

Abstract
Allostery is an important regulatory phenomenon enabling precise control of biological function. Initial understanding of allostery was gained from seminal work on conformational changes exhibited by structured proteins. Within the last decade, protein allostery has also been demonstrated to occur within intrinsically disordered proteins. This emerging concept of disorder-mediated allostery can be usefully understood in the context of a thermodynamic ensemble. The advantage of this ensemble allosteric model is that it unifies the explanations of allostery occurring within both structured and disordered proteins. One central finding from this model is that energetic coupling, the transmission of a signal between separate regions (or domains) of a protein, is maximized when one or more domains are disordered. This is due to a disorder–order transition that contributes additional coupling energy to the allosteric system through formation of a molecular interaction surface or interface. A second key finding is that multiple interfaces may constructively or destructively interfere with each other, resulting in a new form of allosteric regulation called ‘energetic frustration’. Articulating protein allostery in terms of the thermodynamic ensemble permits formulation of experimentally testable hypotheses which can increase fundamental understanding and direct drug-design efforts. These ideas are illustrated here with the specific case of human glucocorticoid receptor, a medically important multi-domain allosteric protein that contains both structured and disordered regions and exemplifies ‘energetic frustration’.
This article is part of a discussion meeting issue ‘Allostery and molecular machines’.
Keywords: allostery, intrinsic disorder, thermodynamic coupling, frustration
1. Introduction
Allostery, a type of signal transmission, is the process whereby binding of a ligand at one site of a macromolecule (often protein) affects a distal site [1–3], resulting in a change of conformation or binding affinity. Early examples of allosteric proteins, such as haemoglobin [4], protein kinase A [5] and aspartate transcarbamoylase [6], identified rigid-body conformational changes between structural conformers that were important for function. All of these examples motivated, and could be described by, classic models of allostery, which were first articulated more than 50 years ago [7–9].
However, it has become clear that structure-based descriptions of allostery lack the ability to quantitatively explain important experimental observations [10]. For example, a growing set of allosteric proteins do not seem to conform to the paradigm of exhibiting well-defined structural endpoints. Such proteins are capable of allosteric events despite substantial intrinsically disordered (ID) regions or conformationally dynamic domains (e.g. E1A, Phd/Doc, PDZ domain, TetR [11–14]). These cases were anticipated decades ago through the pioneering work of Cooper and colleagues, demonstrating the theoretical importance of thermodynamic fluctuations in proteins and that order–disorder transitions might easily result [15,16]. Understanding these proteins and their ‘disorder-mediated allostery’ will be critical to understanding thermodynamic regulation of biology [17].
Because recent work from this laboratory [18–21] has revealed detailed insight into the disorder-mediated allostery within glucocorticoid receptor (GR), GR will be used as a case study throughout this review [19]. GR is a member of the steroid hormone receptor (SHR) family [22], whose members all share some degree of sequence similarity and a similar domain architecture (outlined in figure 1, top). Although structures of the steroid-binding and DNA-binding domains (DBD) are known, a striking property of the SHR family is the presence of disordered N-terminal domains (NTD) of variable length (figure 1, bottom). The DBD exhibits the highest sequence conservation of all the SHR domains, while the NTD exhibits little conservation, making this region difficult to align and transitively infer its structure or function (figure 1). Despite the lack of sequence conservation, these disordered domains are functionally indispensable, being required for transcriptional activity [25]. Moreover, the ID NTD of GR has been shown to go through coupled binding and folding events [26–30]. What is described in the following work likely applies to the broader nuclear receptor (NR) superfamily [31], of which the SHR family is a part, and the general thermodynamic concepts can be applied to other ID, or structured, allosteric systems [17,32].
Figure 1.

Domain architecture of glucocorticoid receptor and sequence conservation within the human steroid hormone receptor family. The domains are depicted as boxes relative to their amino-acid lengths. The DNA-binding and steroid-binding domains are dynamic and shown as wavy boxes. The N-terminal domains, regulatory (R) and functional (F), are intrinsically disordered and shown as a fragmented box that makes up the N-terminus. Thermodynamic interactions are shown as lines with arrows (stabilizing interaction) or with bars (destabilizing interaction) [18,19]. Amino-acid sequence conservation of DBD, the most conserved region of the family, and conservation of the R-domain (residues 1–97), the least conserved region, are explicitly shown [23]. Stronger colour density indicates a higher degree of conservation, yellow denotes hydrophobic side chains, red/blue indicates charged and polar side chains [24]. ER, Estrogen Receptor; PR, Progesterone Receptor; GR, Glucocorticoid Receptor; MR, Mineralocorticoid Receptor; AR, Androgen Receptor. The cartoon molecule denotes the Protein Data Bank structure of GR DBD bound to PAL DNA, accession number 3g99.
2. The protein ensemble: structure and disorder
Every protein in general, and GR in particular, cycles through a large number of possible conformational states at equilibrium [33]. This collection of conformers, ranging from folded, to partly collapsed, to completely denatured, is termed the ‘protein ensemble’. Depending on solution conditions and temperature, these ‘microstates’ may be more or less populated according to their individual stabilities. The formal statistical–mechanical relationship between equilibrium stability (ΔG) and population (P) is expressed as:
| 2.1 |
In the above equation, Pi is the population of an individual microstate i with characteristic stability ΔGi, R is the gas constant, T is absolute temperature and Q is the partition function of the ensemble, defined as
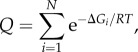 |
2.2 |
where N is the total number of microstates in the ensemble.
Owing to the exponent in equation (2.1), a trace of population versus stability is sigmoidal in nature. In the neighbourhood of the inflection, a relatively small change in stability, perhaps a small multiple of RT, can result in a large population change [34]. If the population change in question was a disorder–order transition, such as a protein folding reaction, a small input of energy would result in a dramatic ‘appearance’ of structure and, perhaps, function. Such an input of energy may come from different sources, such as a temperature shift or a coupled folding–binding reaction (for an example involving ID protein, see NTail [35]).
