

Abstract
The past two decades have witnessed remarkable advances in oxidative stress research, particularly in the context of ischemic brain injury. Oxidative stress in ischemic tissues compromises the integrity of the genome, resulting in DNA lesions, cell death in neurons, glial cells, and vascular cells, and impairments in neurological recovery after stroke. As DNA is particularly vulnerable to oxidative attack, cells have evolved the ability to induce multiple DNA repair mechanisms, including base excision repair (BER), nucleotide excision repair (NER) and non-homogenous endpoint jointing (NHEJ). Defective DNA repair is tightly correlated with worse neurological outcomes after stroke, whereas upregulation of DNA repair enzymes, such as APE1, OGG1, and XRCC1, improves long-term functional recovery following stroke. Indeed, DNA damage and repair are now known to play critical roles in fundamental aspects of stroke recovery, such as neurogenesis, white matter recovery, and neurovascular unit remodeling. Several DNA repair enzymes are essential for comprehensive neural repair mechanisms after stroke, including Polβ and NEIL3 for neurogenesis, APE1 for white matter repair, Gadd45b for axonal regeneration, and DNA-PKs for neurovascular remodeling. This review discusses the emerging role of DNA damage and repair in functional recovery after stroke and highlights the contribution of DNA repair to regenerative elements after stroke.
Keywords: Cerebral ischemia, Oxidative stress, DNA damage, DNA repair, Stroke recovery, Neurovascular remodeling, White matter repair, Axonal regeneration, Neurogenesis
1. Introduction
Cerebral ischemic stroke is a leading cause of long-term disability and mortality, posing an enormous burden on patients and their communities (Feigin et al., 2014). According to the Centers for Disease Control and Prevention, stroke kills approximately 140,000 Americans each year (Yang et al., 2017), costing the United States an estimated $34 billion per annum (Benjamin et al., 2017). Intravenous tissue plasminogen activator and endovascular thrombectomy are the only currently available stroke treatments approved by the FDA. However, both treatments are limited by a narrow therapeutic window and must be administered within 4.5 and 12 h post-stroke, respectively (Gori et al., 2017). These limitations leave the vast majority of stroke patients inadequately treated and at risk for severe neurological deficits. Therefore, enhancing functional recovery after ischemic stroke remains an important priority for stroke researchers and clinicians (Etherton et al., 2017).
Oxidative stress is a hallmark of cerebral ischemic stroke. It is induced by elevated production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which cause damage to all components of the cell, including proteins, lipids, and DNA (Zhao et al., 2016). Oxidative DNA damage is one of the most detrimental consequences of increased oxidative stress in cerebral ischemic stroke (Cui et al., 2000; Li et al., 2011). When left unrepaired, DNA damage triggers multiple pro-death signaling pathways that induce cell apoptosis and jeopardize functional recovery following stroke (Li et al., 2011). However, cells also combat the accumulation of DNA damage by induction of endogenous DNA repair mechanisms, such as base excision repair (BER), nucleotide excision repair (NER) and non-homologous endpoint jointing (NHEJ). Oxidative DNA damage and repair transpire within minutes after cerebral ischemic stroke (Cui et al., 2000; Li et al., 2011), and may also persist even to six months after stroke (Pascotini et al., 2015). Recent studies highlight the important role of DNA repair as a critical element of the endogenous brain repair process during stroke recovery (Li et al., 2006; Liu et al., 2011). DNA repair has a profound impact on a wide range of recovery efforts, including neurogenesis (Jalland et al., 2016; Shimada et al., 2015), angiogenesis (Liu et al., 2015b), axonal outgrowth (Liu et al., 2015a), and remyelination (Stetler et al., 2016), all of which work in concert to orchestrate neurological recovery. Targeting DNA damage and endogenous DNA repair mechanisms after stroke holds promise for accelerating brain tissue repair and functional recovery. In this review, we provide an update on new insights into DNA damage and repair in the context of ischemic stroke and discuss novel targets that might be therapeutically modulated to improve functional recovery.
2. Stroke elicits DNA damage in multiple neuronal cell types
Cerebral ischemia leads to a robust increase in oxidative stress followed by DNA damage and ischemic injury in gray and white matter (Basso and Ratan, 2013). Oxidative DNA damage occurs soon after ischemic stroke (Chen et al., 1997; Cui et al., 2000; Lan et al., 2003). Oxidative DNA damage is usually reversible. This feature is particularly important because it opens an avenue for therapeutic intervention. Therefore, oxidative DNA damage and repair have become primary foci of interest in stroke research.
Oxidative DNA damage in the ischemic brain has been most commonly studied in neuronal cells. Seminal studies in this area suggest that cerebral ischemia induces formation of apurinic/apyrimidinic (AP) sites, base modifications (e.g., 8-oxo-dG, 8-OHdG, and 8-oxo-Gua), single-strand breaks (SSBs), and double-strand breaks (DSBs) in neurons, all of which may trigger pro-death signaling pathways and hasten neuronal demise (Fig. 1). Hsieh et al. demonstrated that urine 8-OHdG levels correlate negatively with improvements in functional outcomes after stroke and are significantly decreased after rehabilitation (Hsieh et al., 2014). These observations suggest that increased urine 8-OHdG levels may serve as a biomarker for poor functional recovery after stroke, whereas reduced 8-OHdG levels after therapeutic interventions may be associated with better prognosis, although more work needs to be done to confirm this. Furthermore, a recent postmortem study revealed more DNA breaks in cortical samples from stroke victims compared to control subjects (Huttner et al., 2014), suggesting that oxidative DNA damage is indeed increased after stroke in the human brain. Taken together, these observations support the notion that DNA damage evolution-profiles, such as 8-OHdG formation, may serve as a biomarker for estimating stroke recovery and that DNA repair may be an attractive therapeutic target in stroke.
Fig. 1. Ischemic stroke elicits DNA damage in neurons.
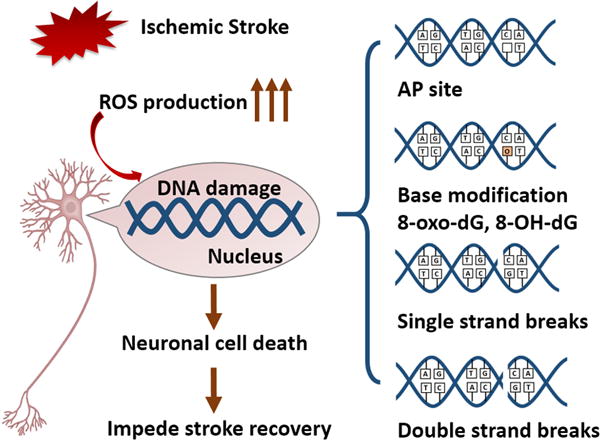
Cerebral ischemic stroke promotes robust ROS production, which can attack and damage DNA. There are four common types of DNA damage: 1) AP sites, 2) base modifications, such as 8-oxo-dG and 8-OHdG formation, 3) single-strand breaks, and 4) double-strand breaks. Each of these forms of DNA damage may contribute to neuronal cell death and impede stroke recovery. ROS, reactive oxygen species; AP site, apurinic/apyridinic abasic site.
In addition to neurons, glial cells, including microglia, astrocytes and oligodendrocytes, are also susceptible to ischemic injury, although glial DNA damage is less well studied than neuronal DNA damage. Microglia are dynamic cells that are exquisitely responsive to an ischemic challenge and produce ROS when activated by injury. After ischemic injury, microglia exhibit oxidative DNA damage (Li et al., 2011), including 8-OHdG formation (Matsuda et al., 2009). Mouse and human brain microglia, but not neurons or astrocytes, express voltage-gated proton channel Hv1, which is required for NADPH oxidase-dependent ROS generation (Wu et al., 2012). Proton currents have been shown to play important auxiliary roles in the function of NADPH oxidases (Seredenina et al., 2015). Proton channels transport excess protons to mitigate acidification and depolarization of cells (DeCoursey et al., 2003; El Chemaly et al., 2010). NADPH oxidase produces more superoxide when proton channel function is preserved because its function is inhibited by depolarization (DeCoursey et al., 2003; El Chemaly et al., 2010). Proton channels are thought to facilitate ROS production through this mechanism. Recent studies suggest that proton channels in microglia from newborn animals might decrease oxidative stress whereas proton channels in microglia of aged animals increase oxidative stress (Kawai et al., 2017). Thus, knockout of voltage-gated proton channel VSOP/Hv1 in old but not young mice reduces infarct sizes (Kawai et al., 2017). Microglia in aged animals might increase oxidative stress through this mechanism, thereby promoting DNA damage. It is worth mentioning that microglia display a dual-faced role in the context of ischemic stroke, with pro-inflammatory phenotypes jeopardizing neuronal survival and a reparatory phenotype supporting neuronal survival (Hu et al., 2012; Liu et al., 2016; Zhao et al., 2015). Modulating the microglial phenotypic switch may facilitate stroke recovery (Hu et al., 2012). However, it is not known whether DNA damage in microglia contributes to their phenotype shifts after stroke. Further studies in this area are warranted, as they may yield novel strategies to improve stroke recovery and advance our understanding of microglial responses to oxidative DNA damage.
