

Abstract
The c-Myb transcription factor is required for adult hematopoiesis, yet little is known about c-Myb function during lineage-specific differentiation due to the embryonic lethality of Myb-null mutations. We previously used tissue-specific inactivation of the murine Myb locus to demonstrate that c-Myb is required for differentiation to the pro-B cell stage, survival during the pro-B cell stage and the pro-B to pre-B cell transition during B-lymphopoiesis. However, few downstream mediators of c-Myb-regulated function have been identified. We demonstrate that c-Myb regulates the intrinsic survival of CD19+ pro-B cells in the absence of IL-7 by repressing expression of the pro-apoptotic proteins Bmf and Bim and that levels of Bmf and Bim mRNA are further repressed by IL-7 signaling in pro-B cells. c-Myb regulates two crucial components of the IL-7 signaling pathway, the IL-7Rα chain and the negative regulator SOCS3 in CD19+ pro-B cells. Bypassing IL-7R signaling through constitutive activation of Stat5b largely rescues survival of c-Myb-deficient pro-B cells, while constitutively active Akt is much less effective. However, rescue of pro-B cell survival is not sufficient to rescue proliferation of pro-B cells or the pro-B to small pre-B cell transition and we further demonstrate that c-Myb-deficient large pre-B cells are hypoproliferative. Analysis of genes crucial for the pre-BCR checkpoint demonstrates that, in addition to IL-7Rα, the genes encoding λ5, cyclin D3 and CXCR4 are downregulated in the absence of c-Myb and λ5 is a direct c-Myb target. Thus, c-Myb coordinates survival with the expression of genes that are required during the pre-BCR checkpoint.
Introduction
B cell development, like the development of each hematopoietic lineage, initiates from a multipotent, self-renewing hematopoietic stem cell and is defined by the sequential expression of cell surface markers and V(D)J recombination events at the immunoglobulin heavy and light chain loci (1). Hematopoietic stem cells (HSCs) give rise to progenitor cells that gradually lose alternative lineage fate potential and gain B-lineage potential as they differentiate to the CD19+ pro-B cell stage, which is the first B-lineage committed progenitor. During the pro-B cell stage, productive rearrangement at the immunoglobulin heavy chain locus results in expression of an immunoglobulin μ-heavy chain protein, which pairs with the surrogate light chain and signaling components Igα and Igβ to form the pre-BCR. These cells differentiate into the large pre-B cell stage and undergo a limited proliferative burst, then exit the cell cycle, differentiate to the small pre-B cell stage and initiate V(D)J rearrangement at the kappa light chain locus (2, 3). Upon productive V(D)J rearrangement at one of the immunoglobulin light chain loci, light chain protein can pair with the μ-heavy chain to form membrane IgM and initiate differentiation to the immature B cell stage.
Control of survival during the pro-B cell stage is crucial as cells must have sufficient time to complete successful V(D)J rearrangements at the heavy chain locus but not so much time that pro-B cells with failed V(D)J recombination accumulate or for potentially oncogenic chromosome translocations to occur (4). The balance of pro-apoptotic and anti-apoptotic Bcl-2 family members mediates the intrinsic survival pathway during the pro-B cell stage (5). Oligomerization of the pro-apoptotic proteins Bak and Bax at the mitochondrial membrane leads to release of cytochrome c and initiation of apoptosis (6). The oligomerization of Bak and Bax is inhibited by interaction with a group of anti-apoptotic proteins that includes Bcl-2 and Mcl-1 and gain and loss of function mouse models have demonstrated that these proteins are important for survival at different stages of B cell development (7–9). The anti-apoptotic proteins are opposed by a group of pro-apoptotic BH3-only proteins that includes Bim, Bad, Bid and Bmf, which act as molecular sensors of cellular stress and interfere with the interaction of Bax and Bak with the anti-apoptotic Bcl-2 family members (10). In general, the BH3-only proteins are highly redundant and only Bim-deficient mice are reported to display a phenotype that is characterized by an increased number of pro-B cells (11). While the Bim deficient mouse model demonstrates a role for Bim in pro-B cell survival, the absolute number of pro-B cells in these mice is less than that observed in a Bcl-2 transgenic mouse model, suggesting that additional BH3-only pro-apoptotic proteins contribute to the survival of CD19+ pro-B cells.
The IL-7 signaling pathway is the major mediator of survival during the CD19+ pro-B cell stage and mediates survival by transcriptional and post-translational regulation of the pro-apoptotic and anti-apoptotic Bcl-2 family members (12). Signaling through the IL-7 receptor activates the Jak/STAT and PI3K/Akt signaling pathways (13, 14). Stat mediates survival during the transition from the common lymphoid progenitor stage to the pro-B cell stage by regulating expression of Mcl-1 and is important for proliferation of pro-B cells (8). PI3K/Akt signaling is also crucial for B cell development. However, simultaneous deletion of p110α and p110δ or p85α alone results in a block to B cell development at the large pre-B cell stage and does not result in decreased accumulation or proliferation of pro-B cells (15–17). Thus, the PI3K/Akt signaling pathway appears to be largely important for proliferation of large pre-B cells rather than survival or proliferation of pro-B cells (2, 3, 18). The IL-7 signaling pathway is negatively regulated by the SOCS and CISH proteins, which bind to the IL-7Rα, Jak1 and Jak3 proteins downstream of IL-7 signaling, preventing their interaction and targeting Jak1, Jak3 proteins for proteasomal degradation (12, 19, 20). IL-7R signaling continues to play a crucial role beyond the pro-B cell stage through the pre-BCR checkpoint. The presence of the pre-BCR on the pro-B cell surface lowers the threshold for IL-7 signaling and allows for selective proliferation of pre-BCR-expressing cells in the limiting quantities of IL-7 present in the bone marrow (20). IL-7 receptor and pre-BCR signaling induce expression of cyclin D3 and c-Myc, which are crucial for proliferation during the large pre-B cell stage after which large pre-B cells cease proliferation to transition to the small pre-B cell stage (21–23).
The Myb proto-oncogene encodes a DNA-binding protein that can act as both a transcription activator and repressor (24). c-Myb is abundantly expressed during the immature stages of hematopoiesis and becomes down regulated as progenitor cells undergo terminal differentiation (25). A crucial role for c-Myb during hematopoiesis was first demonstrated by the embryonic lethality of the c-Myb null mutation (26). c-Myb-deficient embryos die at approximately E14.5 due to an inability to perform adult erythropoiesis in the fetal liver and the embryonic lethality of the c-Myb null mutation has been an impediment to understanding the role of c-Myb during lineage-specific differentiation. To circumvent embryonic lethality of Myb null mutations, we have previously used conditional mutagenesis to demonstrate that c-Myb is absolutely required for B cell development and is important during several stages of B lymphopoiesis (27, 28). Mybf/f CD19-cre mice, which initiate deletion at the Myb locus during the pro-B cell stage, display a partial block at the pro-B to pre-B cell transition as well as defects in mature B cell homeostasis (27, 29). Ablation of c-Myb prior to B-lineage commitment in Mybf/f Mb1-cre mice identified a crucial role for c-Myb in the pre-pro-B to pro-B cell transition as well as the survival of pro-B cells (28, 30). In addition, we reported that c-Myb is required for the proper expression of IL-7Rα and Ebf1 during the pro-B cell stage. However, exogenously supplied IL-7Rα or Ebf1 alone was not sufficient to rescue the survival of c-Myb-deficient pro-B cells, suggesting that c-Myb regulates additional genes that mediate survival during the pro-B cell stage (28).
We demonstrate that two pro-apoptotic Bcl-2 family members, Bmf and Bim, are repressed by c-Myb and control the intrinsic survival (survival in the absence of IL-7) of CD19+ pro-B cells. In addition, we demonstrate that levels of Bmf and Bim mRNA are further repressed by IL-7 signaling. Bypassing IL-7 signaling with constitutively active Stat5b (CA-STAT5) suppressed both Bmf and Bim mRNA expression and induced expression of Mcl-1 mRNA in c-Myb deficient pro-B cells while CA-AKT only partially suppressed Bmf and did not induce expression of Mcl-1. c-Myb regulates IL-7Rα expression but forced expression of IL-7Rα on the surface of c-Myb-deficient pro-B cells is not sufficient for repression of Bmf and Bim expression, suggesting that c-Myb controls the expression of other components of the IL-7 receptor signaling pathway. We further demonstrate that c-Myb represses the expression of SOCS3, a negative regulator of IL-7 signaling. Overexpression of SOCS3 in pro-B cells inhibits their ability to accumulate in response to IL-7, suggesting that upregulation of SOCS3 in the absence of c-Myb can impede proper IL-7 signaling in pro-B cells. Overexpression of Bcl-2 is able to rescue the survival of c-Myb-deficient pro-B cells but is not sufficient to rescue transition from the pro-B to pre-B cell stage of differentiation in the absence of c-Myb and c-Myb-deficient large pre-B cells are hypoproliferative. We demonstrate several proteins that are crucial during the pro-B to pre-B cell transition, in addition to IL-7Rα, including λ5, cyclin D3 and CXCR4, are downregulated in the absence of c-Myb and the promoter of Igll1 (λ5) is a direct c-Myb target. Thus, c-Myb coordinates the survival of pro-B cells with the expression of genes that are necessary for proliferation and differentiation into the pre-B cell compartment.
Materials and Methods
Mice
Mybf/f, Bmf−/−, CD19-cre and Bcl2Tg mice have been described previously (29, 31–33). Bmf−/− mice were a generous gift from Dr. Roger Davis (University of Massachusetts Medical School). Rag2−/− (Taconic Farms), Mybf/f CD19-cre Bcl2Tg, Mybf/f Rag2−/− Bcl2Tg, Mybf/f Rag2−/− Bmf−/− and Rag2−/− Bmf−/− mice were bred at the University of Virginia. Mice were 6-10 weeks old when used for experiments and were housed in a barrier facility at the University of Virginia. These studies were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Virginia.
Cell Culture and Retrovirus Transduction
Pro-B cells from Rag2−/− and Rag2−/− Bmf−/− mice were positively selected from bone marrow using anti-CD19-labeled magnetic beads (Miltenyi Biotec) and cultured for 24 hours in Opti-MEM supplemented with 15% (v/v) FBS (Life Technologies), 100 U/ml penicillin-streptomycin, 2 mM L-glutamine, 50 μM β-mercaptoethanol and 5 ng/ml IL-7 (PeproTech). Cells were then washed, plated in 96-well plates in Opti-MEM supplemented with 15% (v/v) FBS (Life Technologies), 100 U/ml penicillin-streptomycin, 2 mM L-glutamine and 50 μM β-mercaptoethanol and total CD19+ cells per well were analyzed by flow cytometry. For transduction of pro-B cells from Mybf/f Rag2−/−, Mybf/f Rag2−/− Bcl2Tg and Mybf/f Rag2−/− Bmf−/− mice, cells were positively selected from bone marrow using anti-CD19-labeled magnetic beads (Miltenyi Biotec), cultured for 24 hours in Opti-MEM supplemented with 15% (v/v) FBS (Life Technologies), 100 U/ml penicillin-streptomycin, 2 mM L-glutamine, 50 μM β-mercaptoethanol and 5 ng/ml IL-7 (PeproTech) and transduced with retroviral vectors as previously described (28, 34). Following transduction, pro-B cells were cultured in Opti-MEM supplemented with 15% (v/v) FBS (Life Technologies), 100 U/ml penicillin-streptomycin, 2 mM L-glutamine and 50 μM β-mercaptoethanol and were analyzed 24, 48, and 72 hours later by flow cytometry. The pan-caspase inhibitor Q-VD-OPH (SM Biochemicals) was used at 100 μM.