The key insight from our studies is that multi-domain ID proteins, such as GR, are composed of regulatory and functional domains, which can undergo a collection of energetically coupled folding–binding reactions in response to ligand or cofactor binding. If there are at least two domains that can cooperatively interact, the resulting population changes in the GR ensemble can output a repertoire of non-intuitive functional phenotypes. These phenotypes can explain the origins of disorder-mediated allostery, as well as many puzzling aspects of GR biology [19,36].
3. The DNA-binding domains ensemble
A more concrete illustration of the ensemble nature of proteins is given by focusing on the structured GR DBD. In addition to the high overall sequence conservation, the fold of this domain is found in many other protein families besides the SHR family, testifying to its functional utility throughout evolution [37–39]. Surveying only the multiple known structures of GR DBD, of which there are more than 60 with identical amino-acid sequences to the human [40], a surprisingly diverse set of conformers are observed (figure 2 and [42]). These conformers can be considered as a ‘sub-ensemble’, relative to any additional degrees of freedom available to the molecule outside the crystal. Despite the fact that all these DBD snapshots are ‘structured’, the individual structures exhibit a striking degree of variability: extensive loop regions occupy different conformations, corresponding helices are of variable lengths and the N-terminal beta hairpin clearly shows different degrees of foldedness (figure 2).
Figure 2.
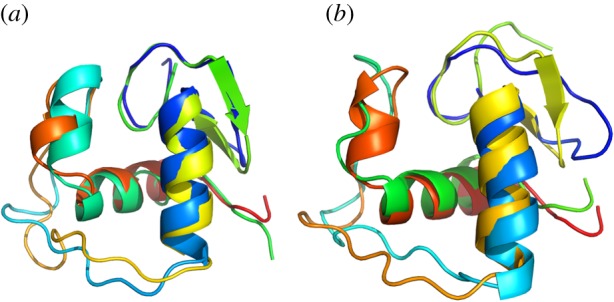
Representative conformational diversity of GR DBD. These four structures (1r4r, 1gdc, 1rgd, 1glu) represent the extremes of known structural differences in GR DBD. Note that all structures are composed of the identical amino-acid sequence that of human (identical to rat) GR. (a) Optimal superposition [41] of 1r4r:1gdc, 2.5 Å RMSD. (b) Optimal superposition of 1rgd:1glu, 2.0 Å RMSD. Individual secondary structure elements show varying degrees of length and folded state.
Many of the pairwise comparisons of different structures result in minimum root mean squared deviations (RMSDs) of more than 2 Å, which is a significantly large variance for such a small, well-conserved, domain [43]. These different conformations are a physical manifestation of one extreme mode of ensemble sampling that domains experience in solution. That this sampling is so evident, even in the most highly conserved sequence region of GR, suggests that these conformational fluctuations may be similarly conserved in other SHRs. Indeed, several studies indicate that conformational plasticity is probably a general attribute of DNA-binding proteins [44–46].
Further indications of conformational plasticity within the DBD are evident when one considers the effects of DNA binding on the isolated DBD. Canonical glucocorticoid response elements (GREs) are essential for the regulation of many genes [47]. GREs contain two tandem 6 bp DNA-binding sites for GR that bind one GR each and are separated by 3 bp. Though crystallographic structures suggest that the interface for interactions between the DNA-bound GR DBDs involves only a few amino acids, large structural perturbations, as measured by NMR spectroscopy, are induced throughout the entire DBD upon any alteration in sequence of the DNA substrate [48]. These canonical GREs allow for pairs of GR to bind, yet the majority of the increased affinity for GR to GREs can be attributed to cooperativity of binding, rather than an energetically favourable dimerization prior to binding [49,50]. The DNA-binding activity of GR implies a very complicated relationship between structure and function that can only partly be explained by the available structural information.
A number of response elements have also been identified that diverge from the canonical sequences; these non-canonical GREs can function both to positively and negatively regulate GR activity through binding to the same sites on the DBD [51,52]. Complicating the conformational landscape of the GR DBD even further are the myriad possibilities for homo- and hetero-dimerization for GR: not only can GR bind to DNA in conjunction with other GR molecules, it can also hetero-dimerize and bind DNA in tandem with some other transcription factors [53,54]. For example, GR can bind to DNA in complex with the androgen receptor (AR) to negatively regulate transcription of the downstream gene. This interaction directly depends on a physical interaction between the dimerization interfaces of each SHR's DBD [55]. Though these interfaces may have been conserved through evolutionary history, it is nonetheless indicative of conformational flexibility that these DBDs are capable of interacting with one another despite having evolved to bind to different DNA sequences. The existence of different binding sites and the variety of transcriptional outcomes that result from binding to those sites indicate that the DBD itself can and does populate a number of different conformational states in response to binding different DNA sites. In order to achieve such a variety of complicated biological responses, the DBD must populate an ensemble of DNA-binding-competent microstates to fully perform in its biologically relevant capacity. Though the DBD is a small, well-ordered domain, it can be viewed as a compelling example of the importance of an ensemble-based approach in even the most classic systems.
4. The disordered ensemble
The other extreme of conformational sampling is that occurring within an unstructured molecule. This mode is biologically relevant for function, because disorder is observed (and predicted) more often than expected in signalling proteins and transcription factors [56–58]. Unlike the DBD, the conformational space of an ID domain, like GR NTD, is much more difficult to visualize. Fortunately, in the context of protein allostery, the exact conformations need not be known in order to productively understand ensemble behaviour. If a domain undergoes a disorder–order transition, any energetic perturbation of a system, such as ligand binding, can effectively redistribute the populations of functional microstates in the ensemble [21,59]. There are three reasons for this: first, folding of ID proteins is often coupled to ligand binding, the energy (ΔGbind) of which will affect microstate populations in the partition function (equation (2.2)) [59]; second, folding may create an interface for a protein–protein interaction that similarly will change the partition function through an interaction energy (Δgint) [19]; third, the folding of the domain alone (ΔG) affects the populations of other microstates in the partition function by its mere presence, through so-called ‘silent states’ detailed below.
