

Abstract
Calcium sensing receptor (CaSR) positive allosteric modulators (PAMs) are therapeutically important. However, few are approved for clinical use, in part due to complexities in assessing allostery at a receptor where the endogenous agonist (extracellular calcium) is present in all biologic fluids. Such complexity impedes efforts to quantify and optimize allosteric drug parameters (affinity, cooperativity, and efficacy) that dictate PAM structure-activity relationships (SARs). Furthermore, an underappreciation of the structural mechanisms underlying CaSR activation hinders predictions of how PAM SAR relates to in vitro and in vivo activity. Herein, we combined site-directed mutagenesis and calcium mobilization assays with analytical pharmacology to compare modes of PAM binding, positive modulation, and agonism. We demonstrate that 3-(2-chlorophenyl)-N-((1R)-1-(3-methoxyphenyl)ethyl)-1-propanamine (NPS R568) binds to a 7 transmembrane domain (7TM) cavity common to class C G protein-coupled receptors and used by (αR)-(−)-α-methyl-N-[3-[3-[trifluoromethylphenyl]propyl]-1-napthalenemethanamine (cinacalcet) and 1-benzothiazol-2-yl-1-(2,4-dimethylphenyl)-ethanol (AC265347); however, there are subtle distinctions in the contribution of select residues to the binding and transmission of cooperativity by PAMs. Furthermore, we reveal some common activation mechanisms used by different CaSR activators, but also demonstrate some differential contributions of residues within the 7TM bundle and extracellular loops to the efficacy of the PAM-agonist, AC265347, versus cooperativity. Finally, we show that PAMS potentiate the affinity of divalent cations. Our results support the existence of both global and ligand-specific CaSR activation mechanisms and reveal that allosteric agonism is mediated in part via distinct mechanisms to positive modulation.
Introduction
The human calcium sensing receptor (CaSR) is a class C G protein-coupled receptor (GPCR) that negatively regulates parathyroid hormone secretion in response to serum extracellular calcium (Ca2+o) concentrations. As such, the small-molecule positive allosteric modulator (PAM), (αR)-(−)-α-methyl-N-[3-[3-[trifluoromethylphenyl]propyl]-1-napthalenemethanamine (cinacalcet), is approved for the treatment of secondary hyperparathyroidism, and some instances for the treatment of primary hyperparathyroidism (Nemeth and Goodman, 2016). Cinacalcet has also been used off-label to correct disorders caused by naturally occurring loss-of-function CaSR mutations (Mayr et al., 2016). Although various CaSR small-molecule PAM chemotypes have been discovered, cinacalcet remains the only one on the market. Failure to translate newer CaSR PAMs to the clinic may reflect complexities associated with detecting and quantifying in vitro allosteric modulator effects at class C GPCRs (Leach and Gregory, 2017), thus hampering structure-activity relationship (SAR) optimization efforts and predictions of in vivo drug efficacy, and impeding dissection of allosteric mechanisms of action. For instance, despite cinacalcet being the first clinically approved GPCR allosteric modulator to reach the market, surprisingly little is known about how cinacalcet and other PAMs mediate positive allosteric modulation and/or agonism at the CaSR. Understanding GPCR activation mechanisms can help guide SAR optimization of ligands with desired activity.
Structural studies of class A and B GPCRs indicate that during receptor activation a rotational and vertical movement of transmembrane (TM) 6 and TM7 relative to TM3 occurs, resulting in conformational changes at the intracellular face of the receptor that promote transducer coupling (reviewed in de Graaf et al., 2017; Manglik and Kruse, 2017). For class C GPCRs, most endogenous agonists predominantly bind in the large extracellular Venus flytrap (VFT) domain, stabilizing activating conformational rearrangements that transmit through the 7 transmembrane domain (7TM) spanning regions and/or extracellular loops (ECLs) to the intracellular side of the receptor. Many synthetic molecules that target class C GPCRs bind to allosteric sites in the 7TM domain and/or ECL regions. Furthermore, for the CaSR, the 7TM and/or ECL domains also contain additional binding sites for endogenous agonists including polyamines and Ca2+o (Ray and Northup, 2002; Leach et al., 2016). However, the 7TM molecular mechanisms that underlie class C GPCR allosteric (and indeed orthosteric) activation are largely unknown. This is due to a complete lack of active-state class C GPCR 7TM structures, but also, in part, due to the difficulties in interpreting mutation-based structure-function studies to delineate residue contributions to the three key molecular properties that govern allosteric activity; ligand affinity, cooperativity, and efficacy.
In light of the difficulties associated with studying GPCR allosteric modulator effects, we developed pharmacological and analytical methods to robustly quantify allosteric modulation of GPCRs (Leach et al., 2007), and recently employed these methods to probe class C GPCR allostery (Gregory et al., 2012, 2013, 2014; Leach et al., 2016). We demonstrated unprecedented insights into class C GPCR drug actions by combining pharmacological, analytical, and detailed structure-function analyses. For instance, we identified a CaSR 7TM domain cavity that accommodates small-molecule CaSR PAMs and negative allosteric modulators, and showed that structurally diverse modulators bind to distinct regions in this cavity; the arylalkylamine PAM, cinacalcet, binds toward the top of the 7TM domain cavity, whereas the structurally distinct CaSR PAM-agonist, 1-benzothiazol-2-yl-1-(2,4-dimethylphenyl)-ethanol (AC265347), binds deeper in the pocket (Leach et al., 2016). Furthermore, we mapped residues that contribute to PAM potentiation of the CaSR’s endogenous agonist, Ca2+o, revealing both shared and distinct activation mechanisms used by cinacalcet and AC265347 (Leach et al., 2016).
Given the importance of understanding the structural basis underlying ligand binding and activation mechanisms at class C GPCRs, the current study sought to build on our prior analysis of CaSR allostery by examining the structural basis of positive allosteric modulation mediated by the small-molecule PAM, 3-(2-chlorophenyl)-N-((1R)-1-(3-methoxyphenyl)ethyl)-1-propanamine (NPS R568). The binding of NPS R568 and other arylalkylamine PAMs is predicted to overlap with the cinacalcet binding site. Given that arylalkylamine PAMs display comparable in vitro pharmacology, we hypothesized that NPS R568 would demonstrate similar structure-function relationships to cinacalcet. We also hypothesized that the PAM-agonist, AC265347, mediates agonism via engagement of residues that transmit positive modulation. Thus, we used mutagenesis to compare mechanisms of positive allosteric modulation and direct allosteric agonism mediated by NPS R568 and AC265347 (Fig. 1A). We identify both global and ligand-specific activation mechanisms at the CaSR, which could inform future drug discovery efforts seeking to rationally optimize the activity of CaSR activators and/or potentiators.
Fig. 1.

Studying positive allosteric modulation at the CaSR. (A) Structures of the arylalkylamine PAM, NPS R568, and the PAM-agonist, AC265347, investigated in this study. (B) According to the operational model of allosterism, the CaSR PAM effects on orthosteric agonist signaling are governed by PAM affinity (KB), binding and/or efficacy cooperativity between the PAM and the orthosteric agonist (α and β, respectively), and the efficacy of the PAM (τB); αβ governs the magnitude of agonist potentiation, reflected by an increase in agonist Emax and/or potency, and enhanced vehicle-mediated signaling plus a reduction in the slope of the transducer function, n, if ambient orthosteric agonists are present in the buffer. If the PAM is also an agonist, τB will dictate PAM-mediated receptor signaling in the presence of vehicle. (C) Snake diagram of the primary CaSR 7TM domain and ECL sequence, indicating residues that were mutated in the current study. Mutations at residues previously shown to be important for the binding of the PAMs, cinacalcet, and/or AC265347, their cooperativity with Ca2+o, or both (Leach et al., 2016), are shown in blue, red, and orange, respectively. All engineered mutations were substituted with alanine, with the exception of A6151.42, A7725.39, A8407.35, and A8447.3, which were substituted with valine, and E8377.32, which was mutated to aspartic acid and isoleucine. The residue highlighted in green indicates where a naturally occurring mutation alters the activity of some PAMs (Cook et al., 2015). Arrows point to the ×.50 conserved class C amino acid residues based on a modified Ballesteros-Weinstein numbering system (Doré et al., 2014).