Flow cytometry
Single cell suspensions from bone marrow were prepared from 6- to 10-week old mice and 2 × 106 cells were stained with optimal amounts of fluorochrome-conjugated antibodies as previously described (27). Cells were subsequently analyzed on a FACSCalibur, FACSCanto (BD Immunocytometry Systems) or Cytoflex (Beckman Coulter Life Sciences). Total cells were determined using AccuCount Blank Particles, 5.27 μm (Spherotech). Flow cytometric data was analyzed using FlowJo software (Tree Star). Cell sorting was performed on a FACSVantage SE Turbo Sorter with DIVA option (BD Immunocytometry Systems). Antibodies and reagents were purchased as follows: eBioscience: anti-B220 PECy7 (RA3-6B2), anti-CD19 PE (6D5), anti-CD19 PerCPCy5.5 (6D5), anti-CD25 PE (PC61.5), anti-CD117 APC (2B8), anti-CD127 PE (A7R34), anti-IgM FITC (R6-60.2); BioLegend: anti-NGFR APC (ME20.4); Molecular Probes: 7-aminoactinomycin D (7AAD); Sigma-Aldrich: 4′-6-diamidino-2-phenylindole (DAPI). For DRAQ5 staining, cells were stained for expression of surface markers followed by incubation with 50 μM DRAQ5 (eBioscience) for 20 minutes at 37°C prior to analysis.
Retrovirus vectors
The retrovirus vectors pMIG-R1, pMSCV-IRES-tNGFR, ptNGFR-Cre, pMIG-cMyb, pMIG-Bcl2, pMIG-Cre, pMIG-CA-STAT5B, pMIG-CA-Akt, and pMIG-IL-7Rα have been previously described (28, 35–40). pMIG-Bcl2 was provided by Dr. Motonari Kondo (Duke University Medical Center). pMIG-CA-STAT5B and pMIG-CA-Akt were provided by Dr. Marcus Clark (University of Chicago). To generate pMIG-Bmf, a Bmf encoding cDNA was isolated from pBabe-3XFLAG-mouseBmf (41) and cloned into the BglII/EcoRI site of pMIG-R1. pBabe-3XFLAG-mouse Bmf was a provided Dr. Joan Brugge (AddGene plasmid #17243). To generate pMIG-BimEL, BimEL cDNA was isolated from pCMV-Tag2b-Flag-BimEL (42) and cloned into the BglII/EcoRI site of pMIG-R1. pCMV-Tag2b-Flag-BimEL was provided by Dr. Roger David (AddGene plasmid #23090). To generate pMIG-SOCS3, SOCS3 cDNA was isolated from pCMV-SOCS3 (43) and cloned into the EcoRI site of pMIG-R1. pCMV-SOCS3 was provided by Dr. Ronald Kahn (AddGene plasmid #11486). To generate pSUPER-Puro-IRES-GFP-shLuc and pSUPER-Puro-IRES-GFP-shBim, the IRES-GFP from pMIG-R1 was cloned into the NsiI site of pSUPER-shLuc and pSUPER-shBim (44). pSUPER-shLuc and pSUPER-shBim were gifts of Dr. Emily Cheng (Memorial Sloan-Kettering Cancer Center). Retrovirus supernatants were generated by transient transfection of HEK-293T cells and titered on NIH-3T3 cells by flow cytometry as previously described (28).
Quantitative RT-PCR
Retrovirus transduced CD19+ pro-B cells were electronically sorted based on the expression of NGFR and GFP and total cellular RNA was isolated using TRIZOL (Invitrogen) according to the manufacturer’s protocol. Contaminating genomic DNA was removed by treatment with RNase-free DNase I (Invitrogen) and cDNA was prepared with the Superscript III First-Strand Synthesis System (Invitrogen). Quantitative RT-PCR (qPCR) was performed on cDNA with Titanium Taq Polymerase (BD Clonetech) with 1X SYBR Green (Molecular Probes) and 0.4 μM of the primer set of interest in 25 μl reaction mixtures in a MyiQ Single Color Real-Time PCR Detection System (Bio-Rad). Conditions for qPCR were as follows: 95°C for 3 minutes, then 40 cycles of 95°C for 40 seconds, 66°C for 20 seconds and 72°C for 30 seconds, followed by an extension at 72°C for 1 minute. Melting curve analysis was then performed to ensure equivalent and appropriate melting temperatures. Each sample was normalized to the expression of Hprt. Primers used are listed in Supplemental Table I.
Western Blotting
Pro-B cells were harvested from cell culture and lysed in 20 mM Tris (pH 7.4), 100 mM NaCl, 10 mM EDTA, 1 mM EGTA and 1% Triton X-100 (45) containing EDTA-free protease inhibitor mixture (Roche) and 1 mM PMSF (Sigma-Aldrich). Ten micrograms of protein was loaded on 15% SDS-polyacrylamide gels and transferred to Protran nitrocellulose transfer membranes (Whatman). Membranes were blocked in PBS plus 0.05% Tween-20 (PBS-T) with 5% nonfat dry milk for 1 hour and then incubated with the appropriate primary antibody overnight at 4°C. Membranes were washed three times with PBS-T and probed with anti-rat-HRP- or anti-rabbit-HRP-conjugated antibodies (GE Healthcare Bioscience) in PBS-T for 1 hour at room temperature. After washing the membranes three times with PBS-T, the proteins were detected by ECL (Amersham). Protein expression was quantified using ImageQuant TL 2005 software. Western blotting primary antibodies: Epitomics: anti-cMyb (EP769Y); Cell Signaling Technology: anti-Bim (2819); Enzo Life Sciences: anti-Bmf (17A9); Sigma-Aldrich: anti-β-actin (AC-15).
Chromatin Immunoprecipitation
CD19+ pro-B cells from Rag2−/− mice were harvested and cultured for 72 hours in Opti-MEM supplemented with 15% (v/v) FBS (Life Technologies), 100 U/ml penicillin-streptomycin, 2 mM L-glutamine, 50 μM β-mercaptoethanol and 5 ng/ml IL-7 (PeproTech). Protein was crosslinked to chromatin by adding 1% formaldehyde to each culture dish at room temperature for 10 minutes. The reaction was stopped by addition of 125 mM glycine and incubated at room temperature for 5 minutes while rocking. Cells were harvested, pelleted and washed with cold PBS. Cells were resuspended at 107 cells/ml in cold cytoplasmic lysis buffer (20 mM Tris-HCl pH 8, 85 mM KCl, 0.5% NP-40, 1 mM PMSF and EDTA-free protease inhibitor mixture (Roche)) and incubated on ice for 10 minutes. Nuclei were centrifuged, resuspended at 107 cells/ml in cold sonication buffer (10mM Tris-HCl pH 8, 0.1 mM EDTA, 1% NP-40, 0.01% SDS, 1 mM PMSF and EDTA-free protease inhibitor mixture) and sonicated using a W-375 cell disruptor (Ultrasonics) to generate chromatin fragment. Debris was cleared by centrifugation and chromatin was supplemented with 5% glycerol and 127 mM NaCl. Chromatin was pre-cleared with protein A/G PLUS-Agarose (Santa Cruz Biotechnology, Inc.) for 1 hour and immunoprecipitated overnight with 5 μg either anti-c-Myb clone 1-1 (Millipore), anti-c-Myb clone EP769Y (Epitomics), normal mouse IgG (Santa Cruz) or normal rabbit IgG (Santa Cruz) with rotation at 4°C. Immune complexes were collected on protein A/G PLUS-Agarose for 1 hour with rotation at 4°C. Bound immune complexes were washed for 3 minutes on ice with low salt wash buffer (10 mM Tris-HCl pH 8, 2 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100), high salt wash buffer (10 mM Tris-HCl pH 8, 2 mM EDTA, 500 mM NaCl, 0.1% SDS, 1% Triton X-100), LiCl wash buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 250 mM LiCl, 1% sodium deoxycholate, 1% NP-40) and TE wash buffer (10 mM Tris-HCl pH 8, 1 mM EDTA). All wash buffers were supplemented with PMSF and EDTA-free protease inhibitor mixture. Bound immune complexes were eluted from agarose in elution buffer (0.1 M NaHCO3, 1% SDS) for 30 minutes with rotation at room temperature. Formaldehyde crosslinking was reversed in the presence of 200 mM NaCl at 65°C overnight. Chromatin was RNase A and proteinase K treated, then DNA was isolated by phenol/chloroform extraction and analyzed by qRT-PCR. Primers are listed in Supplemental Table I.
Statistical Analysis
Differences between data sets were analyzed using the two-tailed Student t test and at confidence level of 95% for all experiments and error bars represent SEM. Data sets were analyzed and figures prepared with Prism v5.01 and v7.0 (GraphPad Software, Inc.).
Results
c-Myb represses Bmf and Bim expression in CD19+ pro-B cells
We previously reported that c-Myb is absolutely required for B cell development and the survival of CD19+ pro-B cells (28). During the initial analysis of c-Myb-deficient pro-B cells we did not detect changes in the expression of Bcl-2, Mcl-1 or Bim mRNA, which are known to be involved in the control of c-Myb-deficient pro-B cell survival (28). However, additional Bcl-2 family members, such as Bak, Bax, Bid, Bad and Bmf, are expressed during B cell development (4). To examine the expression of these Bcl-2 family members in the absence of c-Myb, Mybf/f Rag2−/− pro-B cells were cultured in the presence of IL-7 for 24 hours, transduced with a retrovirus that encodes Cre and GFP (MIG-Cre) to inactivate the Myb locus and subsequently cultured in the absence of IL-7 for 24 hours (Fig. 1A). Deletion efficiency by MIG-Cre is >95% in these cultures (Supplemental Figure 1A). GFP+ cells were electronically sorted 24 hours post-transduction and expression of mRNAs encoding Bcl-2 family members was quantified by qRT-PCR (Fig. 1B). As previously reported (28), we did not detect decreased expression of Bcl-2, Bcl-xL, Mcl-1 or Bim mRNA. However, the amount of steady state mRNA encoding the pro-apoptotic BH3-only family member Bmf was increased 5-fold in the absence of c-Myb, suggesting that Bmf expression is repressed by c-Myb in CD19+ pro-B cells.
