

Abstract
Purpose
Cell-based therapies are showing great promise for a variety of diseases, but remain hindered by the limited information available regarding the biological fate, migration routes and differentiation patterns of infused cells in trials. Previous studies have demonstrated the feasibility of using positron emission tomography (PET) to track single cells utilising an approach known as positron emission particle tracking (PEPT). The radiolabel hexadecyl-4-[18F] fluorobenzoate ([18F]HFB) was identified as a promising candidate for PEPT, due to its efficient and long-lasting labelling capabilities. The purpose of this work was to characterise the labelling efficiency of [18F]HFB in vitro at the single-cell level prior to in vivo studies.
Procedures
The binding efficiency of [18F]HFB to MDA-MB-231 and Jurkat cells was verified in vitro using bulk gamma counting. The measurements were subsequently repeated in single cells using a new method known as radioluminescence microscopy (RLM) and binding of the radiolabel to the single cells was correlated with various fluorescent dyes.
Results
Similar to previous reports, bulk cell labelling was significantly higher with [18F]HFB (18.75 ± 2.47 dpm/cell) than [18F]FDG (7.59 ± 0.73 dpm/cell). However, single-cell imaging using RLM revealed that [18F]HFB accumulation in live cells (8.35 ± 1.48 cpm/cell, n = 9) was not significantly higher than background levels (4.83 ± 0.52 cpm/cell, n = 12) and was 1.7 fold lower than [18F]FDG uptake in the same cell line (14.09 ± 1.90 cpm/cell, n = 13; p < 0.01). Instead, [18F]HFB was found to bind significantly to fragmented membranes associated with dead cell nuclei, suggesting an alternative binding target for [18F]HFB.
Conclusion
This study demonstrates that bulk analysis alone does not always accurately portray the labelling efficiency, highlighting the need for more routine screening of radiolabels using RLM to identify heterogeneity at the single-cell level.
Keywords: Cell tracking, radioluminescence microscopy (RLM), single-cell analysis and hexadecyl-4-[18F]fluorobenzoate ([18F]HFB)
INTRODUCTION
Cell-based treatment strategies—for example, stem cell [1,2], T cell [3,4] and dendritic cell [5,6] therapy—are continuing to emerge as potent therapeutics for refractory malignancies that do not respond well to conventional radiotherapy and chemotherapy. Nevertheless, little is known about the biological mechanisms and local regional effects of infused cells in vivo. Current approaches are restricted to isolated time point measurements of released cytokines associated with activation, estimations of infused cells in the peripheral circulation and postmortem histological assessments. Furthermore, the migration path of transplanted cells remains unknown, which is problematic in light of the diverse cellular phenotypes of hematopoietic cell subsets that are localised in distinct niches, depending on their differentiation state [7]. The development of noninvasive imaging methods for sequential kinetic measurements of therapeutic cells is a highly sought-after goal for detecting differentiation patterns of stem cells, identifying occult sites of inflammation or infection [8] and monitoring specialised interactions with tumour antigens [9].
Positron emission tomography (PET) has shown great translation potential for monitoring cells by enabling highly sensitive, sequential, quantitative and longitudinal measurements of bulk cell populations in vivo [10–13]. A variety of PET radiolabels have been used for cell tracking to visualise labelled cells directly [10,14,15] or indirectly [16–18]. The lipophilic long chain ester, hexadecyl-4-[18F]fluorobenzoate ([18F]HFB) has shown particular promise for direct labelling. The labelling agent is designed to be absorbed in cellular membranes (Fig. 1a) without requiring a specific enzyme, receptor or transporter [19].
Fig. 1. Bulk analysis of [18F]HFB binding in MDA-MB-231 and Jurkat cells.
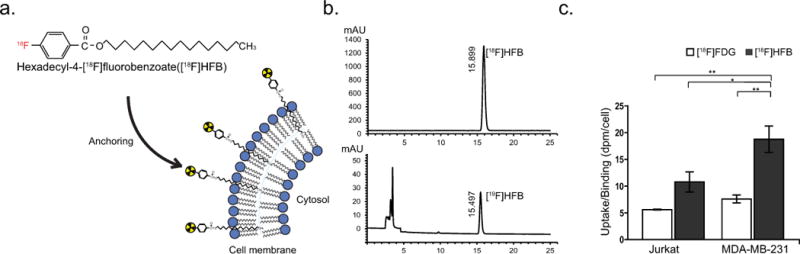
(a) Chemical structure and illustration of the presumed mechanism of binding of [18F]HFB. The lipophilic long chain ester is rapidly absorbed into the cellular membrane. (b) Analytical HPLC indicating >99% radiochemical purity. (c) Bulk analysis of [18F]HFB labelling and [18F]FDG uptake in two cell lines (Jurkat and MDA-MB-231), measured by gamma counting. Cells were incubated with 250 µCi/mL of [18F]FDG (n=5) or [18F]HFB (n = 13) at 37°C for 30 min. Data is presented as disintegrations per minute (dpm) per cell. Experiments were repeated ≥ twice. Error bars are Mean ± SEM, *p ≤ 0.05 and **p ≤ 0.01
The focus of this work on [18F]HFB stems from current development of methods for tracking single cells using PET instrumentation. Although, many strategies are available for monitoring bulk populations of cells, non-invasive methods for detecting single cells remain elusive. Single-cell sensitivity is especially important in cell-based therapy because individual cells distributed throughout the body could have specialised therapeutic effects. To achieve single-cell sensitivity with PET, a novel tracking method was recently developed [20]. This method builds on a previous technique, known as positron emission particle tracking (PEPT), used primarily for measuring liquid and particulate flows in opaque industrial systems [21–25]. Unlike conventional PET, PEPT uses list-mode data to directly localise discrete sources, without the intermediate step of reconstructing the image. While PEPT is used exclusively for studying chemical and industrial processes, the potential of PEPT for biomedical investigations, such as cell tracking, is increasingly being recognised [26]. To assess the feasibility of tracking a single cell, a novel trajectory reconstruction algorithm was developed in-house and validated using computer simulation and phantom models [20,27]. These simulations predict that labelling a single cell with 20 - 25 Bq (0.54-0.68 nCi) of activity is sufficient for tracking the cell in vivo using small animal PET. To meet this requirement, [18F]HFB was identified as a promising candidate due to its reported ability to label cells efficiently (18-30 Bq/cell) with minimal efflux[19,28].