Aside from microglia, astrocytes are also vulnerable to oxidative DNA damage. Astrocyte reactivity induced by ionizing radiation is accompanied by DNA damage and increased DNA repair activity compared to non-reactive astrocytes (Yong et al., 2014). Cerebral ischemia is also known to induce astrocyte activation, and this reactivity may contribute to DNA lesions in ischemic astrocytes. Activated astrocytes in infarcted tissue display oxidative DNA damage such as base modifications and single and double-strand breaks three days after stroke (Chen et al., 1997; Matsuda et al., 2009). Astrocyte activation has a profound impact on post-stroke glial scar formation and remodeling of the neurovascular unit, a structural and functional unit composed of neurons, pericytes, endothelial cells forming the blood-brain barrier, and multiple types of glial cells, including astrocytes and the perivascular end feet contacts they make with neighboring endothelial cells. Recent studies have revealed a critical role of the neurovascular unit in the repair of the ischemic brain (Nahirney et al., 2016; Yanev et al., 2017). Given the integral role of astrocytes in the neurovascular unit, repair of DNA damage in astrocytes is likely to facilitate restoration of the neurovascular unit and improve functional recovery after ischemic stroke.
Oligodendrocytic glial cells are poorly understood compared to neuronal cells and other glial cells. As oligodendrocytes are critical for myelination, DNA damage in oligodendrocytes can elicit demyelination and axonal injury in multiple sclerosis (Madsen et al., 2017). The vital importance of DNA damage and repair in oligodendrocytes has been noted in recent studies of multiple sclerosis, major depressive disorder, and ischemic stroke (Madsen et al., 2017; Stetler et al., 2016; Szebeni et al., 2017).
Oxidative DNA damage also surfaces in vascular endothelial cells. Experiments employing double staining for DNA polymerase I-mediated biotin-dATP nick-translation and a vascular cell marker confirm that endothelial cells in small blood vessel walls exhibit single-strand breaks within 16–72 h after reperfusion onset (Chen et al., 1997). As DNA damage in vascular endothelial cells is associated with cognitive impairments (Jadavji et al., 2015) and the integrity of the brain endothelial lining influences edema and peripheral cell infiltration, oxidative DNA damage in vascular endothelial cells after cerebral ischemia may contribute to deterioration of neurological outcomes following stroke.
Considered together, the evidence outlined above strongly suggests that ischemic stroke elicits oxidative DNA damage in multiple cell types of the brain, including neurons, glial cells, and vascular cells, which may synergistically impair neurological function after stroke. Thus, halting oxidative DNA damage in these cell types and rebuilding neurovascular units in both gray and white matter may be critical for successful stroke therapy. In addition, further studies are warranted to identify CSF or plasma biomarkers of DNA damage and/or repair that correlate strongly with neurological outcomes after brain injury and that could be used to predict the degree of functional recovery.
3. Signaling cascades underlying DNA damage after stroke
In order to develop new therapies targeting DNA damage after stroke, one course of action centers on elucidation of signaling path-ways that initiate DNA damage and its pro-death effects. Below we discuss four major cellular events related to DNA damage: 1) Phosphoinositide 3-kinase (PI3K)-related kinase (PIKK)-mediated DNA damage recognition, 2) poly (ADP-ribose) (PAR) polymerase-1 (PARP-1)-mediated AIF translocation, 3) MIF nucleus translocation, and 4) matrix metalloproteinase (MMP)-mediated repair enzyme degradation (Fig. 2).
Fig. 2. DNA damage-induced cell death signaling.
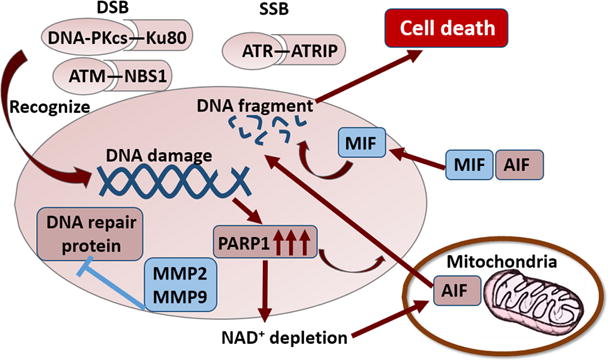
PIKKs recognize DNA damage in response to ischemia. DNA-PKcs and ATM are recruited to DSBs after binding Ku80 and NBS1, respectively. ATR is recruited to SSBs after binding ATRIP. DNA dam-age causes PARP1 overactivation, thereby leading to NAD+ depletion and mitochondrial dysfunction. PARP1 overactivation also promotes AIF translocation from the mitochondrion to the nucleus, resulting in DNA fragmentation. MIF can be recruited into the nucleus in the presence of AIF and functions as a nuclease to cleave genomic DNA. MMP2 and MMP9 can degrade the DNA repair proteins in the nucleus and further promote DNA damage. PIKK, phosphoinositide 3-kinase (PI3K)-related kinase; DSB, double-strand breaks; SSB, single-strand breaks; MIF, migration inhibitory factor; AIF, apoptosis inducing factor; PARP1, poly (ADP-ribose) (PAR) polymerase-1; MMP, matrix metallopeptidase; NAD, nicotinamide adeninedinucleotide; ATM, ataxia-telangiectasia mutated; ATR, ataxia-telangiectasia and Rad-3-related; MMP, matrix metalloproteinase.
Shortly after injury, DNA lesions elicit DNA damage responses, in which three phosphoinositide 3-kinase (PI3K)-related kinases (PIKKs) drive activation of DNA damage cascades via protein phosphorylation. A variety of downstream targets can be phosphorylated by PIKKs, including c-Abl, p53, checkpoint kinase Chk1, Chk2 and H2AX (An et al., 2010; Chou et al., 2015; Moumen et al., 2013; Smits et al., 2006; Wang et al., 2011). DNA-dependent protein kinase, catalytic subunit (DNA-PKcs), ATM, and ATR are the three PIKKs recruited to DNA damage sites (Blackford and Jackson, 2017). The recruitment of these kinases requires distinct factors—Ku80 for DNA-PKcs (Gell and Jackson, 1999; Zou and Elledge, 2003), NBS1 for ATM (Falck et al., 2005), and ATRIP for ATR. DNA-PKcs respond primarily to DSBs, and DSBs directly increase ATM kinase activity. SSBs and stalled DNA replication forks activate ATR (Blackford and Jackson, 2017).
Second, cerebral ischemia-induced DNA damage triggers overactivation of poly (ADP-ribose) (PAR) polymerase-1 (PARP-1), followed by NAD+ depletion (Vosler et al., 2009). This leads to depletion of energy stores and impairments in mitochondrial function. Apoptosis-inducing factor (AIF) is a mitochondrial protein that elicits caspase-independent apoptotic cell death following its translocation to the nucleus after cerebral ischemia (Cao et al., 2003, 2007; Plesnila et al., 2004). Translocation of AIF from the mitochondrion to the nucleus is a critical component of the PARP-1 pro-death process (Yu et al., 2006). Nuclear translocation of AIF during ischemia-induced apoptosis of cerebral endothelial cells is also PARP-1 dependent (Zhang et al., 2005). Therefore, inhibiting PARP-1 activation is a promising strategy to protect against ischemia/reperfusion-induced and oxidative DNA damage-mediated cell apoptosis. Peroxiredoxin 2 is a member of the peroxide-controlling peroxiredoxin family and is expressed in neurons (Hattori and Oikawa, 2007). Peroxiredoxin 2 has been shown to ameliorate PARP-1 activation and attenuate DNA damage–mediated pro-death signaling after cerebral ischemia (Leak et al., 2013).
Third, inflammation is linked to oxidative DNA damage-induced cell death by the PARP1-dependent AIF-associated nuclease (PAAN) function of macrophage migration inhibitory factor (MIF) (Wang et al., 2016a). MIF is an inflammatory cytokine that can be released from white blood cells into the bloodstream in response to bacterial antigens. Circulating MIF can bind to CD74 on other immune cells and trigger acute immune responses (Leng et al., 2003). Recent studies show that MIF can be recruited into neuronal nuclei in the presence of AIF and cleave genomic DNA into large fragments, thereby resulting in chromatinolysis and cell death (Wang et al., 2016a). These novel findings have reshaped our understanding of the interaction between inflammation and DNA damage-induced cell death.