Figure 1. c-Myb represses Bmf and Bim expression in pro-B cells.
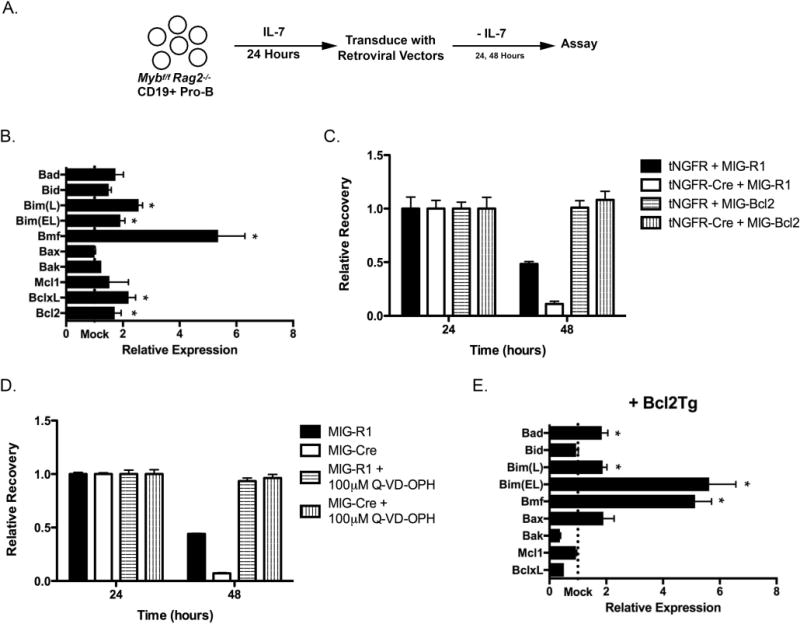
(A) The experimental system to analyze the role of c-Myb during the pro-B cell stage. Pro-B cells from Mybf/f Rag2−/− mice were positively selected using anti-CD19 coated magnetic beads and cultured for 24 hours in the presence of IL-7. These cells were subsequently transduced with retroviruses that produce a bicistronic message that encodes the gene of interest followed by an internal ribosome entry site (IRES) and a reporter gene, either GFP (MIG-R1) or a truncated human nerve growth factor receptor (tNGFR). Following retrovirus transduction, pro-B cells were placed in culture in the absence of exogenous IL-7 to measure the intrinsic survival of these cells. Every 24 hours, cells were analyzed for tNGFR and/or GFP expression as well as total cells per well. The number of tNGFR+ GFP+ cells per well at 24 hours post-transduction was set as 1. The relative recovery of tNGFR+ GFP+ cells at subsequent time points was determined as a ratio compared to the total number of tNGFR+ GFP+ cells present at 24 hours. (B) Mybf/f Rag2−/− CD19+ pro-B cells were transduced with MIG-R1 or MIG-Cre, cultured for 24 hours in the absence of IL-7 and electronically sorted based on GFP expression. Total cellular RNA was harvested and specific mRNA expression was analyzed by quantitative RT-PCR. Gene expression was normalized to the expression of Hprt. “Mock” represents the expression of each gene in MIG-R1-transduced cells. n=4 *, p < 0.05 (C) Mybf/f Rag2−/− CD19+ pro-B cells were cotransduced with tNGFR or tNGFR-Cre and MIG-R1 or MIG-Bcl2 and cultured in the absence of IL-7. Cells were analyzed 24 and 48 hours post-transduction by flow cytometry and relative recovery was determined. Retrovirus transductions were done in triplicate. Retrovirus transductions were done in triplicate. Data are representative of 3 independent experiments. (D) Mybf/f Rag2−/− CD19+ pro-B cells were transduced with MIG-R1 or MIG-Cre and cultured in the absence of IL-7 and the presence of 100 μM Q-VD-OPH. Cells were analyzed 24 and 48 hours post-transduction by flow cytometry and relative recovery was determined. Data are representative of 2 independent experiments. (E) Mybf/f Rag2−/− Bcl2Tg CD19+ pro-B cells were transduced with MIG-R1 or MIG-Cre, cultured for 48 hours in the absence of IL-7 and electronically sorted based on GFP expression. Total cellular RNA was harvested and analyzed by quantitative RT-PCR. Gene expression was normalized the expression of Hprt. “Mock” represents the expression of each gene in MIG-R1-transduced cells. n=4 *, p < 0.05
Lymphocyte progenitor cells very rapidly undergo apoptotic cell death in the absence of c-Myb (28, 46) and we have found that it is often necessary to rescue survival of c-Myb-deficient lymphocyte progenitor cells to accurately determine changes in gene expression as well as identify potential c-Myb mediated activities besides maintaining survival (46). To rescue survival in c-Myb-deficient pro-B cells, Mybf/f Rag2−/− pro-B cells were cotransduced with a retrovirus that encodes Cre recombinase and a truncated nerve growth factor receptor that serves as a marker of transduction (tNGFR-Cre) as well as a retrovirus that encodes Bcl-2 and GFP (MIG-Bcl2). Forced expression of Bcl-2 was able to rescue the survival of c-Myb-deficient pro-B cells (Fig. 1C) and comparable results were obtained using pro-B cells isolated from Mybf/f Rag2−/− Bcl2Tg mice in which the Bcl-2 transgene expression is driven by an SV40 promoter and Eμ immunoglobulin heavy chain enhancer that is constitutively expressed in B- and T-lineage progenitor cells (47) (Supplemental Figure 1B). Survival of c-Myb-deficient Rag2−/− pro-B cells could also be rescued using the pharmacological caspase inhibitor Q-VD-OPH (Fig. 1D). To determine if additional differences in the expression of mRNA encoding Bcl-2 family members could be detected in c-Myb-deficient Rag2−/− pro-B cells when the underlying survival defect was overcome, Mybf/f Rag2−/− Bcl2Tg CD19+ pro-B cells were transduced with MIG-R1 or MIG-Cre and cultured for 48 hours in the absence of IL-7. GFP+ cells were electronically sorted and the expression of mRNA encoding Bcl-2 family members was determined by qRT-PCR (Fig. 1E). Similar to c-Myb-deficient pro-B cells that lack a Bcl-2 transgene, the expression of Bmf mRNA was increased approximately 5-fold in the absence of c-Myb. Furthermore, we also detected a 5-fold increase in the amount of Bim(EL) mRNA in the absence of c-Myb. Comparable results were obtained in c-Myb-deficient pro-B cells rescued by Q-VD-OPH (data not shown). Thus, the steady state level of mRNAs that encode the pro-apoptotic Bcl-2 family proteins Bmf and Bim is repressed by c-Myb in CD19+ pro-B cells, demonstrating that c-Myb is important for setting the base line levels of Bmf and Bim in pro-B.
Rag2−/− CD19+ pro-B cells rapidly died after transduction with retroviruses encoding either Bmf or Bim, demonstrating that either pro-apoptotic protein can induce apoptosis when overexpressed in pro-B cells (Supplemental Figure 2A). To determine if the increased expression of Bmf contributes to poor survival in c-Myb-deficient pro-B cells, we bred Mybf/f Rag2−/− Bmf−/− mice. The Rag2 and Bmf loci are 17 Mb apart on Chromosome 2 (31) and a cross over event was required to produce Mybf/f Rag2−/− Bmf−/− mice. CD19+ pro-B cells from Mybf/f Rag2−/− Bmf−/− mice were transduced with MIG-Cre and cultured for 72 hours in the absence of IL-7. The Bmf loss of function mutation provided a 3-fold increase in the survival of c-Myb-deficient CD19+ pro-B cells in culture 48 hours post-transduction and a 10-fold increase in survival 72 hours post-transduction with MIG-Cre as compared to Mybf/f Rag2−/− pro-B cells (Figure 2A). Thus, Bmf contributes to the intrinsic survival of pro-B cells.
Figure 2. Knockdown or knockout of Bmf and Bim rescues survival in c-Myb-deficient pro-B cells.
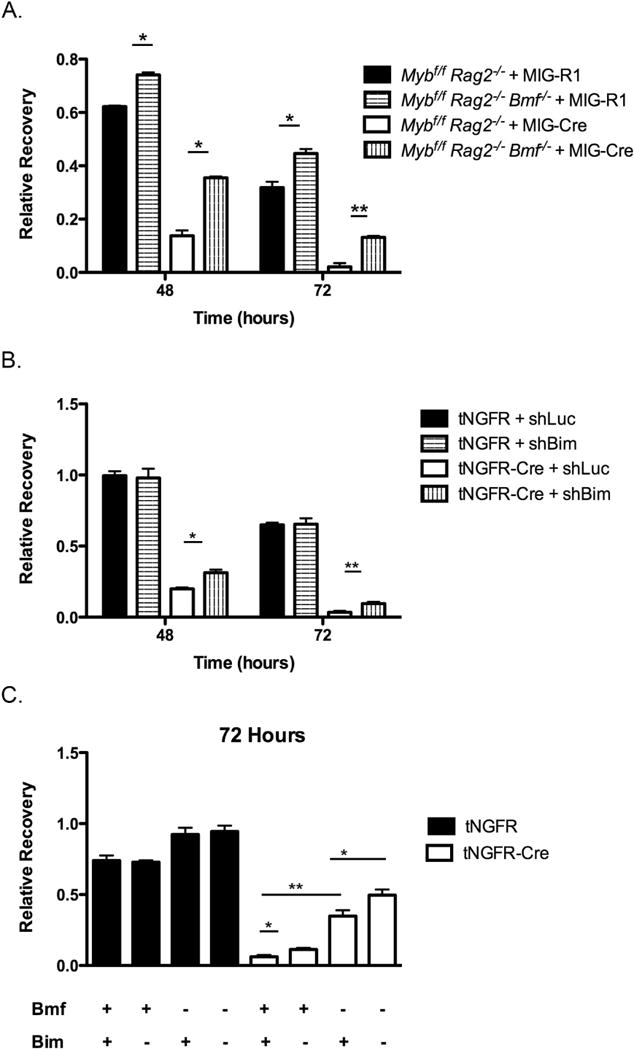
(A) Mybf/f Rag2−/− and Mybf/f Rag2−/− Bmf−/− CD19+ pro-B cells were transduced with MIG-R1 or MIG-Cre and cultured in the absence of IL-7 for 72 hours. The total number of transduced cells was analyzed every 24 hours post-transduction and relative recovery was determined by normalization to the total number of transduced cells present 24 hours post-transduction. Data are representative of 3 independent experiments. *, p < 0.005 **, p < 0.001. (B) Mybf/f Rag2−/− CD19+ pro-B cells were transduced with the pSUPER-Puro-IRES-GFP-shLuc or pSUPER-Puro-IRES-GFP-shBim retroviruses and cultured in the presence of IL-7 for 72 hours. Cells were then transduced with tNGFR or tNGFR-Cre and cultured in the absence of IL-7 for 72 hours. The total number of cotransduced cells was analyzed every 24 hours post-transduction and relative recovery was determined by normalization to the total number of cotransduced cells present 24 hours post-transduction. Data are representative of 3 independent experiments. *, p < 0.05 **, p < 0.005. (C) Mybf/f Rag2−/− and Mybf/f Rag2−/− Bmf−/− CD19+ pro-B cells were transduced with the pSUPER-Puro-IRES-GFP-shLuc or pSUPER-Puro-IRES-GFP-shBim retroviruses and cultured in the presence of IL-7 for 72 hours. Cells were then transduced with tNGFR or tNGFR-Cre and cultured in the absence of IL-7 for 72 hours. The total number of cotransduced cells was analyzed at 24 and 72 hours post-IL-7 withdrawal and relative recovery was determined by normalization to the total number of cotransduced cells present at 24 hours post-transduction. “+” indicates the presence of the gene, while “-“ indicates the absence of the gene either by knockout or shRNA-mediated knockdown. Data are representative of 2 independent experiments. *, p < 0.05 **, p < 0.005.