Materials and Methods
Materials.
Dulbecco’s modified Eagle’s medium, blasticidin S HCl, and fetal bovine serum were obtained from Invitrogen (Carlsbad, CA), while hygromycin B was obtained from Roche (Mannheim, Germany). The Flp-In TREx human embryonic kidney 293 cells and Fluo-4 AM acetoxymethyl ester were purchased from Invitrogen. Quikchange mutagenesis kits were purchased from Agilent Technologies (Santa Clara, CA). NPS R568 was prepared as described previously (Davey et al., 2012). AC265347 and all other chemicals were from Sigma Aldrich (St Louis, MO).
Generation and Maintenance of Cell Lines Expressing Wild-Type (WT) and Mutant CaSRs.
The generation of DNA and cells stably expressing c-myc-tagged WT and mutant human CaSRs in pcDNA5/frt/TO have been described previously (Davey et al., 2012; Leach et al., 2016). Cells were maintained in Dulbecco’s modified Eagle’s medium containing 5%–10% fetal bovine serum, 200 μg/ml hygromycin B, and 5 μg/ml blasticidin S HCl.
Intracellular Calcium (Ca2+i) Mobilization Assays.
Cells were seeded in clear 96-well plates coated with poly-D-lysine (50 μg/ml−1) at 80,000 cells/well and incubated overnight in the presence of 100 ng/ml−1 tetracycline. The following day, cells were washed with assay buffer (150 mM NaCl, 2.6 mM KCl, 1.18 mM MgCl2, 10 mM D-Glucose, 10 mM HEPES, 0.1 mM CaCl2, 0.5% bovine serum albumin, and 4 mM probenecid at pH 7.4) and loaded with Fluo-4 AM acetoxymethyl ester (1 μM in assay buffer) for 1 hour at 37°C. Cells were washed with assay buffer prior to the addition of fresh assay buffer. For interaction studies between Ca2+o and PAMs, modulators were coadded with Ca2+o. For assays in the absence of ambient divalent cations, cells were treated as described previously, except that the final assay buffer and the buffer used for washing and loading the cells did not contain CaCl2 or MgCl2.
For all studies, each well was treated with a single agonist and/or modulator concentration. The release of Ca2+i was measured at 37°C using Flexstation 1 or 3 (Molecular Devices, Sunnyvale, CA). Fluorescence was detected for 60 seconds at 485 nm excitation and 525 nm emission, and the peak Ca2+i mobilization response (approximately 12 seconds after agonist addition) was used for the subsequent determination of the agonist response. Relative peak fluorescence units were normalized to the fluorescence stimulated by 1 μM ionomycin to account for differences in cell number and loading efficiency.
Data Analysis.
All nonlinear regression analyses were performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA). Parametric measures of potency, affinity, and cooperativity were estimated as logarithms (Christopoulos, 1998). Interaction experiments between Ca2+o and allosteric modulators were fitted to an operational model of allosterism (Leach et al., 2007; Aurelio et al., 2009; Leach et al., 2010), which was modified to accommodate the presence of ambient agonist (Davey et al., 2012):
| (1) |
where [A] denotes the concentration of exogenously added orthosteric agonist to stimulate the response; [Ae] is the ambient orthosteric agonist concentration in the buffer; [B] is the molar allosteric ligand concentration; KB is the functional affinity of the allosteric ligand; τB denotes allosteric ligand intrinsic efficacy; α and β are the allosteric effects on orthosteric ligand binding affinity and efficacy, respectively (estimated as a composite αβ value); Emax is the maximal possible system response; and n is the slope factor of the transducer function.
Concentration-response curves for Ca2+o, NPS R568, and AC265347 in the absence of ambient divalent cations were fitted to the following form of an operational model of agonism to obtain estimates of PAM affinity and efficacy:
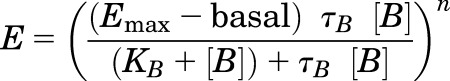 |
(2) |
where all parameters are as described for eq. 1.
Statistics.
An F test (P < 0.001) was used to determine whether eq. 1 best fit interaction data when NPS R568 and AC265347 were assumed to have no agonist efficacy at the WT and mutant CaSRs (i.e., whether the best-fit value for τB was equal to or differed from zero). Statistical differences between the Ca2+o pEC50, or PAM pKB and logαβ at the WT versus the mutant receptors were determined by one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons post-test, where significance was defined as P < 0.05. Statistical differences between PAM pKB at the WT and mutant receptors in the absence versus the presence of ambient divalent cations was determined by two-way ANOVA with Sidak’s comparisons post-test, where significance was considered as P < 0.05.
Molecular Modeling and Docking Studies.
A three-dimensional model of the CaSR 7TM domain was constructed by homology with mGlu5 (PDB: 5CGD) (Christopher et al., 2015) using the ICM software package (Molsoft, San Diego, CA) (Abagyan and Totrov, 1994) as previously described (Leach et al., 2016). The 7TM helices in the model were additionally refined to accommodate irregularities such as nonconserved proline residues (e.g., Pro6823.34) and a one-residue insertion in the extracellular part of TM5 (Ile7775.44). Compound docking relied on the pharmacophore properties of the TM cavity as well as the results of earlier and present mutagenesis studies indicating that mutation of Glu8377.32 abrogates activity of all arylalkylamines. A salt bridge is predicted to occur between the protonated arylalkylamine secondary amine and the Glu8377.32 side chain (Petrel et al., 2003, 2004; Miedlich et al., 2004; Bu et al., 2008; Leach et al., 2016); therefore, the formation of this salt bridge was enforced in our docking studies by imposing a harmonic distance restraint between the corresponding atoms. The arylalkylamine docking proceeded by extensive conformational sampling of the ligands and the residue side chains lining the pocket in the internal coordinates in the presence of this distance restraint. All complexes were further refined by local minimization in the presence of distance restraints maintaining receptor secondary and tertiary structures, and inspected manually.
Results
Rationale for the Study.
We have previously evaluated the effects of CaSR 7TM and ECL mutations on allosteric modulator activity using an interaction paradigm (Davey et al., 2012; Cook et al., 2015; Leach et al., 2016). Here, the effect of a CaSR PAM can manifest as: 1) modulation of the exogenously added agonist to stimulate the response, reflected by a change in the added agonist potency and/or Emax; 2) modulation of ambient agonist present in the buffer, reflected by an increase in the baseline response and/or a shallowing of the agonist concentration-response curve; and 3) direct PAM-mediated agonism, also reflected by an increase in the baseline response (Davey et al., 2012; Cook et al., 2015; Leach et al., 2016) (Fig. 1B). In this work, we have extended our analysis to account for ambient Ca2+o ions present in the assay buffer. Including the ambient Ca2+o concentration in the operational model of allosterism allows delineation of PAM-mediated increases in the vehicle (buffer) response due to agonism versus potentiation of ambient Ca2+o (eq. 1). Accordingly, we are now able to quantify and delineate the effect of CaSR mutations on PAM affinity (pKB), allosteric cooperativity with the orthosteric agonist (αβ), and allosteric agonism (τB).