The initial aims were to verify the binding efficiency of [18F]HFB in vitro using bulk gamma counting and at the single-cell level using radioluminescence microscopy (RLM), prior to in vivo monitoring of individual cells using PET. Bulk gamma counting is a simple, sensitive and inexpensive method for acquiring measurements of average uptake in cell populations. However, the approach lacks the ability to quantify cell-to-cell variations in radioactivity. In particular, bulk counting methods cannot discriminate between uptake in live cells and other elements present in vitro, which is a crucial consideration for radiolabels that do not rely on active processes. To circumvent the limitation of gamma counting, radioluminescence microscopy [29,30] was introduced as a way to explore the heterogeneity of radiotracer uptake within single cells. This method provides an accurate assessment of labelling efficiency at the single-cell level. Radioluminescence microscopy was used to visualise and quantify labelling of individual cells by [18F]HFB and as a reference, [18F-]-labelled fluorodeoxyglucose ([18F]FDG). Single-cell imaging using radioluminescence microscopy demonstrated unexpected preferential accumulation of [18F]HFB in fragments of membranes from dead cells, highlighting the importance of radioluminescence imaging for accurate characterisation of cell labelling efficiency in vitro prior to in vivo models.
MATERIAL & METHODS
Radiochemical synthesis
The precursor as well as the reference compound were synthesised according to a published method [19] and characterised using nuclear magnetic resonance and mass spectrometry (supplementary Fig 1). Briefly, [18F]fluoride (n.c.a.) in [18O]water was passed through a PS-HCO3 cartridge and eluted with 1 mL of a phase transfer catalyst solution containing Kryptofix222 (15 mg) and K2CO3 (3.5 mg) in 9:1 acetonitrile/water. The K222K[18F]fluoride complex was heated and dried under helium at 65 °C (3 min) and at 85 °C (1 min). Following cooling to ~50 °C, a solution containing HFB trimethylammonium triflate precursor (2-3 mg in 1 mL anhydrous DMSO) was added to the reactor and the solution was heated to 95 °C (10 min). The reaction mixture was cooled to ~40 °C, diluted with water (8 mL) and passed through a Sep-Pak SPE C18 light cartridge (Waters). The radiolabelled product was eluted with acetonitrile (2 mL) and sterile water (1 mL) for high-performance liquid chromatography (HPLC) purification. The crude solution was injected to a Phenomenex Luna C5 semi-preparative reversed-phase columns (5 µm, 10 × 250 mm, mobile phase 90% acetonitrile/H2O with 0.1% TFA, flow rate of 5 ml/min). The desired HPLC fraction (Rt ~ 17 min) containing the fluorinated product was collected in a flask containing water (20 mL). The diluted fraction was passed through a Sep-Pak SPE C8 cartridge (Waters), rinsed with water and eluted with ethanol (2 mL). The solvent was evaporated at 75 °C under helium stream. The [18F]HFB was reconstituted with 10% DMSO/saline. The radiochemical yield was 2.5 ± 0.7 % and the molar activity was 2211 ± 961 mCi/µmol, n=5 (decay-corrected to end-of-synthesis, total synthesis time 60 min). Radiochemical purity by analytical HPLC was >99%: Phenomenex Luna C5 (5 µm, 4.6 × 250 mm) column fitted with UV (254 nm) and radioactivity detectors. The mobile phase was 95% acetonitrile/H2O with 0.1% TFA and flow rate of 1 ml/min.
Bulk analysis
Cell culture supplies were obtained from Thermo Fisher Scientific unless otherwise stated. MDA-MB-231 and Jurkat T cells were obtained from the American Type Culture Collection (ATCC) and cultured in DMEM or RPMI media (high glucose; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco), L-glutamine, 4.5 g/L D-glucose, 110 mg/L sodium pyruvate, 10 mM HEPES (Gibco), 100 U/mL penicillin (Gibco) and 100 µg/mL streptomycin (Gibco) (Thermo Fisher Scientific). Cell lines were maintained at 37°C in a humidified 5% CO2 atmosphere. Tryptinized MDA-MB-231 or Jurkat T cells (2 × 105/mL) were incubated with [18F]HFB or [18F]FDG (250 µCi/mL) for 30 min at 37°C. Cells were washed twice with phosphate buffer saline (PBS) and Jurkat T cells were re-suspended in PBS (1 mL). Cell labelling with both [18F]HFB and [18F]FDG was measured using a Cobra II gamma counter (Packard BioScience, now PerkinElmer Life and Analytical Sciences, Boston, MA). All experiments were performed in triplicate and repeated at least twice. Results are represented as mean disintegration per minute per cell (dpm/cell) ± standard error of the mean (SEM).
Single-cell imaging
Adherent MDA-MB-231 cells (5 × 104) were seeded sparsely on a tissue-culture-treated 35 mm glass-bottom dish (ibidi Labware GmbH, Germany). The following day, MDA-MB-231 cells were incubated with [18F]HFB or [18F]FDG (250 µCi/mL) for 30 min at 37°C, whilst suspension MDA-MB-231 cells were obtained by tryptinizing prior to radiolabelling. In addition to [18F]HFB or [18F]FDG, cells were co-incubated with live/dead nuclei fluorescent stain for mammalian cells (NucBlue Live ReadyProbe and NucGreen Dead 488 ReadyProbe; Thermo Fisher Scientific) and/or DiIC18(5) solid (1,1′-Dioctadecyl-3,3,3′,3′ Tetramethylindodicarbocyanine, 4-Chlorobenzenesulfonate Salt) membrane dye (Thermo Fisher Scientific). The cells were washed twice with PBS and suspension MDA-MB-231 as well as Jurkat T cells were re-suspended in PBS (1 mL) and aliquoted in a tissue-culture-treated 35 mm glass-bottom dish. Fluorescence images were initially acquired to confirm cell viability and identify a region containing both live cells, clusters of dead cell nuclei and membrane fragments. The DAPI channel was used for cell nuclei (excitation/emission: 358 nm⁄461 nm), the GFP channel for dead cell nuclei (469 nm/525 nm) and the Texas Red channel for cellular membrane (559 nm/630 nm).
