

Abstract
Cobalamin-dependent radical S-adenosylmethionine (SAM) methylases play vital roles in the de novo biosynthesis of many antibiotics, cofactors, and other important natural products, yet remain an understudied subclass of radical SAM (RS) enzymes. In addition to a [4Fe-4S] cluster that is ligated by three cysteine residues, these enzymes also contain an N-terminal cobalamin-binding domain. In vitro studies of these enzymes have been severely limited because many are insoluble or sparingly soluble upon their overproduction in E. coli. This solubility issue has led a number of groups either to purify the protein from inclusion bodies or to purify soluble protein that often lacks proper cofactor incorporation. Herein, we use TsrM as a model to describe methods that we have used to generate soluble protein that is purified in an active form with both cobalamin and [4Fe-4S] cluster cofactors bound. Additionally, we highlight the methods that we developed to characterize the enzyme following purification.
Keywords: S-adenosylmethionine, radical, methylcobalamin, tryptophan, iron-sulfur cluster, electron paramagnetic resonance spectroscopy, SUMO, methylase, 2-methyltryptophan
1. Introduction
In 2001, a bioinformatics study identified the radical S-adenosylmethionine (SAM) superfamily of enzymes, which, at the time, contained 600 members catalyzing more than 30 different reactions (Sofia, Chen, Hetzler, Reyes-Spindola, & Miller, 2001). To date, the radical SAM (RS) superfamily is composed of almost 114,000 individual protein sequences, catalyzing more than 85 distinct reactions (Akiva et al., 2014). All RS enzymes harbor a [4Fe-4S] cluster coordinated by three conserved cysteines that are most commonly found in a CxxxCxxC motif (Sofia et al., 2001). In almost all cases, the reduced form of the cluster, [4Fe-4S]+, is used to catalyze the reductive cleavage of SAM to methionine and a 5′-deoxyadenosine 5′-radical (5′-dA•) (Frey & Booker, 2001; Frey, Hegeman, & Ruzicka, 2008), which initiates catalysis by abstracting a hydrogen atom from a target substrate. RS enzymes have been predicted and/or shown to catalyze a vast range of biologically important reactions, which include sulfur insertion and thioether bond formation, methylation and methythiolation of unactivated carbon and/or phosphinate phosphorous centers, oxidative decarboxylation of unactivated carbon centers and other C–C bond fragmentations, C–C bond formation, C–P bond fragmentation, cyclopropanation, epimerization, hydroxylation, metallocofactor assembly and organometallic bond formation, oxidation, mutase reactions, and complex rearrangements. These reactions are involved in numerous critical biological pathways, such as DNA/RNA biosynthesis and repair, protection against viral invasion, the biosynthesis of a large number of enzyme cofactors and clinically relevant natural products, nucleic acid and protein maturation, and antibiotic resistance to antibiotic biosynthesis, amongst many others (Booker & Grove, 2010; Broderick, Duffus, Duschene, & Shepard, 2014; Landgraf, McCarthy, & Booker, 2016).
Methylation of unactivated carbon and phosphinate phosphorous centers is uniquely carried out by the subfamily of RS enzymes known as RS methylases. In most instances, the added methyl group is derived from SAM; however, the mechanisms by which the methyl group is added are distinctly different from that of traditional SAM-dependent methyltransferases. RS methylases have been organized into five classes to date: A, B, C, D, and E (Bauerle, Schwalm, & Booker, 2015; Hu & Ribbe, 2016; Zhang, van der Donk, & Liu, 2012). Class A RS methylases are comprised of RlmN and Cfr, which methylate the C2 or C8 amidine carbons of specific adenosine nucleobases in ribosomal RNA (rRNA) or transfer RNA (tRNA) (Kowalak, Bruenger, & McCloskey, 1995; Toh, Xiong, Bae, & Mankin, 2008). Class B methylases require a cobalamin cofactor in addition to the [4Fe-4S] cluster shared by all RS enzymes. These enzymes appear to be the most versatile of the RS methylases, because they methylate sp2- and sp3-hybridized carbon atoms as well as phosphinate phosphorus atoms, while Class A, C, and D RS methylases only methylate sp2-hybridized carbon atoms (Zhang et al., 2012). Class C RS methylases are predicted to use two simultaneously bound SAM molecules to methylate sp2-hybridized carbon centers during the biosynthesis of several complex natural products. Class D methylases also methylate sp2-hybridized carbon centers, but have been suggested to use methylenetetrahydrofolate as the methyl source (Allen, Xu, & White, 2014). The recently annotated Class E RS methylases, by contrast, are involved in the maturation of the M cluster of nitrogenase, and appear to methylate a sulfide ion as part of a metallocluster (Hu & Ribbe, 2016).