For some time now [59], it has been known that site-to-site allosteric coupling is optimized in a multi-domain protein where one or more of the component domains are ID in character. The detailed formalism has been described elsewhere [17,36,59], but the result indicates an alternative, and more general, mechanism of allostery than that proposed by the classical, structure-based models.
GR is a system ideally suited to explore the ramifications of such disorder-mediated allostery: it is composed of several domains (some of which are ID), it contains multiple known binding sites within these domains for DNA and transcriptional cofactors [60], and thermodynamic couplings between these domains, suggesting the presence of transient interfaces, have been discovered and quantified [18,19]. Additional experimental justification for this treatment is that the GR NTD can fold to a globular protein-like structure [18,20], consistent with the generally accepted principle of coupled folding and binding for ID proteins, where the folded conformation is the active conformation.
5. Thermodynamic coupling: ensemble allosteric model
Thermodynamic coupling of domains within a protein can be modelled using statistical thermodynamics. Populations of molecular species (microstates) are directly related to statistical weights, which reflect the relative free energy of each species within the thermodynamic system (equations (2.1) and (2.2)). Thus, the lower the free energy, the higher the population. Although statistical mechanics is not a new idea, the new insight leveraged here is that the thermodynamic coupling energy between protein domains is not negligible and can profoundly affect the partition function, and thus the populations of microstates.
Our model, termed the ensemble allosteric model (EAM) [17], assumes two states, active or inactive, for each domain of a protein. The exact nature of these states is probably important to a detailed atomic-level interpretation of results, but it is irrelevant to the thermodynamic model described. The inactive state could be unfolded, conformationally dynamic or in a folded state that is binding incompetent. The most important aspect is that the states need to be thermodynamically separated by a measurable energy barrier so that their stabilities (ΔG) can be calculated. In the simplified case of a two-domain protein, where the active and inactive states are folded and disordered, respectively, the allosteric coupling energy (Δg1–2) will be the difference in folding free energy of domain 1 in the presence and absence of a folded domain 2 (ΔG1|2 − ΔG1). Of course, if the domains are non-interacting, Δg1–2 = 0.
One caveat in such an analysis is that one must know what constitutes a domain within a protein of interest. Domain prediction algorithms exist [61,62], but in the case of GR, the DBD and steroid-binding domain have been defined structurally and functionally [63,64]. The disordered NTD of GR has been considered one domain for decades, but existence of, and research into, translational isoforms of GR suggested that a more complicated domain architecture existed [65]. Our recent work demonstrates that the NTD of GR is split between a regulatory domain (R) and a functional domain (F) [18,19].
Distinct energetic couplings are observed between R and F, R and DBD, and F and DBD (figure 1). One physical manifestation of an energetic coupling would be a protein–protein interface between pairs of domains (or even the domain triplet). An interface would form most naturally as the result of a coupled folding–binding interaction, as an ostensibly disordered domain folds. Interfaces between GR domains have yet to be completely mapped, but there is evidence that the DBD contains one conserved R-domain interaction surface near its N-terminus [19]. In addition, one naturally occurring isoform, canonically named ‘C3’, that lacks the R-domain may exist to control the absence of the entire R-domain [65]. Truncation of the R-domain simplifies the resulting thermodynamic model (as shown in figure 4), and in the case of GR results in an F-domain that folds more readily.
Figure 4.

The ensembles of GR isoforms upon addition of DNA. The top represents results of the EAM for the A isoform, the bottom left represents results for the C isoform. Legend is located at the bottom right. Positively coupled (functional) states are collected within green boxes and negatively coupled (repressing) states are collected within red boxes. Shown also are silent states that have a low affinity for DNA and transcriptionally inactive. Silent states dominate the pre-DNA ensembles in both cases. Parameters for the EAM were obtained from an optimized fit to experimental data, as described in our previous publications [18,19]. These parameters are as follows (all values in kcal mol−1): ΔGDBD = −6.304, ΔGF = −7.600, ΔGR = 0.552, ΔGDBD-R = 0.447, ΔgR-F = −3.052, ΔgDBD-F = 7.204, ΔgGRE = −8.331.
For the three-domain fragment of GR described (analogous to isoform A), the R-, F- and DBD domains would be separately parametrized in the EAM. A schematic of this ensemble is given in figure 3, which is merely a list of permutations representing all possible thermodynamic states of each domain. For this three-domain system, each domain may adopt one of two thermodynamic states (‘inactive’ or ‘active’, as mentioned above), resulting in a total of 23 = 8 possible energy levels. Again, the key advantage of this treatment, made clear in the second column of figure 3, is that the coupling energies, Δg, are ‘invisible’ when a domain is studied in isolation, but explicitly contribute to the populations of each microstate when the entire system is considered. (Additional energy from ligand binding, ΔgLigand, further perturbs the system by expanding the number of possible microstates, and is elaborated below.)
Figure 3.

Diagram of microstates and populations for the ensemble of a three-domain allosteric model of GR. The inactive states are schematically depicted as curved lines and the active states as filled shapes. The three domains, modelled after the GR A isoform, are labelled DBD (DNA-binding domain, blue), R (Regulatory domain, red) and F (functional domain, grey). The fully active protein (first row) is used as a reference state for the values of ΔG. To get the probability of any microstate, one must first calculate the statistical weight of all the microstates. The statistical weight of any given state is the natural exponential of an ΔG describing that state (equation (2.1)); these are the numerators of the third column. The probabilities are then the statistical weight of the desired state, divided by the sum, Q, of all statistical weights (equation (2.2)). Values of Δg set in lower case represent interaction energies between domains. The GR A isoform may access eight microstates in the ensemble, as depicted here. By contrast, the truncated C3 isoform (composed only of the F-domain and the DBD) can be represented by only four microstates (i.e. those containing R-domain would be missing, not shown). In the EAM, the experimental observable of transcriptional activity is represented by the probability of states whose F-domain is folded (i.e. rows 1, 2, 4, 7 of this figure). The experimental observable of DNA binding affinity is represented by the probability of states whose DBD exhibits the high-affinity DBD (folded) conformation (i.e. rows 1, 3, 4, 6). In the presence of DNA (figure 4), these particular states may access an additional energy level due to DNA binding, this possibility is accounted for by the parenthetical (+ΔgLigand) term.