Single-cell resolution radioluminescence imaging was performed on cells labelled with [18F]HFB or [18F]FDG using a previously described protocol [31]. Briefly, a transparent scintillating crystal (1 cm × 1 cm × 0.5 mm CdWO4, both sides polished; MTI Corp., Richmond, CA) was submerged in an imaging dish and gently positioned on top of the radiolabelled cells. Scintillation was generated by emission of positrons from the radiolabelled cells and captured using a low-light microscope with an effective magnification of 5×. The low-light microscope was comprised of a 20× magnification and 0.75 NA microscope objective lens (CFI Plan Apochromat λ; Nikon Instruments Inc., Melville, NY), a 4× magnification, 0.2 NA tube lens (CFI Plan Apochromat λ; Nikon Instruments Inc., Melville, NY) and deep-cooled electron-multiplying charge-coupled device (EMCCD) camera (ImageEM C9100-13, Hamamatsu Photonics K.K., Japan). Fluorescence and brightfield images were captured with 5× effective magnification and no pixel binning (3.2 × 3.2 µm/px resolution). The microscope was first focused on the cell nucleus using the DAPI channel. The focus was then shifted ~5 µm towards the scintillator edge. The radioluminescence images were acquired using an EM gain of 1200×, 4×4 pixel binning (12.8 × 12.8 µm/px resolution), an exposure time of 9 - 160 ms and 10,000 image frames. To compare different radioluminescence images, the radioactive decay counts were normalised by the total acquired exposure time, i.e., the number of frames multiplied by the exposure time per frame.
Image reconstruction and analysis
Reconstruction of the radioluminescence images was performed using a MATLAB (R2012b; The MathWorks) toolbox called ORBIT (Optical Reconstruction of the Beta-ionization Track) [30]. Briefly, the algorithm processes the images one at a time, identifying the dim scintillation light resulting from incoming 18F positrons. The positions of the individual events are aggregated into a single reconstructed image.
Fluorescence images of the membrane, dead or live nuclei were obtained to guide the manual placing of circular regions of interest (ROIs; radius = 22 μm) on individual cells. ROIs were placed on cell clusters where membrane fragments and dead cell nuclei were in close proximity. Additionally, similar ROIs were manually placed on background control regions. Quantitative radioactivity measurements were obtained by summing the number of counts per minute (cpm) measured in each ROI. These values did not account for the sensitivity of the microscope (≈30%), the decay of the tracer over the course of the acquisition, and the positron yield of 18F (97%). One way analysis of variance (ANOVA) followed by the Tukey’s HSD test were used to perform statistical testing between 3 or more datasets.
RESULTS
Synthesis of [18F]-HFB with high radiochemical yield
A two-step reaction consisting of esterification of 4-dimethylaminobenzoyl chloride, followed by the use of methyl triflate for quaternization of the aniline (Scheme 1) was performed. Synthesis of the fluorine-18 labelled long chain benzoic acid ester was achieved by nucleophilic aromatic displacement (Scheme 2). HPLC purification yielded a final product co-eluted with non-labelled ‘cold’ HFB with radiochemical purity of > 99% (corrected for decay) over a synthesis time of 60 min and molar activity of 1211 ± 96 mCi/µmol (Fig. 1b).
Scheme 1.
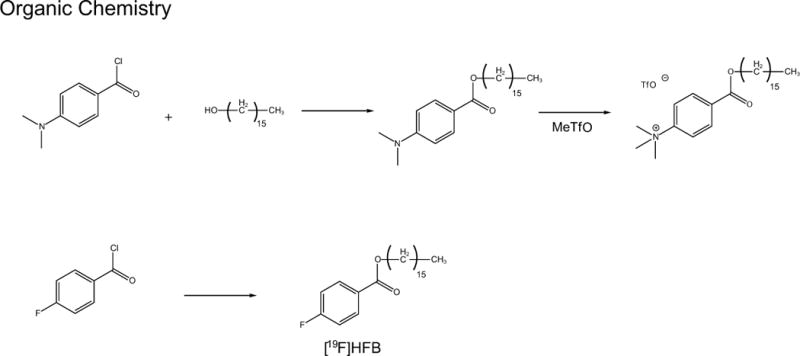
Esterification of 4-dimethylalminobenzoyl chloride followed by quaternization of the aniline using methyl trifilate
Scheme 2.
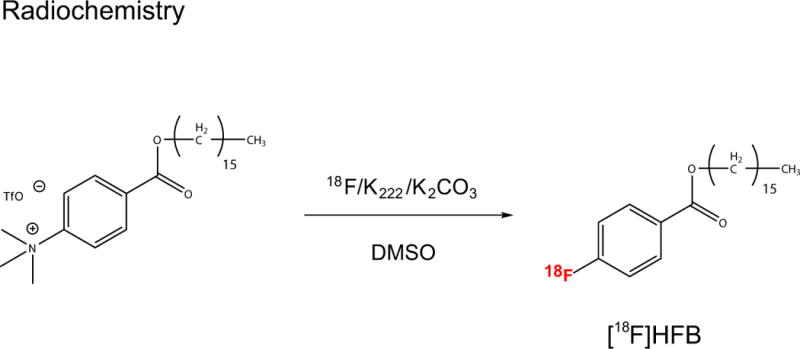
Nucleophilic aromatic displacement reaction to obtain [18F]HFB
Bulk analysis indicated high binding efficiency of [18F]HFB
Bulk gamma counting was used to compare the labelling efficiency of [18F]HFB and [18F]FDG. T lymphocyte (Jurkat) and breast adenocarcinoma (MDA-MB-231) cells were incubated with [18F]HFB or [18F]FDG for 30 minutes (Fig. 1c). Labelling of MDA-MB-231 cells with [18F]HFB (18.75 ± 2.47 dpm/cell, n = 6) was 2.5 fold greater than labelling with [18F]FDG (7.59 ± 0.73 dpm/cell, n=7, p ≤ 0.01). Additionally, in Jurkat cells, labelling with [18F]HFB (10.78 ± 1.89 dpm/cell, n=13) was 1.9 fold greater than labelling with [18F]FDG (5.60 ± 0.10 dpm/cell, n = 5). Labelling of [18F]HFB was 1.7 fold greater (p < 0.05) in MDA-MB-231 cells than in Jurkat cells, whereas uptake of [18F]FDG in MDA-MB-231 and Jurkat cells was not significantly different (p > 0.05). In summary, bulk analysis demonstrated that [18F]HFB was more efficient than [18F]FDG for labelling both Jurkat and MDA-MB-231 cells, suggesting that [18F]HFB may be suitable for in vivo detection of single labelled cells using PET.