Class B RS methylases catalyze key steps in the biosynthesis of a number of biomolecules, including protein cofactors, antibiotics, herbicides, and other natural products, such as bacteriochlorophyll (Chew & Bryant, 2007), pactamycin (Kudo, Kasama, Hirayama, & Eguchi, 2007), mitomycin C (Mao, Varoglu, & Sherman, 1999), moenomycin A (Ostash, Saghatelian, & Walker, 2007), fosfomycin (Hidaka, Goda, et al., 1995; Kuzuyama, Hidaka, Kamigiri, Imai, & Seto, 1992; Woodyer et al., 2006), thienamycin (Nunez, Mendez, Brana, Blanco, & Salas, 2003), gentamicin (J. Y. Kim et al., 2008), clorobiocin (Anderle et al., 2007), fortimicin (Dairi, Ohta, Hashimoto, & Hasegawa, 1992; Kuzuyama, Seki, Dairi, Hidaka, & Seto, 1995), thiostrepton (Kelly, Pan, & Li, 2009), chondrochloren (Rachid, Scharfe, Blocker, Weissman, & Muller, 2009), polytheonamide (Parent et al., 2016), cystobactamids (Baumann et al., 2014; Wang, Schnell, Baumann, Muller, & Begley, 2017), watasemycin (Inahashi et al., 2017), and bialaphos (Hidaka, Hidaka, Kuzuyama, & Seto, 1995; Kamigiri, Hidaka, Imai, Murakami, & Seto, 1992). However, mechanistic investigations of these enzymes have been limited, due in large part to their inherent insolubility upon overproduction in E. coli. Seven Class B RS methylases have been studied in vitro; however, their mechanistic characterization has been scant. Fom3, GenK, and PhpK were solubilized from inclusion bodies by treatment with urea and then refolded and reconstituted with their iron-sulfur (Fe-S) cluster and cobalamin cofactors. However, this strategy resulted in enzymes with poor cofactor incorporation and low activity (Allen & Wang, 2014; H. J. Kim et al., 2013; Werner et al., 2011). Three other Class B methylases, PoyC, ThnK, and TsrM, were amenable to purification from soluble crude lysate in an active form that contained both cofactors (Blaszczyk et al., 2016; Marous et al., 2015; Parent et al., 2016), but only when overproduced in a specialized medium containing ethanolamine, which has been suggested to increase cobalamin uptake in E. coli (Bandarian & Matthews, 2004). Lastly, another Class B RS methylase, CysS, has been purified from soluble crude lysate and reconstituted with cobalamin to yield an active form of the enzyme that contains the Fe-S cluster as well as cobalamin (Wang et al., 2017).
In this chapter, we describe strategies that we have developed for overproducing, purifying, and characterizing RS methylases, with a particular focus on TsrM (Blaszczyk et al., 2016), which methylates C2 of tryptophan (Trp) as part of the biosynthesis of the quinaldic acid moiety of the thiopeptide natural product thiostrepton (Li & Kelly, 2010; Pierre et al., 2012). A previous paper in Methods in Enzymology described our strategies for overproducing and characterizing Fe-S containing enzymes, with a particular focus on those in the RS superfamily (Lanz et al., 2012). In this work, we combine these two strategies, but focus on characterization of the cobalamin cofactor.
2. Overproduction and Purification of TsrM
Our initial attempts to purify TsrM focused on using the same overproduction and purification strategies previously shown to be successful for other RS enzymes (Lanz et al., 2012); however, these methods resulted in poor quantities of protein that was typically less than 10% pure. Almost all of the protein was produced in inclusion bodies. The tsrM gene was eventually cloned into a pSUMO vector, wherein TsrM would be overproduced as a C-terminal fusion with SUMO, a small protein-based tag (~12 kDa) used to modify proteins post-translationally during various cellular processes. SUMO has been shown to increase the solubility of proteins to which it is fused, and can be excised from the fusion construct by a very specific protease, Ulp1, which does not leave residual amino acids on the target protein (Jeffrey G. Marblestone et al., 2006). The N-terminal hexahistidine (His6) tag on the SUMO protein allows for facile purification by immobilized metal affinity chromatography (IMAC). Upon cleavage of the fusion construct by Ulp1, the affinity resin can be used to capture SUMO and other proteins that bind nonspecifically to it. However, only after overproduction of the SUMO-TsrM fusion protein in the RosettaBlue (DE3) pLysS strain of E. coli, was TsrM able to be purified to homogeneity in reasonable yields (10 mg per 16 L of original culture). Below is a detailed description of the methods used to overproduce TsrM and purify it to homogeneity.
2.1 Gene cloning and expression strategy
The tsrM gene from Streptomyces laurentii was cloned into the pSUMO vector (LifeSensors) to generate a construct that encodes a SUMO-TsrM fusion protein with an N-terminal hexahistidine (His6) tag (Blaszczyk et al., 2016). The pSUMO vector is constructed from a pET24 vector. The pET cloning system, coupled with the strong T7 promoter, allows for facile cloning and robust overproduction of proteins (Panavas, Sanders, & Butt, 2009). The pSUMO vector also contains a kanamycin (Kan) resistance marker for in vivo selection, which, in addition, allows for coexpression of genes contained on plasmid pDB1282, because this plasmid confers ampicillin (Amp) resistance. pDB1282 harbors the isc operon from Azotobacter vinelandii, which encodes proteins involved in Fe-S cluster assembly (Cicchillo et al., 2004; Zheng, Cash, Flint, & Dean, 1998). The RosettaBlue (DE3) pLysS strain of E. coli was cotransformed with pDB1282 and pSUMO-TsrM, giving rise to a strain that confers resistance to Kan, Amp, and chloramphenicol (Cam), which is encoded on the plasmid. The RosettaBlue (DE3) pLysS strain is similar to the commonly used BL21 (DE3) strain of E. coli, but encodes additional tRNAs that are rarely used in E. coli. In addition, pLysS expresses low levels of T7 lysozyme, which inhibits T7 RNA polymerase, thereby reducing its basal expression levels (Dubendorf & Studier, 1991).