The Bim locus is also located on Chromosome 2, 9Mb away from the Bmf locus, 26 Mb away from the Rag2 locus and a cross over event would have been required to produce Mybf/f Rag2−/− Bim−/− mice. As an alternative, we used an shRNA mediated knock down approach to determine if increased Bim expression in c-Myb-deficient pro-B cells contributes to poor survival (44). Mybf/f Rag2−/− CD19+ pro-B cells were transduced with a retrovirus encoding an shRNA against Bim (shBim) and cultured for 72 hours in the presence of IL-7 to allow for knockdown of Bim expression. Subsequently, the Mybf/f Rag2−/− pro-B cells were transduced with tNGFR-Cre to inactivate the Myb locus and then cultured for an additional 72 hours in the absence of IL-7. Knockdown of Bim provided a 1.5-fold increase in the survival of c-Myb-deficient pro-B cells 48 hours post transduction and a 3-fold rescue 72 hours post-transduction compared to Mybf/f Rag2−/− pro-B cells cotransduced with tNGFR-Cre and shLuc (Fig. 2B). Bim may contribute more to the intrinsic survival of CD19+ pro-B cells than is apparent from these results due to the poor knock down efficiency of Bim that was achieved in c-Myb-deficient pro-B cells (see Discussion). To determine if there was an additive effect of Bmf and Bim in c-Myb-regulated pro-B cell survival, Mybf/f Rag2−/− Bmf−/− pro-B cells were transduced with the shBim retrovirus and cultured for 72 hours in the presence of IL-7. Mybf/f Rag2−/− Bmf−/− pro-B cells were subsequently transduced with tNGFR-Cre and cultured for an additional 72 hours in the absence of exogenous IL-7. The rescue of survival provided by knockdown of Bim in c-Myb-deficient Rag2−/− Bmf−/− pro-B cells was greater than that provided by deficiency of Bim or Bmf alone (Fig. 2C), demonstrating that suppression of both Bmf and Bim expression by c-Myb is important for the intrinsic survival (survival in the absence of IL-7) of CD19+ pro-B cells.
IL-7 signaling in pro-B cells represses expression of Bmf and Bim mRNA
The IL-7 signaling pathway is the major survival pathway in CD19+ pro-B cells and regulates the expression of Bcl-2 family members at the level of transcription and post-translational modification (12). Bim is expressed at each stage of B cell development (48) and Bmf expression has been reported in pre-B, immature B and mature B cells (49). However, we also detect Bmf protein in freshly isolated Rag2−/− CD19+ pro-B cells (Supplemental Figure 2B). Suppression of Bim expression is important for the maintenance of the pro-B cell compartment downstream of IL-7 (11, 48). In contrast, little is known about the function of Bmf during B cell development, although it plays a role in the survival of large pre-B cells (50). To determine if suppression of Bmf expression is important for the maintenance of pro-B cell survival downstream of IL-7, Rag2−/− and Rag2−/− Bmf−/− pro-B cells were cultured for 24 hours in the presence of IL-7 followed by withdrawal of IL-7 for 24 to 72 hours. Throughout the time course of the experiment, a greater proportion of Rag2−/− Bmf−/− pro-B cells survived in the absence of IL-7 compared to Rag2−/− pro-B cells (Fig. 3A), demonstrating that suppression of Bmf expression, like Bim expression, is an important regulator of pro-B cell survival downstream of IL-7.
Figure 3. Expression of Bmf and Bim mRNA is repressed by IL-7 signaling.

(A) Rag2−/− Bmf−/− CD19+ pro-B cells were cultured in the presence of IL-7 for 24 hours and then without IL-7 for 72 hours. Relative recovery was normalized to the number of CD19+ cells present in the culture at the beginning of the IL-7 withdraw. Data are representative of 3 independent experiments and 3 replicates per condition within each experiment. *, p < 0.005. (B-C) Rag2−/− CD19+ pro-B cells (B) and Rag2−/− Bcl2Tg CD19+ pro-B cells (C) were cultured in the presence or absence of IL-7 for 24 and 48 hours. Expression of mRNA encoding Bmf, Bim(EL) and Bim(L) was analyzed by quantitative RT-PCR. Expression was normalized to the expression of HPRT. Dotted line represents the expression in Rag2−/− pro-B cells or Rag2−/− Bcl2Tg pro-B cells cultured in the presence of IL-7 for the indicated time points. Data are representative of 2 independent experiments and 3 replicates per condition within each experiment. (D) Rag2−/− Bcl2Tg CD19+ pro-B cells were cultured in the presence or absence of IL-7 for 48 hours and Bmf and Bim protein were analyzed by Western blot. β-actin serves as a loading control. Data are representative of 2 independent experiments. (E) Rag2−/− CD19+ pro-B wells were transduced with MIG-R1, MIG-CA-STAT5 or MIG-CA-Akt and cultured for 48 hours in the absence of IL-7. GFP+ cells were electronically sorted and total cellular RNA was analyzed by quantitative real time PCR. Expression was normalized to the expression of Hprt. Data are representative of 2 independent experiments and 3 replicates per condition within each experiment. (F) Mybff Rag2−/− CD19+ pro-B wells were co-transduced with tNGFR or NGFR-Cre and MIG-R1, MIG-CA-STAT5 or MIG-CA-Akt and cultured for 48 hours in the absence of IL-7. tNGFR+ GFP+ cells were electronically sorted and total cellular RNA was analyzed by quantitative real time PCR. Expression was normalized to the expression of Hprt. Data are representative of 2 independent experiments and 3 replicates per condition within each experiment. *, p < 0.05 **, p < 0.005
To determine if expression of Bmf and Bim mRNA is regulated downstream of IL-7 signaling in pro-B cells, Rag2−/− pro-B cells were cultured in the presence or absence of IL-7 for 24 and 48 hours and expression of Bmf and Bim mRNA was determined by qRT-PCR. The amount of Bmf mRNA detected in Rag2−/− CD19+ pro-B cells increased 20-fold after 24 hours and 40-fold after 48 hours in culture in the absence of IL-7 compared to Rag2−/− pro-B cells cultured in the presence of IL-7 (Figure 3B). In addition, the amount of Bim(EL) mRNA detected in c-Myb-deficient pro-B cells increased 7-fold after 24 hours and 10-fold after 48 hours in culture in the absence of IL-7 while Bim(L) mRNA was upregulated 3-fold after 48 hours in culture in the absence of IL-7 compared to Rag2−/− CD19+ pro-B cells cultured in the presence of IL-7.
Pro-B cells die quickly in culture in the absence of IL-7 and we were unable to measure Bmf and Bim protein levels in pro-B cells by Western blot after removal of IL-7 from the growth medium (data not shown). To circumvent this problem, we examined Bmf and Bim mRNA and protein expression in Rag2−/− Bcl2Tg pro-B cells. Similar to Rag2−/− pro-B cells, the amount of Bmf, Bim(EL) and Bim(L) mRNA increased in Rag2−/− Bcl2Tg CD19+ pro-B cells cultured in the absence of IL-7 compared to Rag2−/− Bcl2Tg pro-B cells cultured in the presence of IL-7 (Fig. 3C). The amount of Bmf protein increased 3-fold and Bim protein increased 5-fold after 48 hours in culture in the absence of IL-7 compared to Rag2−/− Bcl2Tg pro-B cells cultured in the presence of IL-7 (Fig. 3D). It is important to note that IL-7 signaling does not regulate c-Myb expression in pro-B cells. c-Myb mRNA is not regulated downstream of IL-7 signaling in B-lineage progenitors (51). Similarly, c-Myb protein expression was not altered in Rag2−/− Bcl2Tg pro-B cells cultured for 48 hours in the presence or absence of IL-7 (Supplemental Figure 2C). Thus, IL-7 signaling represses the expression of Bmf and Bim mRNA and protein in CD19+ pro-B cells.
Signaling through the IL-7 receptor activates the Jak/STAT and the PI3K/Akt signaling pathways in pro-B cells. These pathways regulate survival and proliferation during the pro-B cell stage as well as inhibit differentiation into the pre-B cell compartment (2, 12). To determine if either of these pathways contribute to the regulation of Bmf and Bim mRNA expression, Rag2−/− pro-B cells were transduced with retroviruses encoding either a constitutively active Stat5b protein and GFP (MIG-CA-STAT5) or a constitutively active Akt protein and GFP (MIG-CA-AKT) (40) and cultured in the absence of IL-7 for 48 hours. Transduced pro-B cells were isolated by electronic cell sorting based on GFP expression and Bmf and Bim(EL) mRNA expression was determined by qRT-PCR. The amount of Bmf mRNA was decreased 50% by CA-STAT5 and 87% by the CA-AKT compared to MIG-R1-transduced pro-B cells. In contrast, expression of Bim(EL) mRNA was decreased 70% by CA-STAT5 but CA-AKT did not appear to alter expression of Bim(EL) mRNA in CD19+ pro-B cells (Fig. 3E), consistent with the finding that Foxo1 represses the expression of Bim (34). Similarly, we determined if CA-STAT5 or CA-AKT could suppress expression of Bmf and Bim mRNA in c-Myb deficient pro-B cells. For this purpose we cotransduced Mybf/f Rag2−/− pro-B cells with retroviruses encoding NGFR-Cre and MIG- CA-STAT5 or NGFR-Cre and MIG-CA-AKT (Fig. 3F). As we observed upon IL-7 withdrawal in c-Myb sufficient Rag2−/− pro-B cells, repression of Bmf mRNA was restored by both CA-STAT5 and CA-AKT while repression of Bim mRNA was restored by CA-STAT5 but not CA-AKT in c-Myb deficient Rag2−/− pro-B cells. In addition, IL-7 signaling through Stat5 is the major regulator of Mcl-1 expression in B-lineage progenitor cells (8) and, as expected, we found that Mcl-1 mRNA was decreased in Mybf/f Rag2−/− pro-B cells 48 hours after transduction with NGFR-Cre and that CA-STAT5, but not CA-AKT, was able to restore expression of Mcl-1 mRNA after cotransduction with NGFR-Cre (Fig. 3E). Thus, steady state levels of Bmf and Bim are suppressed by IL-7. Furthermore, CA-STAT5 is able to suppress expression of both Bmf and Bim as well as induce expression of Mcl-1mRNA. In contrast, CA-AKT can only suppress Bmf mRNA and does not induce expression of Mcl-1.