Single cell imaging demonstrated poor binding of [18F]HFB to MDA-MB-231 cells
Radioluminescence microscopy was used to determine the labelling efficiency of [18F]HFB in single cells (Fig. 2a). Bright-field and fluorescence micrographs were acquired to localise MDA-MB-231 cells and their nuclei. Initial assessments of single-cell images comparing [18F]HFB binding and [18F]FDG uptake showed robust cellular uptake of [18F]FDG, but minimal binding of [18F]HFB in MDA-MB-231 cells (Fig. 2b). Subsequent quantification was performed by region of interest (ROI) analysis on the radioluminescence images (Fig. 2c). Contrary to bulk analysis, accumulation of [18F]FDG in MDA-MB-231 cells (14.09 ± 1.90 cpm, n = 13) was 1.7 fold greater than [18F]HFB in the same cell line (8.35 ± 1.48 cpm, n = 9; p ≤ 0.01). Furthermore, [18F]HFB labelling was not significantly different from background levels (4.83 ± 0.52 cpm, n = 12; p > 0.05), indicating [18F]HFB does not bind efficiently to MDA-MB-231 cells.
Fig. 2. Single-cell imaging using radioluminescence microscopy.
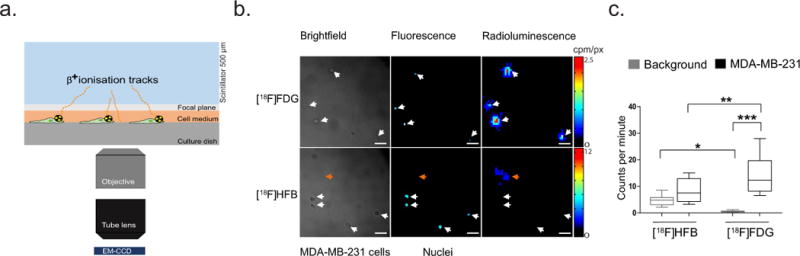
(a) Schematic diagram of the radioluminescence microscope. A CdWO4 scintillator was placed above the radiolabelled cells and excited by β+ particles emitted from decaying radionuclides. The resulting optical photons were captured using a high numerical-aperture objective (20×, 0.75 NA) coupled to a 50 mm focal-length tube lens and an EM-CCD camera. (b) Representative brightfield and fluorescence images revealed the localisation of MDA-MB-231 cells, and radioluminescence images indicated [18F]HFB accumulated sporadically in regions (orange arrows, bottom panel) unrelated to the localisation of MDA-MB-231 cells (white arrows, bottom panel). (c) In contrast to bulk gamma counting, singe-cell analysis showed significantly greater binding of [18F]FDG to MDA-MB-231 cells in comparison to [18F]HFB. The accumulation of [18F]HFB in MDA-MB-231 cells was not significantly different from background. Experiments repeated ≥ twice. Error bars are mean ± SEM, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 and scale bar is 50 µm
Accumulation of [18F]HFB in fragments adjacent to dead cell nuclei
Fluorescent labelling of dead cell nuclei followed by radioluminescence microscopy was performed to ascertain whether [18F]HFB was binding to dead cells. Previous analyses demonstrated poor binding of [18F]HFB in MDA-MB-231 cells (Fig. 2b/c). Fluorescence images showing MDA-MB-231 cell nuclei (Fig. 3a i/ii/iii) and dead cell nuclei (Fig. 3a iv/v/vi) were acquired to survey the imaging dish and identify regions containing clusters of dead cell nuclei, prior to radioluminescence microscopy imaging of [18F]HFB or [18F]FDG (Fig. 3a vii/viii/ix). Quantitative ROI analysis (Fig. 3b) found the uptake of [18F]FDG in adherent MDA-MB-231 cells to be significantly greater in live cells (28.13 ± 2.07 cpm, n = 37) than near dead cell nuclei (3.16 ± 1.04 cpm, n = 7; p ≤ 0.001), whereas [18F]HFB binding was 2.3 fold greater in regions adjacent to dead cell nuclei (230.36 ± 25.50 cpm, n = 16) compared to live cell nuclei (99.55 ± 13.09 cpm, n = 6; p < 0.05) and 1.7 fold greater compared to background (136.45 ± 22.45 cpm, n = 29; p < 0.05).
Fig. 3. Dual fluorescence and radioluminescence microscopy reveals an unexpected binding pattern for [18F]HFB.