Finally, matrix metalloproteinase (MMP), an important mediator of blood-brain barrier disruption, may trigger oxidative DNA damage-induced neuronal cell death. Cerebral ischemia can induce abnormal expression of MMP2 (gelatinase A) and MMP9 (gelatinase B), which contributes to infarct expansion and blood-brain barrier breakdown (Fujimura et al., 1999; Planas et al., 2001; Rosenberg et al., 1996). A recent study demonstrates that MMP2 and MMP9 not only function as gelatinases that destroy the integrity of the blood-brain barrier, but also function as nucleases that enter the neuronal nucleus after stroke and degrade nuclear DNA repair proteins, such as XRCC1 and OGG1, thereby causing the accumulation of oxidative DNA damage in neurons after ischemic stroke (Hill et al., 2012). These findings support the existence of an intrinsic DNA damage-induced cell death pathway in neurons after stroke.
4. Endogenous DNA repair mechanisms after ischemic stroke
In order to reverse oxidative DNA damage and prevent subsequent cell death, cells possess multiple DNA repair capabilities. Decades of intense research efforts have greatly improved our understanding of DNA repair and identified three major DNA repair mechanisms: 1) direct reversal of DNA damage, 2) non-homologous endpoint jointing (NHEJ) and 3) DNA excision repair (Li et al., 2011). Direct reversal DNA repair includes three major mechanisms: 1)photolyases that reverse UV light-induced photo-lesions, 2) O6-alkylguanine-DNA alkyltransferases (AGTs) that reverse O-alkylated DNA damage, and (iii) the AlkB family dioxygenases, which reverse N-alkylated base adducts (Yi and He, 2013). Direct reversal DNA repair has not been widely studied as part of the DNA repair response after stroke. The NHEJ mechanism repairs double-stranded breaks, which constitute a limited amount of the active DNA damage that unfolds after ischemic injury, but is devastating if it does transpire. The progression of the NHEJ pathway relies on a number of proteins, such as Ku70, Ku80, DNA ligase IV, and X-ray cross-complementing group 4 (XRCC4). DNA excision repair is subdivided into two major pathways: base excision repair (BER), the primary repair pathway to fix oxidative DNA damage, and nucleotide excision repair (NER), the primary pathway for removal of bulky adducts after exposure to UV radiation or other carcinogens. NER may also be important in repairing oxidative damage because it may modulate the formation of 8-oxo-dG and thymine glycol (Kuraoka et al., 2000; Melis et al., 2013; Reardon et al., 1997).
As oxidative DNA damage is a major component of ischemic stroke, BER is the primary DNA repair pathway engaged after ischemia/reperfusion injury. BER is initiated by various DNA glycosylases, which excise the damaged bases and generate an AP site in DNA. The central glycosylases that are engaged following ischemic injury include 8-oxoguanine DNA glycosylase (OGG1), endonuclease III-like 1 (NTH1), endonuclease VIII-like 1 (NEIL1) (Canugovi et al., 2013) and endonuclease VIII-like 3 (NEIL3) (Sejersted et al., 2011). The resulting AP site is then recognized by AP endonuclease 1 (APE1), which cleaves the phosphodiester backbone 5′ to the AP site, leaving a nick with a 3′-OH group and a 5′-deoxyribose phosphate (dRP) residue. Next, the dRP is removed and filled in by “short-patch” repair or “long-patch” repair. Short-patch repair involves sequential single-nucleotide gap-filling steps and repairs regular AP sites with unmodified dRP residues. The gap-filling step is initiated by the enzymes Polβ, which synthesizes a single nucleotide to fill the gap. The nick between the synthesized nucleotide and the DNA template is sealed by DNA ligase III-XRCC1 complex, as shown in Fig. 3. Long-patch repair produces a repair tract of at least two, but most frequently four, nucleotides. It is initiated by flap endonuclease-1 (FEN1) when the AP site or the dRP group are not efficiently removed, to avoid generating a nick that is refractory to the action of a DNA ligase. The major enzymes and proteins involved in BER and other DNA repair processes are depicted in Fig. 3.
Fig. 3. DNA repair mechanisms.
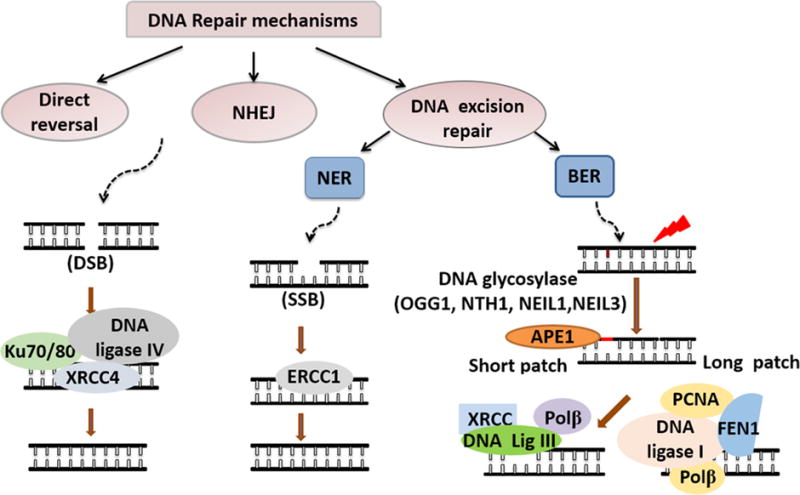
DNA repair mechanisms fall into three categories—direct reversal, NHEJ, and DNA excision repair. NHEJ mainly repairs DSBs and is mediated by Ku70, Ku80, DNA ligase IV, and XRCC4. DNA excision repair is divided into NER and BER. NER is the major repair mechanism for SSBs and is mediated by ERCC1. BER is the primary pathway to repair oxidative DNA damage. The downstream steps of BER, including base recognition, incision, gap filling and ligation, is achieved through two distinct sub-pathways—short patch and long patch BER. The BER pathway is initiated by DNA glycosylases (OGG1, NTH1, NEIL1 and NEIL3) and APE1 then incises DNA to produce AP sites, creating dRP in the process. In short patch BER, the dRP is removed by Polβ and the gap is filled by DNA ligase IV-XRCC complex. In long patch BER, FEN1 cleaves the dRP, Polβ/PCNA then fills the gap, and DNA ligase I finally seals the nick.
After cerebral ischemic stroke, a robust DNA repair response is initiated and upregulation of DNA repair-related proteins can be detected in injured brain tissue. In human chronic ischemic stroke samples, evidence of ATM/ATR activity (i.e., phosphorylation of H2AX to γ-H2AX) and upregulation of APE1 are evident in the penumbra of cortical neurons 7–20 days after ischemic stroke (Huttner et al., 2014). In animal stroke studies, many critical repair enzymes have been linked to functional recovery, including APE1, OGG1, NEIL1, XRCC1, and ERCC1. For example, conditional knockout of APE1 markedly increases post-ischemic neuronal and oligodendrocyte degeneration and therefore aggravates myelin loss and impairs neuronal communication, leaving ischemic stroke animals with dramatically impaired sensorimotor and cognitive functions (Stetler et al., 2016). Loss of glycosylases such as OGG1 and NEIL1, which remove FapyAde, 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua) DNA lesions, exacerbates motor dysfunction in mice after cerebral ischemic stroke (Canugovi et al., 2012; Liu et al., 2011). Partial loss of X-ray repair cross-complementing protein 1 (XRCC1), a nonenzymatic scaffold protein, also impedes recovery after cerebral ischemic stroke (Ghosh et al., 2015). NER contributes to the removal of oxidative DNA lesions and SSBs and is also affected by ischemic stroke (Maddukuri et al., 2007). The expression of ERCC1, the rate-limiting component of NER, increases in the ipsilateral cortex and striatum 3–14 days after middle cerebral artery occlusion. Knocking out ERCC1 expression by injecting antisense plasmids increases DNA damage in neurons and promotes infarct sizes after stroke (He et al., 2009). These findings support the view that coordinated DNA repair is essential to complete ischemic recovery.
In addition to neuronal cells, DNA repair enzymes in non-neuronal cells are also crucial for functional recovery after stroke. Deficiency in a vascular endothelial DNA repair enzyme known as uracil-DNA glycosylase (UNG) is associated with vascular cognitive impairment (Jadavji et al., 2015). Astrocytes in the ischemic penumbra display an increase in ring finger protein 146 (Iduna), which facilitates DNA repair and promotes neurological protection after ischemic stroke (Belayev et al., 2017).
All of the above evidence suggests that DNA repair plays an important role in neurological recovery after stroke. Multiple DNA repair proteins in neurons as well as non-neuronal cells have been identified in the ischemic brain. Loss of their expression impairs DNA repair capacity and is typically associated with deterioration of neurological function after stroke.