c-Myb controls expression of two critical components of IL-7 signaling pathway
c-Myb regulates the expression of IL-7Rα during the pro-B cell stage but forced expression of IL-7Rα is not sufficient to rescue survival in c-Myb-deficient pro-B cells in either the presence or absence of IL-7 (28, 30). Thus, we sought to determine if c-Myb-deficient pro-B cells provided with an exogenous source of IL-7Rα could repress expression of Bmf and Bim mRNA when cultured in the presence of IL-7. Mybf/f Rag2−/− Bcl2Tg pro-B cells were transduced with MIG-IL7Rα and cultured for an additional 24 hours in the presence of IL-7 to allow for expression of IL-7Rα. These pro-B cells were subsequently transduced with tNGFR-Cre and cultured for an additional 48 hours in the presence of IL-7, after which tNGFR+ GFP+ cotransduced pro-B cells were electronically sorted and the expression of Bmf and Bim(EL) mRNA was measured by qRT-PCR. Despite surface expression of IL-7Rα and the presence of IL-7 in the culture medium, c-Myb-deficient pro-B cells still failed to repress expression of Bmf and Bim(EL) (Fig. 4A), suggesting that c-Myb controls the expression of additional components of the IL-7 signaling pathway. The transcription factors STAT5 and Foxo1 are regulated by IL-7 signaling in pro-B cells and are crucial for survival in the pro-B cell compartment (8, 34). However, the expression of STAT5a, STAT5b and Foxo1 mRNA was not significantly altered in the absence of c-Myb (Fig. 4B), suggesting that c-Myb does not regulate expression of STAT5 or Foxo1 in pro-B cells and instead regulates other components of the IL-7 signaling pathway.
Figure 4. c-Myb regulates the expression of two critical components of the IL-7 signaling pathway.

(A) Mybf/f Rag2−/− Bcl2Tg CD19+ pro-B cells were transduced with MIG-R1 or MIG-IL-7Rα and cultured in the presence of IL-7 for 24 hours. Cells were subsequently transduced with tNGFR or tNGFR-Cre and cultured in the presence of IL-7 for an additional 48 hours. tNGFR+ GFP+ cells were electronically sorted and total cellular RNA was analyzed by quantitative real time PCR. Expression was normalized to the expression of Hprt. Data are representative of 2 independent experiments and 3 replicates per condition within each experiment. (B) Mybf/f Rag2−/− Bcl2Tg CD19+ pro-B cells were transduced with MIG-R1 or MIG-Cre, cultured in the absence of IL-7 for 48 hours and electronically sorted based on GFP expression. Total cellular RNA was harvested and analyzed by quantitative real time PCR. Expression was normalized to the expression of Hprt. “Mock” represents the expression in MIG-R1-transduced cells. n=4 *, p < 0.05 (C) Mybf/f Rag2−/− pro-B cells were transduced with MIG-R1 or MIG-Cre, cultured in the absence of IL-7 and presence of 100 μM Q-VD-OPH for 48 hours and electronically sorted based on GFP expression. Total cellular RNA was harvested and analyzed by quantitative RT-PCR. Expression was normalized to the expression of Hprt. “Mock” represents the expression in MIG-R1-transduced cells. n=4 *, p < 0.05 (D) Rag2−/− pro-B cells were transduced with MIG-R1 or MIG-SOCS3, cultured in the presence of IL-7 and analyzed for GFP expression and total cells per well 24, 48 and 72 hours post-transduction. Relative recovery was determined by normalization to the total number of GFP+ cells present at 24 hours post-transduction. Data are representative of 2 independent experiments and 3 replicates per condition within each experiment.
The IL-7 signaling pathway is negatively regulated by the SOCS family proteins, which act as E3 ubiquitin ligases and prevent the interaction between the IL-7 receptor and Jak and STAT proteins, causing the Jak proteins to be targeted for degradation (19). The expression of mRNA encoding the SOCS family members SOCS1 and SOCS3, as well as CISH, is induced by IL-7 signaling in pro-B and large pre-B cells and inhibits IL-7 signaling during the small pre-B cell stage, which allows for initiation of recombination at the kappa light chain locus (12, 20). The amount of SOCS1 and CISH mRNA in CD19+ pro-B cells modestly increased in the absence of c-Myb but the increase did not reach statistical significance. However, the amount of SOCS3 mRNA increased 4-fold in the absence of c-Myb (Fig. 4C), suggesting that c-Myb represses expression of SOCS3. To determine if increased expression of SOCS3 could inhibit the pro-B cell response to IL-7, we transduced Rag2−/− pro-B cells with a SOCS3-expressing retrovirus (MIG-SOCS3) and cultured these cells for 72 hours in the presence of IL-7. The number of Rag2−/− pro-B cells transduced with MIG-R1 increased 4-fold 48 hours post-transduction and 7-fold 72 hours post-transduction (Fig. 4D). In contrast, Rag2−/− pro-B cells transduced with MIG-SOCS3 did not increase in number over the time course of the experiment (Fig. 4D), demonstrating that overexpression of SOCS3 during the pro-B cell stage can inhibit the pro-B cell response to IL-7. Thus, c-Myb controls the expression of two critical components of the IL-7 signaling pathway. First, c-Myb regulates the expression of the IL-7Rα chain, which pairs with the γc chain to form the IL-7 receptor. Second, c-Myb represses the expression of SOCS3, a negative regulator of IL-7 signaling.
c-Myb proliferation of pro-B and large pre-B cells
Since circumventing IL-7 signaling using CA-STAT5 and CA-AKT restored repression of Bim and Bmf mRNA and resulted in increased expression of Mcl-1 mRNA, we determined if this was sufficient to rescue survival of c-Myb deficient Rag2−/− pro-B cells. For this purpose, Mybf/f Rag2−/− pro-B cells were isolated and cotransduced with retroviruses that produce NGFR-Cre and MIG-CA-STAT5 or NGFR-Cre and MIG-CA-AKT followed by culture in the absence of IL-7. CA-STAT5 was able to increase the relative cell recovery of c-Myb deficient pro-B cells 6-fold after the loss of c-Myb while constitutive activation of Akt provided approximately a 3-fold increase in the relative recovery of c-Myb deficient pro-B cells (Fig. 5A). The decreased amount of rescue by CA-AKT compared to CA-STAT5 is consistent with the finding that CA-AKT repressed expression of Bmf but not Bim mRNA and did not result in increased expression of Mcl-1 mRNA in c-Myb-deficient pro-B cells (Fig. 3D, 3E). However, neither CA-STAT5 nor CA-AKT completely rescued the relative recovery of c-Myb deficient pro-B cells. As Rag2−/− pro-B cells proliferate in response to IL-7 and it was also possible that complete rescue was impeded by a lack of proliferation in c-Myb deficient pro-B cells. To determine if c-Myb is important for the proliferation of pro-B cells, in addition to survival, we cotransduced Mybf/f Rag2−/− pro-B cells with NGFR-Cre and MIG-CA-STAT5 or NGFR-Cre and MIG-CA-AKT as described above, cultured them without IL-7, and 48 hours later stained with DRAQ5 to measure DNA content by flow cytometry. In c-Myb sufficient RAG2−/− pro-B cells, CA-STAT5 resulted in 17.3% of pro-B cells with >2n DNA content after withdrawal of IL-7, while CA-AKT had little ability to stimulate proliferation in the absence of IL-7 (Fig 5B). In contrast, CA-STAT5 induced a much smaller proportion of cells (2.9%) with a >2N DNA content in c-Myb deficient Rag2−/− pro-B cells, a >80% decrease in the proportion of cells with a >2N DNA content detected in c-Myb sufficient Rag2−/− pro-B cells. Thus c-Myb is important for both the survival and proliferation of pro-B cells in response to IL-7.
Figure 5. Rescue of survival but not proliferation by CA-STAT5 and CA-AKT in c-Myb deficient pro-B cells.

(A) Mybf/f Rag2−/− pro-B cells were co-transduced with tNGFR or tNGFR-Cre plus MIG-R1, MIG-CA-STAT5 or MIG-CA-AKT and cultured in the absence of IL-7 for 24 hours and 48 hours. Relative recovery of co-transduced tNGFR+ GFP+ cells was calculated at 48 as described in Fig. 1. Representative of 2 independent experiments and 3 replicates per condition within each experiment *, p < 0.05 **, p < 0.005 (B) Mybf/f Rag2−/− pro-B cells were treated as in (A) and DNA content was determined by flow cytometry following DRAQ5 staining at 48 hours post transduction. Representative of 3 independent experiments.
To determine if c-Myb is important for the proliferative expansion of large pre-B cells during the pro-B to pre-B cell transition in addition to regulating survival, we crossed a Bcl-2 transgene onto the Mybf/f CD19-cre background and analyzed the pro-B, large pre-B and small pre-B cell compartments from Mybf/f CD19-cre Bcl2Tg and control mice using flow cytometry. Mybf/f CD19-cre Bcl2Tg mice contained an increase in the proportion of pro-B cells and a decrease in the proportion of pre-B cells in bone marrow compared to Mybf/f Bcl2Tg control mice (Fig. 6A) but the absolute number of pro-B cells in Mybf/f CD19-cre Bcl2Tg mice was equivalent to that in control mice (Fig. 6B). However, the absolute number of large and small pre-B cells detected in Mybf/f CD19-cre Bcl2Tg mice were reduced 40% and 50%, respectively, compared to the number detected in Mybf/f Bcl2Tg control mice. These results suggest that in addition to regulating survival c-Myb also controls proliferation and/or differentiation during the pre-BCR checkpoint. To determine if c-Myb is important for the proliferation of large pre-B cells, we examined DNA content in freshly isolated large pre-B cells by DRAQ5 staining directly ex vivo. While ~50% of large pre-B cells from Mybf/f and Mybf/f Bcl2Tg mice had a >2N DNA content, less than 20% of large pre-B cells from Mybf/f CD19-cre mice and less than 5% of large pre-B cells from Mybf/f CD19-cre Bcl2Tg mice were >2N DNA content (Fig. 6C). Thus, c-Myb is important for proliferation during the large pre-B cell stage.
Figure 6. c-Myb-deficient large pre-B cells are hypoproliferative.