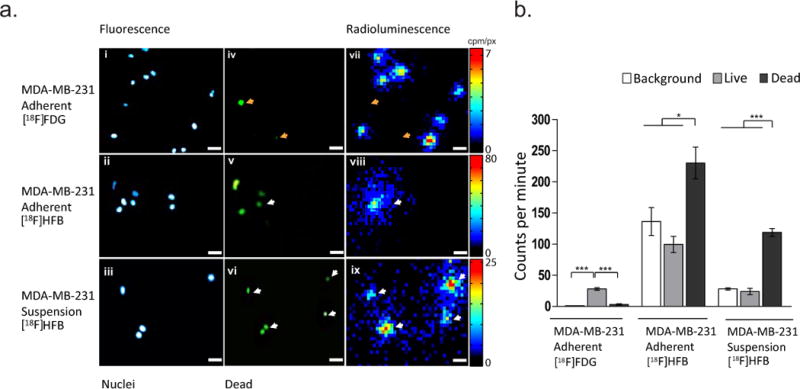
(a) i-ii Representative fluorescence images of adherent and (a) iii suspension MDA-MB-231 nuclei. (a) iv-vi Localisation of dead cell nuclei. (a) vii-ix Representative fluorescence guided radioluminescence images revealed preferential binding of [18F]HFB in regions consisting of dead cell nuclei (white arrows), whilst uptake of [18F]FDG was significantly greater near live MDA-MB-231 nuclei than near dead cell nuclei (orange arrows, top panel). (b) Quantitative ROI analysis showing uptake of [18F]FDG and binding of [18F]HFB in adherent and non-adherent MDA-MB-231 cells. Error bars are mean ± SEM, ***p ≤ 0.001, *p ≤ 0.05 and scale bar is 50 µm
To investigate whether these results could be reproduced in suspension cells, single-cell suspensions of MDA-MB-231 cells were incubated with [18F]HFB and imaged using RLM (Fig. 3a iii/vi/ix). Similar to labelling of adherent MDA-MB-231 cells, binding of [18F]HFB in fragments adjacent to dead cell nuclei (118.84 ± 6.33 cpm, n = 10) was significantly greater in comparison to live cells (24.04 ± 4.98 cpm, n = 11; p ≤ 0.001) and background levels (28.10 ± 1.24 cpm, n = 28; p ≤ 0.001), as shown on Fig. 3b. Increased localisation of [18F]HFB in regions surrounding dead cell nuclei (29.94 ± 4.81 cpm, n = 11) in comparison to live cell nuclei (12.67 ± 1.72 cpm, n = 10) and background (6.11 ± 1.05 cpm, n = 21) was also observed for Jurkat cells (Supplementary Fig. 2).
Co-localisation of radioluminescence and fluorescence images revealed the binding target of [18F]HFB
The proposed mechanism of action for [18F]HFB is rapid absorption in the cellular membrane, therefore fluorescence staining of cellular membranes was performed to determine whether [18F]HFB localised at the cellular membrane. Co-registration of membrane fluorescence and radioluminescence images indicated that [18F]HFB was retained by some cellular membranes, but not others (Fig. 4a/b). An explanation of this phenomena was provided by examining nuclear stain images. Of all membranes associated with dead cells, the vast majority (80%) were positive for [18F]HFB and the remainder were negative. In contrast, none (0%) of the membranes that were adjacent to live MDA-MB-231 nuclei and not surrounded by dead cell nuclei were positive for [18F]HFB (Fig. 4c).
Fig. 4. Preferential accumulation of [18F]HFB in dead cell membranes.
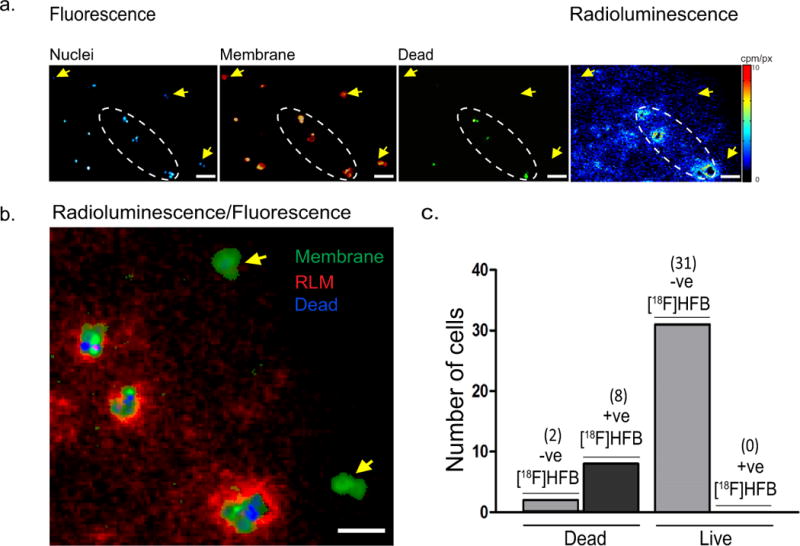
(a) Representative fluorescence and radioluminescence images indicating accumulation of [18F]HFB in relation to localisation of MDA-MB-231 cell nuclei (blue) or dead cell nuclei (green) and membranes (red). No significant binding of [18F]HFB was visualised in live cell membranes (yellow arrows). (b) Co-registered fluorescence and radioluminescence images showed co-localisation of [18F]HFB (red) in regions (white dashed oval) consisting of both membrane fragments (green) and dead cell nuclei (blue). (c) Number of cell membranes, as defined by fluorescence membrane stain, associated with live and dead cell nuclei, according to their level of [18F]HFB labelling (+ve: positive, -ve: negative). Scale bar is 100 µm
DISCUSSION
The ability to visualise the migration patterns of individual cells is of particular importance in cell-based regenerative medicine [32–34]. Strategies for labelling single cells should meet several requirements, such as stable labelling, high retention of activity for detection and applicability in clinical practice [35]. A direct cell labelling approach using [18F]HFB was investigated because the radiotracer has been previously shown to accumulate rapidly in cellular membranes (18-30 Bq/cell) [19,28]. The high labelling efficiency of [18F]HFB was verified using bulk gamma counting, however dual modality fluorescence and radioluminescence microscopy exposed a discordant pattern of [18F]HFB binding to membrane fragments adjacent to dead cell nuclei.
