

Abstract
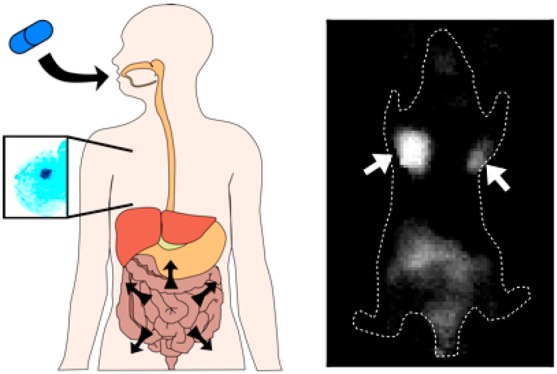
Molecular imaging is advantageous for screening diseases such as breast cancer by providing precise spatial information on disease-associated biomarkers, something neither blood tests nor anatomical imaging can achieve. However, the high cost and risks of ionizing radiation for several molecular imaging modalities have prevented a feasible and scalable approach for screening. Clinical studies have demonstrated the ability to detect breast tumors using nonspecific probes such as indocyanine green, but the lack of molecular information and required intravenous contrast agent does not provide a significant benefit over current noninvasive imaging techniques. Here we demonstrate that negatively charged sulfate groups, commonly used to improve solubility of near-infrared fluorophores, enable sufficient oral absorption and targeting of fluorescent molecular imaging agents for completely noninvasive detection of diseased tissue such as breast cancer. These functional groups improve the pharmacokinetic properties of affinity ligands to achieve targeting efficiencies compatible with clinical imaging devices using safe, nonionizing radiation (near-infrared light). Together, this enables development of a “disease screening pill” capable of oral absorption and systemic availability, target binding, background clearance, and imaging at clinically relevant depths for breast cancer screening. This approach should be adaptable to other molecular targets and diseases for use as a new class of screening agents.
Keywords: noninvasive detection, imaging agent design, anionic fluorophores
Introduction
Molecular imaging has significant potential for disease screening applications by providing both spatial and molecular information to the physician, but so far, a feasible approach has not been developed. The route of administration of a molecular imaging agent for screening large portions of the population is critical for developing a scalable approach. Oral delivery provides several advantages over other avenues of administration that can be grouped into three main categories: safety, cost, and compliance. Oral delivery is generally the safest route of administration1 and can avoid the low risk of anaphylactic shock (e.g., 0.05–0.2% for indocyanine green (ICG)).2 The cost of intravenous (IV) administration is generally higher given the requirement for sterile delivery and medical personnel. For compliance, patients strongly prefer subcutaneous (SC) delivery to IV (91.5%3) and oral delivery to SC injection (93%4), and self-administration would avoid extended or multiple visits for IV delivery and imaging.
One major reason why oral administration of molecular imaging agents has never been previously reported, to our knowledge, is the disparate physicochemical properties required for oral absorption versus efficient targeting. High oral bioavailability for drug-like compounds generally requires low-molecular-weight (<500 Da) and high lipophilicity.5 In contrast, efficient targeting agents often favor higher molecular weights for specific binding and low lipophilicity to reduce off-target interactions. Hydrophilic molecules are generally poorly absorbed, and negatively charged drugs have even worse oral absorption (Figure S1). For example, the FDA approved drug, alendronate, has only 0.7% oral bioavailability in humans (0.2% in mice) despite its small size of 250 Da.6 It is unexpected that a highly charged molecule greater than 1000 Da would have sufficient oral absorption into the bloodstream, and even if it did, the pharmacokinetics following oral delivery are much different than the pharmacokinetics following IV infusion of molecular imaging agents. To be successful, orally delivered molecular imaging agents require a balance of properties including absorption (stability in the gut, intestinal absorption, low first-pass extraction in the liver), efficient targeting, and compatible optical properties for detection at clinical depths (Figure 1, Table S2).
Figure 1.

Ideal properties and structure of imaging agents. (A) Schematic of an orally available systemic imaging agent technique, where the imaging agent is absorbed through the gastrointestinal tract, targets a disease site from the systemic circulation, and is detected noninvasively using near-infrared fluorescence. (B) Venn diagram showing the three main design criteria for developing orally available imaging agents. (C) Structures of the reported agents with varying physicochemical properties.
