression and Ly6C expression patterns, the macrophages were defined as either M1 (pro-inflammatory; classical) or M2 (anti-inflammatory; alternative) by flow cytometry. 12/15LOX-/- mice exhibited higher population of Ly6Clow (13±1%; p<0.05) and CD206+ cells (14±1%; p<0.05) compared to WT at d5 post-MI (Fig. 4C and D) indicative of increased reparative macrophages (Fig. 4C and D). Isolation of CD11b+ cells from infarcted LV confirmed macrophage polarization at d5 post-MI suggesting a resolving (M2) phenotype with increase in Ym-1, Mrc-1/CD206 and Arg-1 in 12/15LOX-/- compared with WT mice (Fig 4E-G). In order to dissect the site of macrophage polarization, spleen or LV, we determined macrophage subpopulations at d5 in splenocytes and the infarcted LV by flow cytometry. Macrophage polarization was minimal in spleen at d5 post-MI in both WT and 12/15LOX-/- mice (Supplementary Fig. 4 and 5A-B). Thus, quantitative flow cytometry data suggest that a macrophage reparative phenotype facilitates the healing process in 12/15LOX-/- mice, thereby accelerating effective resolution of inflammation and improving survival.
Figure 4. 12/15LOX-/- deletion promoted alternative macrophage polarization to resolve inflammation by d5 post-MI.
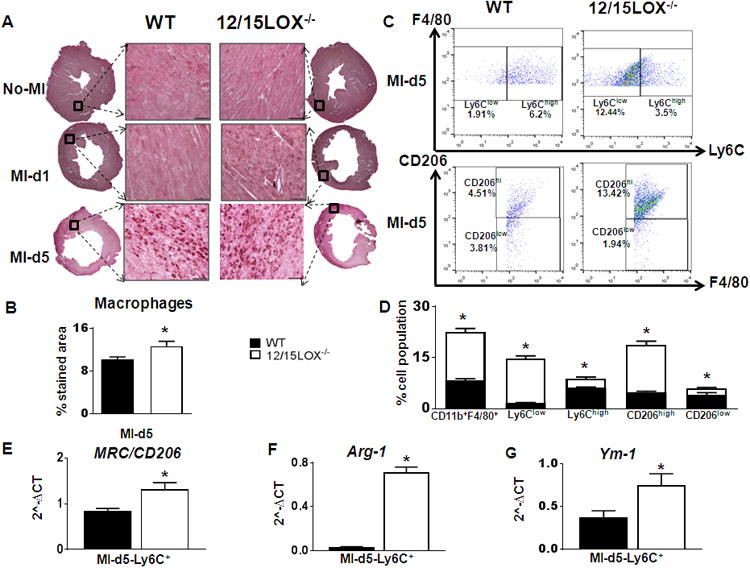
(A) Representative IHC images of LV macrophages (Mac-3 positive) density at d5 post-MI. Representative images at 40× with accompanying 1.25× images scale bar = 50 μm (upper panel). (B) Quantification of % area of macrophages stained area in the LV infarcted zone at d5 post-MI (lower panel). n=4-6 mice; *p<0.05 vs WT. (C) Representative FACS sorting dot plots showing alternative macrophages polarization by F4/80+/Ly6Clow strategy in LV mononuclear cells (upper panel). FACs dot plots showing alternative macrophages polarization by F4/80+/CD206+ strategy in LV mononuclear cells (lower panel). n=4-6 mice. (D) Quantification of CD11b+/F4/80+, F4/80+/Ly6Clow, F4/80+/Ly6Chigh F4/80+/CD206high and F4/80+/CD206low LV mononuclear cells at d5 post-MI. n=4 mice; *p<0.05 vs WT. (E-G) mRNA expression of Mrc-1/CD206, Arg-1, Ym-1 respectively from CD11b+ cells isolated from LV infarct at d5 post-MI. n=4 mice; *p<0.05 vs WT.