5. Enhancing DNA repair capacity promotes functional recovery after ischemic brain injury
If cells display extensive DNA damage, simply inhibiting the pro-death pathways downstream of the DNA damage response is not likely to be an effective treatment strategy. Given the critical importance of genome integrity to survival, DNA itself must be repaired to achieve true and long-lasting protection. Thus, enhancing DNA repair capacity may be key to treating ischemic injury. For example, genetic or pharmacological upregulation of APE1 facilitates the repair of oxidative AP sites and improves stroke recovery as expected (Leak et al., 2015; Yang et al., 2016). Upregulation of APE1 elicits both structural and functional protection of hippocampal neurons after transient global cerebral ischemia, partly by preserving dendritic spine morphology and field excitatory postsynaptic potentials in hippocampal CA1 neurons and leading to superior functional outcomes (Leak et al., 2015). Enhancing APE1 expression by administration of exentin-4, an analogue of glucagon-like peptide-1 (GLP1), also enhances efficient DNA repair in the rat brain after ischemic stroke (Yang et al., 2016).
Ischemic preconditioning is a multimodal neuroprotective paradigm naturally engaged in response to sublethal damage and is associated with markedly increased BER activity. A variety of DNA repair enzymes that function at virtually every level of major DNA repair pathways are upregulated after ischemic preconditioning, including Polβ, APE, and OGG1, XRCC1, and DNA ligase III. Increased expression of these enzymes by ischemic preconditioning dramatically decreases DNA damage in the ischemic brain and improves stroke recovery (Canugovi et al., 2013; Lan et al., 2003; Li et al., 2006).
Several issues require special attention while studying DNA repair for the intention of therapeutic targeting. First, after severe ischemic injury, the protein levels of DNA repair enzymes may be reduced due to impaired protein translation in injured neurons, so that irreparably damaged cells are removed from the tissue. For example, APE1 expression is significantly reduced prior to the onset of neurodegeneration in severely damaged brain regions after cerebral ischemia (Kawase et al., 1999; Lan et al., 2003). Severe cerebral ischemic injury also decreases the expression of Ku, thus inhibiting NHEJ repair (Li et al., 2011). Second, DNA repair enzymes may lead to formation of detrimental byproducts. DNA glycosylases are crucial DNA repair enzymes that initiate the DNA BER process. The 3-alkyladenine DNA glycosylase (Aag) or N-methylpurine DNA glycosylase (Mpg) removes damaged DNA bases and generates toxic abasic sites, which may in turn promote lethal DNA strand breaks (Ebrahimkhani et al., 2014). In contrast to the protection afforded by upregulation of several key repair enzymes, deficiency of Aag/Mpg protects against cerebral ischemic injury (Ebrahimkhani et al., 2014). Third, some of the DNA repair enzymes, including OGG1 and XRCC1, have been shown to be highly polymorphic, which may have an impact on the risk of developing ischemic brain injury (Orhan et al., 2016).
Overall, we must exercise caution with regard to potential conflicting effects of BER in cerebral ischemic recovery. Polymorphism of DNA repair enzyme genes may influence the efficacy of therapeutic strategies that target DNA repair after ischemic stroke. For example, the G-C-T haplotype of the human APE1 gene is more common in patients with a history of stroke than control subjects (Naganuma et al., 2009). Further studies are warranted to improve our comprehension of the nuances of DNA repair in the context of ischemic brain injury and to account for the heterogeneity of the global human population.
6. Promoting endogenous regenerative responses—the role of DNA damage and repair
DNA damage and repair are usually considered early events following cerebral ischemia and reperfusion. The contribution of oxidative stress and DNA damage to long-term stroke outcomes in the late phase of stroke (>6 months) is unclear. In the peripheral blood in stroke patients, markers of oxidative DNA damage and apoptosis are evident even six months after stroke (Pascotini et al., 2015). In the late phase of ischemic stroke, pleiotropic endogenous brain repair mechanisms orchestrate stroke recovery, initiating processes such as neurogenesis, axonal outgrowth, white matter repair, and neurovascular unit remodeling (Chen et al., 2010; Cook et al., 2017; Yang et al., 2013; Zhang et al., 2016) (Fig. 4). As these endogenous brain repair processes extend into the late phase of stroke, during which DNA damage remains elevated, targeting DNA repair might provide a novel therapeutic strategy to promote brain plasticity and functional recovery even at long timeframes (Raefsky and Mattson, 2017). This is an important area to research further and is not addressed by short-term preclinical studies testing therapies against acute stroke injury.
Fig. 4. Endogenous brain repair mechanisms.
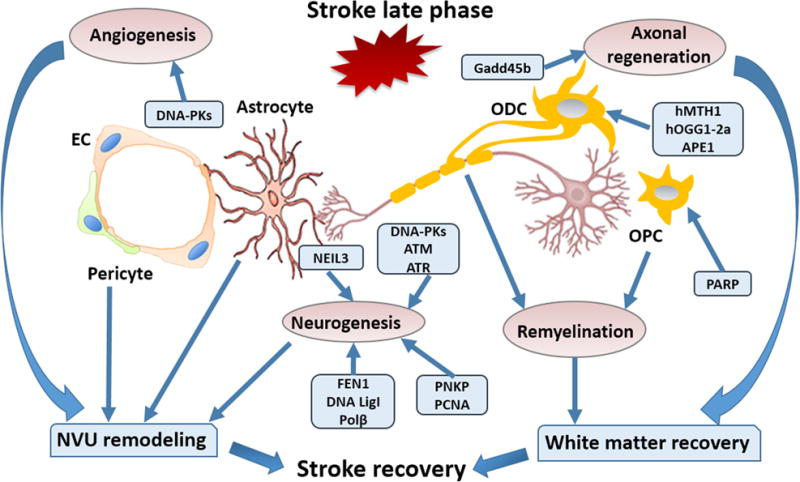
Multiple natural brain repair mechanisms such as neurogenesis, white matter repair, and NVU remodeling orchestrate the stroke recovery process in conjunction with DNA repair proteins. DNA-PKcs, ATM, and ATR are critical for controlling neuronal apoptosis during neurogenesis. BER pathway components such as Polβ, NEIL3, FEN1, PCNA, and DNA Lig1 are critical for neurogenesis. Gadd45b stimulates stroke recovery by enhancing axonal regeneration. DNA repair enzymes such as hMTH1, Hogg1-2a, and APE1 protect white matter after stroke. The repair of the ischemic brain also involves extensive remodeling of the NVU. DNA-PKcs induce angiogenesis and promote NVU remodeling. DNA damage or defective DNA repair in astrocytes and pericytes inhibits NVU remodeling. EC, endothelial cell; NVU, neurovascular unit; Gadd45b, DNA-damage-inducible protein 45 beta; ODC, oligodendrocyte; OPC, oligodendrocyte precursor cell; PNKP, polynucleotide kinase-phosphatase; PCNA, proliferating cell nuclear antigen.
Stroke triggers regenerative responses in neural progenitors to mediate various aspects of tissue repair after neuronal injury. Studies on DNA damage and repair in neural progenitors after stroke are limited. However, developmental studies may guide our understanding of the regeneration of brain tissue following injury. During development, neural progenitors at different stages of differentiation and commitment demonstrate selective sensitivity toward DNA damage, as the early-born cortical progenitors are hypersensitive to replication-associated DNA damage (Lee et al., 2012; McKinnon, 2013). In the developing brain, the DNA damage response plays important roles during neurogenesis (Enriquez-Rios et al., 2017). Three PI3K-like protein kinases, DNA-PKcs, ATM, and ATR are central regulators of DNA damage responses. DNA-PKcs loss may sensitize neuronal progenitors to apoptosis due to excessive damaged DNA. ATM is critical for controlling apoptosis after DNA damage in both proliferative and non-cycling cells, while ATR controls the damage-induced G2/M checkpoint in cortical progenitors independent of ATM and DNA-PKcs (Enriquez-Rios et al., 2017). Thus, multiple DNA damage response elements play critical roles during neurogenesis in the developing brain.
Multiple BER repair enzymes have also been shown to play critical roles during neurogenesis in the developing brain. Targeted depletion of Polβ causes defects in neurogenesis and apoptotic cell death in the developing central and peripheral nervous systems, culminating in lethality at neonatal stages (Sugo et al., 2000). Mice with inactivated polynucleotide kinase-phosphatase (PNKP), a DNA repair factor involved in BER and NHEJ, exhibit general neurodevelopmental defects, indicating an important role for DNA repair in neurogenesis in the developing brain (Shimada et al., 2015).
NEIL3 is a DNA glycosylase in the BER pathway and plays important roles in repairing DNA damage in rapidly proliferating cells. Knockout of NEIL3 incurs profound neuropathy and is associated with fewer microglia and proliferating neuronal progenitors in the striatum after hypoxia-ischemia (Sejersted et al., 2011). Loss of NEIL3 also negatively affects neurogenesis in the subventricular and subgranular zones (Leppo and Meerdink, 1990). NEIL3 deficiency in cultured neural stem/progenitor cells significantly impairs the repair of oxidative DNA base lesions (Sejersted et al., 2011). NEIL3 also promotes neurogenesis and protects against prion disease by reducing oxidative DNA damage (Jalland et al., 2016). Although it is still unknown whether DNA repair can promote neurogenesis after stroke, DNA integrity appears to be crucial for neuron survival in humans; the acute stage of human stroke is characterized by DNA fragmentation and repair in cortical neurons, whereas the chronic stage of stroke is associated with DNA integrity, supporting the clinical importance of genomic integrity for long term viability after stroke (Huttner et al., 2014). One might also speculate that DNA repair promotes the survival of de novo generated neurons after cerebral ischemic stroke. Further studies in this area are warranted.