Previous reports of high labelling efficiency and retention of [18F]HFB over 4 h (77 - 95%) in vitro and in vivo [19,28] suggested the radiolabel may be well suited for monitoring individual cells in vivo using a trajectory reconstruction algorithm developed in-house [20,27].In vitro bulk labelling of rat mesenchymal stem cells (MSCs) (5 × 105/0.5 mL, 1 mCi, 30 min) indicated labelling efficiency of [18F]HFB was 25% after 4h and [18F]HFB-labelled cells were detected in the lungs 55-60 min post-intravenous injection [19]. Subsequent reports comparing [18F]HFB labelling to [18F]FDG uptake, defined the optimal incubation conditions and assessed cytotoxicity of [18F]HFB. The labelling efficiency of [18F]HFB in human cardiac progenitor cells (CPCs) was 12 - 48%, higher than that of [18F]FDG (< 10%). According to bulk gamma counting, optimal [18F]HFB labelling conditions in CPCs (2 × 106/mL, 5-7 mCi or 185-259 MBq) were achieved at 37°C and labelled cells were imaged using PET at 10 min, 2 h and 4 h post injection [28]. Additionally, [18F]HFB has been used to facilitate the detection of injectable biomaterials in vivo. Collagen matrix delivery and biodistribution in a mouse model of myocardial infarction was visualised using PET at 10 min and 2 h post ultrasound guided injections of [18F]HFB labelled matrices [36].
Variants of HFB that incorporate longer-lived radioisotopes such as 124I and 64Cu have been utilised to monitor intramuscularly administered rat adipose derived stem cells (ADSCs, 5 × 106/50 μL, 30 min) for 9 days following injection of [124I] labelled hexadecyl-4-iodobenzoate ([124I]HIB) [37] and for 18 h following injection of 64Cu-labelled hexadecyl-1,4,7,10-tetraazacyclododecane-tetraacetic acid-benzoate (64Cu-DOTA-HB) [38].
A unique property of [18F]FHB is its lipophilic nature that should facilitate anchoring of the radiolabel within the hydrophobic tails of the phospholipid bilayer [37]. However, lack of [18F]FHB binding in live single cells that have an intact cellular membrane implies the radiolabel requires a compromised membrane for binding. In contrast, membranes adjacent to dead cell nuclei likely consist of fragmented phospholipid bilayers that are irregular and positioned in a conformation where the hydrophobic tails are exposed, thus providing a target for the binding of the lipophilic compound. This mechanism may explain the alternative binding pattern observed of preferential accumulation in regions containing compromised membrane fragments and lack of labelling in cells where the cellular membrane integrity was maintained.
In addition to the high level of uptake in fragmented membranes, weak and diffuse accumulation of [18F]HFB but not [18F]FDG was evident in background regions that stained negatively for live, dead nuclei or membrane. The high background activity in the adherent MDA-MB-231 cells may be due to spontaneous adhesion of the lipophilic [18F]HFB to the culture dish during cell labelling. Because the suspension MDA-MB-231 cells were not labelled inside the imaging dish, the background was significantly lower, and was explained by the potential binding of the radiolabel to small membrane fragments present in the suspended sample (Fig. 3b). Finally, the apparent difference in the binding of [18F]HFB to dead adherent cells and dead suspension cells can be explained almost entirely by the higher non-specific background observed for the adherent cells.
Radioluminescence microscopy is a useful tool for quantifying radiolabelling efficiency and visualizing heterogeneity in uptake of PET radiolabels in diverse cell types. The pattern of binding of [18F]HFB, consisting of preferential binding to fragmented membranes of non-viable cells, has not been observed with other PET radiolabels. Previously, radioluminescence microscopy has been applied to imaging of 9-[4-[18F]fluoro-3-(hydroxymethyl)-butyl]guanine ([18F]FHBG) uptake in HeLa cells expressing herpes simplex type 1 virus (HSV1) thymidine kinase [29], binding of [64Cu]rituximab in single Ramos B lymphocytes [39], uptake of [18F]FDG [30,40] and 3′-deoxy-3′-[18F-]fluorothymidine ([18F]FLT) [41] in MDA-MB-231 cells. In these studies, binding of the radiolabel was only observed in viable cells, with minimal background, ruling out the possibility of a technical artefact. This provide additional evidence that the high bulk labelling efficiency of [18F]HFB is primarily the result of the radiolabel binding to fragments of membranes as shown by single-cell imaging using fluorescence-guided radioluminescence microscopy.
An essential consideration when characterising an approach for monitoring cells in vivo is to identify a labelling strategy that does not affect cell viability; yet, in this work the main concern was the preferential binding of the radiolabel to membrane fragments belonging to non-viable cells. This may raise the question whether [18F]HFB was binding to cells that were already dead or binding of large amounts of [18F]HFB resulted in cell death. Previous bulk analyses assessing the effect of [18F]HFB on viability of CPCs [28] and effect of [124I]HIB on ADSCs [37] found that viability was maintained 2 – 4 h post labelling, however viability of CPCs significantly (p < 0.05) decreased at 24 h (65.8 ± 13.3%, control: 81.8 ± 6.9%) and 5 days (62.5 ± 10.4%, control: 84.0 ± 13.3%) post labelling. In this work, fluorescence imaging was performed to identify areas consisting of both live cells and clusters of dead cell nuclei and membrane fragments. Although cell viability was imaged right after labelling with [18F]HFB, single-cell images at later time points to assess the effect on cell viability were not obtained once the unexpected binding pattern of [18F]HFB was revealed. In light of previous reports indicating good cell viability 4h after [18F]HFB labeling, it is likely that [18F]HFB labeled pre-existing fragmented membranes from dead cells within the original sample. The difficulty associated with finding clusters of dead cells also confirms cell viability was high.
The effect of [124I]HIB labelling on ADSCs morphology and the expression of 24 genes involved in cell cycle, transcription, DNA repair and regulation has been previously evaluated. Bulk analysis demonstrated the expression patterns of select genes were not significantly different between [124I]HIB labelled ADSCs (0.3–2.0 Bq/cell) and control non-labelled ADSCs. Non-quantitative morphological assessments of [124I]HIB labelled ADSCs as well as control cells were performed using microscopy and no remarkable differences in morphology were observed [42]. Assessments of whether [124I]HIB behaves in a similar fashion to [18F]HFB have not been conducted. Nevertheless, in the event that [124I]HIB exhibits a pattern of cellular binding similar to that of [18F]HFB, the majority of live ADSCs in the sample incubated with [124I]HIB would not have been labelled and would likely display morphology and expression profile similar to control cells not incubated with [124I]HIB.