An important conceptual difference between the EAM and classic models of allostery is that the EAM treats both structure and disorder as integral components of allostery; the fully unfolded microstate is explicitly considered (the last row of figure 3), while in classic models this microstate is ignored. Also of note in the EAM is the presence of interaction energies (Δgi−j), these are of importance in the newly discovered phenomenon of ‘energetic frustration’ [19], whereby domains in a protein are coupled in an antagonistic manner (the details more fully described later in this work). In figure 3, ‘active’ or ‘binding-competent’ domains are generally indicated by folded structure (i.e. filled shapes), while ‘inactive’ or ‘binding-incompetent’ domains are indicated by unfolded structure (i.e. curved lines). Thus the atomic structure of the molecule is not thermodynamically relevant for explaining the functional consequences of the allosteric mechanism.
6. Glucocorticoid receptor as an exemplar of disorder-mediated allostery
For the specific case of GR, we will just consider the first three domains (R, F and DBD) and two of its translational isoforms [65]. The A isoform has the full N-terminus, including the R- and F-domains. The C3 isoform lacks the R-domain, and its F-domain is thus considerably more stable [18]. Our previous work defined the coupling interactions between these domains (figures 3 and 4 and [18,19]), and thus defined the possible microstates in the GR conformational ensemble. In figure 4, we consider the effect of DNA binding to DBD on this GR ensemble. Some of these microstates have a DBD that is binding competent, and of these states the F-domain is either folded (active) or unfolded (repressed). There are also silent states where the DBD is in a low-affinity state that preferentially disappears upon introduction of DNA. The exact structure of low-affinity DBD is unclear, though it may be a minor conformation that is already captured in the known experimental DBD structures described above or the NMR structure [66]. To model DNA-binding, one need only introduce an additional energy term into the EAM, which has been explicitly coded into a publicly available executable [19]. The energy of binding ligand is described as follows [59,67,68]:
| 6.1 |
where Ka is the association constant for ligand, DNA in the present example. As with the other interaction energies already described, the energy of ligand binding modifies the total energy of states that can bind ligand. In this example, ΔgLigand will be negative and added to the stabilities of the states in rows 1, 3, 4 and 6 of figure 3.
DNA binding provides additional concentration-dependent stabilization (ΔG) to the DBD. Supporting this parametrization is the observation that different DNA sequences, and whether they are ‘half-sites’ or ‘full-sites’, have been experimentally shown to provide different amounts of stabilization [42,69].
To model all these effects, an ensemble of states was constructed for the A and C3 isoforms of GR, enumerating all possible combinations of the DBD being in the high- (folded) or low-affinity (unfolded) states, and the F-domain being in its active (folded) or inactive (unfolded) states. Given the other experimentally measurable inputs of ΔGi and Δgi−j, the EAM computes populations of the various microstates in the GR ensemble, before and after introduction of DNA to the system. Examples of these populations are shown in figure 4 for the A and C3 isoforms. Since a functional phenotype directly depends on the population of functional molecule present, changes in population are of unique value in explaining biological function. As seen in figure 4, population changes, and their functional implications, can be dramatic and non-intuitive. This emphasizes the fundamental probabilistic nature of the allosteric mechanism. Inspection of the resultant ensembles provides both a quantitative description of the allosteric response and unanticipated insight into how GR uses an opposing control mechanism to tune activity.
The non-intuitive experimental observation is that the A isoform binds DNA more tightly than the C3 isoform, but is unexpectedly less transcriptionally active [19,65]. How could this possibly be if the sequences and structures of DBD- and F-domains are identical between the two isoforms?
First, the EAM predicts a large difference between the A and C3 isoforms, both in terms of the DNA binding affinity and transcriptional activity upon binding DNA. Both isoforms exhibit unmeasurable activity in the absence of DNA, and in the presence of DNA the active F-domain is populated at 7.3% in the A isoform, while the C3 isoform is populated more than four times higher to a level of 33.0% (green boxes, figure 4). Simultaneously, the A isoform binds DNA more strongly than the C3 isoform (red and green boxes, figure 4). These outcomes cannot be rationalized by a structure-based model of allostery because the folded DBD itself has the same intrinsic affinity regardless of the isoform. Instead, the outcome is determined by the presence in the A isoform of the R-domain, which is negatively coupled to F-domain and positively coupled to DBD. These ensembles thus provide a quantitative explanation for how the GR A isoform displays higher affinity for DNA, while the GR C3 isoform is more active: the negative coupling between the R- and F-domains produces ensembles wherein states with different combinations of properties (i.e. DNA-binding affinity and transcriptional activity) have different probabilities for each isoform. Because the overall observed value for a given property is an ensemble- (or Boltzmann-)weighted contribution of all states, different combinations of high and low DNA-binding affinity and high and low transcriptional activity will produce ensembles with different average properties. Importantly, these properties cannot be assigned to a unique conformation—instead the effect is statistical.