(A) Bone marrow from Mybf/f, Mybf/f CD19-cre, Mybf/f Bcl2Tg and Mybf/f CD19-cre Bcl2Tg mice was stained for surface expression of B220, CD19, IgM, CD117 and CD25 and analyzed by flow cytometry. Top tier presents B220 versus IgM after gating on B220+ CD19+ cells. Bottom tier presents CD25 versus CD117 after gating on B220+ IgM– cells. Pre-B cells are defined as B220+ CD19+ IgM– CD25+, and pro-B cells are defined as B220+ CD19+ IgM– CD117+. Viable cells were defined as DAPI–. Data are representative of 4 mice per genotype. (B) The absolute number of pro-B, large pre-B and small pre-B cells were determined from Mybf/f and Mybf/f CD19-cre mice (left panel) or Mybf/f Bcl2Tg and Mybf/f CD19-cre Bcl2Tg mice (right panel). Pro-B cells were defined as DAPI– B220+ CD19+ IgM– CD117+, large pre-B cells were defined as DAPI– B220+ CD19+ IgM– CD25+ FSChigh and small pre-B cells were defined as DAPI– B220+ CD19+ IgM– CD25+ FSClow. Data are compiled from 4 mice per genotype. * p < 0.05. (C) DNA content in freshly isolated large pre-B cells (defined as B220+ CD25+ FSChigh) from Mybf/f, Mybf/f CD19-cre, Mybf/f Bcl2Tg and Mybf/f CD19-cre Bcl2Tg mice was analyzed by DRAQ5. Data are representative of 2 mice per genotype.
c-Myb regulates the expression of critical genes required for transition across the pre-BCR checkpoint
The pre-BCR checkpoint requires that pro-B cells express components of signaling pathways that mediate selection into the pre-B cell compartment and allow for proliferative expansion of large pre-B cells (52). Mybf/f Rag2−/− Bcl2Tg pro-B cells were transduced with MIG-R1 or MIG-Cre and GFP+ cells were electronically sorted 48 hours post-transduction and we analyzed expression of mRNAs that encode proteins known to play critical roles during the pre-BCR checkpoint, including the proliferation factor cyclin D3, the cytokine/chemokine receptors IL-7Rα and CXCR4 and components of the pre-BCR (λ5, VpreB, mb1, B29) by qRT-PCR (Figure 7A). Of these genes, mRNAs encoding cyclin D3, IL-7Rα, CXCR4 and λ5 were downregulated in the absence of c-Myb, demonstrating that c-Myb is important for the proper expression of key molecules that are required for proliferation and differentiation during the pre-BCR checkpoint.
Figure 7. Cyclin D3, CXCR4, λ5 and IL-7Rα mRNA expression is decreased in c-Myb-deficient pro-B cells.

(A) Mybf/f Rag2−/− Bcl2Tg pro-B cells were transduced with MIG-R1 or MIG-Cre and cultured for 48 hours in the absence of IL-7. GFP+ cells were electronically sorted and total RNA was prepared. Expression of mRNA encoding cyclin D3, CXCR4, λ5, VpreB, IL-7Rα, B29 and mb1 was analyzed by quantitative RT-PCR and normalized to the expression of HPRT. Expression in MIG-R1-transduced pro-B cells was set as 1 and is represented by the dotted line. Expression in c-Myb-deficient pro-B cells was normalized to the expression in MIG-R1-transduced pro-B cells. n=4 *, p < 0.05 **, p < 0.01 (B) Rag2−/− pro-B cells were cultured for 72 hours in the presence of IL-7 and then a chromatin immunoprecipitation for c-Myb was performed using an antibody against the amino-terminal of c-Myb (EP769Y) (left panel) or the carboxyl-terminal end of c-Myb (1–1) (right panel). Potential c-Myb binding sites within the promoters of Igll1, Bcl2l11, Ccnd3, Cxcr4 and Il7ra and within intron 2 of Bmf were analyzed. The Ccng2 (cyclin G2) promoter was used as a negative control. Data are representative of 2 independent experiments and 3 replicates per condition within each experiment.
c-Myb functions as a transcriptional activator and repressor by directly binding to chromatin (53–55). c-Myb has been reported to directly bind to the Cxcr4 promoter in MCF-7 human breast carcinoma cells and the Bcl2l11 (Bim) promoter in PC12 neuronal cells by chromatin immunoprecipitation (56, 57). The Igll1 (λ5) promoter has been suggested to be a c-Myb target in murine pre-B cell lines based on electrophoretic mobility shift assays and luciferase reporter assays but direct binding of c-Myb to the Igll1 promoter in vivo was not reported (58). In addition, the promoters of the Il7ra and Ccnd3, as well as intron 2 of the Bmf gene, contain potential c-Myb binding sites, although direct binding of c-Myb to these sites has also not been detected (28, 30, 59, 60). To determine if putative c-Myb binding sites in the Bcl2l11, Bmf, Ccnd3, Igll1, Il7ra or Cxcr4 genes are direct c-Myb target genes during the pro-B cell stage, we performed chromatin immunoprecipitation assays using chromatin from Rag2−/− CD19+ pro-B cells cultured for 72 hours in the presence of IL-7. Antibodies directed against the amino-terminal end (EP769Y) and carboxyl-terminal end (1–1) of c-Myb were used for these experiments and immunoprecipitated chromatin was analyzed by qRT-PCR for enrichment of c-Myb binding at the potential c-Myb binding sites in each gene. Of the promoters tested, enrichment for c-Myb binding was only detected on the Igll1 promoter when compared to normal mouse/rabbit IgG and the negative control Ccng2 (cyclin G2) promoter (Figure 7B). Thus, c-Myb controls the expression of critical signaling pathway components that are important for the pre-BCR checkpoint and at least one of these genes, Igll1, is a direct c-Myb target. Taken together, the results reported in this manuscript demonstrate that c-Myb coordinates survival with the expression of genes that are important to drive proliferation and differentiation across the pre-BCR checkpoint.
Discussion
The c-Myb transcription factor is crucial for the regulation of survival, proliferation and differentiation of hematopoietic progenitor cells (24, 25). We previously reported an absolute requirement for c-Myb during B cell differentiation and the control of survival in CD19+ pro-B cells during B cell development (28) but downstream mechanisms and factors that mediate c-Myb-regulated survival are poorly understood. We have now demonstrated that c-Myb regulates the intrinsic survival of pro-B cells (survival in the absence of IL-7) by repression of the pro-apoptotic proteins Bmf and Bim. Thus, c-Myb is crucial to set the basal level of Bmf and Bim expression in pro-B cells. IL-7 signaling via Stat5 is thought to be the major determinant of pro-B cell survival, at least in part, by controlling expression of the anti-apoptotic factor Mcl-1 (8). We have now demonstrated that IL-7 signaling further represses the expression of both Bmf and Bim in pro-B cells largely via Stat5 in both c-Myb sufficient and deficient pro-B cells, although CA-AKT partially suppressed expression of Bmf. The lack of suppression of Bim mRNA and modest decrease in Bmf mRNA expression mediated by CA-AKT is consistent with reports that loss of function mutations in components of PI3K as well as Akt1/2 have little effect on pro-B cell development (15, 17, 61). Thus, in addition to increasing the expression of Mcl-1 (8), IL-7 signaling serves to act on the baseline level of Bmf and Bim expression that is determined by c-Myb in pro-B cells, creating a balance of pro- and anti-apoptotic factors that favors survival.
We note that the rescue of intrinsic survival in c-Myb-deficient pro-B cells by the loss of Bmf and Bim was not to the level of control Rag2−/− pro-B cells and this could be a consequence of incomplete knockdown of Bim (data not shown). We were able to achieve essentially complete knockdown of Bim expression in several Abelson virus transformed pro-B and pre-B cell lines but only achieved very inefficient knockdown of Bim protein expression in c-Myb-deficient CD19+ pro-B cells, likely due to the large amount of Bim encoding mRNA and protein that accumulates in pro-B cells in the absence of c-Myb. It remains possible that additional Bcl2 family members are involved in controlling the intrinsic survival of pro-B cells. We detected a 50% decrease in the expression of Bcl-xL in the absence of c-Myb and Bcl-xL has been described as a mediator of c-Myb-regulated survival during the DP stage of thymopoiesis (46). However, lymphoid-specific deletion of Bcl2l1 suggests that it is important for the survival of pre-B cells but dispensable prior to the pre-B cell stage (8). Bim has previously been described as a direct c-Myb target in neuronal cells during nerve growth factor withdrawal (57). In neuronal cells, c-Myb is reported to activate expression of Bim as opposed to the repression of Bim expression that we observe in pro-B cells, suggesting that c-Myb regulates Bim expression in a cell- and stage-specific manner. In addition to the c-Myb binding site identified within the Bcl2l11 promoter in neuronal cells, analysis of anti-Myb ChIP-Seq data performed in the MCF-7 human breast adenocarcinoma cell line revealed a potential c-Myb binding site within intron 2 of the Bmf locus (56). However, we did not detect direct binding of c-Myb to the Bcl2l11 promoter or the potential c-Myb site in Bmf intron 2 in Rag2−/− CD19+ pro-B cells by chromatin immunoprecipitation (data not shown). It remains possible that c-Myb suppresses Bim and Bmf expression during the pro-B cell stage directly through unidentified regulatory regions, by association with other proteins that tether c-Myb to the Bmf or Bcl2l1 regulatory regions or through an indirect mechanism.
IL-7 signaling provides the major survival signals in pro-B cells (2, 12) and in addition to controlling the basal level of Bmf and Bim in the absence of IL-7, we have demonstrated that c-Myb controls the expression of two key components of the IL-7 signaling pathway. c-Myb is crucial for expression of the IL-7Ra chain (25, 28). However, we hae found that in the absence of c-Myb, exogenously supplied IL7Ra is not sufficient to repress expression of Bmf and Bim and does not rescue survival. Thus, we examined the expression of downstream components of the IL-7 signaling pathway and further demonstrate that c-Myb represses expression of SOCS3, a negative regulator of IL-7 signaling (12). The SOCS family of proteins inhibit signaling downstream of cytokine receptors by binding to the Jak tyrosine kinases and targeting them for proteosomal degradation and, in some cases, interfering with the interaction of Jak kinases with Stat proteins (19). The expression of SOCS1, SOCS3 and CISH mRNA is induced downstream of IL-7 signaling in pro-B and large pre-B cells (12, 20, 62) and upregulation of the SOCS and CISH family members inhibits IL-7 signaling during the small pre-B cell stage, which may allow for the initiation of recombination at the immunoglobulin kappa light chain loci by relieving Stat5 mediated repression (8, 12, 63). We detected increased expression of SOCS3 mRNA in c-Myb-deficient pro-B cells and demonstrate that overexpression of SOCS3 during the pro-B cell stage was able to inhibit accumulation of pro-B cells in response to IL-7. Thus, c-Myb regulates IL-7 signaling in CD19+ pro-B cells through at least two mechanisms. First, c-Myb is required for expression of the IL-7Rα chain, which associates with the γc chain to form the functional IL-7 receptor. Second, c-Myb represses SOCS3, which functions to inhibit signaling through the IL-7 receptor. Therefore, in addition to regulating the baseline levels of Bmf and Bim in pro-B cells, c-Myb is also required for proper expression of the IL-7Rα component of the IL-7 receptor and the negative regulator of IL-7R signaling, SOCS3, which provides a mechanism to further modulate the expression of Bmf and Bim and control the lifespan of CD19+ pro-B cells (see Supplemental Figure 3).