Using this analytical paradigm, we examined two tool compounds; NPS R568 and AC265347. NPS R568 (Fig. 1A) was among the first identified CaSR PAMs (Nemeth et al., 1998) and a precursor to cinacalcet, which is an approved clinical treatment of human hyperparathyroidism. Both PAMs display similar in vitro pharmacology (Cook et al., 2015), despite subtle differences in their chemical structure; although NPS R568 and cinacalcet share a three carbon aliphatic chain linked to a secondary amine, they differ in their terminal aromatics: chlorophenyl and methoxyphenyl groups in NPS R568 were replaced with tri-fluoromethyl-phenyl and naphthyl groups in cinacalcet. With these differences in mind, we aimed to examine the SAR of NPS R568 in comparison with cinacalcet in more detail. Furthermore, whereas cinacalcet and NPS R568 exhibit little agonism, the chemically distinct CaSR PAM, AC265347, is both a PAM and an agonist (a PAM-agonist) (Cook et al., 2015). We were, therefore, interested in elucidating the different activation mechanisms used by these chemically and pharmacologically distinct CaSR activators. Of further interest, AC265347 exhibits biased CaSR modulation when compared with cinacalcet, presumably via stabilizing different active states (Davey et al., 2012; Leach et al., 2013, 2016; Cook et al., 2015).
Therefore, in this work, we used mutagenesis in combination with an operational model of allosterism that explicitly accommodates ambient agonist levels (eq. 1), to characterize important features of NPS R568 binding and positive cooperativity, and to directly probe AC265347 agonism. 7TM and ECL mutations were selected based on those known to alter the affinity and/or cooperativity of cinacalcet and/or AC265347 (Fig. 1C).
NPS R568 Binds to a Common 7TM Allosteric Binding Site.
We first confirmed that NPS R568 bound within the same 7TM cavity used by other arylalkylamine modulators (Leach et al., 2016). We fitted NPS R568 interaction data at the WT and 7TM mutant receptors to an operational model of allosterism with ambient agonist to assess the impact of 7TM amino acid substitutions on NPS R568 affinity (pKB). WT and representative mutants are shown in Fig. 2 that exemplify the different profiles observed. Similar to our previous observations with cinacalcet (Leach et al., 2016), F6682.56A, F6843.36A, F6883.40A, E8377.32I, and I8417.36A mutations abolished NPS R568 activity or reduced NPS R568 pKB, whereas F8216.53A, E8377.32D, and A8447.39V increased NPS R568 pKB (Fig. 2; Fig. 3A; Supplemental Fig. 1; Table 1). Furthermore, although the magnitude of NPS R568 potentiation of Ca2+o potency at the Y8256.57A mutant was reduced (Supplemental Fig. 1), a significant and maximal ∼2-fold increase in Ca2+o potency was reached in the presence of 0.1 μM NPS R568, suggesting the PAM pKB was increased. To confirm this finding, we performed interaction studies between AC265347 and NPS R568. We found that 0.1–10 μM NPS R568 caused a dextral translocation of the AC265347 concentration-response curve at the Y8256.42A mutant, with no apparent decrease in AC265347 Emax (Supplemental Fig. 2); although limitations in AC265347 solubility prevented determination of complete AC265347 concentration-response curves in the presence of the higher NPS R568 concentrations. Nonetheless, these findings suggest that the interaction between NPS R568 and AC265347 is competitive, or an allosteric interaction governed by extremely high negative cooperativity. In support of a competitive or highly negatively cooperative interaction, when AC265347 and NPS R568 interactions at the Y8256.42A mutant were fitted to a modified Hill/Gaddum/Schild equation (Motulsky and Christopoulos, 2004; Langmead et al., 2006), the Schild slope was not significantly different from unity. Under these circumstances, the concentration of NPS R568 that shifts the AC265347 EC50 2-fold is equal to the NPS R568 pKB; this analysis confirmed that the NPS R568 pKB was increased by the Y8256.57A mutation (Table 1). A similar increase in cinacalcet pKB has been observed at this mutant (Leach et al., 2016). Our findings are consistent with NPS R568 binding in the 7TM cavity in a similar pose to cinacalcet, concordant with previous predictions that E8377.32 forms a hydrogen bond with the core secondary amine in arylalkylamine modulators (Petrel et al., 2003, 2004; Miedlich et al., 2004; Bu et al., 2008; Leach et al., 2016). With this interaction, the terminal naphthyl (cinacalcet) or methoxyphenyl (NPS R568) moiety extends downward, π-stacking with F6843.36 and forming hydrophobic interactions with F6682.56, F6883.40, and I8417.36.
Fig. 2.

7TM mutations alter the activity of NPS R568 at the CaSR. Concentration-response curves to Ca2+o in the absence and presence of NPS R568 were determined in Ca2+i mobilization assays to identify mutations that altered NPS R568 activity (A–E). Representative mutants are shown that reduced NPS R568 affinity (F6843.36A), increased NPS R568 affinity (A8447.39V), reduced NPS R568 cooperativity (V8176.49A), or increased NPS R568 affinity while decreasing cooperativity (F8216.53A). Data are mean + S.E.M. pooled from at least four separate experiments performed in duplicate. Curves are the best fit of the data to eq. 1. Vehicle (buffer) is plotted as the smallest [Ca2+o] in accordance with the logarithmic scale.
Fig. 3.

7TM and ECL mutations alter the affinity and cooperativity of NPS R568 at the CaSR. (A) Concentration-response curves to Ca2+o in the absence and presence of NPS R568 determined in Ca2+i mobilization assays were fitted to an operational model of allosterism (eq. 1) to determine the change in affinity (ΔpKB) (A) and cooperativity (Δlogαβ) (C) of NPS R568 at the mutant CaSRs in comparison with the WT receptor. White bars represent no significant change in pKB or logαβ. Colored bars that sit above and below zero represent a significant increase or decrease in pKB or logαβ, respectively. A 1.5-fold or lower reduction in pKB and/or logαβ is represented by yellow bars, a 1.6-fold to 2.5-fold reduction is represented by orange bars, and a greater than 2.5-fold reduction or increase is represented by red and blue bars, respectively. NA, negligible PAM activity. Data are the ΔpKB or Δlogαβ + S.E.M. calculated from WT and mutant pKB or logαβ values and experiment numbers shown in Table 1; *denotes significant difference in comparison with WT (P < 0.05, one-way ANOVA with Dunnett’s multiple comparisons post-test). Affinity (B) and cooperativity (D) residues are shown on a CaSR 7TM domain homology model (gray cartoon), along with the predicted NPS R568 pose from docking studies. Residue positions are shown as ball and stick, and colors correspond to the fold change in affinity or cooperativity shown in (A and C).
TABLE 1.
Functional affinity (pKB) and cooperativity (logαβ) parameters for NPS R568 determined in Ca2+i mobilization assays using an interaction paradigm
Data are mean ± S.E.M. and 95% confidence interval from the indicated number of independent experiments (n).