Despite these challenges for oral delivery of molecular imaging agents, we hypothesized that sufficient targeting could be achieved with appropriately balanced physicochemical properties. The initial design criteria for selecting the targeting ligand are listed in Table S2. We selected a low-molecular-weight targeting ligand against integrin (αvβ3) receptors. This extracellular target has high expression on several cell types (e.g., in cancer-activated endothelium, macrophages, and tumor cells) for robust expression. Importantly, the target internalizes with a sufficient rate for probe trapping over extended periods of time. This target also has a series of affinity ligands developed against it for therapeutic purposes that have been optimized by medicinal chemists for stability in the GI tract, low first-pass metabolism, moderate plasma protein binding, and low toxicity.7−11 The selected ligand maintains targeting following conjugation to several fluorophores,12 allowing us to manipulate the physicochemical properties with the fluorophore for mechanistic studies.13,14
Breast cancer screening is one application where molecular imaging could be beneficial. Cancer therapy has been rapidly advancing toward molecular characterization, but screening technology generally relies on anatomical differences that have several limitations.15 These include the lack of molecular information to identify aggressive tumors versus those that pose no mortality risk (overdiagnosed tumors)16,17 and the lack of contrast in dense breast tissue that carries increased risk, particularly prevalent in younger women.18,19 These limitations have led to an estimated $4 billion being overspent on false-positives and overdiagnosis.20 Near-infrared fluorescence imaging has been investigated as a method to improve sensitivity and specificity,21,22 and current scanners are capable of imaging tumors less than 2 cm in diameter that are imbedded deep in breast tissue at the contrast levels reported here following oral delivery.23,24
Experimental Section
Imaging Agent Synthesis
The agents were generated similar to previous reports.13 Briefly, the targeting ligand (ChemPartner, Waltham, MA), synthesized as an ester, was resuspended in DMSO at a concentration of 300 mg/mL. The ester group on the integrin binder was hydrolyzed to a carboxylic acid by mixing it with 150 μL of ethanol and 7 μL of 1 M NaOH per mg of drug with continuous stirring overnight. This mixture was then neutralized with 1 M HCl, and the solvents were evaporated. IRDye 800CW was obtained from LI-COR (Lincoln, NE)), Alexa Fluor 680 (AF680), BODIPY 650/665-X (BODIPY 650), and CellTrace Far Red DDAO (DDAO) were obtained from Life Technologies (Carlsbad, CA), and Sulfo-Cyanine7 (Sulfo-Cy7) was obtained from Lumiprobe (Hallandale Beach, FL) in the NHS ester form. The hydrolyzed integrin binder was reacted with the fluorescent dyes in a 1:1.5 molar ratio in the presence of 2 μL of triethylamine per mg of drug. The reaction was run overnight and purified using a preparative scale Luna C18(2) column (Phenomenex; Torrance, CA) on a Shimadzu reverse phase HPLC. Full purification methods are provided in the supplementary data in Table S4. The purified products were run on a MALDI-TOF (IRDye800CW agent: m/z calculated 1397, found 1396; AF680 agent: m/z calculated 1253, found 1253; Sulfo-Cy7 agent: m/z calculated 1103, found 1104; DDAO agent: m/z calculated 800, found 798; BODIPY 650 agent: m/z calculated 939, found 940; IRDye800CW agent stereoisomer: m/z calculated 1397, found 1399). The purities of these agents (254 nm) were measured on HPLC (IRDye800CW agent: 95% (99.9% fluorescence purity); AF680 agent: 96.5%; Sulfo-cyanine7 agent: 88% (99.4% fluorescence purity); DDAO agent: 92%; BODIPY 650 agent: 87.5% (99.2% fluorescence purity); IRDye800CW agent stereoisomer: 97.4% (99.9% fluorescence purity)).
Cell Lines
All reagents mentioned below were obtained from Life Technologies (Carlsbad, CA) unless specified otherwise. MDA-MB-231 and HEK-293 cells were purchased from ATCC (Manassas, VA) and grown in DMEM with 10% FBS and 1% penicillin–streptomycin (supplemented with 1% l-Glutamine for MDA-MB-231). HEK-293 cells, which express endogenous αv but not β325 were transfected with the β3 integrin subunit (Addgene plasmid 27289) to generate an αvβ3 positive line. Cells were transfected with Lipofectamine 2000 according to the manufacturer’s instructions and selected with 1 mg/mL G418 in the media.
Characterization (logD, PPB, Affinity)
The binding affinity of all agents was measured using the transfected HEK-293 cells. The cells were harvested and incubated in triplicate with varying concentrations of the agents in suspension for 3 h on ice. The cells were washed with PBS and run on an Attune acoustic focusing cytometer to quantify the fluorescence. Dissociation constants for each of the agents were determined by analyzing the data on Prism (GraphPad Software; La Jolla, CA). The large amount of nonspecific signal generated from receptor-negative cells for the lipophilic BODIPY-650 agent was subtracted from the receptor positive cell line signal for an accurate measurement of the specific receptor dissociation constant.
Transfected and untransfected HEK-293 cells were plated on chamber slides and allowed to attach overnight. The cells were incubated with either 10 nM of the agent (IRDye 800CW and AF 680) or 50 nM (BODIPY 650, DDAO, and Sulfo-Cy7) for 25 min at 37 °C and washed with fresh media. The cells were then imaged on an Olympus FV1200 confocal microscope using the 405, 635, and 748 nm lasers. These cells were also run on an Attune acoustic focusing cytometer to obtain quantitative data. For the blocked samples, the transfected cells were preblocked with either 1 μM (IRDye 800CW and AF 680) or 5 μM (BODIPY 650, DDAO, and Sulfo-Cy7) of the unconjugated integrin binder for 25 min at 37 °C. The cells were then washed and resuspended in either 10 nM of the agent (IRDye 800CW and AF 680) or 50 nM (BODIPY 650, DDAO, and Sulfo-Cy7) in the presence of 100-fold higher unconjugated integrin binder.