Second, the possible states of the ensemble pre-encode the possible allosteric mechanisms. In other words, the overall ensemble is composed of at least three different functionally relevant sub-ensembles (figure 4): (i) the sub-ensemble in which the DBD is positively coupled to the F-domain (figure 4, green boxes), (ii) the one in which it is negatively coupled (figure 4, red boxes) and (iii) the sub-ensemble of functionally ‘silent states’ (figure 4, grey boxes; i.e. states that are neither active nor bind DNA tightly). Importantly, states with high affinity for DNA (i.e. those that will be preferentially populated in presence of DNA) are associated with both active and inactive forms of the F-domain. Whether the binding of DNA will result in an increase or a decrease in the probability of the transcriptionally active F-domain is a consequence of which sub-ensembles dominate the overall ensemble. Perhaps counterintuitively, within the unbound ensembles, the silent sub-ensemble actually dominates the probabilities. Addition of DNA (figure 4, dashed lines) stabilizes (i.e. lowers the energy of) the states with high affinity for DNA, which increases the probability of both the positively and negatively correlated sub-ensembles, and decreases the probability of the silent sub-ensemble. Interestingly, for both isoforms, the negatively correlated sub-ensemble is increased to a greater extent than the positively correlated sub-ensemble, even though the overall impact is to increase the probability of the active state of the F-domain. This unexpected result is a direct consequence of the simultaneous tuning of both activating and repressing sub-ensembles.
7. Energetic frustration: a robust mechanism for allosteric regulation
The realization that different outcomes, activation or repression, can be pre-encoded within any protein's ensemble suggests a flexible allosteric mechanism that many multi-domain proteins, with varying degrees of structure, might use to respond to an array of different effector molecules. A subset of outcomes seems to represent one particularly effective allosteric regulatory mechanism, whereby activation or repression is highly dependent on the specific signs and magnitudes of the inter-domain interactions. We have termed this special mechanism ‘energetic frustration’ [19].
The name of this mechanism was inspired by ‘geometric frustration’ from solid-state physics [70,71], but the two concepts are different. Energetic frustration is based on an equilibrium model that is agnostic to protein folding and just refers to different thermodynamic states (e.g. high versus low affinity to some ligand of interest). Although these states may possibly correspond to order–disorder transitions, energetic frustration contrasts with the application of geometric frustration to protein folding (called ‘minimal frustration’), whereby frustration refers to kinetically trapped partly folded intermediates of a folding protein [72,73].
Energetic frustration is a specific case of the EAM where the domain–domain interaction energies (Δgi−j) destructively interfere with one another to influence the ensemble. This can happen as a natural consequence of negative and positive cooperativity between individual pairs of domains (figure 5). For example, if two domains positively affect a third domain's stability, but destabilize one another, the first two domains will be energetically frustrated. Their direct coupling interactions would simultaneously be indirectly opposed.
Figure 5.
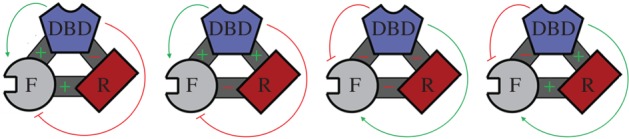
Energetic frustration within a three-domain protein. GR DBD, R- and F- domains are coloured and shaped according to the schemes contained in figures 3 and 4, with inter-domain interactions depicted as dark grey connectors. An inter-domain cooperative interaction (positive Δg) between domains is indicated by a green ‘+’, and an inter-domain negatively cooperative interaction (negative Δg) is indicated by a red ‘−’. Thin lines indicate the ultimate effect of the inter-domain interaction on the functional phenotype at the F-domain, red perpendicular terminations ultimately decreasing activity and green arrows ultimately increasing activity. Thus increased activity may result from two negatively cooperative interactions, and, in general, tunable activity results from two opposing types of cooperativity affecting the F-domain. Although GR corresponds to the second graphic in the series, these cases of energetic frustration could generally apply to any three-domain protein, and the framework is theoretically extendable to arbitrary numbers of domains with varying types of inter-domain interactions.
For the three-domain GR protein discussed, such a situation arises whenever an odd number of domain–domain interactions exhibit negative cooperativity (figure 5). (Even numbers result in constitutive function that is not able to be regulated by the ensemble, e.g. a ‘committed’ activator or repressor.) Recent evidence indicates that the naturally occurring GR conforms to the example of energetic frustration, depicted as case 2 in figure 5. The experiments supporting this conclusion include unfavourable coupling between R- and F-domains by osmolyte folding and protease sensitivity assay [18,19], and favourable coupling between the DBD and F-domain from those same studies. Additional evidence supporting a positive cooperativity between the DBD and F-domain comes from cell-based transcriptional assays that show increased activity in that context [19].
We speculate that this allosteric mechanism is widespread, because many multi-domain proteins appear to contain extensive disorder [74], domain-segregated functions [75] and discrete regulatory regions. Alternative translation starts sites, alternative splicing and post-translational modifications, often used in eukaryotes [76], would broaden the allosteric strategies available to the organism by changing the presence of domains and the cooperativity between domains. Indeed, engineered point mutations that target the interface between GR domains could completely change the cooperativity, and thus change the efficacy of existing therapeutics for this medically important protein. The discovery of energetic frustration opens new vistas to explore how structured and unstructured proteins are regulated.
Acknowledgement
The authors thank the reviewers for insightful comments that greatly improved the original manuscript.
Data accessibility
This article has no additional data.
Authors' contributions
All authors approved the final version of the article. J.T.W. designed experiments, acquired data, analysed data, interpreted results, drafted and critically revised the article. J.L. designed experiments, acquired data, analysed data, interpreted results, drafted and critically revised the article. E.G. interpreted results, drafted and critically revised the article. J.O.W. acquired data, analysed data, interpreted results, drafted and critically revised the article. V.J.H. supervised research, conceived and designed experiments, analysed data, interpreted results, drafted and critically revised the article.
Competing interests
The authors declare no competing interests.
Funding
This work was supported by National Science Foundation grant MCB-1330211 and National Institutes of Health grants GM-126130 and T32-GM-008403.