| NPS R568 |
|||
|---|---|---|---|
| pKB (95% CI) | Logαβ (95% CI) (αβ) | n | |
| WT | 5.99 ± 0.05 (5.89–6.09) | 0.55 ± 0.01 (0.52–0.58) (3.5) | 17 |
| F6121.39A | 6.39 ± 0.15 (6.03–6.75) | 0.35 ± 0.04a (0.28–0.43) (2.2) | 4 |
| A6151.42V | 6.22 ± 0.13 (5.93–6.49) | 0.45 ± 0.04 (0.38–0.53) (2.8) | 6 |
| F6682.56A | <5 | ND | 4 |
| G670ECL1A | 6.39 ± 0.08 (6.23–6.55) | 0.09 ± 0.04a (−0.07–0.25) (1.2) | 4 |
| F6843.36A | 5.28 ± 0.18a (4.79–5.63) | 0.35 ± 0.05a (0.28–0.51) (2.2) | 4 |
| F6883.40A | 5.40 ± 0.16a (5.00–5.73) | 0.52 ± 0.05 (0.44–0.65) (3.3) | 5 |
| E767ECL2A | 6.36 ± 0.22 (5.93–6.77) | 0.25 ± 0.03a (0.18–0.33) (1.8) | 3 |
| A7725.39V | 6.33 ± 0.08 (6.17–6.48) | 0.38 ± 0.02a (0.35–0.42) (2.4) | 4 |
| F7755.42A | 6.11 ± 0.07 (5.95–6.27) | 0.39 ± 0.02a (0.36–0.42) (2.5) | 4 |
| L7765.43A | 6.40 ± 0.09 (6.20–6.58) | 0.46 ± 0.03 (0.40–0.51) (2.9) | 4 |
| V8176.49A | 5.89 ± 0.26 (5.29–6.38) | 0.37 ± 0.05a (0.27–0.50) (2.3) | 4 |
| W8186.50A | 5.88 ± 0.05 (5.77–5.98) | 0.55 ± 0.01 (0.52–0.58) (3.5) | 5 |
| F8216.53A | 7.14 ± 0.18a (6.80–7.58) | 0.18 ± 0.02a (0.14–0.22) (1.5) | 4 |
| Y8256.57A | 6.88 ± 0.11a,b (6.65–7.11) | 0.07 ± 0.02a,c (0.04–0.10) (1.2) | 4 |
| K831ECL3A | 5.60 ± 0.15 (5.26–5.91) | 0.60 ± 0.05 (0.49–0.71) (4.0) | 4 |
| V833ECL3A | 6.07 ± 0.11 (5.77–6.33) | 0.35 ± 0.02a (0.31–0.40) (2.2) | 4 |
| E8377.32D | 7.13 ± 0.16a (6.85–7.52) | 0.26 ± 0.03a (0.22–0.32) (1.9) | 4 |
| E8377.32I | <5 | ND | 7 |
| A8407.35V | 6.31 ± 0.13 (6.05–6.56) | 0.33 ± 0.03a (0.28–0.39) (2.1) | 4 |
| I8417.36A | NA | NA | 4 |
| A8447.39V | 6.69 ± 0.09a (6.49–6.90) | 0.05 ± 0.09a (−0.17–0.26) (1.1) | 5 |
| L8487.43A | 6.04 ± 0.13 (5.79–6.28) | 0.29 ± 0.02a (0.25–0.32) (1.9) | 4 |
CI, confidence interval; NA, negligible PAM activity; ND, not determined due to weak cooperativity or low affinity.
significantly different to WT (P < 0.05, one-way ANOVA with Dunnett’s multiple comparisons post-test).
pKB determined in interaction studies between AC265347 and NPS R568 (n = 3).
pKB was fixed to 6.88 determined in interaction studies between AC265347 and NPS R568.
There were, however, subtle differences in the effects of some mutations on NPS R568 pKB when compared with cinacalcet. For instance, W8186.50A decreased, whereas E767ECL2A, A7725.39V, or V833ECL3A increased, cinacalcet pKB (Leach et al., 2016); in contrast, these mutations had no significant effect on NPS R568 pKB (Fig. 3A; Table 1). To better understand the differences in the pKB effects of these mutations, we constructed a homology model of the CaSR 7TM domain based on the mGlu5 7TM structure (Christopher et al., 2015) and interrogated this model bound to NPS R568 in silico. Since the NPS R568 core consists of an arylalkylamine, whose secondary amine is predicted to interact directly with E8377.32, the distance required for hydrogen bonding between this residue and the secondary amine of NPS R568 was maintained during the docking process. Unsurprisingly, this produced docking poses for NPS R568 that were consistent with previously published predictions for cinacalcet and other arylalkylamine-based modulators (Leach et al., 2016) (Fig. 3B).
Close inspection of docking poses for NPS R568 in the CaSR homology model explained the observed differences in mutational effects on cinacalcet versus NPS R568 pKB. First, our predicted docking poses oriented the methoxyphenyl moiety of NPS R568 toward the hydrophobic-aromatic network that surrounds the napthyl group of cinacalcet (F6843.36, F7755.42, L7765.43, W8186.50, and I8417.36), but crucially, the smaller and more hydrophilic NPS R568 moiety does not interact with W8186.50 (Fig. 3B; Supplemental Fig. 3). This change in binding mechanism likely underpins why W8186.50A does not influence NPS R568 pKB. Second, the predicted NPS R568 pose places the chloro-phenyl group of NPS R568 in a position identical to the tri-fluoromethyl-phenyl group of cinacalcet, but the NPS R568 moiety occupies appreciably less chemical space. Indeed, the increase in cinacalcet pKB observed at the E767ECL2A and V833ECL3A mutants is likely due to an increase in the volume of the top of the binding pocket, which is more favorable for cinacalcet but makes no difference to the binding of the smaller NPS R568 (Supplemental Fig. 3). Collectively, these data suggest that NPS R568 and cinacalcet bind to an overlapping binding site; however, due to key differences in their chemical topology, there are subtle distinctions in the contribution of select residues to the binding of the two PAMs.
Identification of Residues that Mediate Cooperativity between Ca2+o and NPS R568.
By applying an operational model of allosterism to our data, we also revealed nine residues within the CaSR 7TM bundle that are involved in NPS R568 cooperativity (logαβ) but not affinity (pKB), with major contributions from ECL1 and 2 as well as TMs 6 and 7 (WT and representative mutants are shown in Fig. 2, quantification is shown in Fig. 3C). In contrast, five mutations influenced both affinity and cooperativity; F6882.56A decreased pKB and logαβ, whereas F8216.53A, E8377.32D, Y8256.42A, or A8447.39V had differential effects on the two parameters (Fig. 3, A and C; Table 1). Thus, residues within the binding pocket differentially contribute to NPS R568 affinity versus cooperativity with Ca2+o.
Our previous structure-function analysis at the CaSR identified residues that contributed to the transmission of positive cooperativity between Ca2+o and cinacalcet (Leach et al., 2016). Comparisons between cooperativity determinants for cinacalcet and NPS R568 revealed key similarities and differences. Five mutations (E767ECL2A, V8176.49A, Y8256.57A, A8447.39V, and L8487.43A) significantly reduced the cooperativity of both NPS R568 and cinacalcet, suggesting contributions to a common network that transmits positive cooperativity by structurally similar PAMs (Fig. 3, C and D). In contrast, six of the mutations that specifically reduce NPS R568 cooperativity with Ca2+o without impacting affinity (F6121.39A, G670ECL1A, A7725.39V, F7755.42A, V833ECL3A, and A8407.35V) did not influence cinacalcet cooperativity. Furthermore, A6151.42V, W8186.50A, and K831ECL3A, which we previously reported to influence cinacalcet cooperativity, had no significant effect on the cooperativity between NPS R568 and Ca2+o, demonstrating that the contribution of these residues to the transmission of positive cooperativity is ligand specific. Collectively, our data reveal differences in the network of residues that transmit cooperativity mediated by structurally similar PAMs, indicating that distinct conformational states are favored by cinacalcet and NPS R568.
Identification of Global and Ligand-Specific Activation Mechanisms at the CaSR.
Having identified the NPS R568 binding site and differentiated ligand-receptor interactions governing affinity and cooperativity, we next sought to compare activation mechanisms used by different CaSR activators. To do so, we first validated the ability of the operational model of allosterism with ambient agonist (eq. 1) to distinguish between PAMs (e.g., NPS R568) and PAM-agonists (e.g., AC265347). When applied to Ca2+o versus NPS R568 interaction data, we found that the operational model of allosterism with ambient agonist fit the data best when the PAM was presumed to have no efficacy (i.e., the best fit value for τB did not differ from zero, F test; P < 0.001) (Fig. 2A; Tables 1 and 2). This agreed with our previous study showing that cinacalcet, NPS R568, and other structurally related PAMs demonstrate negligible intrinsic agonism (Cook et al., 2015). In contrast, and also consistent with our previous study (Cook et al., 2015), the interaction between Ca2+o and the PAM-agonist AC265347 were fitted best when the model assumed AC265347 was an agonist (i.e., the best fit value for τB differed from zero, F test; P < 0.001) (Fig. 4A; Table 2). Importantly, reanalysis of these previously reported data with eq. 1 yielded similar estimates of AC265347 pKB and logαβ (Supplemental Table 1). Taken together, these findings demonstrate that our operational model of allosterism with ambient agonist can differentiate between two PAMs with different agonist activity. Therefore, we next assessed the impact of mutations on allosteric modulator efficacy, τB, independently of positive cooperativity.