12/15LOX deletion activated HO-1 in macrophages thereby effective resolution post-MI
HO-1 and 5LOX have been shown to facilitate healing post-MI [22-24] and EETs are known to stimulate HO-1. As expected, 12/15LOX deletion increased mRNA expression of HO-1 and 5LOX and protein in the infarcted and remote LV area (Fig. 5A-D). HO-1 is expressed in several cell types including macrophages. To delineate the cellular source of HO-1, macrophages were stained for HO-1 (green) and co-localized with Mac3 (red) using immunofluorescence; yellow staining in the overlay suggested macrophages as a source of HO-1 (Fig. 5E). Increased levels of HO-1 and 5LOX was supported by an increase in the expression of alternative macrophage markers such as CD163, Ym-1, Mrc-1/CD206 and Arg-1 with simultaneous decrease in M1 macrophages, viz;Ccr2, Tnf-α, IL-1β and IL-6 at d5 post-MIin the infarcted LV of 12/15LOX-/- mice compared with WT counterparts (Fig. 5F). Further 5LOX, COX-1, COX-2 and HO-1 protein expression were measured in LV (remote and infarct zones) and the spleen (supplementary Fig. 6 and 7). To delineate the role of HO-1 in 12/15LOX-/- mice, we inhibited HO-1 using Tin Protoporphyrin IX, a specific HO-1 inhibitor (post-MI d1 to d5) in 12/15LOX-/- mice. Of note, HO-1 inhibition led to 100% mortality in 12/15LOX mice by d5 post-MI with no rupture (Fig 5G). Thus, our results indicated macrophage-specific HO-1 expression plays a crucial role in resolution of inflammation and survival of 12/15LOX-/- mice post-MI.
Figure 5. Macrophages derived HO-1 promoted effective resolution of inflammation in 12/15LOX-/- mice.
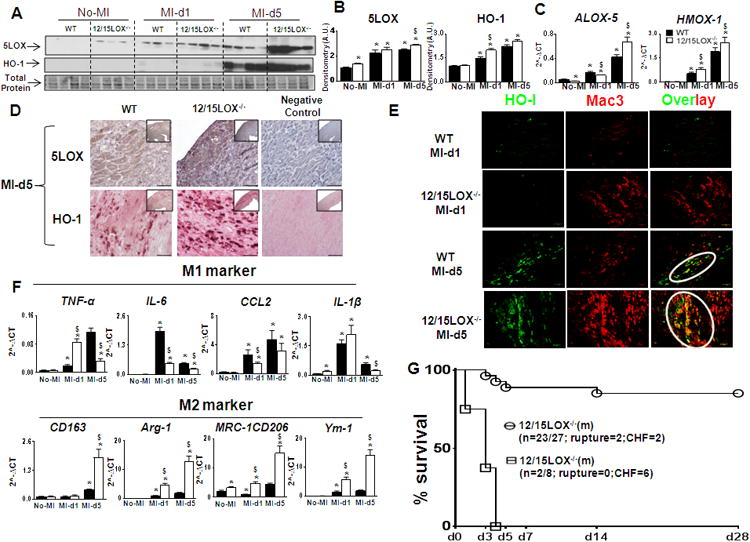
(A) Immunoblot representing 5-LOX and HO-1 expression in LVI. (B) Densitometric analysis showing increase 5LOX and HO-1; n=3 mice for no-MI; n=4 mice post-MI; *p<0.05 vs no-MI, $p<0.05 vs MI-WT at respective day. (C) mRNA expression of Alox-5 and Hmox-1 in LVI. n=4 mice; *p<0.05 vs No-MI, $p<0.05vs MI-WT at respective day. (D) Representative LV IHC images showing 5LOX and HO-1 expression at d5 post-MI. (E) Representative immunofluorescence co-localization images (yellow) of HO-1 (green) with Mac-3 (red) (Magnification 10×). (F) Gene expression of M1 and M2 markers. n=4 mice; *p<0.05 vs no-MI, $p<0.05vs MI-WT at respective day. (G) Survival curve presenting HO-1 inhibitor (tin protoporphyrin IX) increased mortality in 12/15LOX-/- mice bylog-lank test *p<0.01.