Overproduction of TsrM is performed in M9-ethanolamine medium supplemented with cobalamin. This medium is similar to M9-minimal media, but lacks ammonium chloride and glucose, which serve as nitrogen and carbon sources, respectively. It has been shown that culturing E. coli under conditions in which ethanolamine serves as the sole source of nitrogen and carbon leads to upregulation of the adenosylcobalamin (AdoCbl)-dependent ethanolamine ammonium lyase (EAL) (Blackwell & Turner, 1978), which catalyzes the conversion of ethanolamine to acetaldehyde and ammonia (Babior, Moss, Orme-Johnson, & Beinert, 1974). More importantly, when M9-ethanolamine media is supplemented with cobalamin, the intracellular cobalamin concentration is augmented (Bandarian & Matthews, 2004). This increase in cobalamin concentration can be leveraged to overproduce TsrM with a bound cobalamin cofactor.
2.2 Protocol for expression of the tsrM gene
Introduce pDB1282 and pSUMO-TsrM by cotransformation of E. coli RosettaBlue (DE3) pLysS using standard procedures with at least 1 μg of each plasmid. Select transformants on Luria–Bertani (LB) plates containing Kan, Amp, and Cam at final concentrations of 50, 100, and 35 μg/mL, respectively.
Use a single colony to inoculate 200 mL of LB medium containing Kan, Amp, and Cam at 50, 100, and 35 μg/mL, respectively. Incubate at 37 °C and shake at 250 rpm for ~24 h. In addition, place four 6 L flasks, each containing 4 L of M9-ethanolamine medium (Table 1) in an incubator-shaker to allow for equilibration to 37 °C.
Add the following to each flask just prior to inoculation: 50 μg/mL Kan, 100 μg/mL Amp, 35 μg/mL Cam, 54 mL of 10 × cobalamin supplement (Table 1), and 100 mL of 1 M ethanolamine (Table 1).
Inoculate each 6 L flask with ~2 mL of the 200 mL starter culture grown to an OD600 of 2.0.
At an OD600 of 0.3 (~15 h), add L-arabinose to a final concentration of 0.1% (w/v), and FeCl3 and L-cysteine to final concentrations of 25 and 150 μM, respectively.
At an OD600 of 0.6, cool flasks to ~20 °C in an ice bath and adjust the temperature in the incubator to 20 °C. Once the medium is equilibrated (about 20 min), return the flasks to the shaker and induce expression of the tsrM gene by adding isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 800 μM. Add FeCl3 and L-cysteine again to final concentrations of 25 and 150 μM, respectively.
Allow cultures to incubate for ~16 h at 20 °C while shaking at 180 rpm.
Harvest the bacteria by centrifugation and freeze the resulting cell paste (~1.5 g/L) in liquid N2 and store in a liquid N2 dewar.
Table 1.
Composition of M9-ethanolamine Minimal Medium
| 4 L of 5× M9 Saltsa | g |
|---|---|
| Na2HPO4 (Anhydrous) | 136 |
| KH2PO4 | 60 |
| NaCl | 10 |
| 1 L of 10,000× Micronutrient Mixture | mg/L |
| (NH4)2Mo7O2 | 37.1 |
| CoCl2•6H2O | 71.4 |
| H3BO3 | 247 |
| CuSO4•5H2O | 25 |
| MnCl2•4H2O | 198 |
| ZnSO4•7H2O | 28.8 |
| 500 mL of 10 × Cobalamin Supplementb | Quantity |
| 10,000× Micronutrient Mixture | 4 mL |
| MgSO4•7H2O | 9.2 g |
| CaCl2•2H2O | 0.6 g |
| Thiamine | 37 mg |
| Hydroxocobalamin | 126 mg |
| 1 M Ethanolamineb | Volume |
| 12 M Stock HCl | 78 mL |
| 16 M Stock Ethanolamine | 60 mL |
| Water | 862 mL |
| pH to 7.4 with HCl or Ethanolamine | |
| Volumes for 4 L of M9 Ethanolamine Minimal Medium | |
| 20× M9 Salts | 800 mL |
| 10× Cobalamin Supplement | 54 mL |
| 1 M Ethanolamine | 100 mL |
| ddH2Oc | 3046 mL |
After salts dissolve, adjust the pH of the medium to 7.4 with HCl and KOH; adjust volume to 4 L with water.
Solutions should be sterilized by filtration and added just before inoculation.
Medium should be autoclaved after addition to ddH2O in a 6 L Erlenmeyer flask.
2.3 Purification of TsrM
In addition to enhancing the solubility of TsrM during overproduction, the SUMO-TsrM fusion construct allows for an additional purification step following cleavage with Ulp1 (Figure 1). After initial purification by IMAC, the protein is judged to be ~30% pure by SDS-PAGE analysis. Native TsrM is then generated upon cleavage with Ulp1 and can be collected in the flow-through after re-applying the cleavage mixture to the IMAC matrix (J. G. Marblestone et al., 2006). The SUMO tag and proteins that bind to the matrix nonspecifically are recaptured during this step. An SDS-PAGE analysis of the TsrM purification process is displayed in Figure 2.
Figure 1.
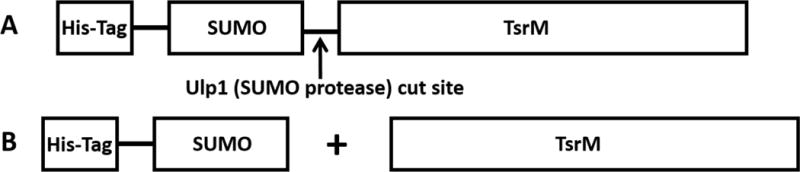
SUMO-TsrM fusion construct (A) before and (B) after cleavage.
Figure 2.