White matter typically occupies a large fraction of the infarct zone in stroke patients (Wang et al., 2016b). White matter recovery includes remyelination and axonal regeneration and is an important component of the recovery phase after cerebral ischemic stroke (Shindo et al., 2016). Oligodendrocytes are the major cells responsible for generating myelin sheaths to support and insulate axons and are therefore critical for remyelination after ischemic stroke (Wang et al., 2016b). Accumulating evidence suggests that DNA damage and repair can affect the myelination of neural tissue (Iida et al., 2004; O’Sullivan et al., 2017; Stetler et al., 2016). Oxidative stress generated by glucose oxidase and catalase induces demyelination in mouse organotypic cerebellar slice cultures (O’Sullivan et al., 2017). DNA repair enzymes geared toward oxidative DNA damage, such as oxidized purine-nucleoside triphosphatase (hMTH1) and a mitochondrial type of 8-oxoguanine DNA glycosylase (hOGG1-2a) are strongly expressed in oligodendrocytes in the acute and subacute stages of ischemic stroke (Iida et al., 2004). Higher levels of expression of DNA repair enzymes in injured oligodendrocytes may help improve their viability and lead to superior white matter recovery after stroke. Recent studies have demonstrated that endogenous APE1 plays an important role in protecting white matter after stroke (Stetler et al., 2016). In those studies, tamoxifen-induced conditional knockout of APE1 markedly increased the accumulation of oxidative DNA damage and activation of pro-death signaling in oligodendrocytes after cerebral ischemic stroke, exacerbating behavioral outcomes in the long recovery phase (Stetler et al., 2016). Administration of the antioxidant L-carnitine may reduce oxidative DNA damage and increase oligodendrocytic myelination of axons, thereby reducing white matter lesions and cognitive impairments in a rat model of cerebral hypoperfusion (Ueno et al., 2015). DNA damage and repair signaling in oligodendrocyte precursor cells (OPCs) also affects white matter integrity. For example, inhibition of the signaling molecule PARP is cytotoxic and impairs OPC maturation into myelinating oligodendrocytes in the fetal brain (Baldassarro et al., 2017). While this may seem difficult to reconcile with the pro-death role of PARP1 and its destructive effects on DNA, it should be noted that these effects were not observed in OPC cultures from adult brain and that PARP activity was higher in fetal than adult OPCs, perhaps because the brain is being actively sculpted by aggressive developmental processes (Baldassarro et al., 2017).
Axonal regeneration and plasticity play important roles in functional recovery after cerebral ischemic stroke (Chen et al., 2010). The growth arrest and DNA-damage-inducible protein 45 beta (Gadd45b) promotes axonal plasticity and this is associated with improved recovery from stroke (Liu et al., 2015a). Decreasing Gadd45b expression by RNA interference significantly inhibits axonal regeneration as revealed by suppression of the growth cone marker growth-associated protein 43 (GAP-43), and is associated with worse recovery of multiple neurological functions (Liu et al., 2015a). L-carnitine is an isoform of carnitine. It is involved in the oxidation of fatty acids and has been shown to protect against DNA damage (Mescka et al., 2015; Thangasamy et al., 2009). Administration of L-carnitine also enhances axonal plasticity in the rat model of chronic cerebral hypoperfusion alluded to above (Ueno et al., 2015).
In an animal model of spinal cord injury involving axonal regeneration, microglia also display DNA damage (Noristani et al., 2017). However, the mechanism underlying the impact of microglial DNA damage on axonal regeneration in neighboring neurons has not been elucidated.
Reactive astrocytes induced by macrophage-derived osteopontin in the ischemic penumbra extend processes toward the center of the infarct to repair the compromised blood-brain barrier (Gliem et al., 2015). As mentioned above, reactive astrocytes also exhibit greater DNA damage than non-reactive astrocytes (Yong et al., 2014). However, it remains unknown whether reducing DNA damage or enhancing DNA repair in reactive astrocytes can impact astrocyte endfeet coverage of the blood-brain barrier, thereby perhaps promoting neurovascular unit remodeling.
Angiogenesis is an integral part of neurovascular unit remodeling (dela Pena et al., 2015). DNA-PKcs, one of the PIKKs important for repair of DSBs, is critical for the increase in vascular endothelial growth factor (VEGF) and angiogenesis in response to ionizing radiation (Liu et al., 2015b). Ischemic injury induces robust angiogenesis to foster nutritional support for the de novo generation of neuronal networks. Enhancing DNA repair capacity may be beneficial for angiogenesis after ischemic brain injury and the promotion of neurovascular unit remodeling, although further studies are needed to confirm this.
Pericytes cover approximately 80% of brain microvessels (Winkler et al., 2012) and respond dynamically to ischemic stroke and contribute to neurovascular unit remodeling (Attwell et al., 2016; ElAli et al., 2014; Neuhaus et al., 2017). Mitochondria-derived ROS production induced by pepstatin A decreases the viability of isolated brain pericytes (Okada et al., 2015). Oxidative DNA damage in brain pericytes increases both vessel diameter and permeability of the blood-brain barrier (Okada et al., 2015). These findings suggest that DNA damage in pericytes may represent a novel target for blood-brain barrier protection after stroke.
Accumulating evidence suggests that multiple non-neuronal cells, such as pericytes, endothelial cells, astrocytes, microglia, and oligodendrocytes play critical roles in post-stroke brain repair and regenerative responses. However, whether and how DNA damage and repair in these non-neuronal cells affects post-stroke brain recovery is poorly understood.
7. Concluding remarks
Recent studies have shed light on cerebral ischemia-induced DNA damage and its contribution to neuronal cell death and slowing of neurological recovery after stroke. The impact of DNA damage accumulation has now been extended to non-neuronal cells and likely extends beyond facilitation of post-ischemic cell death. Rather, DNA damage profoundly affects the repair and restoration of the neurovascular network of the post-ischemic brain. Although additional research is required to determine if the phenotype switching of non-neuronal cells is influenced by DNA damage and repair, prevailing evidence suggests that DNA repair enzymes play critical roles in comprehensive stroke recovery. Deficiency of key enzymes, such as APE1 and OGG1, hinders functional recovery after stroke, while upregulation of these enzymes facilitates stroke recovery as expected. Neural progenitor cells exhibit greater DNA damage than differentiated cells and thus may require increased DNA repair capacities. Enhanced DNA repair is also likely to be critical for white matter recovery after stroke as it supports the viability of oligodendrocytes and OPCs and promotes axonal sprouting. In addition, angiogenesis and activation of astrocytes and pericytes in the neurovascular unit is associated with higher DNA damage and requires enhanced DNA repair capacity to prevent the accumulation of lesions. Recent and historical achievements in determining the therapeutic potential of DNA repair have improved our understanding of stroke recovery and paved the path for additional novel research questions. However, there are only a few clinical reports of DNA repair status in the brains of stroke victims and all studies of pharmacological BER enhancement have been performed in animals or in cell culture. Thus, there is an urgent, unmet need to develop safe and effective BER-enhancing treatments that repair and preserve the integrity of the genome after ischemic injury and promote long-lasting protection in patient populations.
Acknowledgments
This work was supported by,NIH/NINDS grants NS036736 (to Jun Chen, Rehana K. Leak, and Michael V.L. Bennett), NS095671, NS089534 and NS45048 (to Jun Chen), and NS100803 (to R. Anne Stetler), VA merit grants I01BX002495 and I01RX000420 (to Jun Chen), the U.S. Department of Veterans Affairs Senior Research Career Scientist Award (to Jun Chen); and Shanghai Rising-Star Program 16QA1402600 (to Peiying Li), and Chinese Natural Science Foundation grants 81400956 and 81722017 (to Peiying Li).