12/15LOX deletion limited myofibroblast formation and collagen deposition post-MI
Myocardial injury is followed by scar formation. Thus, to evaluate post-MI matrix remodeling (ECM) in 12/15LOX-/- mice, we studied the fibroblast-myofibroblast axis. Analysis of picroSirius Red (PSR) stained area in no-MI control and post-MI d5 revealed lower collagen levels in 12/15LOX-/- mice compared to WT mice (Fig 6A-C). This was complemented by downregulation of MMP-9 at d1 and upregulation of Timp-1 at d5 post-MI in the LV-infarct of 12/15LOX-/- mice. Interestingly, collagen VI level was increased in 12/15LOX-/- mice compared to WT at d5 post-MI in the LV infarct (Fig 6D). Further, to explore post-MI changes in ECM remodeling in 12/15LOX-/- mice, we performed gene array for 84 ECM and adhesion molecules in the LV-infarcted area. Out of 84 genes, col1a1, col2a1 and col3a1 expression were lowered, while col6a1 expression was increased, in 12/15LOX-/- mice compared to WT, supporting reduced ECM remodeling (Fig. 6F, Supplementary figure 8B and supplementary table 5). Post-MI increase in col6a1 expression was associated with an increase Tgf-bi, ctgf, ecm-1 and adamts2 expression. Fibroblast-myofibroblast transition is marked by increase α-smooth muscle actin (α-SMA) expression. 12/15LOX-/- mice had lower α-SMA expression (Fig. 6D, upper panel, 6E; upper panel and 6G) as compared with WT mice at d5 post-MI in the infarct zone. Lower α-SMA expression in 12/15LOX-/- mice suggested diminished fibroblast to myofibroblast differentiation at d5 post-MI.
Figure 6. 12/15LOX deletion attenuated extracellular matrix (ECM) deposition by limiting myofibroblast formation post-MI.
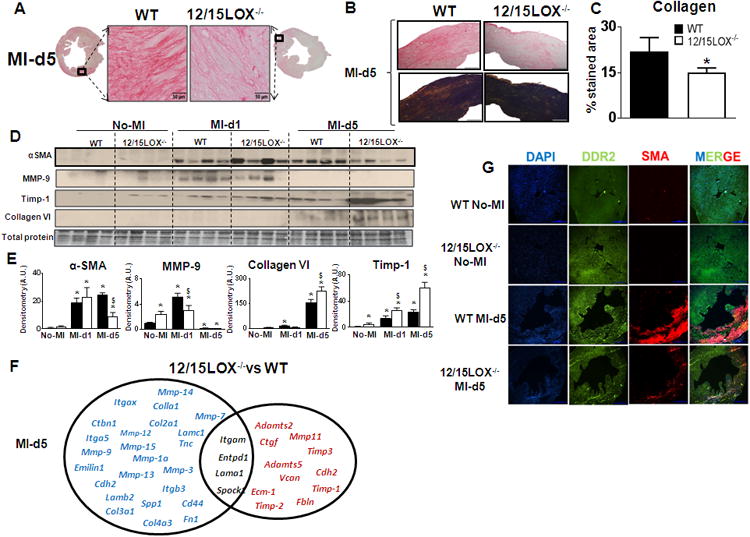
(A) Picrosirus red (PSR) stained LV images (40× magnification; scale bar = 50 μm.) with complete LV acquired (1.25× magnification) at d5 post-MI, (B) Representative LV collagen deposition under plane polarized light (right panel). (C) Quantification of PSR stained collagen levels presented as % stained area in 5-6 images/mouse (left panel), n=4-6 mice; *p<0.05 vs WT. (D) Immunoblot representing α-SMA, MMP-9, TIMP-1 and collagen VI expression. (E) Densitometric analysis of α-SMA, MMP-9, TIMP-1 and collagen VI levels normalized to total protein. n=4-6 mice; *p<0.05 vs no-MI, $p<0.05 vs MI-WT at respective day. (F) Venn diagram representing post-MI ECM remodeling genes in LVI regions of 12/15LOX-/- mice. Post-MI d5 gene expressions are normalized to respective no-MI. Red indicates ↑ed genes. Blue indicates ↓ed genes. Black indicates no change in genes. n=4; *p<0.05 vs WT at d5 post-MI. (G) Representative immunofluorescence images indicating reduced α-SMA (myofibroblast) and no change DDR2 (fibroblast) expression in LV of 12/15LOX-/- at d5 post-MI.
Discussion
Heart failure secondary to MI is characterized by sustained inflammation and ventricular dilation. This pathological feature of cardiac remodeling is associated with decreased LV function and lower survival [2, 3]. The current study focused on understanding the role of 12/15LOX in post-MI LV remodeling and survival. Here, we established that, 12/15LOX deletion in the post-MI setting leads to: 1) improved survival up to 28 d; 2) decreased levels of 12(S)-HETE, and increased levels of CYP2J-derived bioactive EETs; 3) altered leukocyte phenotype with increased N2 type neutrophils and M2 type Ly6Clow and CD206+ macrophages; 4) increased HO-1 expression in alternative macrophages that promotes post-MI healing; and 5) reduced collagen deposition with less myofibroblast differentiation. Thus, 12/15LOX deletion improved LV function by promoting effective resolution of inflammation, modulating leukocytes toward a reparative phenotype, and delaying ECM deposition post-MI.