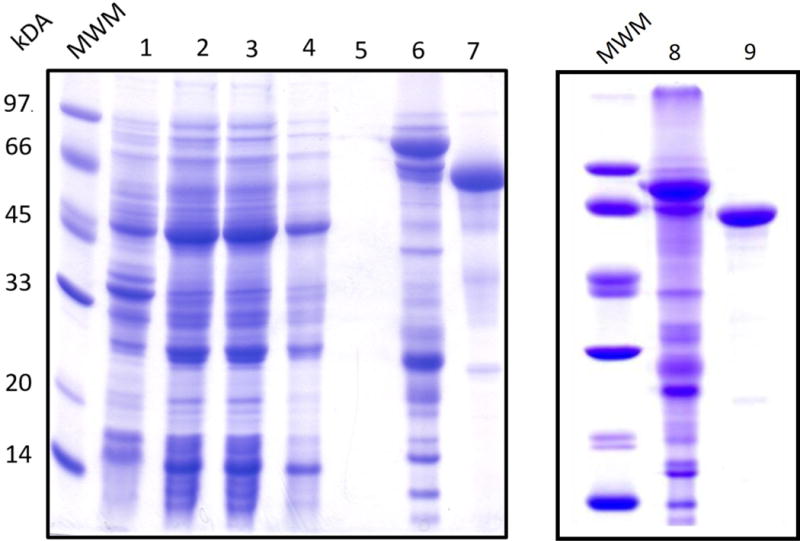
SDS-PAGE gel of TsrM purification. The lanes are as follows: (1) pellet, (2) crude lysate, (3) flow through, (4) wash 1, (5) wash 2, (6) eluate, (7) post-cleavage flow through, (8) eluate, and (9) post-S200.
One of the greatest challenges of isolating RS enzymes is their instability upon exposure to oxygen. To address this issue, purification of TsrM and all subsequent manipulations of the protein are done in a Coy anaerobic chamber to exclude oxygen, as has been previously described (Lanz et al., 2012). Additionally, the purification process is completed without interruption while keeping the protein chilled during each step. The following steps detail the protocol that is used to isolate homogeneous TsrM in its native form.
In an anaerobic chamber, prepare 500 mL each of Lysis Buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 5% glycerol, 10 mM BME, 10 mM imidazole), Wash Buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 5% glycerol, 10 mM BME, 20 mM imidazole), Elution Buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 15% glycerol, 10 mM BME, 300 mM imidazole), Cleavage Buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 15% glycerol, 10 mM BME), and Gel-filtration Buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 15% glycerol, 5 mM DTT) with water that is oxygen free. Except for centrifugation, all subsequent steps are performed in an anaerobic chamber.
In the anaerobic chamber, resuspend 50 g of frozen cell paste in 150 mL Lysis Buffer containing lysozyme (1 mg/mL), and DNase I (0.1 mg/mL) in a 250 mL metal sonication cup. Hydroxocobalamin (OHCbl) at 0.2 mg/mL can also be included if necessary. However, in TsrM, cobalamin incorporation takes place during expression; inclusion of hydroxocobalamin in the lysis buffer has no additional effect.
Once the cell paste is completely resuspended (~30 min), insert the metal sonication cup into a crystallizing dish packed with ice, ensuring not to introduce any ice (which contains oxygen) into the sonication cup.
Sonicate for 45 s at 65% output using a Fisher 1 550 sonic dismembrator. Repeat for at least 6 cycles with a 7 min interval between each cycle to allow for temperature re-equilibration (~8 °C).
Pour the lysate into centrifuge tubes. Cap the tubes tightly and seal the edges with vinyl tape before removing from the anaerobic chamber.
Centrifuge the lysate at 45,000 × g for 1 h at 4 °C. During centrifugation, equilibrate a column of Ni-NTA resin with the chilled Lysis Buffer.
After centrifugation, re-introduce the centrifuge tubes into the anaerobic chamber and carefully decant the supernatant into a clean 250 mL bottle. Remove a 100 μL sample of the supernatant and a sample of the pellet for SDS-PAGE analysis.
Load the supernatant onto the column. Proceed to wash with 150 mL of pre-chilled Wash Buffer. Take 100 μL samples of the flow-through both from the loaded lysate and from the two washes for SDS-PAGE analysis.
Elute the protein from the column with pre-chilled Elution Buffer. Once the colored band begins to elute, collect fractions until all of the protein (brown/black band) elutes from the column. Concentrate the protein by centrifugal ultrafiltration at 3,500 × g at 4 °C in an Amicon centrifugal filter device (10 kDA molecular weight cutoff) to a final volume of < 2.5 mL. While the protein is concentrating, equilibrate a PD-10 column with pre-chilled Cleavage Buffer.
Collect a 20 μL sample of the concentrated protein for SDS-PAGE analysis. Exchange the protein into pre-chilled Cleavage Buffer using the pre-equilibrated PD-10 column. Treat the flow-through with ~400 μg of the commercially available Ulp1 protease. Keep the cleavage reaction overnight on ice to generate native TsrM. Note that a chemical reconstitution step can take place simultaneously at this time by adding FeCl3•6H2O, Na2S•9H2O, and additional BME to the cleavage reaction, as described in detail below.
After allowing the cleavage reaction to proceed for ~15 h, equilibrate a Ni-NTA column with 100 mL of pre-chilled Cleavage Buffer.
Load the pre-chilled cleavage reaction onto the Ni-NTA column and elute TsrM with additional Cleavage Buffer. Collect the red/brown band of native TsrM (may be very faint in color). The SUMO protein and Ulp1 protease, both of which contain hexahistidine tags, should remain bound to the Ni-NTA resin.