References
- 1.Monod J, Changeux JP, Jacob F. 1963. Allosteric proteins and cellular control systems. J. Mol. Biol. 6, 306–329. ( 10.1016/S0022-2836(63)80091-1) [DOI] [PubMed] [Google Scholar]
- 2.Changeux JP, Edelstein SJ. 2005. Allosteric mechanisms of signal transduction. Science 308, 1424–1428. ( 10.1126/science.1108595) [DOI] [PubMed] [Google Scholar]
- 3.Cui Q, Karplus M. 2008. Allostery and cooperativity revisited. Protein Sci. 17, 1295–1307: ( 10.1110/ps.03259908) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perutz MF. 1970. Stereochemistry of cooperative effects in haemoglobin. Nature 228, 726–739. ( 10.1038/228726a0) [DOI] [PubMed] [Google Scholar]
- 5.Taylor SS, Kornev AP. 2011. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77. ( 10.1016/j.tibs.2010.09.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beernink PT, Endrizzi JA, Alber T, Schachman HK. 1999. Assessment of the allosteric mechanism of aspartate transcarbamoylase based on the crystalline structure of the unregulated catalytic subunit. Proc. Natl Acad. Sci. USA 96, 5388–5393. ( 10.1073/pnas.96.10.5388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monod J, Wyman J, Changeux JP. 1965. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118. ( 10.1016/S0022-2836(65)80285-6) [DOI] [PubMed] [Google Scholar]
- 8.Koshland DE Jr, Nemethy G, Filmer D. 1966. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5, 365–385. ( 10.1021/bi00865a047) [DOI] [PubMed] [Google Scholar]
- 9.Changeux JP. 2012. Allostery and the Monod-Wyman-Changeux model after 50 years. Ann. Rev. Biophys. 41, 103–133. ( 10.1146/annurev-biophys-050511-102222) [DOI] [PubMed] [Google Scholar]
- 10.Motlagh HN, Wrabl JO, Li J, Hilser VJ. 2014. The ensemble nature of allostery. Nature 508, 331–339. ( 10.1038/nature13001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferreon AC, Ferreon JC, Wright PE, Deniz AA. 2013. Modulation of allostery by protein intrinsic disorder. Nature 498, 390–394. ( 10.1038/nature12294) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, Lee AL. 2009. Hidden dynamic allostery in a PDZ domain. Proc. Natl Acad. Sci. USA 106, 18 249–18 254. ( 10.1073/pnas.0904492106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reichheld SE, Yu Z, Davidson AR. 2009. The induction of folding cooperativity by ligand binding drives the allosteric response of tetracycline repressor. Proc. Natl Acad. Sci. USA 106, 22 263–22 268. ( 10.1073/pnas.0911566106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Pino A, Balasubramanian S, Wyns L, Gazit E, De Greve H, Magnuson RD, Charlier D, Van Nuland NAJ, Loris R.. 2010. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 142, 101–111. ( 10.1016/j.cell.2010.05.039) [DOI] [PubMed] [Google Scholar]
- 15.Cooper A. 1976. Thermodynamic fluctuations in protein molecules. Proc. Natl Acad. Sci. USA 73, 2740–2741. ( 10.1073/pnas.73.8.2740) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper A. 1984. Protein fluctuations and the thermodynamic uncertainty principle. Prog. Biophys. Mol. Biol. 44, 181–214. ( 10.1016/0079-6107(84)90008-7) [DOI] [PubMed] [Google Scholar]
- 17.Hilser VJ, Wrabl JO, Motlagh HN. 2012. Structural and energetic basis of allostery. Annu. Rev. Biophys. 41, 585–609. ( 10.1146/annurev-biophys-050511-102319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Motlagh HN, Chakuroff C, Thompson EB, Hilser VJ. 2012. Thermodynamic dissection of the intrinsically disordered N-terminal domain of human glucocorticoid receptor. J. Biol. Chem. 287, 26 777–26 787. ( 10.1074/jbc.M112.355651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, et al. 2017. Genetically tunable frustration controls allostery in an intrinsically disordered transcription factor. eLife 12, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motlagh HN, Anderson JA, Li J, Hilser VJ. 2015. Disordered allostery: lessons from glucocorticoid receptor. Biophys. Rev. 7, 257–265. ( 10.1007/s12551-015-0173-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Motlagh HN, Li J, Thompson EB, Hilser VJ. 2012. Interplay between allostery and intrinsic disorder in an ensemble. Biochem. Soc. Trans. 40, 975–980. ( 10.1042/BST20120163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beato M, Herrlich P, Schutz G. 1995. Steroid hormone receptors: many actors in search of a plot. Cell 83, 851–857. ( 10.1016/0092-8674(95)90201-5) [DOI] [PubMed] [Google Scholar]
- 23.Pei J, Kim BH, Grishin NV. 2008. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300. ( 10.1093/nar/gkn072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodstadt L, Ponting CP. 2001. CHROMA: consensus-based coloring of multiple alignments for publication. Bioinformatics 17, 845–846. ( 10.1093/bioinformatics/17.9.845) [DOI] [PubMed] [Google Scholar]
- 25.Giguere V, Hollenberg SM, Rosenfeld MG, Evans RM. 1986. Functional domains of the human glucocorticoid receptor. Cell 46, 645–652. ( 10.1016/0092-8674(86)90339-9) [DOI] [PubMed] [Google Scholar]
- 26.Warnmark A, Wikstrom A, Wright AP, Gustafsson JA, Hard T. 2001. The N-terminal regions of estrogen receptor α and β are unstructured in vitro and show different TBP binding properties. J. Biol. Chem. 276, 45 939–45 944. ( 10.1074/jbc.M107875200) [DOI] [PubMed] [Google Scholar]