Plasma protein binding (PPB) of the agents was measured using a Rapid Equilibrium Dialysis (Thermo Scientific; Rockford, IL) plate according to the manufacturer’s instructions. Mouse plasma (Innovative Research; Novi, MI; Cat. No. C57BL6) was mixed with 1 μM of the agents. After equilibration, the buffer in each chamber was adjusted to 50% mouse plasma in PBS to eliminate the effects of protein binding on fluorescence. The signal was measured using an Odyssey CLx (LI-COR; Lincoln, NE).
The logD7.4 for all the agents was measured using a protocol adapted from Miller et al.26 The agents were prepared at a concentration of 5–50 μM in octanol-saturated phosphate buffered saline (PBS, pH 7.4). A volume of 200 μL of this solution was mixed with 200 μL of PBS-saturated octanol. The mixture was stirred at 700 rpm for 24 h. For lipophilic agents, the aqueous phase concentration was directly measured using fluorescence with the mass balance yielding the logD. Hydrophilic compounds with minimal loss in the aqueous phase were measured using a second extraction step. After the first equilibration, 150 μL of the octanol was mixed with 50 μL of fresh octanol-saturated PBS to extract the agent in the octanol phase and measure the aqueous fluorescence. During the fluorescence read-out, the samples and calibration curves were diluted in 0.5% PBS–BSA to minimize precipitation and sticking to the vessel surface.
In Vivo Oral Absorption and Imaging
C57BL/6 female mice (8 weeks old, n = 3–4 per cohort) were used to measure the oral absorption of the imaging agents. All experiments involving mice were conducted in compliance with the University of Michigan University Institutional Animal Care and Use Committee (IACUC). The mice were dosed with 1 mg/kg of the imaging agent via oral gavage and placed in a metabolic cage for a period of 24 h. Urine was collected from these cages at the end of 24 h. The urine was diluted 10-fold in 0.1% PBS–BSA to prevent the agent from sticking to vessel surfaces. This was then measured on an Odyssey CLx to determine the amount of fluorescent agent present in the urine. This was quantified using a calibration curve of each agent.
MDA MB 231 cells were used for tumor xenografts in female nude mice of 6–8 weeks of age (Jackson Laboratory; Bar Harbor, ME). The cells were harvested using trypsin–EDTA (0.05%) and resuspended in Matrigel (Corning; Corning, NY) at a concentration of 5 000 000 cells/50 μL. Mice were anesthetized using isoflurane at 2% and 1 L/min oxygen, and the cells were injected subcutaneously by the first nipple in the mammary fat pad to avoid fluorescence signal from the gut when imaging. The mice were dosed with the imaging agent once the longest axis of the tumor reached 7–10 mm. Tumor-bearing mice were fed AIN-93 M nonfluorescent chow (Harlan; Indianapolis, IN; Cat. No. TD.94048) for 2 weeks before oral gavage. Mice (n = 3 per cohort) were imaged on an IVIS Spectrum (PerkinElmer; Waltham, MA) at 6, 24, and 48 h after administration of 5 mg/kg of the agent, and the LivingImage software (PerkinElmer; Waltham,MA) was used to measure the signal intensity in the tumor and TBR. The biodistribution protocol is adapted from Oliviera et al.27 In short, the mice were euthanized at 48 h post administration, and all of their organs were resected. The organs (part of the tumor was frozen in OCT for histology slides) were minced with a razor blade and weighed. They were placed in Eppendorf tubes and digested using a collagenase (Worthington Biochemical; Lakewood, NJ; Cat. No. CLS-4) solution (5 mg/mL) in RIPA buffer (Boston BioProducts; Ashland, MA) at 37 °C for 20 min. The organs were sonicated, digested for 30 min at 37 °C using a 50:50 trypsin and RIPA buffer solution, and sonicated again. The resulting mixture was plated in a dilution series in a black walled 96-well plate and imaged using the Odyssey CLx. Absolute quantification was obtained by comparison with a calibration curve. The uptake values were normalized to the average amount of agent that reached the systemic circulation (absorbed dose) per gram of tissue (versus injected dose per gram (%ID/g) used for intravenous delivery).
Histology
Frozen blocks of the tumor in OCT were sectioned into 14 μm slices on a cryostat. The slices were first imaged on the Odyssey CLx in the presence of PBS to prevent the tissue from drying out. This slice was then stained ex vivo with Hoechst 33342 (Thermo Fisher Scientific; Cat. No.H3570), the AF680 agent (due to its higher affinity), anti-Mac3 antibody (BD Biosciences, San Jose, CA: Cat. No. 553322) labeled Alexa Fluor555, and anti-CD31 (BioLegends, San Diego, CA; Cat. No. 102402) labeled Alexa Fluor 488. These slides were then washed in PBS and imaged on an Olympus FV1200 confocal microscope equipped with 405, 488, 543, 633, and 748 nm laser lines.
The integrin image in Figure 4 was post processed to remove stitching artifacts (Figure S2). We used the ImageJ FFT bandpass filter, suppressing horizontal and vertical lines with a cutoff of 2500 pixels and 0 pixels with a 5% tolerance.
Figure 4.

Macroscopic images of a tumor histology slide taken on an Odyssey CLx (agent) and Olympus FV1200 confocal microscope (other channels). The orally delivered agent shows a diffuse pattern in tumors compared to the negative control (Figure S6). The intensity appears to be slightly higher in regions with higher macrophage density versus tumor cells or blood vessels (CD31). The slides were labeled with the AF680 agent ex vivo (integrin) and stained with Hoechst 33342 to show the presence of cells and integrin throughout the tissue. Scale bar = 500 μm.