Concentrate TsrM by centrifugal ultrafiltration at 3,500 × g at 4 °C in an Amicon centrifugal filter device (10 kDA molecular weight cutoff) to a final volume of < 2.5 mL. While the protein is concentrating, equilibrate the PD-10 column with Gel-Filtration Buffer. Elute the nonspecifically bound proteins on the Ni-NTA resin with Elution Buffer.
Collect 20 μL of native TsrM for SDS-PAGE analysis. Exchange the protein into Gel-Filtration Buffer using the equilibrated PD-10 column.
Concentrate the protein by centrifugal ultrafiltration with the Amicon filter devices if desired. Aliquot and flash-freeze the protein in liquid N2 before storing in a liquid N2 dewar.
2.4 Chemical reconstitution of as-isolated TsrM
Although the overproduction and purification methods described above result in TsrM that binds cobalamin stoichiometrically and contains 3.4 and 3.1 moles of iron and sulfide, respectively, reconstitution can enhance Fe-S cluster incorporation. To avoid an additional overnight incubation step, Ulp1 can be added to the chemical reconstitution reaction, such that cleavage and reconstitution take place simultaneously, given that both steps require long incubation periods. Although DTT has been used in the reconstitution of other RS enzymes, this reconstitution method requires an additional IMAC step following cleavage of the SUMO-TsrM construct. During this step, the reductant β-mercaptoethanol (BME) is used, because DTT can reduce the Ni2+ ions in the Ni-NTA resin when present at concentrations greater than 1 mM (Lanz et al., 2012).
Introduce the following into the anaerobic chamber: 27 mg of FeCl3•6H2O (270.3 g/mol), 24 mg of Na2S•9H2O (240.2 g/mol), 17.5 μL of stock BME (14.3 M), and a 15 mL conical tube.
Combine the following into the conical tube: 6.03 mL of Cleavage Buffer, 1 mL of 1 M HEPES buffer, pH 7.5, and the 17.5 μL of BME (14.3 M).
Dissolve the FeCl3•6H2O and Na2S•9H2O in 1 mL of water. Add 50 μL of the Na2S solution followed by 50 μL of the FeCl3 solution to the conical tube.
Place the mixture on ice for ~15 min before adjusting the pH, if necessary, to ~7.0.
Add 2.85 mL of the cleavage reaction, which includes SUMO-TsrM and Ulp1, and incubate on ice overnight for ~12 h.
Centrifuge the reconstituted protein at 10,000 × g to remove any insoluble aggregates.
Apply the supernatant onto the Ni-NTA resin and collect the flow-through, which should contain native TsrM.
Concentrate the flow-through to ≤ 2.5 mL by centrifugal ultrafiltration as described above.
Exchange the concentrated protein into Gel-Filtration Buffer using a PD-10 column.
Load the protein onto an S-200 column equilibrated in Gel-Filtration Buffer and connected to an ÅKTA fast-protein liquid chromatography system (housed in an anaerobic chamber) for further purification by size exclusion chromatography.
3. Determination of Cofactor Stoichiometry
3.1 Strategy for determining the stoichiometry and forms of iron-sulfur clusters
Determining the correct number of Fe-S clusters per polypeptide is critical for accurate characterization of RS enzymes, which is typically performed in concert with Mössbauer and electron paramagnetic resonance (EPR) spectroscopies (Pandelia, Lanz, Booker, & Krebs, 2015). We follow protocols that have been previously described for determining the amount of iron and sulfide per protein in other RS enzymes (Lanz et al., 2012). A brief description of these methods follows.
Following purification, the concentration of enzyme is determined by the Bradford assay using bovine serum albumin, fraction V, (BSA) as a standard (Bradford, 1976).
Amino acid analysis is conducted to determine a correction factor for TsrM when BSA is used as a standard for Bradford analysis.
Quantification of the amount of iron and acid-labile sulfide per protein is done using established procedures (Beinert, 1978) (Beinert, 1983).
Although determining the amount of iron and sulfide per protein is useful, further characterization is required to determine the types of Fe-S clusters bound to an enzyme, which is done using EPR and 57Fe-Mössbauer spectroscopies. Preparation of samples for EPR and Mössbauer spectroscopy is carried out using methods that we have previously described (Lanz et al., 2012).
3.2 Strategy for determining stoichiometry of bound cobalamin
The ability to determine the stoichiometry of cobalamin bound to Class B RS methylases is an important prerequisite for any in vitro mechanistic studies. These enzymes are predicted to bind a methylcobalamin (MeCbl) cofactor, whose methyl moiety is transferred to an sp2- or sp3-hybridized carbon center or phosphinate phosphorous atom (Zhang et al., 2012). In studies of TsrM purified without cobalamin, it has been shown that activity is dependent upon addition of MeCbl to the reaction (Pierre et al., 2012). Although MeCbl is an essential cofactor for Class B RS methylases, information regarding the stoichiometry of bound cobalamin per polypeptide has only been reported for the enzymes PoyC and TsrM (Blaszczyk et al., 2016; Parent et al., 2016). The protocol below describes our strategy for determining the stoichiometry of bound cobalamin per TsrM polypeptide.
Determine the concentration of purified enzyme by Bradford analysis.
Dilute TsrM in water to ~ 11 μM in a final volume of 900 mL.
To the sample, add 100 μL of 1 M KCN to bring the final concentrations of enzyme and KCN to be ~10 μM and 100 mM, respectively. This treatment allows for the formation of the dicyanocobalamin adduct, which can be easily detected and quantified because of its unique UV–vis spectrum.