TABLE 2.
Efficacy (τB) of NPS R568 and AC265347 determined in Ca2+i mobilization assays using an interaction paradigm
Data are mean ± S.E.M. and 95% confidence intervals from the indicated number of independent experiments (n).
| NPS R568 |
AC265347 |
|||
|---|---|---|---|---|
| LogτB (95% CI) (τB) | n | LogτB (95% CI) (τB) | n | |
| WT | ND | 17 | −0.22 ± 0.09 (−0.45 to −0.08) (0.6) | 8 |
| F6121.39A | ND | 4 | 0.12 ± 0.09a (−0.10 to 0.44) (1.3) | 4 |
| A6151.42V | ND | 6 | 0.04 ± 0.03 (−0.01 to 0.09) (1.1) | 4 |
| F6682.56A | ND | 4 | ND | 4 |
| G670ECL1A | −0.25 ± 0.06 (−0.41 to −0.15) (0.6) | 4 | −0.15 ± 0.09 (−0.39 to 0.02) (0.7) | 4 |
| F6843.36A | ND | 4 | ND | 5 |
| F6883.40A | ND | 5 | ND | 4 |
| E767ECL2A | ND | 3 | 0.05 ± 0.03 (−0.02 to 0.12) (1.1) | 4 |
| A7725.39V | ND | 4 | ND | 4 |
| F7755.42A | ND | 4 | −0.26 ± 0.07 (−0.48 to 0.14) (0.5) | 7 |
| L7765.43A | ND | 4 | 0.19 ± 0.12a (−0.11 to 0.32) (1.5) | 6 |
| V8176.49A | ND | 4 | 0.33 ± 0.04a,b (0.26–0.45) (2.1) | 5 |
| W8186.50A | ND | 5 | ND | 4 |
| F8216.53A | ND | 4 | ND | 4 |
| Y8256.57A | ND | 4 | ND | 4 |
| K831ECL3A | ND | 4 | −0.05 ± 0.03 (−0.10 to 0.00) (0.9) | 5 |
| V833ECL3A | ND | 4 | ND | 5 |
| E8377.32D | ND | 4 | 0.09 ± 0.12 (−0.18 to 0.28) (1.2) | 4 |
| E8377.32I | NA | 7 | NA | 5 |
| A8407.35V | ND | 4 | ND | 7 |
| I8417.36A | NA | 4 | ND | 4 |
| A8447.39V | −0.22 ± 0.07 (−0.45 to −0.11) (0.6) | 5 | −0.12 ± 0.03 (−0.14 to −0.09) (0.8) | 6 |
| L8487.43A | ND | 4 | −0.17 ± 0.07 (−0.35 to −0.07) (0.7) | 4 |
CI, confidence interval; NA, negligible PAM activity; ND, not determined due to negligible efficacy.
Significantly different from WT (P < 0.05, one-way ANOVA with Dunnett’s multiple comparisons post-test).
To account for a reduction in cell surface expression caused by V8176.49A compared with the WT CaSR (Leach et al., 2016), logτB was normalized to determine what it would be if this mutant was expressed at the same level as the WT receptor (Gregory et al., 2010).
Fig. 4.

7TM domain mutations alter orthosteric and allosteric agonist activity at the CaSR. Concentration-response curves to Ca2+o determined in Ca2+i mobilization assays in the absence and presence of AC265347 (A–D) have been published previously (Leach et al., 2016), and were reanalyzed with an operational model of allosterism that considered ambient divalent cations to determine the effect of mutations on AC265347 efficacy (τB). WT and representative mutants are shown that caused a simultaneous reduction in Ca2+o potency and loss of AC265347 efficacy (F6883.40A), a simultaneous increase in Ca2+o potency and gain in AC265347 efficacy (L7765.43A), and a selective loss of AC265347 efficacy (A8407.35V). Data are mean + S.E.M. pooled from at least four separate experiments performed in duplicate. Curves through the data points are the best fit of the data to eq. 1. Vehicle (buffer) is plotted as the smallest [Ca2+o] in accordance with the logarithmic scale.
Alanine substitution of F6843.36 or F6883.40 was previously found to reduce Ca2+o potency (Leach et al., 2016), and in the current study also abolished AC265347-mediated Ca2+i mobilization in the presence of vehicle (while positive cooperativity with Ca2+o was retained) (Fig. 4B; Supplemental Fig. 1; Tables 1 and 2). Using a tetracycline-inducible cell line, we titrated F6843.36 and F6883.40 receptor expression levels to establish the Ca2+o pEC50 under conditions where it should more closely reflect its affinity (i.e., in the absence of receptor reserve) (Supplemental Fig. 4). With reduced expression levels, Ca2+o pEC50 values at the F6843.36A (2.90 ± 0.07) or F6883.40A (2.95 ± 0.03) mutants were not significantly different from WT (3.06 ± 0.04), indicating that the reduction in Ca2+o potency at the mutants versus WT under high expression conditions was due to reduced Ca2+o efficacy, not affinity. In contrast, F6121.39A, L7765.43A, or V8176.49A increased Ca2+o potency (Leach et al., 2016) and AC265347 efficacy (Table 2). The changes in Ca2+o potency and AC265347 efficacy at the F6843.36A, F6883.40A, F6121.39, L7765.43, or V8176.49A mutants under conditions where WT and mutant cell surface expression levels are comparable (Leach et al., 2016) suggests these residues are important for the transmission of agonism imparted by both Ca2+o and AC265347.
In contrast, W8186.50A, F6682.56A, A7725.39V, F8216.53A, Y8256.57A, V833ECL3A, A8407.35V, or I8417.36A did not reduce Ca2+o potency (several increased it), or with the exception of F6682.56A did not reduce cell surface expression (Leach et al., 2016), but they all abolished AC265347 efficacy (Fig. 4D; Table 2). Qualitatively, AC265347 enhanced Ca2+i mobilization in the presence of vehicle alone at certain mutations (see, for example, Fig. 4D); however, our analysis is consistent with these baseline increases being attributable to positive modulation of ambient cations in the buffer rather than intrinsic AC265347 efficacy (Supplemental Table 1). Therefore, these eight residues are critical for mediating agonism of AC265347, but not Ca2+o.
Intriguingly, analysis of NPS R568 interactions with Ca2+o revealed two mutations (G670AECL1A and A8447.39V) where there was a gain in allosteric agonism, as demonstrated by a significant increase in τB, but neither mutation enhanced AC265347 efficacy (Table 2). Combined with a comparison of mutations that influence Ca2+o potency (Supplemental Fig. 5) versus AC265347 efficacy (Fig. 5; Table 2), our findings indicate that some 7TM and ECL residues play a global role in agonism imparted by all classes of agonist (i.e., orthosteric and allosteric), whereas others contribute to ligand-specific receptor activation.
Fig. 5.

7TM and ECL mutations alter AC265347 efficacy at the CaSR. (A) Previously published concentration-response curves to Ca2+o in the absence and presence of AC265347 determined in Ca2+i mobilization assays (Leach et al., 2016) were reanalyzed with an operational model of allosterism that accounted for ambient divalent cations (eq. 1) to determine the change in AC265347 efficacy (ΔτB) at the mutant CaSRs in comparison with the WT receptor. White bars represent no significant change in τB. Colored bars that sit above zero represent an increase in τB. A significant 1.6–2.5-fold increase in τB is represented by the purple bar and a greater than 2.5-fold increase is represented by blue bars. ND, not determined due to negligible efficacy; NA, no PAM activity. Data are the ΔτB + S.E.M. calculated from WT and mutant τB values and experiment numbers shown in Table 2; * demotes significant difference in comparison with WT (P < 0.05, one-way ANOVA with Dunnett’s multiple comparisons post-test). Efficacy residues are shown on a CaSR 7TM domain homology model (B), along with the predicted AC265347 pose from docking studies. Blue and purple colors correspond to the fold change in efficacy shown in (A), whereas red corresponds to a complete loss in AC265347 efficacy.