Strong affinity of [18F]HFB for fragmented cellular membranes suggests [18F]HFB could be suitable for imaging cell death in vivo. Predominant uptake of free [18F]HFB in the liver is reported immediately and 100 min post-intravenous injection [19]. Additionally, high liver activity (5.1 ± 2.4% ID/g) was also evident 10 min post echo-guided intramyocardial injection of free [18F]HFB [28]. The majority of previous work has used [18F]HFB for labelling ADCs and CPCs, however future work should investigate its potential for imaging tumour necrosis.
CONCLUSION
This study described the first use of radioluminescence microscopy to quantitatively determine the labelling efficiency of [18F]HFB, a lipophilic long-chain ester rapidly absorbed into the cellular membrane, with single-cell resolution. Initial bulk analysis, which demonstrated superior binding capabilities of [18F]HFB compared to [18F]FDG, were inconsistent with fluorescence-guided radioluminescence microscopy, which revealed poor binding of [18F]HFB to live cells and high binding to membrane fragments from dead cells. These results indicate bulk labelling alone may not accurately portray the labelling efficiency, thereby highlighting the need for routine single-cell imaging using radioluminescence microscopy to identify heterogeneity in radioactivity uptake, particularly for direct cell labelling.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) grants to Dr. Pratx (5R21HL127900 and 5R01CA186275) and by a NIH training grant to Dr. Kim (T32CA11868).
Funding
This work was funded in part by NIH grants to Dr. Pratx (5R21HL127900 and 5R01CA186275) and by a NIH training grant to Dr. Kim (T32CA11868).
Footnotes
COMPLIANCE WITH ETHICAL STANDARDS
Disclosure of potential conflicts of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.Slavin S, Nagler A, Naparstek E, Kapelushnik Y, Aker M, Cividalli G, et al. Nonmyeloablative stem cell transplantation and cell therapy as an alternative to conventional bone marrow transplantation with lethal cytoreduction for the treatment of malignant and nonmalignant hematologic diseases. Blood. 1998;91:756–63. [PubMed] [Google Scholar]
- 2.Koc NO, Gerson LS, Cooper WB, Dyhouse MS, Haynesworth ES, Caplan IA, Lazarus MH. Rapid Hematopoietic Recovery After Coinfusion of Autologous-Blood Stem Cells and Culture-Expanded Marrow Mesenchymal Stem Cells in Advanced Breast. J Clin Oncol. 2016;18:307–16. doi: 10.1200/JCO.2000.18.2.307. [DOI] [PubMed] [Google Scholar]
- 3.Maus MV, Grupp Sa, Porter DL, June CH. Antibody modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123:2625–35. doi: 10.1182/blood-2013-11-492231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19:747–52. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EGLR. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:2338–44. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 6.Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg GSD. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4:201–6. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 7.Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, et al. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92–6. doi: 10.1038/nature07434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doherty WP, Bushberg TJ, Lipton JM, Meares FC, Goodwin A. The use of indium-111-labeled leukocytes for abscess detection. Clin Nucl Med. 1978;3:108–10. doi: 10.1097/00003072-197803000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Sharpe M, Mount N. Genetically modified T cells in cancer therapy : opportunities and challenges. Dis Model Mech. 2015;8:337–50. doi: 10.1242/dmm.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griessinger CM, Kehlbach R, Bukala D, Wiehr S, Bantleon R, Cay F, et al. In Vivo Tracking of Th1 Cells by PET Reveals Quantitative and Temporal Distribution and Specific Homing in Lymphatic Tissue. J Nucl Med. 2014;2:301–7. doi: 10.2967/jnumed.113.126318. [DOI] [PubMed] [Google Scholar]
- 11.Yaghoubi SS, Jensen MC, Satyamurthy N, Paik D, Czernin J, Gambhir SS. NonInvasive Detection Of Therapeutic Cytolytic T Cells With PET in a Glioma Patient. Nat Rev Clin Oncol. 2012;6:53–8. doi: 10.1038/ncponc1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez-porcel M, Kronenberg MW, Henry TD, Jay H, Pepine CJ, Ellis SG, et al. Cell tracking and the development of cell-based therapies: a view from the Cadiovascular Cell Therapty Research Network. JACC Cardiovasc Imaging. 2012;5:559–65. doi: 10.1016/j.jcmg.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kircher MF, Gambhir SS, Grimm J. Noninvasive cell-tracking methods. Nat Rev Clin Oncol. 2011;8:677–88. doi: 10.1038/nrclinonc.2011.141. [DOI] [PubMed] [Google Scholar]
- 14.Adonai NN, Nguyen KN, Walsh J, Iyer M, Toyokuni T, Phelps ME, et al. Ex vivo cell labeling with 64Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography. Proc Natl Acad Sci. 2002;99:3030–5. doi: 10.1073/pnas.052709599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hofmann M, Wollert KC, Meyer GP, Menke A, Arseniev L, Hertenstein B, et al. Monitoring of bone marrow cell homing into the infarcted human myocardium. Circulation. 2005;111:2198–202. doi: 10.1161/01.CIR.0000163546.27639.AA. [DOI] [PubMed] [Google Scholar]
- 16.Gambhir SS, Bauer E, Black ME, Liang Q, Kokoris MS, Barrio JR, et al. A mutant herpes simplex virus type 1 thymidine kinase reporter gene shows improved sensitivity for imaging reporter gene expression with positron emission tomography. Proc Natl Acad Sci. 2000;97:2785–90. doi: 10.1073/pnas.97.6.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang JH, Lee DS, Paeng JC, Lee JS, Kim YH, Lee YJ, et al. Development of a sodium/iodide symporter (NIS)-transgenic mouse for imaging of cardiomyocyte-specific reporter gene expression. J Nucl Med. 2005;46:479–83. [PubMed] [Google Scholar]
- 18.Moroz MA, Serganova I, Zanzonico P, Ageyeva L, Beresten T, Dyomina E, et al. Imaging hNET reporter gene expression with 124I-MIBG. J Nucl Med. 2007;48:827–36. doi: 10.2967/jnumed.106.037812. [DOI] [PubMed] [Google Scholar]