To release the dicyanocobalamin from the enzyme, boil the sample for 5 min.
Centrifuge the sample at 14,000 × g for 5 min to separate the dicyanocobalamin from precipitated protein.
Using UV-vis spectroscopy, determine the concentration of dicyanocobalamin, which has an extinction coefficient of 30,800 M−1 cm−1 at 367 nm (Ljungdahl, LeGall, & Lee, 1973).
Determine the amount of cobalamin per protein by dividing the concentration of dicyanocobalamin by the concentration of protein.
*The absorbance spectrum of TsrM treated with potassium cyanide is displayed in Figure 3.
Figure 3.
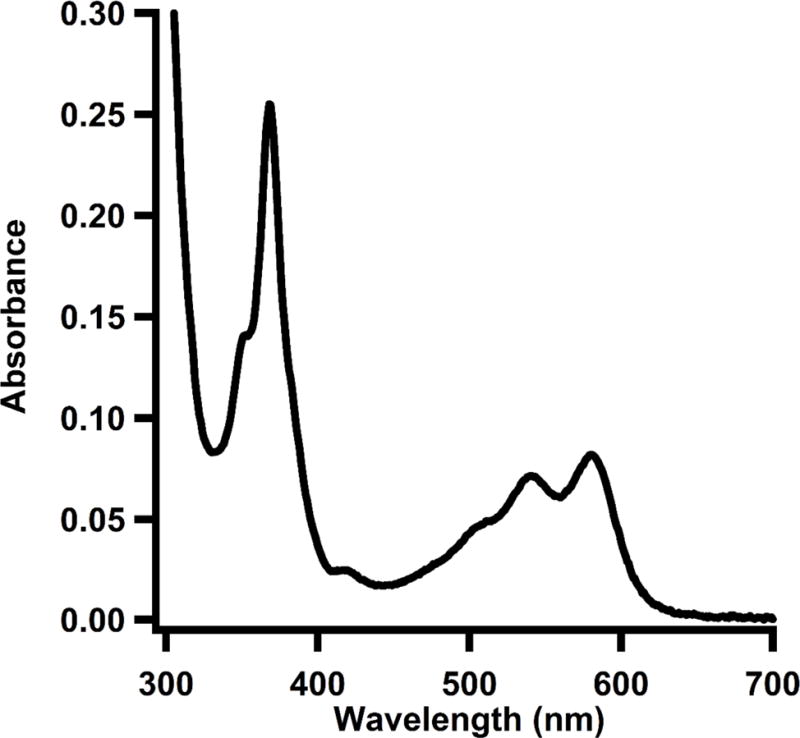
UV-vis spectrum of as-isolated TsrM (10 μM) treated with potassium cyanide.
4. Characterization of Cobalamin Forms
Cobalamin exists in three different oxidation states—cob(I)alamin, cob(II)alamin, or cob(III)alamin—and some Class B RS methylases have been proposed to access each of these states during a reaction cycle (Zhang et al., 2012). In Class B RS methylases, cob(I)alamin is widely accepted to act as a supernucleophile, attacking the methyl group of SAM via an SN2 displacement mechanism to generate S-adenosylhomocysteine (SAH) and methylcob(III)alamin. The next step of the reaction, which involves the transfer of a methyl group from MeCbl to substrate, remains elusive for many of the Class B RS methylases. In the first proposed mechanism, a substrate radical, presumably generated upon abstraction of a hydrogen atom by a 5′dA•, attacks the methyl moiety of MeCbl, with subsequent homolysis of the Co–C bond to yield cob(II)alamin. The cob(II)alamin is then reduced to reform cob(I)alamin, which initiates another round of catalysis by attacking the methyl moiety of SAM. Class B RS methylases also methylate sp2-hybridized carbon centers in pyrrole-containing substrates, albeit via a mechanism that appears to be distinctly different from that employed for enzymes that methylate sp3-hybridized carbon centers. The model system for this reaction is TsrM, which methylates C2 of Trp to form 2-methyltryptophan (2-MeTrp). This enzyme appears to be distinct from all other characterized RS enzymes, because it does not catalyze a reductive cleavage of SAM to give a 5′-dA•. The reaction appears to involve two polar SN2 displacements. In the first step, a cob(I)alamin species attacks the methyl moiety of SAM to generate MeCbl and SAH. In the second step, the pi electrons at C2 of Trp attack the methyl moiety of MeCbl to yield the methylated product upon loss of the C2 proton. This second step appears to be facilitated by proton abstraction from the indole nitrogen of Trp (Blaszczyk et al). The ability to monitor different forms of cobalamin throughout the reaction is critical in elucidating the mechanisms employed by these enzymes. Furthermore, when characterizing cobalamin during a reaction, it is necessary to remove excess cobalamin from reactions to ensure that any changes observed reflect enzyme-bound forms of the cofactor.
4.1 Strategies for characterizing cobalamin
Cob(I)alamin has a d8 electron configuration at its Co+1 metal center, and when monitored by UV-vis spectroscopy, displays a sharp absorbance peak at 390 nm (R. Banerjee & Ragsdale, 2003; R. V. Banerjee, Frasca, Ballou, & Matthews, 1990). Cob(I)alamin is extremely prone to oxidation, so UV-vis analysis must be performed in an anaerobic cuvette or in an anaerobic chamber to retain the reduced form of the cofactor. Because the redox potential for the cob(II)alamin/cob(I)alamin couple is −610 mV, a low potential reductant is generally required to generate cob(I)alamin (R. V. Banerjee, Harder, Ragsdale, & Matthews, 1990; Lexa & Saveant, 1983). However, mild reductants such as BME and DTT have also been shown to reduce cob(II)alamin in the presence of OHCbl (Jarrett, Goulding, Fluhr, Huang, & Matthews, 1997).