References
- An J, Huang YC, Xu QZ, Zhou LJ, Shang ZF, Huang B, Wang Y, Liu XD, Wu DC, Zhou PK. DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol Biol. 2010;11:18. doi: 10.1186/1471-2199-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T. What is a pericyte? J Cereb Blood Flow Metab. 2016;36:451–455. doi: 10.1177/0271678X15610340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldassarro VA, Marchesini A, Giardino L, Calza L. PARP activity and inhibition in fetal and adult oligodendrocyte precursor cells: effect on cell survival and differentiation. Stem Cell Res. 2017;22:54–60. doi: 10.1016/j.scr.2017.05.011. [DOI] [PubMed] [Google Scholar]
- Basso M, Ratan RR. Transglutaminase is a therapeutic target for oxidative stress, excitotoxicity and stroke: a new epigenetic kid on the CNS block. J Cereb Blood Flow Metab. 2013;33:809–818. doi: 10.1038/jcbfm.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Mukherjee PK, Balaszczuk V, Calandria JM, Obenaus A, Khoutorova L, Hong SH, Bazan NG. Neuroprotectin D1 upregulates Iduna expression and provides protection in cellular uncompensated oxidative stress and in experimental ischemic stroke. Cell Death Differ. 2017;24:1091–1099. doi: 10.1038/cdd.2017.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics, C., Stroke Statistics, S Heart disease and stroke Statistics-2017 update: a report from the American heart association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Canugovi C, Misiak M, Ferrarelli LK, Croteau DL, Bohr VA. The role of DNA repair in brain related disease pathology. DNA Repair (Amst) 2013;12:578–587. doi: 10.1016/j.dnarep.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canugovi C, Yoon JS, Feldman NH, Croteau DL, Mattson MP, Bohr VA. Endonuclease VIII-like 1 (NEIL1) promotes short-term spatial memory retention and protects from ischemic stroke-induced brain dysfunction and death in mice. Proc Natl Acad Sci U S A. 2012;109:14948–14953. doi: 10.1073/pnas.1204156109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, Graham SH, Chen J. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003;23:1137–1150. doi: 10.1097/01.WCB.0000087090.01171.E7. [DOI] [PubMed] [Google Scholar]
- Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA, Simon RP. Early detection of DNA strand breaks in the brain after transient focal ischemia: implications for the role of DNA damage in apoptosis and neuronal cell death. J Neurochem. 1997;69:232–245. doi: 10.1046/j.1471-4159.1997.69010232.x. [DOI] [PubMed] [Google Scholar]
- Chen J, Zacharek A, Cui X, Shehadah A, Jiang H, Roberts C, Lu M, Chopp M. Treatment of stroke with a synthetic liver X receptor agonist, TO901317, promotes synaptic plasticity and axonal regeneration in mice. J Cereb Blood Flow Metab. 2010;30:102–109. doi: 10.1038/jcbfm.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WC, Hu LY, Hsiung CN, Shen CY. Initiation of the ATM-Chk2 DNA damage response through the base excision repair pathway. Carcinogenesis. 2015;36:832–840. doi: 10.1093/carcin/bgv079. [DOI] [PubMed] [Google Scholar]
- Cook DJ, Nguyen C, Chun HN, I LL, Chiu AS, Machnicki M, Zarembinski TI, Carmichael ST. Hydrogel-delivered brain-derived neurotrophic factor promotes tissue repair and recovery after stroke. J Cereb Blood Flow Metab. 2017;37:1030–1045. doi: 10.1177/0271678X16649964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Holmes EH, Greene TG, Liu PK. Oxidative DNA damage precedes DNA fragmentation after experimental stroke in rat brain. FASEB J. 2000;14:955–967. doi: 10.1096/fasebj.14.7.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE, Morgan D, Cherny VV. The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature. 2003;422:531–534. doi: 10.1038/nature01523. [DOI] [PubMed] [Google Scholar]
- dela Pena IC, Yoo A, Tajiri N, Acosta SA, Ji X, Kaneko Y, Borlongan CV. Granulocyte colony-stimulating factor attenuates delayed tPA-induced hemorrhagic transformation in ischemic stroke rats by enhancing angiogenesis and vasculogenesis. J Cereb Blood Flow Metab. 2015;35:338–346. doi: 10.1038/jcbfm.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimkhani MR, Daneshmand A, Mazumder A, Allocca M, Calvo JA, Abolhassani N, Jhun I, Muthupalani S, Ayata C, Samson LD. Aagnitiated base excision repair promotes ischemia reperfusion injury in liver, brain, and kidney. Proc Natl Acad Sci U S A. 2014;111:E4878–E4886. doi: 10.1073/pnas.1413582111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Chemaly A, Okochi Y, Sasaki M, Arnaudeau S, Okamura Y, Demaurex N. VSOP/Hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J Exp Med. 2010;207:129–139. doi: 10.1084/jem.20091837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElAli A, Theriault P, Rivest S. The role of pericytes in neurovascular unit remodeling in brain disorders. Int J Mol Sci. 2014;15:6453–6474. doi: 10.3390/ijms15046453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriquez-Rios V, Dumitrache LC, Downing SM, Li Y, Brown EJ, Russell HR, McKinnon PJ. DNA-PKcs, ATM, and ATR interplay maintains genome integrity during neurogenesis. J Neurosci. 2017;37:893–905. doi: 10.1523/JNEUROSCI.4213-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherton MR, Rost NS, Wu O. Infarct topography and functional outcomes. J Cereb Blood Flow Metab. 2017:271678X17700666. doi: 10.1177/0271678X17700666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA, Connor M, Bennett DA, Moran AE, Sacco RL, Anderson L, Truelsen T, O’Donnell M, Venketasubramanian N, Barker-Collo S, Lawes CM, Wang W, Shinohara Y, Witt E, Ezzati M, Naghavi M, Murray C, Global Burden of Diseases, I., Risk Factors, S., the, G. B. D. S. E. G Global and regional burden of stroke during 1990-2010: findings from the global burden of disease study 2010. Lancet. 2014;383:245–254. doi: 10.1016/s0140-6736(13)61953-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- Gell D, Jackson SP. Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Res. 1999;27:3494–3502. doi: 10.1093/nar/27.17.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Canugovi C, Yoon JS, Wilson DM, 3rd, Croteau DL, Mattson MP, Bohr VA. Partial loss of the DNA repair scaffolding protein, Xrcc1, results in increased brain damage and reduced recovery from ischemic stroke in mice. Neurobiol Aging. 2015;36:2319–2330. doi: 10.1016/j.neurobiolaging.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliem M, Krammes K, Liaw L, van Rooijen N, Hartung HP, Jander S. Macrophage-derived osteopontin induces reactive astrocyte polarization and pro motes re-establishment of the blood brain barrier after ischemic stroke. Glia. 2015;63:2198–2207. doi: 10.1002/glia.22885. [DOI] [PubMed] [Google Scholar]
- Gori AM, Giusti B, Piccardi B, Nencini P, Palumbo V, Nesi M, Nucera A, Pracucci G, Tonelli P, Innocenti E, Sereni A, Sticchi E, Toni D, Bovi P, Guidotti M, Tola MR, Consoli D, Micieli G, Tassi R, Orlandi G, Sessa M, Perini F, Delodovici ML, Zedde ML, Massaro F, Abbate R, Inzitari D. Inflammatory and metalloproteinases profiles predict three-month poor outcomes in ischemic stroke treated with thrombolysis. J Cereb Blood Flow Metab. 2017:271678X17695572. doi: 10.1177/0271678X17695572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori F, Oikawa S. Peroxiredoxins in the central nervous system. Subcell Biochem. 2007;44:357–374. doi: 10.1007/978-1-4020-6051-9_17. [DOI] [PubMed] [Google Scholar]
- He KY, Yang SZ, Shen DH, Zhang LM, Lu SD, Sun FY. Excision repair cross-complementing 1 expression protects against ischemic injury following middle cerebral artery occlusion in the rat brain. Gene Ther. 2009;16:840–848. doi: 10.1038/gt.2009.48. [DOI] [PubMed] [Google Scholar]
- Hill JW, Poddar R, Thompson JF, Rosenberg GA, Yang Y. Intranuclear matrix metalloproteinases promote DNA damage and apoptosis induced by oxygen-glucose deprivation in neurons. Neuroscience. 2012;220:277–290. doi: 10.1016/j.neuroscience.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YW, Lin KC, Korivi M, Lee TH, Wu CY, Wu KY. The reliability and predictive ability of a biomarker of oxidative DNA damage on functional outcomes after stroke rehabilitation. Int J Mol Sci. 2014;15:6504–6516. doi: 10.3390/ijms15046504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- Huttner HB, Bergmann O, Salehpour M, Racz A, Tatarishvili J, Lindgren E, Csonka T, Csiba L, Hortobagyi T, Mehes G, Englund E, Solnestam BW, Zdunek S, Scharenberg C, Strom L, Stahl P, Sigurgeirsson B, Dahl A, Schwab S, Possnert G, Bernard S, Kokaia Z, Lindvall O, Lundeberg J, Frisen J. The age and genomic integrity of neurons after cortical stroke in humans. Nat Neurosci. 2014;17:801–803. doi: 10.1038/nn.3706. [DOI] [PubMed] [Google Scholar]