Lipid mediators diversity and leukocyte phenotypes
Pro-inflammatory lipid mediators such as 12(S)-HETE contribute to non-resolving inflammation in cardiac pathology [25]. Thus, reduced plasma levels of 12(S)-HETE in 12/15LOX-/- mice may improve post-MI survival secondary to the expedited resolution of inflammation. The post-MI wound healing process involves an acute inflammatory response followed by a resolution phase, and is accompanied by the entry of immune cells from the splenic reservoir [19]. Deletion of the 12/15LOX gene resulted in an attenuation of pathological LV remodeling with a decrease in the pro-inflammatory lipid mediator 12(S)-HETE at d1 post-MI. 12(S)-HETE is known to increase pro-inflammatory markers such as MCP-1, IL-6 [26, 27] and the recruitment of monocytes, thereby facilitating a systemic pro-inflammatory milieu. Earlier reports have shown that 5LOX deletion impairs wound healing and promotes cardiac rupture, indicating a crucial role in resolving post-MI inflammation [22]. This is consistent with our study where 12/15LOX deletion reduced rupture rate in males and females together with an increase in 5LOX and 5-HETE. Thus, these data are consistent with an altered microenvironment due to the decreased level of 12(S)-HETE and other pro-inflammatory mediators in the 12/15LOX-/- mice that reduced the rate of rupture and improved post-MI survival.
A timely and well-orchestrated immune response is essential for cardiac healing [28, 29], and the infiltration of neutrophils, and neutrophil subtypes, have been recognized to play pathophysiologic roles. Reprogramming of neutrophils toward and N2 phenotype imparts beneficial effects, similar to those attributed to M2 reparative macrophages. The reprogramming of neutrophils has been established recently in stroke, and found to be driven by transforming growth factor-β (TGF-β) [30]. Neutrophils in 12/15LOX-/- mice displayed higher levels of Tnf-α and IL-6 at d1 post-MI (N1 phenotype), but greater expression of Ym-1, Mrc-1/CD206 and Arg-1 at d5 post-MI indicating that deletion of 12/15LOX supports polarization of neutrophils towards an N2 phenotype. Alternative activation of macrophages is also essential for optimal cardiac repair post-MI, requiring a balance between classically activated M1 and alternatively activated M2 macrophages as previously reported [21, 31]. Phenotypically, mouse Ly6Chigh and Ly6Clow cells are analogous to human CD14+ and CD14lowCD16+ monocytes, respectively [17]. The prevalence of CD11b+/F4/80+/Ly6chigh/CD206low cells marks the prolonged inflammatory phase post-MI in WT mice compared to presence of CD11b+/F4/80+/Ly6Clow/CD206high population in 12/15LOX-/- mice indicating an increase in alternative (M2) macrophage polarization. Therefore, we report first time that 12/15LOX deletion reprograms neutrophil towards an N2 phenotype and macrophages towards an M2 phenotype, leading to improved healing, increased survival and better LV function post-MI.
Splenocardiac axis in the resolution of inflammation
Splenic mononuclear cells heighten the immune response and promote LV remodeling and dysfunction in chronic ischemic heart failure [19, 20]. Previous studies have explained the recruitment of splenic monocytes and change in the architecture of spleen in response to MI [19, 32]. We examined whether splenic monocytes/macrophages post-MI express a unique pattern of pro-inflammatory and anti-inflammatory markers. The 12/15LOX-/- spleen exhibited an increase in F4/80+/CD206+ macrophages with an elevated mRNA levels of Ym-1. Our previous study indicates that the splenic proresolving lipid mediator RvD1 contributes to the resolution of inflammation [33]. Metabololipidomic analysis suggested an increase in CYP-derived EETs, termed as C epoxins, with an increase in CYP2J expression in 12/15LOX-/- mouse spleens. The deletion of 12/15LOX shifted AA metabolism towards the CYP (cytochrome P450 epoxygenase) pathway, leading to the formation of EETs [34]. The biosynthesis of EETs depends on activation of epoxygenase (CYP enzymes) and fatty acids substrate. In arachidonate cascade, the P450 epoxygenase is unique in regio- and stereochemical selectivity to biosynthesize EET mediators. EETs are known to reduce infarct size, preserve LV function and limit cardiac hypertrophy [35, 36]. CYP2J subfamilies of CYP are abundantly expressed in the cardiovascular system and metabolize AA to produce four biologically active EETs: 5,6 (cypoxins; CExn1)-, 8,9 (CExn2)-, 11,12 (CExn3)-, and 14,15 (CExn4)-EETs [37]. Here we report a reciprocal relationship between EETs and DHETs in spleens of 12/15LOX-/- mice. The increased C epoxins such as 5,6-EET; 8,9-EET; 11,12-EET and 14,15-EET is accompanied by a decrease in dihydroxyeicosatrienoic acids (DiHETEs), i.e., 5,6-: 8,9-; 11,12- and 14,15-DiHETE. This was consistent with a decrease in Ephx2 mRNA levels in 12/15 LOX-/- mice indicating a decrease in soluble epoxide hydrolase (sEH). These results the align with the results from Yang et al. [35] where a decrease in EETs generated an inflammatory response in coronary heart disease (CHD) patients. Other clinical studies have also shown obstructive coronary artery disease (CAD) is associated with lower EET metabolite levels [38]. Furthermore, studies have shown that EETs upregulate HO-1 and ameliorate cardiac fibrosis and improve angiogenesis and cardiac function post-MI [39]. Further, HO-1 expression was increased in spleen, similar to observations in cardiac pro-resolving leukocytes. Thus, 12/15LOX deletion shifts lipid metabolism towards CYP2J generated EETs post-MI which actively promotes the resolution of inflammation.