Unlike cob(I)alamin and cob(III)alamin, cob(II)alamin has a non-integer spin state (S = ½) and can be observed by EPR spectroscopy. Cob(II)alamin typically contains a five-coordinate cobalt center that has one occupied and one unoccupied axial ligand. Usually, either a conserved histidine (His) or the dimethylbenzimidazole moiety of cobalamin binds as the lower axial ligand to generate a His-on or base-on form of cob(II)alamin, respectively. His-on and base-on cob(II)alamin have a distinguishable EPR spectrum that exhibits superhyperfine coupling from the 14N (I = 1) that is coordinated to the cobalt ion. However, as-isolated TsrM does not show this superhyperfine coupling, but exhibits a unique EPR spectrum that is consistent with an atom in the lower-axial position. Based on these observations, as well as similarities with two cobalamin-dependent dehalogenases, which have been shown by x-ray crystallography to have water molecules bound to their lower-axial positions (Bommer et al., 2014; Payne et al., 2015), an oxygen atom (I = 0) from a water molecule was proposed to be coordinated in the lower-axial position (Blaszczyk et al., 2016). The other common form of cob(II)alamin is referred to as base-off, which typically has a water molecule bound to the upper-axial ligand. This form of the cofactor also has a distinct EPR spectrum that can easily be distinguished.
Cob(III)alamin is most commonly found as MeCbl, AdoCbl, or OHCbl in Nature. All three of these forms can be distinguished by UV-vis spectroscopy; however, their UV-vis spectra can be perturbed from that exhibited in free solution when the cobalamin form is bound to an enzyme. Free MeCbl, AdoCbl, and OHCbl display absorption features at 520 nm (ε520 = 8,600 M−1 cm−1), 262 nm (ε262 = 35,100 M−1 cm−1), and 351 (ε351 =26,500 M−1 cm−1) nm, respectively, in aqueous solutions (Wood, 1987). As is the case in TsrM, MeCbl possessing neither a His nor a dimethylbenzimidazole moiety in its lower-axial position loses its absorption feature at 520 nm and displays a new peak at 450 nm (Menon & Ragsdale, 1998). Figure 4 displays a UV-vis spectrum of TsrM with a reduced cobalamin cofactor, as evident by the absorption peak at 390 nm, which corresponds to cob(I)alamin. The addition of SAM to the sample results in a UV-vis spectrum with a new peak at ~450 nm, which corresponds to the formation of methylcobalamin. Liquid chromatography with detection by tandem mass spectrometry (LC-MS/MS) is an alternative method that allows for characterization of MeCbl, AdoCbl, and OHCbl. By using standards with known concentrations of all three forms of cob(III)alamin, LC-MS/MS can be used for their accurate quantification.
Figure 4.
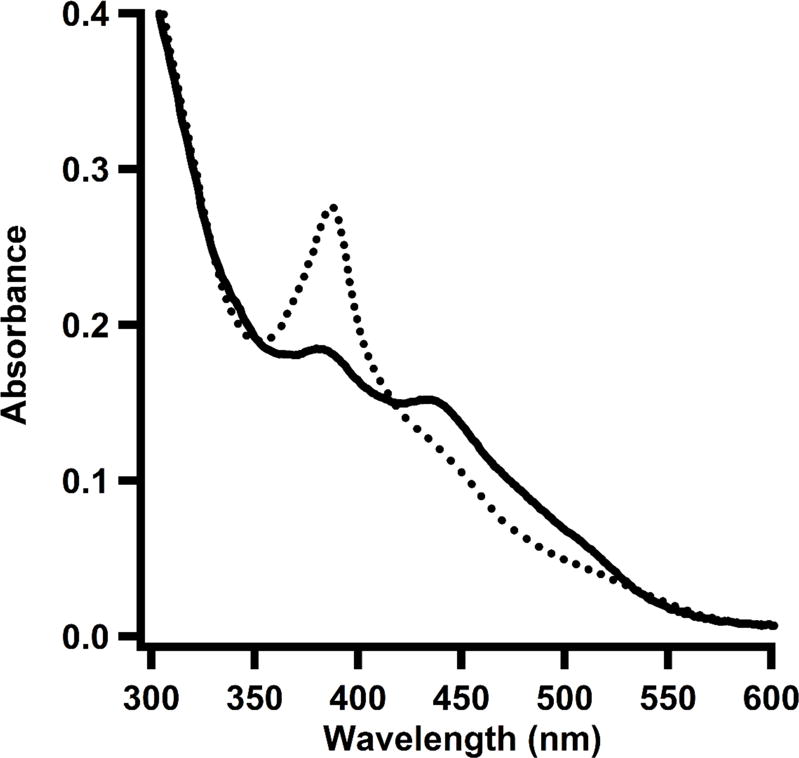
UV-vis spectrum of TsrM treated with dithionite. Reduced form of enzyme (dotted line) shows a predominant peak at 390 nm, corresponding to cob(I)alamin. Upon addition of SAM (solid line), the cob(I)alamin peak decreases and a new peak arises at 450 nm, which corresponds to the formation of methylcobalamin in its base-off/His-off form.
4.2 Protocol for preparation of samples for EPR analysis of cob(II)alamin
Freeze a small container of isopentane in liquid N2 until it becomes a slush.