Results
Imaging Agent Properties
The integrin-binding peptidomimetic with high affinity to αvβ314 was conjugated with several near-infrared dyes to create imaging agents (Figure 1) with a range properties that were tested in vitro and in vivo. Overall, the oral absorption and urinary excretion of the polyanionic conjugates was relatively high compared to other anionic molecules (Figure S1). Following oral gavage of 1 mg/kg of agent formulated in water, 2.5% of the IRDye800CW agent was collected intact in the urine (Figures 2 and S3). The molecular imaging agents had binding affinities that ranged from subnanomolar to double digit nanomolar and moderate to high plasma protein binding as well as a range in logD (lipophilicity) (Figures 2 and S4). However, the BODIPY-650 agent, with lipophilicity most similar to higher-molecular-weight oral therapeutics such as cyclosporine A (logP = 2.92,28 oral bioavailability = 30%29), resulted in high nonspecific sticking to antigen-negative and blocked cells (Figure 2C). The fluorophore on the lowest-molecular-weight compound, DDAO, has a pKa of 5, resulting in intracellular fluorescence quenching following internalization, which is an important mechanism for long-term retention. In contrast, the sulfated cyanine dyes (IRDye800CW, AF680, and Sulfo-Cy7) had excellent targeting specificity and showed high oral absorption given their size and charge (Figure 2D). Although the Alexa Fluor 680 agent had higher affinity, the IRDye800CW agent was selected for studies in the orthotopic breast cancer model due to its higher oral absorption and ideal optical properties (lower background autofluorescence and higher penetration in tissue than 680 nm dyes).30
Figure 2.

Specificity of in vitro cell labeling. (A, top) HEK-293 cells with the transfected αvβ3 receptor show specific extracellular labeling for all four agents. (A, bottom) HEK-293 cells without the receptor show significant nonspecific labeling with the BODIPY 650 agent and little to no signal for all the other agents. The tabulated data (B) shows the physicochemical properties for the agents with varying lipophilicity (increasing from left to right). PPB = Plasma Protein Binding. (C) Quantitative data for nonspecific interactions of the imaging agents using antigen-negative (αvβ3(−)) and blocked controls normalized to the antigen positive cells (αvβ3(+)). (D) Absorbed dose of the negatively charged imaging agents along with their formal charge.
High Uptake and Specificity of Imaging Agent in Breast Cancer
Figure 3 shows images of the orthotopic xenograft model of breast cancer at 6, 24, and 48 h post administration of the IRDye800CW agent. The tumors had significant uptake by 6 h, and the intestinal signal cleared at the later time points to yield high contrast by 24 and 48 h (tumor to background ratios of ∼4 at 48 h). The image intensity shows that the amount of agent in the tumor stays relatively constant over a period of 48 h owing to the residualizing nature of the NIR fluorophore.31 To demonstrate specificity, we used a low-affinity stereoisomer of the IRDye800CW agent, since the two most common techniques, blocking specific uptake or using an antigen-negative tumor, were not feasible. Complete blocking of the targeting receptor over the course of several hours while the agent is absorbed orally would require an impractical amount of the rapidly cleared nonfluorescent ligand, and antigen-negative xenografts still express integrins on the neovasculature and macrophages. The plasma signals were similar between the stereoisomers (Figure S5), but the whole animal fluorescence intensity and biodistribution showed dramatically lower uptake for the low-affinity stereoisomer in all organs (Figure 3). Integrins are expressed at high levels on breast cancer cells, tumor-associated macrophages, and neovasculature relative to healthy tissue, providing robust detection by diversifying the cellular targets (Table S3). The rapid tumor penetration of this low-molecular-weight compound and intermixed cell types in this model32 resulted in relatively uniform signal throughout the tumor with potentially higher uptake by tumor-associated macrophages (Figure 4).
Figure 3.

Application of orally available imaging agents for breast cancer screening. (A) The high-affinity IRDye800CW agent shows increasing contrast for orthotopic breast cancer xenografts using MDA-MB-231 cells from 6 to 48 h with the arrow indicating the tumor. (B) The low-affinity stereoisomer of the same agent shows low uptake in the tumor highlighting the target specificity of the agent. The high signal from the gut is due to unabsorbed agent. (C) The data (n = 3 mice) shows the difference in the tumor uptake and target to background ratios (TBR ± SD) between the two stereoisomers at 6, 24, and 48 h. (D) The 48 h biodistribution data from the mice shows the selectivity of the agent in targeting the tumor and emphasizes the difference in uptake between the two stereoisomers despite similar plasma concentrations.
Discussion
Orally delivered molecular imaging agents have the promise to provide spatial and molecular information to a physician using a method less invasive than a blood test. Near-infrared fluorescence imaging is intrinsically less expensive than other modalities, such as nuclear imaging and MRI, due to the comparatively lower costs of the imaging equipment and molecular imaging agents with the main limitation being the depth of imaging. Since this system employs near-infrared imaging, the light scattering and absorption limits applications to several millimeters for epifluorescence imaging or several centimeters (e.g., through the human breast) using tomography.