In response to cardiac injury, 12/15LOX deletion in mice leads to increase expression of CYP2J along with a remarkable increase of HO-1 expression at d5 in LV and spleen, indicating enzymatic flexibility to process fatty acids substrate. HO-1 has been shown to improve ventricular remodeling [24, 40]. Our findings that post-MI inhibition of HO-1 in 12/15LOX-/- lead to 100% mortality by 5 days post-MI indicates an essential role for HO-1 in post-MI cardiac healing. Further, the direct links between CYPJ2 and HO-1 have been reported [41]. Many previous reports of gene expression profiling showed that HO-1 is co-expressed by M2 macrophages and confers cardioprotective anti-inflammatory responses [23]. Co-expression of HO-1 and Mac-3 in 12/15LOX-/- mice suggested macrophages as the prime source of HO-1. Deletion of 12/15LOX enhanced macrophage-specific HO-1 expression in 12/15LOX-/- mice that promoted an M2 phenotype. HO-1 expression was also observed in WT mice, but it appeared to be derived from smooth muscle cells or other cell types and was not co-localized with macrophages. The simultaneous increase in an M2 macrophage gene profile and HO-1 expression in 12/15LOX-/- mice indicated that M2 macrophages expedited the resolution of inflammation.
Post-MI, cardiac fibroblasts undergo a series of changes to form a reparative scar [29]. Myofibroblast formation is essential for wound healing and repair. Myofibroblasts express large amounts of α-SMA, which marks myofibroblast contractility [42]; uncontrolled myofibroblast formation leads to cardiac fibrosis and LV dysfunction [43]. In the present report, 12/15LOX deletion reduced total collagen levels at d5 post-MI compared to WT. Reduced ECM was supported by the decrease in α-SMA expression in 12/15LOX-/- mice. Increase in collagen VI was observed in 12/15LOX-/- mice compared to WT that indicates matrix complexity in heart failure pathology [44]. Post-MI, MMP-9 is known to increase within the first 24 h, together with increased neutrophil infiltration [15]. A higher level of MMP-9 at d1 post-MI with a marked decrease by d5 post-MI in 12/15LOX-/- mice provides one of the potential mechanisms for the attenuated ECM remodeling in 12/15LOX-/- mice.
Our results imply that 12/15LOX-mediated local microenvironment polarizes macrophages to resolving or non-resolving functions post-MI. CYP2J directly impacted HO-1, which modified the local milieu to define the plasticity of resolving neutrophils and macrophages. In summary, the 12/15LOX deletion improved post-MI survival by promoting the effective resolution of inflammation and by reducing post-MI rupture. Our study proposes the novel paradigm of a 12/15LOX-dependent balance between the effective and defective resolution of inflammation, which, in the absence of 12/15LOX, channels immune cells towards a reparative phenotype, leading to better wound healing, improved LV function, and increased survival post-MI.