In addition to restoring repression of Bim and Bmf, we found that forced activation of IL-7 signaling pathways through Stat5 (CA-STAT5) and, to a lesser extent, Akt (CA-AKT), led to significantly increased survival of c-Myb deficient pro-B cells. However, the relative cell recovery of c-Myb-deficient-pro-B cells was not completely rescued by exogenous CA-STAT5 or CA-AKT expression in c-Myb-deficient pro-B cells following IL-7 withdrawal, suggesting that processes downstream of IL-7 signaling besides survival were impacted by loss of c-Myb. Signals transmitted through the IL-7 receptor are important for the proliferation of pro-B and large pre-B cells (2, 12) and we found that neither CA-STAT5, despite inducing expression of Cyclin D3 in c-Myb deficient Rag2−/− pro-B cells, nor CA-AKT are able to rescue proliferation in c-Myb deficient pro-B cells. The inability of CA-AKT to drive proliferation in pro-B cells is consistent with reports that demonstrate activation of PI3K is not important for the proliferation of pro-B cells (15–17, 64). We conclude that control of pro-B cell survival by c-Myb is mainly mediated by IL-7 driven activation of Stat5. c-Myb controls the expression of Cyclin D3, which is crucial for the proliferation of pro-B cells, via IL-7 mediated activation of Stat5. However, the control of pro-B cell proliferation by c-Myb is also mediated by genes that are regulated by c-Myb independently of IL-7 signaling via Stat5 or PI3K/Akt.
We previously identified a critical role for c-Myb during the pro-B to pre-B cell transition in Mybf/f CD19-cre mice (27). CD19-cre is produced late during the pro-B cell stage and we did not detect decreased production or turnover of pro-B cells in Mybf/f CD19-cre mice However, the number of pre-B cells was reduced in Mybf/f C19-cre mice and we detected counter selection of the deleted Myb allele in pre-B cells compared to pro-B cells, suggesting that c-Myb was important for the proliferation, survival or differentiation of large pre-B cells (27, 28). In principle, decreased survival of large and small pre-B cells could explain the decreased number of pre-B cells in these mice. However, when we crossed a Bcl-2 producing transgene onto the Mybf/f CD19-cre background, which rescued survival of c-Myb deficient pro-B cells, we found that it failed to rescue the number of large and small pre-B cells, suggesting that c-Myb is important beyond controlling survival during the transition from the pro-B cell to pre-B cell compartment. Indeed, we found that c-Myb-deficient large pre-B cells are hypoproliferative compared to c-Myb-sufficient large pre-B cells, demonstrating that c-Myb is crucial for proliferation of large pre-B cells during the pro-B to small pre-B cell transition.
To gain insight into changes in gene expression that underlie the failure of c-Myb deficient large pre-B cells to proliferate we examined the expression of critical genes that are required for proliferation during the large pre-B cell stage and found that mRNAs encoding IL-7Rα (Il7r), Lambda-5 (Igll1), Cxcr4 (Cxcr4) and cyclin D3 (Ccnd3) were down regulated in the absence of c-Myb. Importantly, Lambda-5, which is a component of the pre-BCR surrogate light chain, and cyclin D3 are both crucial for the development and proliferation of large pre-B cells and progress across the pre-BCR checkpoint (21, 65–67). In addition, mRNA encoding CXCR4 (Cxcr4), which is thought to guide the migration of pre-B cells within the bone marrow microenvironment (68, 69), was also down regulated in the absence of c-Myb, suggesting that c-Myb may play a crucial role in guiding the migration of pre-B cells away from IL-7 producing stromal cells with the consequence of effectively dampening IL-7 signaling, which is important for differentiation to the small pre-B cell stage (2, 3). Of these genes, Igll1 and Cxcr4 have previously been implicated as direct c-Myb targets (56, 58), although direct binding of c-Myb to the Igll1 promoter was not been reported. Direct c-Myb binding to the Cxcr4 promoter region was reported in MCF-7 breast carcinoma cells (56). Chromatin immunoprecipitation of c-Myb in Rag2−/− pro-B cells revealed that the Igll1 promoter is a direct c-Myb target during the pro-B cell stage. However, we were unable to detect direct interaction of c-Myb with the CXCR4, cyclin D3 or IL-7Rα promoters in pro-B cells. It remains possible that c-Myb directly regulates expression of these genes in pro-B cells through unknown regulatory elements or indirect mechanisms. Our results make clear that c-Myb is important beyond the maintenance of survival during B cell development and coordinates survival with the expression of genes that are important for differentiation to the next developmental stage. In addition, these findings suggest that c-Myb is important at stages of B cell development after the pro-B cell stage.
Supplementary Material
Acknowledgments
The authors thank Drs. Ulrike Lorenz, Kodi Ravichandran and Loren Erickson for advice and valuable discussions. The authors are grateful to the Flow Cytometry Core Facility at the University of Virginia and in particular thank Joanne Lannigan, Michael Solga, Claude Chew and Sebastien Coquery for their expert help and advice.
Abbreviations in this paper
- c-Myb
myeloblastosis oncogene
- HSC
hematopoietic stem cell
- Stat
signal transducer and activator of transcription
- SOCS
suppressor of cytokine signaling
- CISH
cytokine-inducible SH2-containing protein
- tNGFR
truncated nerve growth factor receptor
- shRNA
short hairpin RNA
- CA
constitutively active
- qRT-PCR
quantitative real time PCR
- ChIP
chromatin immunoprecipitation
Footnotes
This work was supported in part by National Institutes of Health (NIH) grant GM100776 (to TPB) and NIH Training Grant AI07496 (to SPF and ARD).
References
- 1.Hardy RR, Kincade PW, Dorshkind K. The protean nature of cells in the B lymphocyte lineage. Immunity. 2007;26:703–714. doi: 10.1016/j.immuni.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Clark MR, Mandal M, Ochiai K, Singh H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat Rev Immunol. 2014;14:69–80. doi: 10.1038/nri3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reth M, Nielsen P. Signaling circuits in early B-cell development. Adv Immunol. 2014;122:129–175. doi: 10.1016/B978-0-12-800267-4.00004-3. [DOI] [PubMed] [Google Scholar]
- 4.Opferman JT. Apoptosis in the development of the immune system. Cell Death Differ. 2008;15:234–242. doi: 10.1038/sj.cdd.4402182. [DOI] [PubMed] [Google Scholar]
- 5.Tischner D, Woess C, Ottina E, Villunger A. Bcl-2-regulated cell death signalling in the prevention of autoimmunity. Cell Death Dis. 2010;1:e48. doi: 10.1038/cddis.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ. Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci U S A. 2005;102:11272–11277. doi: 10.1073/pnas.0504783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 8.Malin S, McManus S, Cobaleda C, Novatchkova M, Delogu A, Bouillet P, Strasser A, Busslinger M. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat Immunol. 2010;11:171–179. doi: 10.1038/ni.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merino R, Ding L, Veis DJ, Korsmeyer SJ, Nunez G. Developmental regulation of the Bcl-2 protein and susceptibility to cell death in B lymphocytes. EMBO J. 1994;13:683–691. doi: 10.1002/j.1460-2075.1994.tb06307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strasser A. The role of BH3-only proteins in the immune system. Nat Rev Immunol. 2005;5:189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- 11.Erlacher M, Labi V, Manzl C, Bock G, Tzankov A, Hacker G, Michalak E, Strasser A, Villunger A. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med. 2006;203:2939–2951. doi: 10.1084/jem.20061552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corfe SA, Paige CJ. The many roles of IL-7 in B cell development; mediator of survival, proliferation and differentiation. Semin Immunol. 2012;24:198–208. doi: 10.1016/j.smim.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Venkitaraman AR, Cowling RJ. Interleukin-7 induces the association of phosphatidylinositol 3-kinase with the alpha chain of the interleukin-7 receptor. Eur J Immunol. 1994;24:2168–2174. doi: 10.1002/eji.1830240935. [DOI] [PubMed] [Google Scholar]
- 14.Corcoran AE, Smart FM, Cowling RJ, Crompton T, Owen MJ, Venkitaraman AR. The interleukin-7 receptor alpha chain transmits distinct signals for proliferation and differentiation during B lymphopoiesis. EMBO J. 1996;15:1924–1932. [PMC free article] [PubMed] [Google Scholar]
- 15.Ramadani F, Bolland DJ, Garcon F, Emery JL, Vanhaesebroeck B, Corcoran AE, Okkenhaug K. The PI3K Isoforms p110{alpha} and p110{delta} Are Essential for Pre-B Cell Receptor Signaling and B Cell Development. Science Signaling. 2010;3:ra60. doi: 10.1126/scisignal.2001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, Cantley LC. Impaired B Cell Development and Proliferation in Absence of Phosphoinositide 3-Kinase p85. Science. 1999;283:393–397. doi: 10.1126/science.283.5400.393. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki H, Terauchi Y, Fujiwara M, Aizawa S, Yazaki Y, Kadowaki T, Koyasu S. Xid-like immunodeficiency in mice with disruption of the p85alpha subunit of phosphoinositide 3-kinase. Science. 1999;283:390–392. doi: 10.1126/science.283.5400.390. [DOI] [PubMed] [Google Scholar]
- 18.Herzog S, Reth M, Jumaa H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat Rev Immunol. 2009;9:195–205. doi: 10.1038/nri2491. [DOI] [PubMed] [Google Scholar]
- 19.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–422. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertolino E, Reddy K, Medina KL, Parganas E, Ihle J, Singh H. Regulation of interleukin 7-dependent immunoglobulin heavy-chain variable gene rearrangements by transcription factor STAT5. Nat Immunol. 2005;6:836–843. doi: 10.1038/ni1226. [DOI] [PubMed] [Google Scholar]
- 21.Cooper AB, Sawai CM, Sicinska E, Powers SE, Sicinski P, Clark MR, Aifantis I. A unique function for cyclin D3 in early B cell development. Nat Immunol. 2006;7:489–497. doi: 10.1038/ni1324. [DOI] [PubMed] [Google Scholar]