We selected integrin (αvβ3) targeting agents as a model system due to their physicochemical properties, safety even at large doses,33 and the importance of integrin expression in breast cancer detection. Many integrin-targeted imaging agents are currently being investigated in clinical trials.34 Women with dense breast tissue, which makes detection particularly difficult using mammography, have a markedly increased risk for invasive breast cancer35 and have a higher risk of dying from their cancer.36 Because the detection reported here is based on a molecularly targeted agent with fluorescence and not tissue density differences, the sensitivity and specificity is not expected to be highly dependent on breast tissue density37 like it is for nonspecific NIR probes.38 This benefit has been demonstrated with radiolabeled integrin targeting agents in the clinic.39 The high level of integrin expression on multiple distinct cell types, including stromal tissue, is advantageous for early screening for robust detection sensitivity and the decreased risk of saturating the target, which would reduce contrast. The optimal 800 nm imaging channel and high target expression provide a large window for achieving efficient contrast even with significant variability in absorption since both signal and background intensity vary in tandem resulting in relatively constant contrast. In addition to detection sensitivity, molecular imaging agents have the potential to better differentiate aggressive lesions versus benign conditions and reduce the rate of overdiagnosis through better detection of at-risk tumors. This is particularly poignant as analyses of large data sets indicate that the current use of mammography is overdiagnosing and treating women.40,41
We hypothesized that balanced physicochemical properties could help achieve high contrast following oral delivery of a near-infrared fluorescent molecular imaging agent. Results from this study demonstrated that molecules can overcome the significant physical and kinetic barriers for sufficient oral delivery and targeting of molecular imaging agents in living subjects. Hydrophilic molecules, particularly anionic drugs, have low oral bioavailability and high patient-to-patient variability in oral absorption. However, several polyanionic molecules, including heparins and chondroitin sulfate, have shown measurable absorption following oral delivery in animals and the clinic.42−44 Because of the variability in absorption and rapid renal excretion, a metabolic cage was used to collect urine to estimate absorption and avoid interpolation errors from measure plasma concentrations. HPLC and mass spectrometry verified excretion of the intact probe following oral gavage (Figure S3).
Besides total oral absorption, there are significant hurdles in imaging agent targeting due to the slower absorption rate following oral delivery compared to intravenous injection. Due to rapid plasma clearance following oral absorption, a dissociation rate of days at 37 °C would be required to maintain significant signal until the agent is fully absorbed and cleared from background tissue. However, the anionic charge on the internalizing agents in this study can aid in trapping the dye within cells for days.31 Despite the long time period between oral administration and imaging, this would still be favorable from a patient compliance standpoint, since intravenous delivery of low-molecular-weight agents requires a minimum of hours to develop contrast. This would necessitate extended or multiple clinical visits—the first to receive an intravenous dose and monitoring for adverse reactions followed by an imaging visit several hours to days later. While this may not be an impediment for life-saving surgery with intraoperative imaging,45 it could have a major impact on patient compliance in screening large healthy populations. Because material costs for small molecules are often low (e.g., $1 per dose for small molecule drugs46), the associated costs of medical personnel and sterile intravenous delivery would likely be higher than any cost savings from improved bioavailability from more invasive routes.
Plasma protein binding mediated by the high anionic charge also plays a role in enabling orally delivered molecular imaging agents. In the current approach, contrary to radiolabeled probes,13 some plasma protein binding is beneficial by slowing clearance relative to absorption, increasing the plasma concentration, and improving target signal relative to background autofluorescence. The plasma concentrations were similar between the low- and high-affinity stereoisomers (Figure S5), indicating similar absorption, protein binding, and clearance. However, the biodistribution values were significantly higher in all the organs (and tumor) for the high-affinity stereoisomer (Figure 3), indicative of binding to integrins within the tumor and healthy tissue. While healthy tissue uptake lowers the absolute contrast between the target and background tissue, it can actually reduce variation in tumor contrast. If the background signal originated solely from a constant level of tissue autofluorescence, variable oral absorption would result in large changes in measured target to background ratios (TBR). However, when the background signal is from absorbed probe, both the target and background signal are proportional to absorbed dose, resulting in a more constant TBR.
There are several current limitations and areas for improvement with this approach. First, the 2.5% absorbed dose results in variability between mice. While this variability does not impact the contrast significantly, since both tumor signal and background are approximately linear with absorbed dose, mice at the very low end of the optical absorption profile risk having autofluorescence lower the contrast to noise ratio. Improved formulation could increase the oral absorption and lower the variability, thereby reducing the dose needed. Second, clinical trials with intravenously delivered (nonspecific) contrast agents have highlighted the difficulty in imaging tumors in the breast near the chest wall (similar to mammography). Improvements in alternative detection methods, such as optoacoustic imaging, may provide better detection in this region. Extensive work has been conducted in NIR tomographic imaging of the breast, but in general, NIR fluorescence is suitable for diseases in superficial tissues (e.g., within several millimeters of the surface for planar epifluorescence imaging47). Finally, although the current agent provides molecular information, which is an improvement over anatomical images, a single biomarker will not be able to differentiate all tumor types. Additional biomarkers, e.g., from dual-channel imaging at a second NIR wavelength with a different agent, could significantly increase the diagnostic specificity. The use of negatively charged agents should be adaptable to other small molecule targeting ligands against different extracellular markers.