- 27.Kumar R, Moure CM, Khan SH, Callaway C, Grimm SL, Goswami D, Griffin PR, Edwards DP. 2013. Regulation of the structurally dynamic N-terminal domain of progesterone receptor by protein-induced folding. J. Biol. Chem. 288, 30 285–30 299. ( 10.1074/jbc.M113.491787) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar R, Betney R, Li J, Thompson EB, McEwan IJ. 2004. Induced α-helix structure in AF1 of the androgen receptor upon binding transcription factor TFIIF. Biochemistry 43, 3008–3013. ( 10.1021/bi035934p) [DOI] [PubMed] [Google Scholar]
- 29.Kumar R, Lee JC, Bolen DW, Thompson EB. 2001. The conformation of the glucocorticoid receptor af1/tau1 domain induced by osmolyte binds co-regulatory proteins. J. Biol. Chem. 276, 18 146–18 152. ( 10.1074/jbc.M100825200) [DOI] [PubMed] [Google Scholar]
- 30.Khan SH, Awasthi S, Guo C, Goswami D, Ling J, Griffin PR, Simons SS, Kumar R.. 2012. Binding of the N-terminal region of coactivator TIF2 to the intrinsically disordered AF1 domain of the glucocorticoid receptor is accompanied by conformational reorganizations. J. Biol. Chem. 287, 44 546–44 560. ( 10.1074/jbc.M112.411330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helsen C, Claessens F. 2014. Looking at nuclear receptors from a new angle. Mol. Cell. Endocrinol. 382, 97–106. ( 10.1016/j.mce.2013.09.009) [DOI] [PubMed] [Google Scholar]
- 32.Freiburger L, Miletti T, Zhu S, Baettig O, Berghuis A, Auclair K Mittermaier A. 2014. Substrate-dependent switching of the allosteric binding mechanism of a dimeric enzyme. Nat. Chem. Biol. 10, 937–942. ( 10.1038/nchembio.1626) [DOI] [PubMed] [Google Scholar]
- 33.Freire E. 2001. The thermodynamic linkage between protein structure, stability, and function. Methods Mol. Biol. 168, 37–68. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Wrabl JO, Hilser VJ. 2014. Intrinsically disordered protein: a thermodynamic perspective. In: Computational approaches to protein dynamics: from quantum to coarse-grained methods, 1st edn (ed. Fuxreiter M.), pp. 207–231. Boca Raton, FL: CRC Press. [Google Scholar]
- 35.Bonetti D, Troilo F, Toto A, Brunori M, Longhi S, Gianni S. 2017. Analyzing the folding and binding steps of an intrinsically disordered protein by protein engineering. Biochemistry 56, 3780–3786. ( 10.1021/acs.biochem.7b00350) [DOI] [PubMed] [Google Scholar]
- 36.Hilser VJ, Thompson EB. 2011. Structural dynamics, intrinsic disorder, and allostery in nuclear receptors as transcription factors. J. Biol. Chem. 286, 39 675–39 682. ( 10.1074/jbc.R111.278929) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaeffer RD, Liao Y, Cheng H, Grishin NV. 2017. ECOD: new developments in the evolutionary classification of domains. Nucleic Acids Res 45, D296–D302. ( 10.1093/nar/gkw1137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freedman LP, Luisi BF, Korszun ZR, Basavappa R, Sigler PB, Yamamoto KR. 1988. The function and structure of the metal coordination sites within the glucocorticoid receptor DNA binding domain. Nature 334, 543–546. ( 10.1038/334543a0) [DOI] [PubMed] [Google Scholar]
- 39.Low LY, Hernandez H, Robinson CV, O'Brien R, Grossmann JG, Ladbury JE, Luisi B. 2002. Metal-dependent folding and stability of nuclear hormone receptor DNA-binding domains. J. Mol. Biol. 319, 87–106. ( 10.1016/S0022-2836(02)00236-X) [DOI] [PubMed] [Google Scholar]
- 40.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Boume PE. 2000. The Protein Data Bank. Nucleic Acids Res. 28, 235–242. ( 10.1093/nar/28.1.235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Skolnick J. 2005. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309. ( 10.1093/nar/gki524) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. 2009. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324, 407–410. ( 10.1126/science.1164265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monzon AM, Zea DJ, Fornasari MS, Saldano TE, Fernandez-Alberti S, Tosatto SC, Parisi G. 2017. Conformational diversity analysis reveals three functional mechanisms in proteins. PLoS Comput. Biol. 13, e1005398 ( 10.1371/journal.pcbi.1005398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adhikary R, Tan YX, Liu J, Zimmermann J, Holcomb M, Yvellez C, Dawson PE, Romesberg FE. 2017. Conformational heterogeneity and DNA recognition by the morphogen bicoid. Biochemistry 56, 2787–2793. ( 10.1021/acs.biochem.7b00255) [DOI] [PubMed] [Google Scholar]
- 45.Kalodimos CG, Biris N, Bonvin AM, Levandoski MM, Guennuegues M, Boelens R, Kaptein R. et al. 2004. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science 305, 386–389. ( 10.1126/science.1097064) [DOI] [PubMed] [Google Scholar]
- 46.Spolar RS, Record MT Jr. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science 263, 777–784. ( 10.1126/science.8303294) [DOI] [PubMed] [Google Scholar]
- 47.Schoneveld OJ, Gaemers IC, Lamers WH. 2004. Mechanisms of glucocorticoid signalling. Biochim. Biophys. Acta 1680, 114–128. ( 10.1016/j.bbaexp.2004.09.004) [DOI] [PubMed] [Google Scholar]
- 48.Schone S, et al. 2016. Sequences flanking the core-binding site modulate glucocorticoid receptor structure and activity. Nat. Commun. 7, 12621 ( 10.1038/ncomms12621) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lundback T, Cairns C, Gustafsson JA, Carlstedt-Duke J, Hard T. 1993. Thermodynamics of the glucocorticoid receptor-DNA interaction: binding of wild-type GR DBD to different response elements. Biochemistry 32, 5074–5082. ( 10.1021/bi00070a015) [DOI] [PubMed] [Google Scholar]
- 50.Lundback T, Zilliacus J, Gustafsson JA, Carlstedt-Duke J, Hard T. 1994. Thermodynamics of sequence-specific glucocorticoid receptor-DNA interactions. Biochemistry 33, 5955–5965. ( 10.1021/bi00185a037) [DOI] [PubMed] [Google Scholar]