- Iida T, Furuta A, Nakabeppu Y, Iwaki T. Defense mechanism to oxidative DNA damage in glial cells. Neuropathology. 2004;24:125–130. doi: 10.1111/j.1440-1789.2003.00540.x. [DOI] [PubMed] [Google Scholar]
- Jadavji NM, Farr TD, Lips J, Khalil AA, Boehm-Sturm P, Foddis M, Harms C, Fuchtemeier M, Dirnagl U. Elevated levels of plasma homocysteine, deficiencies in dietary folic acid and uracil-DNA glycosylase impair learning in a mouse model of vascular cognitive impairment. Behav Brain Res. 2015;283:215–226. doi: 10.1016/j.bbr.2015.01.040. [DOI] [PubMed] [Google Scholar]
- Jalland CM, Scheffler K, Benestad SL, Moldal T, Ersdal C, Gunnes G, Suganthan R, Bjoras M, Tranulis MA. Neil3 induced neurogenesis protects against prion disease during the clinical phase. Sci Rep. 2016;6:37844. doi: 10.1038/srep37844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Okochi Y, Ozaki T, Imura Y, Koizumi S, Yamazaki M, Abe M, Sakimura K, Yamashita T, Okamura Y. Unconventional role of voltage-gated proton channels (VSOP/Hv1) in regulation of microglial ROS production. J Neurochem. 2017 doi: 10.1111/jnc.14106. [DOI] [PubMed] [Google Scholar]
- Kawase M, Fujimura M, Morita-Fujimura Y, Chan PH. Reduction of apurinic/apyrimidinic endonuclease expression after transient global cerebral ischemia in rats: implication of the failure of DNA repair in neuronal apoptosis. Stroke. 1999;30:441–448. doi: 10.1161/01.str.30.2.441. discussion 449. [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Bender C, Romieu A, Cadet J, Wood RD, Lindahl T. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc Natl Acad Sci U S A. 2000;97:3832–3837. doi: 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J, Li W, Zhang F, Sun FY, Nagayama T, O’Horo C, Chen J. Inducible repair of oxidative DNA lesions in the rat brain after transient focal ischemia and reperfusion. J Cereb Blood Flow Metab. 2003;23:1324–1339. doi: 10.1097/01.WCB.0000091540.60196.F2. [DOI] [PubMed] [Google Scholar]
- Leak RK, Li P, Zhang F, Sulaiman HH, Weng Z, Wang G, Stetler RA, Shi Y, Cao G, Gao Y, Chen J. Apurinic/apyrimidinic endonuclease 1 upregulation reduces oxidative DNA damage and protects hippocampal neurons from ischemic injury. Antioxid Redox Signal. 2015;22:135–148. doi: 10.1089/ars.2013.5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leak RK, Zhang L, Luo Y, Li P, Zhao H, Liu X, Ling F, Jia J, Chen J, Ji X. Peroxiredoxin 2 battles poly(ADP-ribose) polymerase 1- and p53-dependent prodeath pathways after ischemic injury. Stroke. 2013;44:1124–1134. doi: 10.1161/STROKEAHA.111.680157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Katyal S, Downing SM, Zhao J, Russell HR, McKinnon PJ. Neurogenesis requires TopBP1 to prevent catastrophic replicative DNA damage in early progenitors. Nat Neurosci. 2012;15:819–826. doi: 10.1038/nn.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–1476. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppo JA, Meerdink DJ. Comparative myocardial extraction of two technetium-labeled BATO derivatives (SQ30217, SQ32014) and thallium. J Nucl Med. 1990;31:67–74. [PubMed] [Google Scholar]
- Li P, Hu X, Gan Y, Gao Y, Liang W, Chen J. Mechanistic insight into DNA damage and repair in ischemic stroke: exploiting the base excision repair pathway as a model of neuroprotection. Antioxid Redox Signal. 2011;14:1905–1918. doi: 10.1089/ars.2010.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Luo Y, Zhang F, Signore AP, Gobbel GT, Simon RP, Chen J. Ischemic preconditioning in the rat brain enhances the repair of endogenous oxidative DNA damage by activating the base-excision repair pathway. J Cereb Blood Flow Metab. 2006;26:181–198. doi: 10.1038/sj.jcbfm.9600180. [DOI] [PubMed] [Google Scholar]
- Liu B, Li LL, Tan XD, Zhang YH, Jiang Y, He GQ, Chen Q, Li CQ. Gadd45b mediates axonal plasticity and subsequent functional recovery after experimental stroke in rats. Mol Neurobiol. 2015a;52:1245–1256. doi: 10.1007/s12035-014-8909-0. [DOI] [PubMed] [Google Scholar]
- Liu D, Croteau DL, Souza-Pinto N, Pitta M, Tian J, Wu C, Jiang H, Mustafa K, Keijzers G, Bohr VA, Mattson MP. Evidence that OGG1 glycosylase protects neurons against oxidative DNA damage and cell death under ischemic conditions. J Cereb Blood Flow Metab. 2011;31:680–692. doi: 10.1038/jcbfm.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai M, Ji X, Leak RK, Gao Y, Chen J, Hu X. Interleukin-4 is essential for microglia/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke. 2016;47:498–504. doi: 10.1161/STROKEAHA.115.012079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang L, Liu Y, Sun C, Zhang H, Miao G, Di CX, Zhou X, Zhou R, Wang Z. DNA-PKcs deficiency inhibits glioblastoma cell-derived angiogenesis after ionizing radiation. J Cell Physiol. 2015b;230:1094–1103. doi: 10.1002/jcp.24841. [DOI] [PubMed] [Google Scholar]
- Maddukuri L, Dudzinska D, Tudek B. Bacterial DNA repair genes and their eukaryotic homologues: 4. The role of nucleotide excision DNA repair (NER) system in mammalian cells. Acta Biochim Pol. 2007;54:469–482. [PubMed] [Google Scholar]
- Madsen PM, Pinto M, Patel S, McCarthy S, Gao H, Taherian M, Karmally S, Pereira CV, Dvoriantchikova G, Ivanov D, Tanaka KF, Moraes CT, Brambilla R. Mitochondrial DNA double-strand breaks in oligodendrocytes cause demyelination, axonal injury, and CNS inflammation. J Neurosci. 2017;37:10185–10199. doi: 10.1523/JNEUROSCI.1378-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Umeda M, Kato H, Araki T. Glial damage after transient focal cerebral ischemia in rats. J Mol Neurosci. 2009;38:220–226. doi: 10.1007/s12031-008-9165-4. [DOI] [PubMed] [Google Scholar]
- McKinnon PJ. Maintaining genome stability in the nervous system. Nat Neurosci. 2013;16:1523–1529. doi: 10.1038/nn.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis JP, van Steeg H, Luijten M. Oxidative DNA damage and nucleotide excision repair. Antioxid Redox Signal. 2013;18:2409–2419. doi: 10.1089/ars.2012.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mescka CP, Guerreiro G, Hammerschmidt T, Faverzani J, de Moura Coelho D, Mandredini V, Wayhs CA, Wajner M, Dutra-Filho CS, Vargas CR. L-Carnitine supplementation decreases DNA damage in treated MSUD patients. Mutat Res. 2015;775:43–47. doi: 10.1016/j.mrfmmm.2015.03.008. [DOI] [PubMed] [Google Scholar]
- Moumen A, Magill C, Dry KL, Jackson SP. ATM-dependent phosphorylation of heterogeneous nuclear ribonucleoprotein K promotes p53 transcriptional activation in response to DNA damage. Cell Cycle. 2013;12:698–704. doi: 10.4161/cc.23592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma T, Nakayama T, Sato N, Fu Z, Yamaguchi M, Soma M, Aoi N, Usami R, Doba N, Hinohara S. Haplotype-based case-control study between human apurinic/apyrimidinic endonuclease 1/redox effector factor-1 gene and cerebral infarction. Clin Biochem. 2009;42:1493–1499. doi: 10.1016/j.clinbiochem.2009.07.016. [DOI] [PubMed] [Google Scholar]
- Nahirney PC, Reeson P, Brown CE. Ultrastructural analysis of blood-brain barrier breakdown in the peri-infarct zone in young adult and aged mice. J Cereb Blood Flow Metab. 2016;36:413–425. doi: 10.1177/0271678X15608396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus AA, Couch Y, Sutherland BA, Buchan AM. Novel method to study pericyte contractility and responses to ischaemia in vitro using electrical impedance. J Cereb Blood Flow Metab. 2017;37:2013–2024. doi: 10.1177/0271678X16659495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noristani HN, Gerber YN, Sabourin JC, Le Corre M, Lonjon N, Mestre-Frances N, Hirbec HE, Perrin FE. RNA-seq analysis of microglia reveals time-dependent activation of specific genetic programs following spinal cord injury. Front Mol Neurosci. 2017;10:90. doi: 10.3389/fnmol.2017.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan SA, Velasco-Estevez M, Dev KK. Demyelination induced by oxidative stress is regulated by sphingosine 1-phosphate receptors. Glia. 2017;65:1119–1136. doi: 10.1002/glia.23148. [DOI] [PubMed] [Google Scholar]