In conclusion, the high negative charge density on the agent enabled sufficient oral absorption, reduced nonspecific internalization, facilitated targeting by extending the clearance half-life, and increased retention through cellular trapping to yield efficient targeting contrast in a small animal model of breast cancer for detection at clinically relevant depths. To our knowledge, this is the first demonstration of a disease screening approach using oral administration of a molecular imaging agent, and these mechanisms should be applicable to additional agents and disease targets for developing a series of molecular imaging agents for noninvasive screening.
Acknowledgments
We thank Liang Zhang for processing the histology slides. Funding: Support for this work was provided in part by the Foundation for Studying and Combating Cancer (G.M.T.), a National Science Foundation CAREER Award (G.M.T.), and NIH R01GM115481 (to D.E.S.). Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA046592 by the use of the following Cancer Center Shared Resource(s): histology and imaging. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.7b00994.
Oral absorption of anionic molecules; intact excretion of probe in urine; binding affinity of imaging agents; plasma clearance curves; negative controls for confocal images; serum stability of the agent; clinical breast imaging studies; target and ligand selection criteria; integrin expression levels; purification methods and purity of imaging agents (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Goldberg M.; Gomez-Orellana I. Challenges for the oral delivery of macromolecules. Nat. Rev. Drug Discovery 2003, 2 (4), 289–295. 10.1038/nrd1067. [DOI] [PubMed] [Google Scholar]
- Hope-Ross M.; Yannuzzi L. A.; Gragoudas E. S.; Guyer D. R.; Slakter J. S.; Sorenson J. A.; Krupsky S.; Orlock D. A.; Puliafito C. A. Adverse reactions due to indocyanine green. Ophthalmology 1994, 101 (3), 529–33. 10.1016/S0161-6420(94)31303-0. [DOI] [PubMed] [Google Scholar]
- Jackisch C.; Muller V.; Maintz C.; Hell S.; Ataseven B. Subcutaneous Administration of Monoclonal Antibodies in Oncology. Geburtshilfe Frauenheilkd. 2014, 74 (4), 343–349. 10.1055/s-0034-1368173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utz K. S.; Hoog J.; Wentrup A.; Berg S.; Lammer A.; Jainsch B.; Waschbisch A.; Lee D. H.; Linker R. A.; Schenk T. Patient preferences for disease-modifying drugs in multiple sclerosis therapy: a choice-based conjoint analysis. Ther. Adv. Neurol. Disord. 2014, 7 (6), 263–75. 10.1177/1756285614555335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46 (1–3), 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Porras A. G.; Holland S. D.; Gertz B. J. Pharmacokinetics of alendronate. Clin. Pharmacokinet. 1999, 36 (5), 315–28. 10.2165/00003088-199936050-00002. [DOI] [PubMed] [Google Scholar]
- Coleman P. J.; Brashear K. M.; Askew B. C.; Hutchinson J. H.; McVean C. A.; Duong L. T.; Feuston B. P.; Fernandez-Metzler C.; Gentile M. A.; Hartman G. D.; Kimmel D. B.; Leu C. T.; Lipfert L.; Merkle K.; Pennypacker B.; Prueksaritanont T.; Rodan G. A.; Wesolowski G. A.; Rodan S. B.; Duggan M. E. Nonpeptide alpha(v)beta(3) antagonists. Part 11: Discovery and preclinical evaluation of potent alpha v beta(3) antagonists for the prevention and treatment of osteoporosis. J. Med. Chem. 2004, 47 (20), 4829–4837. 10.1021/jm049874c. [DOI] [PubMed] [Google Scholar]
- Coleman P. J.; Brashear K. M.; Hunt C. A.; Hoffman W. F.; Hutchinson J. H.; Breslin M. J.; McVean C. A.; Askew B. C.; Hartman G. D.; Rodan S. B.; Rodan G. A.; Leu C. T.; Prueksaritanont T.; Fernandez-Metzler C.; Ma B.; Libby L. A.; Merkle K. M.; Stump G. L.; Wallace A. A.; Lynch J. J.; Lynch R.; Duggan M. E. Non-peptide alpha(v)beta(3) antagonists. Part 3: identification of potent RGD mimetics incorporating novel beta-amino acids as aspartic acid replacements. Bioorg. Med. Chem. Lett. 2002, 12 (1), 31–4. 10.1016/S0960-894X(01)00666-7. [DOI] [PubMed] [Google Scholar]
- Han X.; Nabors L. B. Integrins - a Relevant Target in Glioblastoma. European Journal of Clinical and Medical Oncology 2010, 2 (1), 59–64. [Google Scholar]