- 51.Burnstein KL, Cidlowski JA. 1992. The down side of glucocorticoid receptor regulation. Mol. Cell. Endocrinol. 83, C1–C8. ( 10.1016/0303-7207(92)90187-B) [DOI] [PubMed] [Google Scholar]
- 52.Dostert A, Heinzel T. 2004. Negative glucocorticoid receptor response elements and their role in glucocorticoid action. Curr. Pharm. Des. 10, 2807–2816. ( 10.2174/1381612043383601) [DOI] [PubMed] [Google Scholar]
- 53.Diamond MI, Miner JN, Yoshinaga SK, Yamamoto KR. 1990. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science 249, 1266–1272. ( 10.1126/science.2119054) [DOI] [PubMed] [Google Scholar]
- 54.Trapp T, Rupprecht R, Castren M, Reul JM, Holsboer F. 1994. Heterodimerization between mineralocorticoid and glucocorticoid receptor: a new principle of glucocorticoid action in the CNS. Neuron 13, 1457–1462. ( 10.1016/0896-6273(94)90431-6) [DOI] [PubMed] [Google Scholar]
- 55.Chen S, Wang J, Yu G, Liu W, Pearce D. 1997. Androgen and glucocorticoid receptor heterodimer formation. A possible mechanism for mutual inhibition of transcriptional activity. J. Biol. Chem. 272, 14 087–14 092. ( 10.1074/jbc.272.22.14087) [DOI] [PubMed] [Google Scholar]
- 56.Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, Dunker AK. 2006. Intrinsic disorder in transcription factors. Biochemistry 45, 6873–6888. ( 10.1021/bi0602718) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krasowski MD, Reschly EJ, Ekins S. 2008. Intrinsic disorder in nuclear hormone receptors. J. Proteome Res. 7, 4359–4372. ( 10.1021/pr8003024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uversky VN, Oldfield CJ, Dunker AK. 2005. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 18, 343–384. ( 10.1002/jmr.747) [DOI] [PubMed] [Google Scholar]
- 59.Hilser VJ, Thompson EB. 2007. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl Acad. Sci. USA 104, 8311–8315. ( 10.1073/pnas.0700329104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hermanson O, Glass CK, Rosenfeld MG. 2002. Nuclear receptor coregulators: multiple modes of modification. Trends Endocrinol. Metab. 13, 55–60. ( 10.1016/S1043-2760(01)00527-6) [DOI] [PubMed] [Google Scholar]
- 61.Alexandrov N, Shindyalov I. 2003. PDP: protein domain parser. Bioinformatics 19, 429–430. ( 10.1093/bioinformatics/btg006) [DOI] [PubMed] [Google Scholar]
- 62.Holland TA, Veretnik S, Shindyalov IN, Bourne PE. 2006. Partitioning protein structures into domains: why is it so difficult? J. Mol. Biol. 361, 562–590. ( 10.1016/j.jmb.2006.05.060) [DOI] [PubMed] [Google Scholar]
- 63.Luisi BF, Xu WX, Otwinowski Z, Freedman LP, Yamamoto KR, Sigler PB. 1991. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 352, 497–505. ( 10.1038/352497a0) [DOI] [PubMed] [Google Scholar]
- 64.Kauppi B, et al. 2003. The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J. Biol. Chem. 278, 22 748–22 754. ( 10.1074/jbc.M212711200) [DOI] [PubMed] [Google Scholar]
- 65.Lu NZ, Cidlowski JA. 2005. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol. Cell. 18, 331–342. ( 10.1016/j.molcel.2005.03.025) [DOI] [PubMed] [Google Scholar]
- 66.Baumann H, Paulsen K, Kovacs H, Berglund H, Wright APH, Gustafsson JA, Haerd T. 1993. Refined solution structure of the glucocorticoid receptor DNA-binding domain. Biochemistry 32, 13 463–13 471. ( 10.1021/bi00212a011) [DOI] [PubMed] [Google Scholar]
- 67.Motlagh HN, Hilser VJ. 2012. Agonism/antagonism switching in allosteric ensembles. Proc. Natl Acad. Sci. USA 109, 4134–4139. ( 10.1073/pnas.1120519109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.White JT, Motlagh HN, Li J, Thompson EB, Hilser VJ. 2015. Allosteric regulation and intrinsic disorder in nuclear hormone receptors. In Nuclear receptors: from structure to the clinic (ed. McEwan IJ, Kumar R), part 1, pp. 73–91. Switzerland: Springer International Publishing. [Google Scholar]
- 69.Gewirth DT, Sigler PB. 1995. The basis for half-site specificity explored through a non-cognate steroid receptor-DNA complex. Nat. Struct. Biol. 2, 386–394. ( 10.1038/nsb0595-386) [DOI] [PubMed] [Google Scholar]
- 70.Vannimenus J, Toulouse G. 1977. Theory of frustration effect. II. Ising spins on a square lattice. J. Phys. C 10, L537–LL42. ( 10.1088/0022-3719/10/18/008) [DOI] [Google Scholar]
- 71.Villain J. 1977. Two-level systems in a spin-glass model. I. General formalism and two-dimensional model. J. Phys. C 10, 4793–4803. ( 10.1088/0022-3719/10/23/013) [DOI] [Google Scholar]
- 72.Bryngelson JD, Wolynes PG. 1987. Spin glasses and the statistical mechanics of protein folding. Proc. Natl Acad. Sci. USA 84, 7524–7528. ( 10.1073/pnas.84.21.7524) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ferreiro DU, Komives EA, Wolynes PG. 2014. Frustration in biomolecules. Q Rev. Biophys. 47, 285–363. ( 10.1017/S0033583514000092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Uversky VN. 2013. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 22, 693–724. ( 10.1002/pro.2261) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cesareni G, Gimona M, Sudol M, Yaffe M (eds). 2005. Modular protein domains. Weinheim, Germany: Wiley-VCH. [Google Scholar]
- 76.Buljan M, Chalancon G, Eustermann S, Wagner GP, Fuxreiter M, Bateman A, Babu MM.. 2012. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 46, 871–883. ( 10.1016/j.molcel.2012.05.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.