The finding that the G670A and A844V mutations significantly reduced NPS R568 cooperativity yet converted NPS R568 to a PAM-agonist was intriguing. Therefore, we next sought to compare the global and AC265347-specific activation residues with the AC265347 cooperativity networks previously established (Leach et al., 2016) to determine whether there were also distinctions between cooperativity versus agonism networks used by other PAMs. Two mutations (F6121.39A and L7765.43A) that increased AC265347 efficacy (Fig. 5, A and B) did not change positive cooperativity, whereas V8176.49A increased AC265347 efficacy while decreasing cooperativity. In contrast, the loss in AC265347 efficacy at F6682.56A was opposed by increases in cooperativity. Four mutations that decreased AC265347 cooperativity (A6151.42V, E767ECL2A, K831ECL3A, and A8447.39V) had no effect on AC265347 efficacy. Collectively, these data reveal differential contributions of residues within the 7TM bundle and ECLs to efficacy versus cooperativity.
CaSR PAMs Enhance the Affinity of Orthosteric Agonists.
To confirm mutational effects on PAM efficacy predicted from our analysis of interaction data, we assessed the ability of NPS R568 or AC265347 to stimulate Ca2+i mobilization in the absence of ambient Ca2+o or Mg2+o at WT and select mutant receptors. We evaluated G670ECL1A and A8447.39V mutations, where NPS R568 was predicted to gain agonist activity, and A8407.35V as a representative mutation that decreased AC265347 efficacy (Table 2). Furthermore, we evaluated E8377.32D as a control, because this mutation had no effect on the efficacy of NPS R568 or AC265347. Where possible, we fit agonist concentration response data to an operational model of agonism (eq. 2) to determine agonist affinity and efficacy at WT and/or mutant CaSRs (Table 3).
TABLE 3.
Functional affinity (pKB) and efficacy (logτB) parameters for NPS R568 and AC265347 determined in Ca2+i mobilization assays in the absence of ambient divalent cations
Data are mean ± S.E.M. and 95% confidence intervals from the indicated number of independent experiments (n).
| NPS R568 |
AC265347 |
|||||
|---|---|---|---|---|---|---|
| pKB (95% CI) | LogτB (95% CI) (τB) | n | pKB (95% CI) | LogτB (95% CI) (τB) | n | |
| WT | ND | ND | 8 | 5.43 ± 0.10a (5.23–5.63) | 0.05 ± 0.10 (0.01–0.11) (1.1) | 8 |
| G670ECL1A | 5.71 ± 0.18a (5.28–6.13) | −0.19 ± 0.04 (−0.27 to −0.11) (0.6) | 7 | 6.13 ± 0.08b (5.96–6.31) | 0.07 ± 0.03 (0.04–0.11) (1.2) | 7 |
| E8377.32D | ND | ND | 8 | 5.90 ± 0.14a,b (5.60–6.19) | −0.03 ± 0.03 (−0.10 to 0.03) (0.9) | 8 |
| A8407.35V | ND | ND | 7 | ND | ND | 7 |
| A8447.39V | 6.48 ± 0.32 (5.78–7.21) | −0.27 ± 0.06 (−0.42 to −0.17) (0.5) | 6 | 6.89 ± 0.20b (6.51–7.31) | −0.16 ± 0.04b (−0.25 to −0.09) (0.7) | 6 |
CI, confidence interval; ND, not determined due to negligible efficacy and/or low affinity.
Significantly lower than in the presence of ambient divalent cations (P < 0.05, two-way ANOVA with Sidak’s comparisons post-test).
Significantly different from WT (P < 0.05, one-way ANOVA with Dunnett’s multiple comparisons post-test).
In agreement with our previous evaluation of PAM agonism (Cook et al., 2015), up to 30 μM NPS R568 did not stimulate Ca2+i mobilization in the absence of ambient cations (Fig. 6A); due to limited solubility, it was not possible to test higher concentrations. Nonetheless, these findings confirm that NPS R568 exhibits negligible agonism at the CaSR. In contrast to WT CaSR, NPS R568 stimulated concentration-dependent Ca2+i mobilization in the absence of ambient cations at G670ECL1A and A8447.39V (Fig. 6, B and C), suggestive of increased NPS R568 efficacy, as predicted by our operational model analysis that incorporated the presence of ambient cations (Table 2). However, the estimated NPS R568 pKB at the G670ECL2A mutant was significantly lower in the absence of ambient cations (Table 3) than in their presence (Table 1) (P < 0.05, two-way ANOVA with Sidak’s multiple comparisons). A similar trend toward a lower NPS R568 pKB in the absence of ambient cations was also observed at the A8447.39V mutant, but this did not reach significance. At E8377.32D and A8407.35V, NPS R568 exhibited negligible agonist activity, similar to WT CaSR and as predicted from our operational model of allosterism analysis.
Fig. 6.

CaSR PAMs bind with lower affinity in the absence vs. presence of ambient divalent cations, and exhibit differential efficacy at the 7TM domain and ECL mutants. Concentration-response curves to Ca2+o, NPS R568, and AC265347 were determined in Ca2+i mobilization assays in the absence of ambient divalent cations to evaluate PAM efficacy (A–E). Representative mutants were chosen based on their ability to provoke NPS R568 efficacy (G670ECL2A and A8447.39V), reduce AC265347 efficacy (A8407.35V), or have no effect on PAM efficacy (E8377.32D). Data are mean + S.E.M. pooled from at least four separate experiments performed in duplicate. Curves are the best fit of the data to eq. 2. Vehicle (buffer) is plotted as the smallest [Ca2+o] in accordance with the logarithmic scale.
In contrast to NPS R568, AC265347 stimulated relatively robust Ca2+i mobilization at the WT CaSR in the absence of ambient cations (Fig. 6A; Table 3), indicative of intrinsic agonism. AC265347 was also an agonist at the G670ECL2A, E8377.32D, and A8447.39V mutants in the absence of ambient cations, although there was a small but significant decrease in its efficacy at A8447.39V (Fig. 6; Table 3). Consistent with our operational model of allosterism analysis, AC265347 efficacy was reduced at the A8407.35V mutant.
Similar to NPS R568 at WT CaSR and G670ECL2A, A8447.39V, E8377.32D, or A8407.35V mutants, AC265347 bound with lower affinity in the absence of ambient cations than in their presence (P < 0.05, two-way ANOVA with Sidak’s multiple comparisons). The difference in affinities suggests that CaSR PAMs bind preferentially when an orthosteric agonist simultaneously occupies the receptor. Since cooperative allosteric effects on affinity are reciprocal (and, therefore, affect both agonist and modulator) (May et al., 2007), the elevated PAM affinity in the presence of ambient cations is expected if the PAM also enhances the affinity of the ambient cations. Thus, NPS R568 and AC265347 likely act, at least in part, by potentiating the affinity of divalent cations. This could occur if the PAMs stabilized the closed form of the VFT via allosteric interactions between the 7TM and VFT domains.
Discussion
The current study has used a structure-function approach combined with analytical pharmacology to probe CaSR 7TM domain and ECL residues that contribute to receptor activation and positive allosteric modulation. Importantly, by incorporating the presence of ambient agonist into an operational model of allosterism, we were able to delineate the contribution of residues to PAM intrinsic efficacy (agonism) independently of positive cooperativity (potentiation of the orthosteric agonist) and affinity. We found both ligand-specific and global activation networks within the CaSR ECLs and 7TM bundle. The fact that mutations in these regions of the receptor have substantial effects on both orthosteric and allosteric ligands highlights the pivotal role that the 7TM domain and ECLs play in transmitting activating stimuli, irrespective of the location of the ligand binding site. Importantly, our ability to delineate mutation effects on cooperativity and efficacy reveals that positive cooperativity appears to be mediated in part by residues that are distinct to those that transmit allosteric agonism. For instance, A6151.42, E767ECL2, and K831ECL3 all contributed to the transmission of AC265347 cooperativity yet played no role in AC265347 efficacy. Conversely, F6121.39 and L7765.43 constrained AC265347 efficacy but not positive cooperativity. In other instances, there were opposing contributions of residues to cooperativity versus efficacy, as was observed for G670ECL2 and A8447.39; mutation of these residues decreased NPS R568 cooperativity yet converted NPS R568 to an agonist. Similarly, V8176.49A increased AC265347 efficacy while decreasing cooperativity, where the opposite was true for F6682.56A.