- Millard M.; Odde S.; Neamati N. Integrin targeted therapeutics. Theranostics 2011, 1, 154–88. 10.7150/thno/v01p0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder R. L. Integrin alpha V beta 3 as a target for treatment of rheumatoid arthritis and related rheumatic diseases. Ann. Rheum. Dis. 2002, 61 (Suppl 2), ii96–ii99. 10.1136/ard.61.suppl_2.ii96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossodo S.; Pickarski M.; Lin S. A.; Gleason A.; Gaspar R.; Buono C.; Ho G.; Blusztajn A.; Cuneo G.; Zhang J.; et al. Dual in vivo quantification of integrin-targeted and protease-activated agents in cancer using fluorescence molecular tomography (FMT). Mol Imaging Biol 2010, 12 (5), 488–499. 10.1007/s11307-009-0279-z. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Bhatnagar S.; Deschenes E.; Thurber G. M. Mechanistic and quantitative insight into cell surface targeted molecular imaging agent design. Sci. Rep. 2016, 6, 25424. 10.1038/srep25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurber G. M.; Reiner T.; Yang K. S.; Kohler R. H.; Weissleder R. Effect of Small-Molecule Modification on Single-Cell Pharmacokinetics of PARP Inhibitors. Mol. Cancer Ther. 2014, 13 (4), 986–95. 10.1158/1535-7163.MCT-13-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamimoto R.; Senda M.; Jinnouchi S.; Terauchi T.; Yoshida T.; Inoue T. Detection of breast cancer in an FDG-PET cancer screening program: results of a nationwide Japanese survey. Clin. Breast Cancer 2015, 15 (2), e139–46. 10.1016/j.clbc.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Lannin D. R.; Wang S. Are Small Breast Cancers Good because They Are Small or Small because They Are Good?. N. Engl. J. Med. 2017, 376 (23), 2286–91. 10.1056/NEJMsr1613680. [DOI] [PubMed] [Google Scholar]
- Welch H. G.; Prorok P. C.; O’Malley A. J.; Kramer B. S. Breast-Cancer Tumor Size, Overdiagnosis, and Mammography Screening Effectiveness. N. Engl. J. Med. 2016, 375 (15), 1438–1447. 10.1056/NEJMoa1600249. [DOI] [PubMed] [Google Scholar]
- Gill J. K.; Maskarinec G.; Pagano I.; Kolonel L. N. The association of mammographic density with ductal carcinoma in situ of the breast: the Multiethnic Cohort. Breast Cancer Res. 2006, 8 (3), R30. 10.1186/bcr1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E.; Velentgas P.; Mandelson M. T.; Lehman C. D.; Elmore J. G.; Porter P.; Yasui Y.; Taplin S. H. Variation in Mammographic Breast Density by Time in Menstrual Cycle Among Women Aged 40–49 Years. JNCI: Journal of the National Cancer Institute 1998, 90 (12), 906–910. 10.1093/jnci/90.12.906. [DOI] [PubMed] [Google Scholar]
- Ong M. S.; Mandl K. D. National expenditure for false-positive mammograms and breast cancer overdiagnoses estimated at $4 billion a year. Health affairs 2015, 34 (4), 576–83. 10.1377/hlthaff.2014.1087. [DOI] [PubMed] [Google Scholar]
- Hawrysz D. J.; Sevick-Muraca E. M. Developments toward diagnostic breast cancer imaging using near-infrared optical measurements and fluorescent contrast agents. Neoplasia 2000, 2 (5), 388–417. 10.1038/sj.neo.7900118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos V.; Yodh A. G.; Schnall M.; Chance B. Concurrent MRI and diffuse optical tomography of breast after indocyanine green enhancement. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (6), 2767–72. 10.1073/pnas.040570597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Chance B.; Yodh A. G. Fluorescent heterogeneities in turbid media: limits for detection, characterization, and comparison with absorption. Appl. Opt. 1998, 37 (28), 6833–44. 10.1364/AO.37.006833. [DOI] [PubMed] [Google Scholar]
- Davis S. C.; Pogue B. W.; Dehghani H.; Paulsen K. D. Tissue drug concentration determines whether fluorescence or absorption measurements are more sensitive in diffuse optical tomography of exogenous contrast agents. Appl. Opt. 2009, 48 (10), D262–72. 10.1364/AO.48.00D262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E.; Brown S. L.; Stupack D. G.; Puente X. S.; Cheresh D. A.; Nemerow G. R. Integrin αvβ1 Is an Adenovirus Coreceptor. Journal of virology 2001, 75 (11), 5405–5409. 10.1128/JVI.75.11.5405-5409.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. M.; Dahan A.; Gupta D.; Varghese S.; Amidon G. L. Enabling the intestinal absorption of highly polar antiviral agents: ion-pair facilitated membrane permeation of zanamivir heptyl ester and guanidino oseltamivir. Mol. Pharmaceutics 2010, 7 (4), 1223–34. 10.1021/mp100050d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira S.; Cohen R.; Stigter-van Walsum M.; van Dongen G.; Elias S. G.; van Diest P. J.; Mali W.; van Bergen en Henegouwen P. M. A novel method to quantify IRDye800CW fluorescent antibody probes ex vivo in tissue distribution studies. EJNMMI Res. 2012, 2 (1), 50. 10.1186/2191-219X-2-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czogalla A. Oral cyclosporine A - the current picture of its liposomal and other delivery systems. Cellular and Molecular Biology Letters 2009, 14 (1), 139–152. 10.2478/s11658-008-0041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert M. F.; Roberts J. P.; Prueksaritanont T.; Benet L. Z. Bioavailability of cyclosporine with concomitant rifampin administration is markedly less than predicted by hepatic enzyme induction. Clin. Pharmacol. Ther. 1992, 52 (5), 453–457. 10.1038/clpt.1992.171. [DOI] [PubMed] [Google Scholar]