- 19.Ma B, Hankenson KD, Dennis JE, Caplan AI, Goldstein SA, Kilbourn MR. A simple method for stem cell labeling with fluorine 18. Nucl Med Biol. 2005;32:701–5. doi: 10.1016/j.nucmedbio.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 20.Lee KS, Kim TJ, Pratx G. Single-cell tracking with PET using a novel trajectory reconstruction algorithm. IEEE Trans Med Imaging. 2015;34:994–1003. doi: 10.1109/TMI.2014.2373351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langford ST, Wiggins CS, Santos R, Hauser M, Becker JM, Ruggles AE. Three-dimensional spatiotemporal tracking of fluorine-18 radiolabeled yeast cells via positron emission particle tracking. PLoS One. 2017;12:7. doi: 10.1371/journal.pone.0180503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiggins C, Santos R, Ruggles A. A novel clustering approach to positron emission particle tracking. Nucl Instruments Methods Phys Res Sect A. 2016;811:18–24. [Google Scholar]
- 23.Parker DJ, Broadbent CJ, Fowles P, Hawkesworth MR, McNeil P. Positron emission particle tracking - a technique for studying flow within engineering equipment. Nucl Inst Methods Phys Res A. 1993;326:592–607. [Google Scholar]
- 24.Bemrose CR, Fowles P, Hawkesworth MR, O’Dwyer MA. Application of positron emission tomography to particulate flow measurement in chemical engineering processes. Nucl Instruments Methods Phys Res A. 1988;273:874–80. [Google Scholar]
- 25.Patel N, Wiggins C, Ruggles A. Positron emission particle tracking in pulsatile flow. Exp Fluids. 2017;58:42. [Google Scholar]
- 26.Langford ST, Wiggins CS, Santos R, Hauser M, Becker JM, Ruggles AE. Three-dimensional spatiotemporal tracking of fluorine-18 radiolabeled yeast cells via positron emission particle tracking. PLoS One. 2017;12:7. doi: 10.1371/journal.pone.0180503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ouyang Y, Kim TJ, Pratx G. Evaluation of a BGO-Based PET System for Single-Cell Tracking Performance by Simulation and Phantom Studies. Mol Imaging. 2016;15:1–8. doi: 10.1177/1536012116646489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, DaSilva JN, Hadizad T, Thorn S, Kuraitis D, Renaud JM, et al. F-18-FDG Cell Labeling May Underestimate Transplanted Cell Homing: More Accurate, Efficient, and Stable Cell Labeling With Hexadecyl-4-[F-18]Fluorobenzoate for In Vivo Tracking of Transplanted Human Progenitor Cells by Positron Emission Tomography. Cell Transplant. 2012;21:1821–35. doi: 10.3727/096368911X637416. [DOI] [PubMed] [Google Scholar]
- 29.Pratx G, Chen K, Sun C, Martin L, Carpenter CM, Olcott PD, et al. Radioluminescence Microscopy: Measuring the Heterogeneous Uptake of Radiotracers in Single Living Cells. PLoS One. 2012;7:1–9. doi: 10.1371/journal.pone.0046285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pratx G, Chen K, Sun C, Axente M, Sasportas L, Carpenter C, et al. High-resolution radioluminescence microscopy of 18F-FDG uptake by reconstructing the β-ionization track. J Nucl Med. 2013;54:1841–6. doi: 10.2967/jnumed.112.113365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim TJ, Türkcan S, Pratx G. Modular low-light microscope for imaging cellular bioluminescence and radioluminescence. Nat Protoc. 2017;12:1055–76. doi: 10.1038/nprot.2017.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu M, Kwon HY, Rattis F, Blum J, Zhao C, Ashkenazi R, et al. Imaging Hematopoietic Precursor Division in Real Time. Cell Stem Cell. 2017;1:541–54. doi: 10.1016/j.stem.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–7. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ravin R, Hoeppner DJ, Munno DM, Carmel L, Sullivan J, Levitt DL, et al. Potency and Fate Specification in CNS Stem Cell Populations In Vitro. Cell Stem Cell. 2008;3:670–80. doi: 10.1016/j.stem.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 35.Naumova AV, Modo M, Moore A, Murry CE, Frank JA. Clinical imaging in regenerative medicine. Nat Biotechnol. 2014;32:804–18. doi: 10.1038/nbt.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmadi A, Thorn SL, Alarcon EI, Kordos M, Padavan DT, Hadizad T, et al. PET imaging of a collagen matrix reveals its effective injection and targeted retention in a mouse model of myocardial infarction. Biomaterials. 2015;49:18–26. doi: 10.1016/j.biomaterials.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 37.Kim MH, Woo SK, Lee KC, An G, II, Pandya D, Park NW, et al. Longitudinal monitoring adipose-derived stem cell survival by PET imaging hexadecyl-4-124I-iodobenzoate in rat myocardial infarction model. Biochem Biophys Res Commun. 2015;456:13–9. doi: 10.1016/j.bbrc.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Kim MH, Woo S-K, Kim K, II, Lee TS, Kim CW, Kang JH, et al. Simple Methods for Tracking Stem Cells with 64 Cu-Labeled DOTA-hexadecyl-benzoate. ACS Med Chem Lett. 2015;6:528–30. doi: 10.1021/acsmedchemlett.5b00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Natarajan A, Türkcan S, Gambhir SS, Pratx G. Multiscale Framework for Imaging Radiolabeled Therapeutics. Mol Pharm. 2015;12:4554–60. doi: 10.1021/acs.molpharmaceut.5b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim TJ, Tuerkcan S, Ceballos A, Pratx G. Modular platform for low-light microscopy. Biomed Opt Express. 2015;6:4585–98. doi: 10.1364/BOE.6.004585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sengupta D, Pratx G. Single-cell characterization of FLT uptake with radioluminescence microscopy. J Nucl Med. 2016;2764:1136–41. doi: 10.2967/jnumed.115.167734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim MH, Lee KC, An G, II, Woo S-K, Park NW, Kim B, II, et al. Evaluation of safety and efficacy of adipose-derived stem cells in rat myocardial infarction model using hexadecyl-4-[(124)I]iodobenzoate for cell tracking. Appl Radiat Isot. 2015;108:116–23. doi: 10.1016/j.apradiso.2015.12.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.