Using mutagenesis to dissect the contribution of 7TM and ECL residues to NPS R568 affinity, we confirm that NPS R568 binds to an extended cavity in the CaSR’s 7TM domain, much like cinacalcet, with key interactions between F6682.56, F6843.36, F6883.40, E8377.32, and I8417.36. We did, however, identify some subtle differences in the binding of NPS R568 and cinacalcet, likely attributed to their different terminal aromatic groups, permitting cinacalcet to occupy more space at the top and bottom of the binding pocket. Despite this, and the identification of a number of residues that differentially contribute to NPS R568 and cinacalcet cooperativity, these two PAMs from the arylalkylamine family are pharmacologically very similar (Cook et al., 2015). The differences in NPS R568 and cinacalcet SAR appear to be overcome by shared contributions of E767ECL2, V8176.49, Y8256.57, A8447.39, and L8487.43 to their transmission of positive cooperativity. Importantly, E767ECL2, V8176.49, and A8447.39 are also critical for positive cooperativity mediated by the structurally distinct PAM-agonist AC265347, suggesting that these residues may contribute to a global allosteric activation network. Furthermore, mutation of these residues also increases Ca2+o potency, suggesting a reduction in the energy barrier to transition to an active CaSR 7TM domain that mirrors the effects of PAMs. In fact, greater than 70% of the 7TM/ECL mutations studied herein increase Ca2+o potency (Leach et al., 2016), supporting the idea that substitution of residues in the allosteric binding cavity lowers the activation barrier for Ca2+o. Mutations that reduce Ca2+o potency (e.g., F6843.36A, F6883.40A, and E8377.32I) were at residues critical to the binding of allosteric drugs. Collectively, the influence of these 7TM/ECL residues on both orthosteric agonist activity and small-molecule binding and agonism indicates that the 7TM allosteric binding site has evolutionary importance and raises the possibility that an endogenous allosteric modulator may act via this site.
With regards to mutational effects on agonism, it was of particular note that aside from L7765.43 and V8176.49, which were important for the efficacy of both Ca2+o and AC265347, the majority of residues differentially contributed to orthosteric versus allosteric efficacy. This was demonstrated by the opposing effect of many mutations, which increased Ca2+o potency yet decreased AC265347 efficacy. We cannot exclude the possibility that changes in Ca2+o potency were due to altered Ca2+o affinity, but we did show this was not the case for F6843.36A and F6883.40A mutations (Supplemental Fig. 4). With the exception of E8377.32, which we postulate may bind Ca2+o, it is unlikely that mutation of other residues in this pocket significantly alter Ca2+o affinity, given that four key Ca2+o binding sites are located in the receptor’s VFT domain (Geng et al., 2016). Thus, our data support the existence of global and ligand-specific activation mechanisms in the CaSR’s 7TM and ECL regions.
Another key finding of this study is the demonstration that CaSR PAMs act by potentiating the affinity of Ca2+o. This is in contrast to other class C GPCR PAMs, which enhance orthosteric agonist-mediated efficacy but do not appear to potentiate their binding (Gregory et al., 2012). The significant loss in PAM affinity in the absence of ambient cations versus in their presence is reminiscent of the reciprocal interaction between Ca2+o and L-amino acids (Conigrave et al., 2000, 2004); structural studies suggest both ligands are bound to the CaSR’s VFT domain to achieve a fully active receptor conformation (Geng et al., 2016; Zhang et al., 2016). However, unlike L-amino acids, our current and previous studies (Leach et al., 2016) demonstrate that small-molecule PAMs bind in an extended CaSR 7TM domain cavity.
Our findings have important implications for future drug discovery efforts at the CaSR. Cinacalcet use is restricted due to an adverse risk of hypocalcemia (Chonchol et al., 2009), in part caused by oversuppression of parathyroid hormone secretion along with stimulation of thyroid CaSRs and consequent calcitonin secretion, and/or suppression of Ca2+o reabsorption via activation of renal CaSRs (reviewed in Leach et al., 2014, 2015). There is consequently continued interest in developing CaSR activators with greater tissue selectivity. Indeed, weak (partial) agonists or PAMs would be expected to stimulate robust pharmacological responses only in tissues where strong stimulus-response coupling exists, such as high CaSR-expressing parathyroid cells. To date, no small-molecule CaSR agonists devoid of PAM activity have been identified, and there have been no reported efforts to examine the physiologic consequences of PAMs with different degrees of cooperativity. Furthermore, as demonstrated by AC265347, some PAMs also exhibit agonism, which may heighten adverse effects by further amplifying receptor signaling. A greater appreciation of the drivers of CaSR agonism versus allosteric potentiation may afford an opportunity to rationally tune in or tune out PAM and agonist activity.
In conclusion, the current study provides new insight into 7TM and ECL CaSR activation mechanisms. In the future, this information could facilitate rational structure-based design of novel CaSR activators whose pharmacological and consequent therapeutic properties could be optimized by fine-tuning their PAM and/or agonist activity.
Abbreviations
- AC265347
1-benzothiazol-2-yl-1-(2,4-dimethylphenyl)-ethanol
- ANOVA
analysis of variance
- Ca2+i
intracellular calcium
- Ca2+o
extracellular calcium
- CaSR
calcium sensing receptor
- cinacalcet
(αR)-(−)-α-methyl-N-[3-[3-[trifluoromethylphenyl]propyl]-1-napthalenemethanamine
- ECL
extracellular loop
- GPCR
G protein-coupled receptor
- NPS R568
3-(2-chlorophenyl)-N-((1R)-1-(3-methoxyphenyl)ethyl)-1-propanamine
- PAM
positive allosteric modulator
- SAR
structure-activity relationship
- TM
transmembrane
- 7TM
7 transmembrane domain
- VFT
Venus flytrap
- WT
wild type
Authorship Contributions
Participated in research design: Gregory, Leach.
Conducted experiments: Keller, Kufareva, Josephs, Diao, Mai, Gregory, Leach.
Performed data analysis: Gregory, Leach.
Wrote or contributed to the writing of the manuscript: Keller, Kufareva, Conigrave, Christopoulos, Gregory, Leach.
Footnotes
This work was funded by the Australian National Health and Medical Research Council [Grants APP1026962 and APP1085143] and the National Institutes of Health National Institute of General Medical Sciences and National Institute of Allergy and Infectious Diseases [Grants GM117424 and AI118985, respectively].