- Weissleder R. A clearer vision for in vivo imaging. Nat. Biotechnol. 2001, 19 (4), 316–317. 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- Cilliers C.; Liao J.; Atangcho L.; Thurber G. Residualization Rates of Near-Infrared Dyes for the Rational Design of Molecular Imaging Agents. Molecular Imaging and Biology 2015, 17, 757–762. 10.1007/s11307-015-0851-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurber G. M.; Yang K. S.; Reiner T.; Kohler R. H.; Sorger P.; Mitchison T.; Weissleder R. Single-cell and subcellular pharmacokinetic imaging allows insight into drug action in vivo. Nat. Commun. 2013, 4, 1504. 10.1038/ncomms2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabors L. B.; Mikkelsen T.; Rosenfeld S. S.; Hochberg F.; Akella N. S.; Fisher J. D.; Cloud G. A.; Zhang Y.; Carson K.; Wittemer S. M.; Colevas A. D.; Grossman S. A. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J. Clin. Oncol. 2007, 25 (13), 1651–7. 10.1200/JCO.2006.06.6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Niu G.; Wu H.; Chen X. Clinical Application of Radiolabeled RGD Peptides for PET Imaging of Integrin alphavbeta3. Theranostics 2016, 6 (1), 78–92. 10.7150/thno.13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd N. F.; Martin L. J.; Yaffe M. J.; Minkin S. Mammographic density and breast cancer risk: current understanding and future prospects. Breast Cancer Res. 2011, 13 (6), 223. 10.1186/bcr2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habel L. A.; Dignam J. J.; Land S. R.; Salane M.; Capra A. M.; Julian T. B. Mammographic density and breast cancer after ductal carcinoma in situ. J. Natl. Cancer Inst 2004, 96 (19), 1467–72. 10.1093/jnci/djh260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques S. L. Optical properties of biological tissues: a review. Phys. Med. Biol. 2013, 58 (11), R37–61. 10.1088/0031-9155/58/11/R37. [DOI] [PubMed] [Google Scholar]
- van de Ven S.; Wiethoff A.; Nielsen T.; Brendel B.; van der Voort M.; Nachabe R.; Van der Mark M.; Van Beek M.; Bakker L.; Fels L.; Elias S.; Luijten P.; Mali W. A novel fluorescent imaging agent for diffuse optical tomography of the breast: first clinical experience in patients. Molecular imaging and biology: MIB: the official publication of the Academy of Molecular Imaging 2010, 12 (3), 343–8. 10.1007/s11307-009-0269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.; Ouyang Z.; Wang F.; Wu H.; Jia B.; Chordia M. D. Evaluation of Tc-99m-3PRGD2 Integrin Receptor Imaging in the Differential Diagnosis of Breast Lesions and Comparison With Mammography. Cancer Invest. 2017, 35 (2), 108–115. 10.1080/07357907.2016.1270957. [DOI] [PubMed] [Google Scholar]
- Narod S. A.; Iqbal J.; Giannakeas V.; Sopik V.; Sun P. Breast Cancer Mortality After a Diagnosis of Ductal Carcinoma In Situ. JAMA oncology 2015, 1 (7), 888–96. 10.1001/jamaoncol.2015.2510. [DOI] [PubMed] [Google Scholar]
- Esserman L.; Yau C. Rethinking the Standard for Ductal Carcinoma In Situ Treatment. JAMA oncology 2015, 1 (7), 881–3. 10.1001/jamaoncol.2015.2607. [DOI] [PubMed] [Google Scholar]
- Ralay-Ranaivo B.; Desmaele D.; Bianchini E. P.; Lepeltier E.; Bourgaux C.; Borgel D.; Pouget T.; Tranchant J. F.; Couvreur P.; Gref R. Novel self assembling nanoparticles for the oral administration of fondaparinux: synthesis, characterization and in vivo evaluation. J. Controlled Release 2014, 194, 323–31. 10.1016/j.jconrel.2014.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbit E.; Goldberg M.; Gomez-Orellana I.; Majuru S. Oral heparin: status review. Thromb. J. 2006, 4, 6. 10.1186/1477-9560-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpi N. Oral bioavailability of chondroitin sulfate (Condrosulf) and its constituents in healthy male volunteers. Osteoarthritis Cartilage 2002, 10 (10), 768–77. 10.1053/joca.2002.0824. [DOI] [PubMed] [Google Scholar]
- van Dam G. M.; Themelis G.; Crane L. M.; Harlaar N. J.; Pleijhuis R. G.; Kelder W.; Sarantopoulos A.; de Jong J. S.; Arts H. J.; van der Zee A. G.; Bart J.; Low P. S.; Ntziachristos V. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-alpha targeting: first in-human results. Nat. Med. 2011, 17 (10), 1315–9. 10.1038/nm.2472. [DOI] [PubMed] [Google Scholar]
- McCamish M.; Woollett G. Worldwide experience with biosimilar development. MAbs 2011, 3 (2), 209–17. 10.4161/mabs.3.2.15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurber G. M.; Figueiredo J. L.; Weissleder R. Detection Limits of Intraoperative Near Infrared Imaging for Tumor Resection. J. Surg. Oncol. 2010, 102 (7), 758–764. 10.1002/jso.21735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.