- 22.Habib T, Park H, Tsang M, de Alboran IM, Nicks A, Wilson L, Knoepfler PS, Andrews S, Rawlings DJ, Eisenman RN, Iritani BM. Myc stimulates B lymphocyte differentiation and amplifies calcium signaling. J Cell Biol. 2007;179:717–731. doi: 10.1083/jcb.200704173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yasuda T, Sanjo H, Pages G, Kawano Y, Karasuyama H, Pouyssegur J, Ogata M, Kurosaki T. Erk kinases link pre-B cell receptor signaling to transcriptional events required for early B cell expansion. Immunity. 2008;28:499–508. doi: 10.1016/j.immuni.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 24.Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer. 2008;8:523–534. doi: 10.1038/nrc2439. [DOI] [PubMed] [Google Scholar]
- 25.Greig KT, Carotta S, Nutt SL. Critical roles for c-Myb in hematopoietic progenitor cells. Semin Immunol. 2008;20:247–256. doi: 10.1016/j.smim.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Mucenski ML, Mclain K, Kier AB, Swerdlow SH, Schreiner CM, Miller TA, Pietryga DW, Scott WJ, Potter SS. A Functional C-Myb Gene Is Required for Normal Murine Fetal Hepatic Hematopoiesis. Cell. 1991;65:677–689. doi: 10.1016/0092-8674(91)90099-k. [DOI] [PubMed] [Google Scholar]
- 27.Thomas MD, Kremer CS, Ravichandran KS, Rajewsky K, Bender TP. c-Myb is critical for B cell development and maintenance of follicular B cells. Immunity. 2005;23:275–286. doi: 10.1016/j.immuni.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Fahl SP, Crittenden RB, Allman D, Bender TP. c-Myb is required for pro-B cell differentiation. J Immunol. 2009;183:5582–5592. doi: 10.4049/jimmunol.0901187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bender TP, Kremer CS, Kraus M, Buch T, Rajewsky K. Critical functions for c-Myb at three checkpoints during thymocyte development. Nat Immunol. 2004;5:721–729. doi: 10.1038/ni1085. [DOI] [PubMed] [Google Scholar]
- 30.Greig KT, de Graaf CA, Murphy JM, Carpinelli MR, Pang SH, Frampton J, Kile BT, Hilton DJ, Nutt SL. Critical roles for c-Myb in lymphoid priming and early B-cell development. Blood. 2010;115:2796–2805. doi: 10.1182/blood-2009-08-239210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hubner A, Cavanagh-Kyros J, Rincon M, Flavell RA, Davis RJ. Functional cooperation of the proapoptotic Bcl2 family proteins Bmf and Bim in vivo. Mol Cell Biol. 2010;30:98–105. doi: 10.1128/MCB.01155-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strasser A, Harris AW, Cory S. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 1991;67:889–899. doi: 10.1016/0092-8674(91)90362-3. [DOI] [PubMed] [Google Scholar]
- 33.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 34.Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, DePinho RA, Rickert RC. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol. 2008;9:1388–1398. doi: 10.1038/ni.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- 36.DeKoter RP, Lee HJ, Singh H. PU.1 regulates expression of the interleukin-7 receptor in lymphoid progenitors. Immunity. 2002;16:297–309. doi: 10.1016/s1074-7613(02)00269-8. [DOI] [PubMed] [Google Scholar]
- 37.Izon DJ, Punt JA, Xu L, Karnell FG, Allman D, Myung PS, Boerth NJ, Pui JC, Koretzky GA, Pear WS. Notch1 regulates maturation of CD4+ and CD8+ thymocytes by modulating TCR signal strength. Immunity. 2001;14:253–264. doi: 10.1016/s1074-7613(01)00107-8. [DOI] [PubMed] [Google Scholar]
- 38.Kikuchi K, Lai AY, Hsu CL, Kondo M. IL-7 receptor signaling is necessary for stage transition in adult B cell development through up-regulation of EBF. J Exp Med. 2005;201:1197–1203. doi: 10.1084/jem.20050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mandal M, Crusio KM, Meng F, Liu S, Kinsella M, Clark MR, Takeuchi O, Aifantis I. Regulation of lymphocyte progenitor survival by the proapoptotic activities of Bim and Bid. Proc Natl Acad Sci U S A. 2008;105:20840–20845. doi: 10.1073/pnas.0807557106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mandal M, Powers SE, Ochiai K, Georgopoulos K, Kee BL, Singh H, Clark MR. Ras orchestrates exit from the cell cycle and light-chain recombination during early B cell development. Nat Immunol. 2009;10:1110–1117. doi: 10.1038/ni.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmelzle T, Mailleux AA, Overholtzer M, Carroll JS, Solimini NL, Lightcap ES, Veiby OP, Brugge JS. Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. Proc Natl Acad Sci U S A. 2007;104:3787–3792. doi: 10.1073/pnas.0700115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hubner A, Barrett T, Flavell RA, Davis RJ. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell. 2008;30:415–425. doi: 10.1016/j.molcel.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol. 2004;24:5434–5446. doi: 10.1128/MCB.24.12.5434-5446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harada H, Quearry B, Ruiz-Vela A, Korsmeyer SJ. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci U S A. 2004;101:15313–15317. doi: 10.1073/pnas.0406837101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emambokus N, Vegiopoulos A, Harman B, Jenkinson E, Anderson G, Frampton J. Progression through key stages of haemopoiesis is dependent on distinct threshold levels of c-Myb. EMBO J. 2003;22:4478–4488. doi: 10.1093/emboj/cdg434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan J, Crittenden RB, Bender TP. c-Myb promotes the survival of CD4+CD8+ double-positive thymocytes through upregulation of Bcl-xL. J Immunol. 2010;184:2793–2804. doi: 10.4049/jimmunol.0902846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, Harris AW. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci U S A. 1991;88:8661–8665. doi: 10.1073/pnas.88.19.8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oliver PM, Wang M, Zhu Y, White J, Kappler J, Marrack P. Loss of Bim allows precursor B cell survival but not precursor B cell differentiation in the absence of interleukin 7. J Exp Med. 2004;200:1179–1187. doi: 10.1084/jem.20041129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Labi V, Erlacher M, Kiessling S, Manzl C, Frenzel A, O’Reilly L, Strasser A, Villunger A. Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates gamma irradiation-induced thymic lymphoma development. J Exp Med. 2008;205:641–655. doi: 10.1084/jem.20071658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Labi V, Woess C, Tuzlak S, Erlacher M, Bouillet P, Strasser A, Tzankov A, Villunger A. Deregulated cell death and lymphocyte homeostasis cause premature lethality in mice lacking the BH3-only proteins Bim and Bmf. Blood. 2014;123:2652–2662. doi: 10.1182/blood-2013-11-537217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morrow MA, Lee G, Gillis S, Yancopoulos GD, Alt FW. Interleukin-7 induces N-myc and c-myc expression in normal precursor B lymphocytes. Genes Dev. 1992;6:61–70. doi: 10.1101/gad.6.1.61. [DOI] [PubMed] [Google Scholar]
- 52.Geier JK, Schlissel MS. Pre-BCR signals and the control of Ig gene rearrangements. Semin Immunol. 2006;18:31–39. doi: 10.1016/j.smim.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 53.Biedenkapp H, Borgmeyer U, Sippel AE, Klempnauer KH. Viral myb oncogene encodes a sequence-specific DNA-binding activity. Nature. 1988;335:835–837. doi: 10.1038/335835a0. [DOI] [PubMed] [Google Scholar]
- 54.Weston K, Bishop JM. Transcriptional activation by the v-myb oncogene and its cellular progenitor, c-myb. Cell. 1989;58:85–93. doi: 10.1016/0092-8674(89)90405-4. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Y, Ness SA. Myb proteins: angels and demons in normal and transformed cells. Front Biosci (Landmark Ed) 2011;16:1109–1131. doi: 10.2741/3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quintana AM, Liu F, O’Rourke JP, Ness SA. Identification and regulation of c-Myb target genes in MCF-7 cells. BMC Cancer. 2011;11:30. doi: 10.1186/1471-2407-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biswas SC, Liu DX, Greene LA. Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J Neurosci. 2005;25:8349–8358. doi: 10.1523/JNEUROSCI.1570-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martensson A, Xie XQ, Persson C, Holm M, Grundstrom T, Martensson IL. PEBP2 and c-myb sites crucial for lambda5 core enhancer activity in pre-B cells. Eur J Immunol. 2001;31:3165–3174. doi: 10.1002/1521-4141(200111)31:11<3165::aid-immu3165>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 59.Brooks AR, Shiffman D, Chan CS, Brooks EE, Milner PG. Functional analysis of the human cyclin D2 and cyclin D3 promoters. J Biol Chem. 1996;271:9090–9099. doi: 10.1074/jbc.271.15.9090. [DOI] [PubMed] [Google Scholar]
- 60.Quintana AM, Zhou YE, Pena JJ, O’Rourke JP, Ness SA. Dramatic repositioning of c-Myb to different promoters during the cell cycle observed by combining cell sorting with chromatin immunoprecipitation. PLoS One. 2011;6:e17362. doi: 10.1371/journal.pone.0017362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, Cantley LC. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science. 1999;283:393–397. doi: 10.1126/science.283.5400.393. [DOI] [PubMed] [Google Scholar]
- 62.Corfe SA, Rottapel R, Paige CJ. Modulation of IL-7 thresholds by SOCS proteins in developing B lineage cells. J Immunol. 2011;187:3499–3510. doi: 10.4049/jimmunol.1100424. [DOI] [PubMed] [Google Scholar]
- 63.Mandal M, Powers SE, Maienschein-Cline M, Bartom ET, Hamel KM, Kee BL, Dinner AR, Clark MR. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat Immunol. 2011;12:1212–1220. doi: 10.1038/ni.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Powers SE, Mandal M, Matsuda S, Miletic AV, Cato MH, Tanaka A, Rickert RC, Koyasu S, Clark MR. Subnuclear cyclin D3 compartments and the coordinated regulation of proliferation and immunoglobulin variable gene repression. The Journal of Experimental Medicine. 2012 doi: 10.1084/jem.20120800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kitamura D, Kudo A, Schaal S, Muller W, Melchers F, Rajewsky K. A critical role of lambda 5 protein in B cell development. Cell. 1992;69:823–831. doi: 10.1016/0092-8674(92)90293-l. [DOI] [PubMed] [Google Scholar]
- 66.Ohnishi K, Melchers F. The nonimmunoglobulin portion of [lambda]5 mediates cell-autonomous pre-B cell receptor signaling. Nat Immunol. 2003;4:849–856. doi: 10.1038/ni959. [DOI] [PubMed] [Google Scholar]
- 67.Bankovich AJ, Raunser S, Juo ZS, Walz T, Davis MM, Garcia KC. Structural Insight into Pre-B Cell Receptor Function. Science. 2007;316:291–294. doi: 10.1126/science.1139412. [DOI] [PubMed] [Google Scholar]
- 68.Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20:707–718. doi: 10.1016/j.immuni.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 69.Johnson K, Hashimshony T, Sawai CM, Pongubala JM, Skok JA, Aifantis I, Singh H. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity. 2008;28:335–345. doi: 10.1016/j.immuni.2007.12.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.