K.L. and K.J.G. are Australian Research Council Future Fellowship recipients. A.C. is a Senior Principal Research Fellow of the Australian National Health and Medical Research Council.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Abagyan R, Totrov M. (1994) Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol 235:983–1002. [DOI] [PubMed] [Google Scholar]
- Aurelio L, Valant C, Flynn BL, Sexton PM, Christopoulos A, Scammells PJ. (2009) Allosteric modulators of the adenosine A1 receptor: synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes. J Med Chem 52:4543–4547. [DOI] [PubMed] [Google Scholar]
- Bu L, Michino M, Wolf RM, Brooks CL., III (2008) Improved model building and assessment of the calcium-sensing receptor transmembrane domain. Proteins 71:215–226. [DOI] [PubMed] [Google Scholar]
- Chonchol M, Locatelli F, Abboud HE, Charytan C, de Francisco AL, Jolly S, Kaplan M, Roger SD, Sarkar S, Albizem MB, et al. (2009) A randomized, double-blind, placebo-controlled study to assess the efficacy and safety of cinacalcet HCl in participants with CKD not receiving dialysis. Am J Kidney Dis 53:197–207. [DOI] [PubMed] [Google Scholar]
- Christopher JA, Aves SJ, Bennett KA, Doré AS, Errey JC, Jazayeri A, Marshall FH, Okrasa K, Serrano-Vega MJ, Tehan BG, et al. (2015) Fragment and structure-based drug discovery for a class C GPCR: discovery of the mGlu5 negative allosteric modulator HTL14242 (3-chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile). J Med Chem 58:6653–6664. [DOI] [PubMed] [Google Scholar]
- Christopoulos A. (1998) Assessing the distribution of parameters in models of ligand-receptor interaction: to log or not to log. Trends Pharmacol Sci 19:351–357. [DOI] [PubMed] [Google Scholar]
- Conigrave AD, Mun HC, Delbridge L, Quinn SJ, Wilkinson M, Brown EM. (2004) L-amino acids regulate parathyroid hormone secretion. J Biol Chem 279:38151–38159. [DOI] [PubMed] [Google Scholar]
- Conigrave AD, Quinn SJ, Brown EM. (2000) L-amino acid sensing by the extracellular Ca2+-sensing receptor. Proc Natl Acad Sci USA 97:4814–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook AE, Mistry SN, Gregory KJ, Furness SG, Sexton PM, Scammells PJ, Conigrave AD, Christopoulos A, Leach K. (2015) Biased allosteric modulation at the CaS receptor engendered by structurally diverse calcimimetics. Br J Pharmacol 172:185–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, Christopoulos A. (2012) Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology 153:1232–1241. [DOI] [PubMed] [Google Scholar]
- de Graaf C, Song G, Cao C, Zhao Q, Wang MW, Wu B, Stevens RC. (2017) Extending the structural view of class B GPCRs. Trends Biochem Sci 42:946–960. [DOI] [PubMed] [Google Scholar]
- Doré AS, Okrasa K, Patel JC, Serrano-Vega M, Bennett K, Cooke RM, Errey JC, Jazayeri A, Khan S, Tehan B, et al. (2014) Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511:557–562. [DOI] [PubMed] [Google Scholar]
- Geng Y, Mosyak L, Kurinov I, Zuo H, Sturchler E, Cheng TC, Subramanyam P, Brown AP, Brennan SC, Mun HC, et al. (2016) Structural mechanism of ligand activation in human calcium-sensing receptor. eLife 5:e13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Hall NE, Tobin AB, Sexton PM, Christopoulos A. (2010) Identification of orthosteric and allosteric site mutations in M2 muscarinic acetylcholine receptors that contribute to ligand-selective signaling bias. J Biol Chem 285:7459–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Nguyen ED, Malosh C, Mendenhall JL, Zic JZ, Bates BS, Noetzel MJ, Squire EF, Turner EM, Rook JM, et al. (2014) Identification of specific ligand-receptor interactions that govern binding and cooperativity of diverse modulators to a common metabotropic glutamate receptor 5 allosteric site. ACS Chem Neurosci 5:282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Nguyen ED, Reiff SD, Squire EF, Stauffer SR, Lindsley CW, Meiler J, Conn PJ. (2013) Probing the metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a “molecular switch” in PAM pharmacology. Mol Pharmacol 83:991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Noetzel MJ, Rook JM, Vinson PN, Stauffer SR, Rodriguez AL, Emmitte KA, Zhou Y, Chun AC, Felts AS, et al. (2012) Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol Pharmacol 82:860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead CJ, Fry VA, Forbes IT, Branch CL, Christopoulos A, Wood MD, Herdon HJ. (2006) Probing the molecular mechanism of interaction between 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine (AC-42) and the muscarinic M1 receptor: direct pharmacological evidence that AC-42 is an allosteric agonist. Mol Pharmacol 69:236–246. [DOI] [PubMed] [Google Scholar]
- Leach K, Conigrave AD, Sexton PM, Christopoulos A. (2015) Towards tissue-specific pharmacology: insights from the calcium-sensing receptor as a paradigm for GPCR (patho)physiological bias. Trends Pharmacol Sci 36:215–225. [DOI] [PubMed] [Google Scholar]
- Leach K, Gregory KJ. (2017) Molecular insights into allosteric modulation of class C G protein-coupled receptors. Pharmacol Res 116:105–118. [DOI] [PubMed] [Google Scholar]
- Leach K, Gregory KJ, Kufareva I, Khajehali E, Cook AE, Abagyan R, Conigrave AD, Sexton PM, Christopoulos A. (2016) Towards a structural understanding of allosteric drugs at the human calcium-sensing receptor. Cell Res 26:574–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Loiacono RE, Felder CC, McKinzie DL, Mogg A, Shaw DB, Sexton PM, Christopoulos A. (2010) Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology 35:855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci 28:382–389. [DOI] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A, Conigrave AD. (2014) Engendering biased signalling from the calcium-sensing receptor for the pharmacotherapy of diverse disorders. Br J Pharmacol 171:1142–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Wen A, Cook AE, Sexton PM, Conigrave AD, Christopoulos A. (2013) Impact of clinically relevant mutations on the pharmacoregulation and signaling bias of the calcium-sensing receptor by positive and negative allosteric modulators. Endocrinology 154:1105–1116. [DOI] [PubMed] [Google Scholar]
- Manglik A, Kruse AC. (2017) Structural basis for G protein-coupled receptor activation. Biochemistry 56:5628–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51. [DOI] [PubMed] [Google Scholar]
- Mayr B, Schnabel D, Dörr HG, Schöfl C. (2016) Genetics in endocrinology: gain and loss of function mutations of the calcium-sensing receptor and associated proteins: current treatment concepts. Eur J Endocrinol 174:R189–R208. [DOI] [PubMed] [Google Scholar]
- Miedlich SU, Gama L, Seuwen K, Wolf RM, Breitwieser GE. (2004) Homology modeling of the transmembrane domain of the human calcium sensing receptor and localization of an allosteric binding site. J Biol Chem 279:7254–7263. [DOI] [PubMed] [Google Scholar]
- Motulsky H, Christopoulos A. (2004) Fitting Models to Biological Data Using Linear and Nonlinear Regression. A Practical Guide to Curve Fitting, Oxford University Press, New York. [Google Scholar]
- Nemeth EF, Goodman WG. (2016) Calcimimetic and calcilytic drugs: feats, flops, and futures. Calcif Tissue Int 98:341–358. [DOI] [PubMed] [Google Scholar]
- Nemeth EF, Steffey ME, Hammerland LG, Hung BC, Van Wagenen BC, DelMar EG, Balandrin MF. (1998) Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc Natl Acad Sci USA 95:4040–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrel C, Kessler A, Dauban P, Dodd RH, Rognan D, Ruat M. (2004) Positive and negative allosteric modulators of the Ca2+-sensing receptor interact within overlapping but not identical binding sites in the transmembrane domain. J Biol Chem 279:18990–18997. [DOI] [PubMed] [Google Scholar]
- Petrel C, Kessler A, Maslah F, Dauban P, Dodd RH, Rognan D, Ruat M. (2003) Modeling and mutagenesis of the binding site of Calhex 231, a novel negative allosteric modulator of the extracellular Ca2+-sensing receptor. J Biol Chem 278:49487–49494. [DOI] [PubMed] [Google Scholar]
- Ray K, Northup J. (2002) Evidence for distinct cation and calcimimetic compound (NPS 568) recognition domains in the transmembrane regions of the human Ca2+ receptor. J Biol Chem 277:18908–18913. [DOI] [PubMed] [Google Scholar]
- Zhang C, Zhang T, Zou J, Miller CL, Gorkhali R, Yang JY, Schilmiller A, Wang S, Huang K, Brown EM, et al. (2016) Structural basis for regulation of human calcium-sensing receptor by magnesium ions and an unexpected tryptophan derivative co-agonist. Sci Adv 2:e1600241. [DOI] [PMC free article] [PubMed] [Google Scholar]