Clinical Implications
Emerging evidence has established that the non-resolving inflammation after MI contributes to the heart failure. 12/15LOX increases post-MI increase in 12(S)-HETE levels creates pro-inflammatory environment. Preclinical study has shown that LOX inhibitor attributes its significance because it maintains the human 12/15LOX selectivity, but is also effective in a mouse stroke model and can be of clinical relevance [45]. The strategic manipulation of specific 12/15LOX to control defective inflammatory disorders including diabetes, asthma, and atherosclerosis has been under investigation. Thus, an understanding of complex arachidonic acidlipid derivatives and their metabolites in resolving post-MI inflammation could help in developing specialized therapeutics to delay heart failure.
Translational outlook
The presented report not only highlights 12/15LOX as a potential target for treating heart failure but provides evidence that delicate balance of lipid mediators i.e. 12(S)-HETE (pro-inflammatory) and EETs (proresolving) generated through arachidonic acid metabolism. These pathways can be clinically implicated in the resolution of inflammation post-MI.
Supplementary Material
Figure 7.
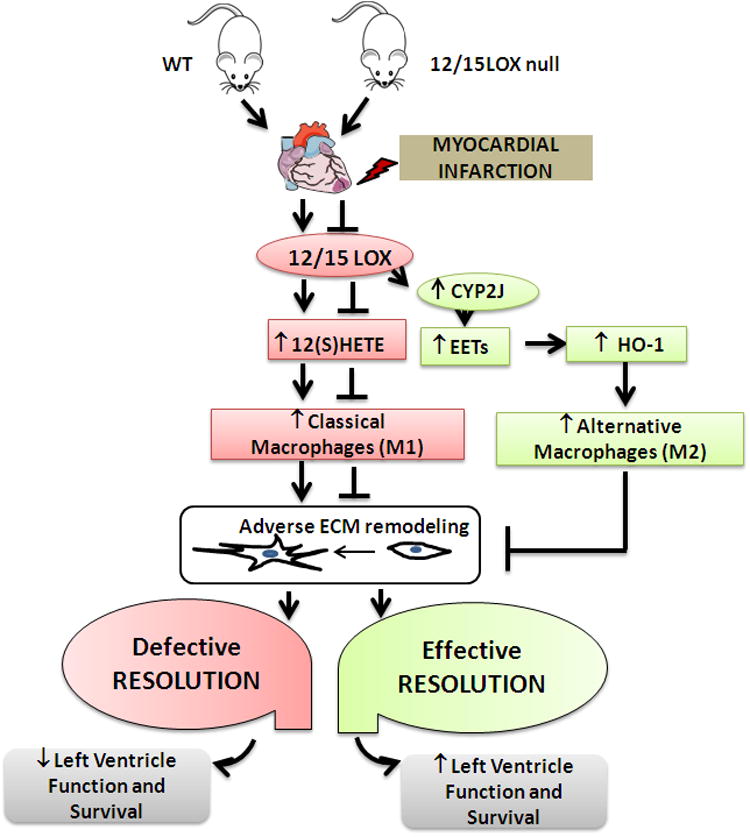
Schematic design indicating lack of 12/15LOX promotes effective resolution of inflammation post-MI in heart failure pathology.
Highlights.
12/15LOX deletion improved post-MI survival by reducing cardiac rupture and ameliorated cardiac function.
12/15LOX deletion accelerated resolution of inflammation by promoting N2 neutrophil polarization and M2 macrophage polarization post-MI.
12/15LOX deletion decreased pro-inflammatory 12(S)-HETE and increased CYP2J-derived proresolving epoxyeicosatrienoic acid (EETs) thereby limiting inflammation and adverse cardiac remodeling.
Acknowledgments
This work was supported in part by the National Institutes of Health [AT006704], [HL132989] and Pittman Scholar Award to G.V.H., NIH R01 HL125735 and VA I01 BX002706 to S.D.P, and American Heart Association postdoctoral fellowship [POST31000008] to V. K. Funds for the mass spectrometer used in this study were provided by the UAB Health Services Foundation General Endowment Fund to S. B. Authors are thankful to Dr. Darryl C. Zeldin, NEIHS for proving CYP2J antibody and recombinant protein for the present study.
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest.