- Okada R, Wu Z, Zhu A, Ni J, Zhang J, Yoshimine Y, Peters C, Saftig P, Nakanishi H. Cathepsin D deficiency induces oxidative damage in brain pericytes and impairs the blood-brain barrier. Mol Cell Neurosci. 2015;64:51–60. doi: 10.1016/j.mcn.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Orhan G, Elkama A, Mungan SO, Eruyar E, Karahalil B. The impact of detoxifying and repair gene polymorphisms on oxidative stress in ischemic stroke. Neurol Sci. 2016;37:955–961. doi: 10.1007/s10072-016-2524-y. [DOI] [PubMed] [Google Scholar]
- Pascotini ET, Flores AE, Kegler A, Gabbi P, Bochi GV, Algarve TD, Prado AL, Duarte MM, da Cruz IB, Moresco RN, Royes LF, Fighera MR. Apoptotic markers and DNA damage are related to late phase of stroke: involvement of dyslipidemia and inflammation. Physiol Behav. 2015;151:369–378. doi: 10.1016/j.physbeh.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Planas AM, Sole S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8:834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- Plesnila N, Zhu C, Culmsee C, Groger M, Moskowitz MA, Blomgren K. Nuclear translocation of apoptosis-inducing factor after focal cerebral ischemia. J Cereb Blood Flow Metab. 2004;24:458–466. doi: 10.1097/00004647-200404000-00011. [DOI] [PubMed] [Google Scholar]
- Raefsky SM, Mattson MP. Adaptive responses of neuronal mitochondria to bioenergetic challenges: roles in neuroplasticity and disease resistance. Free Radic Biol Med. 2017;102:203–216. doi: 10.1016/j.freeradbiomed.2016.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc Natl Acad Sci U S A. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab. 1996;16:360–366. doi: 10.1097/00004647-199605000-00002. [DOI] [PubMed] [Google Scholar]
- Sejersted Y, Hildrestrand GA, Kunke D, Rolseth V, Krokeide SZ, Neurauter CG, Suganthan R, Atneosen-Asegg M, Fleming AM, Saugstad OD, Burrows CJ, Luna L, Bjoras M. Endonuclease VIII-like 3 (Neil3) DNA glycosylase promotes neurogenesis induced by hypoxia-ischemia. Proc Natl Acad Sci U S A. 2011;108:18802–18807. doi: 10.1073/pnas.1106880108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seredenina T, Demaurex N, Krause KH. Voltage-gated proton channels as novel drug targets: from NADPH oxidase regulation to sperm biology. Antioxid Redox Signal. 2015;23:490–513. doi: 10.1089/ars.2013.5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada M, Dumitrache LC, Russell HR, McKinnon PJ. Polynucleotide kinase-phosphatase enables neurogenesis via multiple DNA repair pathways to maintain genome stability. EMBO J. 2015;34:2465–2480. doi: 10.15252/embj.201591363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo A, Liang AC, Maki T, Miyamoto N, Tomimoto H, Lo EH, Arai K. Subcortical ischemic vascular disease: roles of oligodendrocyte function in experimental models of subcortical white-matter injury. J Cereb Blood Flow Metab. 2016;36:187–198. doi: 10.1038/jcbfm.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol. 2006;16:150–159. doi: 10.1016/j.cub.2005.11.066. [DOI] [PubMed] [Google Scholar]
- Stetler RA, Gao Y, Leak RK, Weng Z, Shi Y, Zhang L, Pu H, Zhang F, Hu X, Hassan S, Ferguson C, Homanics GE, Cao G, Bennett MV, Chen J. APE1/Ref-1 facilitates recovery of gray and white matter and neurological function after mild stroke injury. Proc Natl Acad Sci U S A. 2016;113:E3558–E3567. doi: 10.1073/pnas.1606226113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 2000;19:1397–1404. doi: 10.1093/emboj/19.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebeni A, Szebeni K, DiPeri TP, Johnson LA, Stockmeier CA, Crawford JD, Chandley MJ, Hernandez LJ, Burgess KC, Brown RW, Ordway GA. Elevated DNA oxidation and DNA repair enzyme expression in brain white matter in major depressive disorder. Int J Neuropsychopharmacol. 2017;20:363–373. doi: 10.1093/ijnp/pyw114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangasamy T, Jeyakumar P, Sittadjody S, Joyee AG, Chinnakannu P. L-carnitine mediates protection against DNA damage in lymphocytes of aged rats. Biogerontology. 2009;10:163–172. doi: 10.1007/s10522-008-9159-1. [DOI] [PubMed] [Google Scholar]
- Ueno Y, Koike M, Shimada Y, Shimura H, Hira K, Tanaka R, Uchiyama Y, Hattori N, Urabe T. L-carnitine enhances axonal plasticity and improves white-matter lesions after chronic hypoperfusion in rat brain. J Cereb Blood Flow Metab. 2015;35:382–391. doi: 10.1038/jcbfm.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosler PS, Sun D, Wang S, Gao Y, Kintner DB, Signore AP, Cao G, Chen J. Calcium dysregulation induces apoptosis-inducing factor release: cross-talk between PARP-1- and calpain-signaling pathways. Exp Neurol. 2009;218:213–220. doi: 10.1016/j.expneurol.2009.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zeng L, Wang J, Chau JF, Lai KP, Jia D, Poonepalli A, Hande MP, Liu H, He G, He L, Li B. A positive role for c-Abl in Atm and Atr activation in DNA damage response. Cell Death Differ. 2011;18:5–15. doi: 10.1038/cdd.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, Kim B, Bao L, Harraz MM, Chang C, Chen R, Wang JE, Kam TI, Jeong JS, Xie Z, Neifert S, Qian J, Andrabi SA, Blackshaw S, Zhu H, Song H, Ming GL, Dawson VL, Dawson TM. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. 2016a;354 doi: 10.1126/science.aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu G, Hong D, Chen F, Ji X, Cao G. White matter injury in ischemic stroke. Prog Neurobiol. 2016b;141:45–60. doi: 10.1016/j.pneurobio.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler EA, Sengillo JD, Bell RD, Wang J, Zlokovic BV. Blood-spinal cord barrier pericyte reductions contribute to increased capillary permeability. J Cereb Blood Flow Metab. 2012;32:1841–1852. doi: 10.1038/jcbfm.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LJ, Wu G, Akhavan Sharif MR, Baker A, Jia Y, Fahey FH, Luo HR, Feener EP, Clapham DE. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci. 2012;15:565–573. doi: 10.1038/nn.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanev P, Seevinck PR, Rudrapatna US, Bouts MJ, van der Toorn A, Gertz K, Kronenberg G, Endres M, van Tilborg GA, Dijkhuizen RM. Magnetic resonance imaging of local and remote vascular remodelling after experimental stroke. J Cereb Blood Flow Metab. 2017;37:2768–2779. doi: 10.1177/0271678X16674737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JL, Chen WY, Chen YP, Kuo CY, Chen SD. Activation of GLP-1 receptor enhances neuronal base excision repair via PI3K-AKT-induced expression of apurinic/apyrimidinic endonuclease 1. Theranostics. 2016;6:2015–2027. doi: 10.7150/thno.15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Tong X, Schieb L, Vaughan A, Gillespie C, Wiltz JL, King SC, Odom E, Merritt R, Hong Y, George MG. Vital signs: recent trends in stroke death rates - United States, 2000-2015. MMWR Morb Mortal Wkly Rep. 2017;66:933–939. doi: 10.15585/mmwr.mm6635e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Thompson JF, Taheri S, Salayandia VM, McAvoy TA, Hill JW, Yang Y, Estrada EY, Rosenberg GA. Early inhibition of MMP activity in ischemic rat brain promotes expression of tight junction proteins and angiogenesis during recovery. J Cereb Blood Flow Metab. 2013;33:1104–1114. doi: 10.1038/jcbfm.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, He C. DNA repair by reversal of DNA damage. Cold Spring Harb Perspect Biol. 2013;5:a012575. doi: 10.1101/cshperspect.a012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong RL, Yang C, Lu J, Wang H, Schlaff CD, Tandle A, Graves CA, Elkahloun AG, Chen X, Zhuang Z, Lonser RR. Cell transcriptional state alters genomic patterns of DNA double-strand break repair in human astrocytes. Nat Commun. 2014;5:5799. doi: 10.1038/ncomms6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Zhang Z, Chopp M. Function of neural stem cells in ischemic brain repair processes. J Cereb Blood Flow Metab. 2016;36:2034–2043. doi: 10.1177/0271678X16674487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang X, Park TS, Gidday JM. Cerebral endothelial cell apoptosis after ischemia-reperfusion: role of PARP activation and AIF translocation. J Cereb Blood Flow Metab. 2005;25:868–877. doi: 10.1038/sj.jcbfm.9600081. [DOI] [PubMed] [Google Scholar]
- Zhao H, Han Z, Ji X, Luo Y. Epigenetic regulation of oxidative stress in ischemic stroke. Aging Dis. 2016;7:295–306. doi: 10.14336/AD.2015.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal Interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci. 2015;35:11281–11291. doi: 10.1523/JNEUROSCI.1685-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]