References
- 1.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Journal of the American College of Cardiology. 2000;35:569–82. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 2.Kain V, Prabhu SD, Halade GV. Inflammation revisited: inflammation versus resolution of inflammation following myocardial infarction. Basic research in cardiology. 2014;109:444. doi: 10.1007/s00395-014-0444-7. [DOI] [PubMed] [Google Scholar]
- 3.Reina-Couto M, Carvalho J, Valente MJ, Vale L, Afonso J, Carvalho F, et al. Impaired resolution of inflammation in human chronic heart failure. European journal of clinical investigation. 2014;44:527–38. doi: 10.1111/eci.12265. [DOI] [PubMed] [Google Scholar]
- 4.Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA, et al. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634–42. doi: 10.1161/CIRCULATIONAHA.106.181424. [DOI] [PubMed] [Google Scholar]
- 5.Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circulation research. 2010;107:1170–84. doi: 10.1161/CIRCRESAHA.110.223883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chemical reviews. 2011;111:5866–98. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 7.Shamina MGM, Sarah AT, Banumathi KC, Kaiwen M, Norine SK, Tina Duong C, et al. Deletion of 12/15-Lipoxygenase Alters Macrophage and Islet Function in NOD-Alox15null Mice, Leading to Protection against Type 1 Diabetes Development. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0056763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, et al. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. The Journal of clinical investigation. 1999;103:1597–604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuhn H, O'Donnell VB. Inflammation and immune regulation by 12/15-lipoxygenases. Progress in lipid research. 2006;45:334–56. doi: 10.1016/j.plipres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Soberman RJ, Harper TW, Betteridge D, Lewis RA, Austen KF. Characterization and separation of the arachidonic acid 5-lipoxygenase and linoleic acid omega-6 lipoxygenase (arachidonic acid 15-lipoxygenase) of human polymorphonuclear leukocytes. The Journal of biological chemistry. 1985;260:4508–15. [PubMed] [Google Scholar]
- 11.Kayama Y, Minamino T, Toko H, Sakamoto M, Shimizu I, Takahashi H, et al. Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. The Journal of experimental medicine. 2009;206:1565–74. doi: 10.1084/jem.20082596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sellers RS, Radi ZA, Khan NK. Pathophysiology of cyclooxygenases in cardiovascular homeostasis. Veterinary pathology. 2010;47:601–13. doi: 10.1177/0300985810364389. [DOI] [PubMed] [Google Scholar]
- 13.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature reviews Immunology. 2008;8:349–61. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tourki B, Halade G. Leukocyte diversity in resolving and nonresolving mechanisms of cardiac remodeling. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2017;31:4226–39. doi: 10.1096/fj.201700109R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, et al. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circulation research. 2013;112:675–88. doi: 10.1161/CIRCRESAHA.111.300502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halade GV, Kain V, Ingle KA. Heart functional and structural compendium of cardiosplenic and cardiorenal networks in acute and chronic heart failure pathology. American journal of physiology Heart and circulatory physiology. 2018;314:H255–H67. doi: 10.1152/ajpheart.00528.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nature reviews Immunology. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 18.Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. American journal of physiology Cell physiology. 2014;307:C39–54. doi: 10.1152/ajpcell.00024.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circulation research. 2014;114:266–82. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, et al. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circulation Heart failure. 2017;10:e003688. doi: 10.1161/CIRCHEARTFAILURE.116.003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, et al. Foxp3+ CD4+ T Cells Improve Healing After Myocardial Infarction by Modulating Monocyte/Macrophage Differentiation. Circulation research. 2014;115:55–67. doi: 10.1161/CIRCRESAHA.115.303895. [DOI] [PubMed] [Google Scholar]
- 22.Blomer N, Pachel C, Hofmann U, Nordbeck P, Bauer W, Mathes D, et al. 5-Lipoxygenase facilitates healing after myocardial infarction. Basic research in cardiology. 2013;108:367. doi: 10.1007/s00395-013-0367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi KM, Kashyap PC, Dutta N, Stoltz GJ, Ordog T, Shea Donohue T, et al. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology. 2010;138:2399–409, 409 e1. doi: 10.1053/j.gastro.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang G, Hamid T, Keith RJ, Zhou G, Partridge CR, Xiang X, et al. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation. 2010;121:1912–25. doi: 10.1161/CIRCULATIONAHA.109.905471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopez EF, Kabarowski JH, Ingle KA, Kain V, Barnes S, Crossman DK, et al. Obesity superimposed on aging magnifies inflammation and delays the resolving response after myocardial infarction. American journal of physiology Heart and circulatory physiology. 2015;308:H269–80. doi: 10.1152/ajpheart.00604.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen Y, Gu J, Vandenhoff GE, Liu X, Nadler JL. Role of 12/15-lipoxygenase in the expression of MCP-1 in mouse macrophages. American journal of physiology Heart and circulatory physiology. 2008;294:H1933–8. doi: 10.1152/ajpheart.00260.2007. [DOI] [PubMed] [Google Scholar]
- 27.Dwarakanath RS, Sahar S, Lanting L, Wang N, Stemerman MB, Natarajan R, et al. Viral vector-mediated 12/15-lipoxygenase overexpression in vascular smooth muscle cells enhances inflammatory gene expression and migration. Journal of vascular research. 2008;45:132–42. doi: 10.1159/000109966. [DOI] [PubMed] [Google Scholar]
- 28.Frantz S, Bauersachs J, Ertl G. Post-infarct remodelling: contribution of wound healing and inflammation. Cardiovascular research. 2009;81:474–81. doi: 10.1093/cvr/cvn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction. From Inflammation to Fibrosis. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA, et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke. 2013;44:3498–508. doi: 10.1161/STROKEAHA.113.002470. [DOI] [PubMed] [Google Scholar]
- 31.Fujiu K, Nagai R. Contributions of cardiomyocyte-cardiac fibroblast-immune cell interactions in heart failure development. Basic research in cardiology. 2013;108:357. doi: 10.1007/s00395-013-0357-x. [DOI] [PubMed] [Google Scholar]
- 32.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–45. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, et al. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol. 2015;84:24–35. doi: 10.1016/j.yjmcc.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halade GV, Kain V, Ingle KA, Prabhu SD. Interaction of 12/15-lipoxygenase with fatty acids alters the leukocyte kinetics leading to improved postmyocardial infarction healing. American journal of physiology Heart and circulatory physiology. 2017;313:H89–H102. doi: 10.1152/ajpheart.00040.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang T, Peng R, Guo Y, Shen L, Zhao S, Xu D. The role of 14,15-dihydroxyeicosatrienoic acid levels in inflammation and its relationship to lipoproteins. Lipids Health Dis. 2013;12:151. doi: 10.1186/1476-511X-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tacconelli S, Patrignani P. Inside epoxyeicosatrienoic acids and cardiovascular disease. Front Pharmacol. 2014;5:239. doi: 10.3389/fphar.2014.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu X, Zhang XA, Wang DW. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv Drug Deliv Rev. 2011;63:597–609. doi: 10.1016/j.addr.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 38.Oni-Orisan A, Edin ML, Lee JA, Wells MA, Christensen ES, Vendrov KC, et al. Cytochrome P450-derived epoxyeicosatrienoic acids and coronary artery disease in humans: a targeted metabolomics study. Journal of lipid research. 2016;57:109–19. doi: 10.1194/jlr.M061697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao J, Tsenovoy PL, Thompson EA, Falck JR, Touchon R, Sodhi K, et al. Agonists of epoxyeicosatrienoic acids reduce infarct size and ameliorate cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway. Prostaglandins & other lipid mediators. 2015;116-117:76–86. doi: 10.1016/j.prostaglandins.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yet SF, Tian R, Layne MD, Wang ZY, Maemura K, Solovyeva M, et al. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circulation research. 2001;89:168–73. doi: 10.1161/hh1401.093314. [DOI] [PubMed] [Google Scholar]
- 41.Elmarakby AA, Faulkner J, Pye C, Rouch K, Alhashim A, Maddipati KR, et al. Role of haem oxygenase in the renoprotective effects of soluble epoxide hydrolase inhibition in diabetic spontaneously hypertensive rats. Clin Sci (Lond) 2013;125:349–59. doi: 10.1042/CS20130003. [DOI] [PubMed] [Google Scholar]
- 42.Jain M, DerSimonian H, Brenner DA, Ngoy S, Teller P, Edge AS, et al. Cell therapy attenuates deleterious ventricular remodeling and improves cardiac performance after myocardial infarction. Circulation. 2001;103:1920–7. doi: 10.1161/01.cir.103.14.1920. [DOI] [PubMed] [Google Scholar]
- 43.Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:437–42. doi: 10.1073/pnas.0408704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luther DJ, Thodeti CK, Shamhart PE, Adapala RK, Hodnichak C, Weihrauch D, et al. Absence of type VI collagen paradoxically improves cardiac function, structure, and remodeling after myocardial infarction. Circulation research. 2012;110:851–6. doi: 10.1161/CIRCRESAHA.111.252734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rai G, Joshi N, Jung JE, Liu Y, Schultz L, Yasgar A, et al. Potent and selective inhibitors of human reticulocyte 12/15-lipoxygenase as anti-stroke therapies. Journal of medicinal chemistry. 2014;57:4035–48. doi: 10.1021/jm401915r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.