Dilute the protein sample to a final concentration of ~200 μM in 200 μL. In addition to an as-isolated sample, also make samples with 1 mM substrate and/or SAM to monitor spectral perturbations that reflect changes in cobalamin states.
Remove sample, place in an EPR tube, and immediately freeze by immersing in isopentane that is cooled with liquid N2.
Record an EPR spectrum of the sample at ~70 K.
To quantify the amount of cob(II)alamin present, make a 1 mM copper (II) sulfate standard and record an EPR spectrum under identical conditions. Calculate the double integral and determine the concentration of cob(II)alamin in each sample using protocols previously described (Aasa & Vänngård, 1975).
*EPR spectra of TsrM are displayed in Figure 5.
Figure 5.
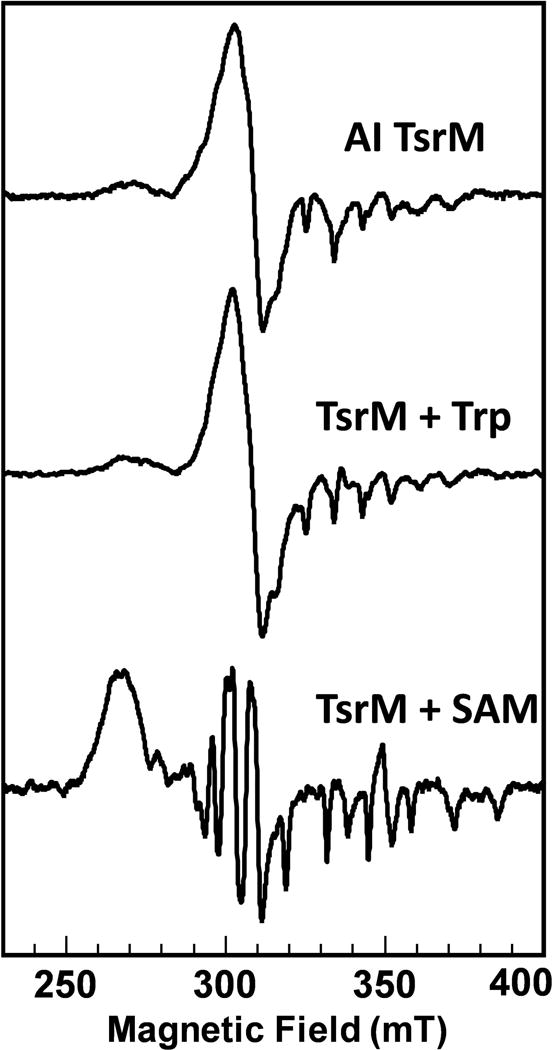
EPR spectra of as-isolated TsrM, TsrM in the presence of Trp, and TsrM in the presence of SAM.
4.3 Sample preparation protocol for analysis of methylcobalamin, adenosylcobalamin, and hydroxocobalamin in reactions
Because of the susceptibility of MeCbl and AdoCbl to photolysis, reactions should be performed both in the dark and in an anaerobic chamber. To protect the cobalamin from light, all samples should be prepared in amber tubes.
To release enzyme-bound cobalamin from the protein, quench samples in H2SO4 at a final concentration of 150 mM.
Centrifuge samples for 15 min at 14,000 × g to remove precipitated protein.
In the dark, remove the soluble fraction and pipette at least 40 μL into amber MS vials.
After preparing samples, LC-MS/MS analysis should be performed immediately to avoid breakdown of cob(III)alamin forms.
4.3 Protocol for quantification of cobalamin by HPLC coupled to electrosprayionization mass spectrometry
Prepare standards of MeCbl, AdoCbl, and OHCbl in the dark in an anaerobic chamber. Note that the concentration of each form of cobalamin can be determined using its known extinction coefficient or by treating with potassium cyanide and boiling to form the dicyanocobalamin adduct, which is not light or oxygen sensitive.
In the dark, create a standard curve containing 0.031–64 μM MeCbl, AdoCbl, and OHCbl and 100 μM Tyr (internal standard). Generate a standard curve by making a serial dilution starting from a sample containing 64 μM of each form of cobalamin that is diluted in two-fold increments, as previously described (Lanz et al., 2012).
Centrifuge samples for 15 min at 14,000 × g and, in the dark, aliquot 60 μL into amber MS vials.
Analyze the samples on an Agilent Technologies Zorbax Extend-C18 RRHD column (2.1 mm × 50 mm, 1.8 μm particle size) equilibrated in 95% solvent A (0.1% formic acid, pH 2.6) and 5% solvent B (acetonitrile) at a flow rate of 0.35 mL/min. Apply a gradient of 5-20% solvent B from 0.4 to 0.8 min, and then a gradient of 20-41% solvent B from 0.8 to 1.6 min before returning to 5% solvent B from 1.6 to 1.9 min. Allow the column to re-equilibrate for 0.5 min under initial conditions before subsequent sample injections. MeCbl, AdoCbl, and OHCbl elute at approximately 1.70, 1.53, and 1.17 min, respectively.
Use MS/MS to detect MeCbl, AdoCbl, and OHCbl by electrospray-ionization in positive mode (ESI+). MeCbl and AdoCbl are detected in the +2 charge state with m/z values of 673.0 and 790.6, respectively, with an optimal fragmentor voltage of 104 V for the former and 108 V for the latter. OHCbl is detected in its +2 charge state after the loss of a water molecule using a 120 V fragmentor voltage. The transitions and collision
*Elution profile of cobalamin standards analyzed by MS/MS is displayed in Figure 6.
Figure 6.
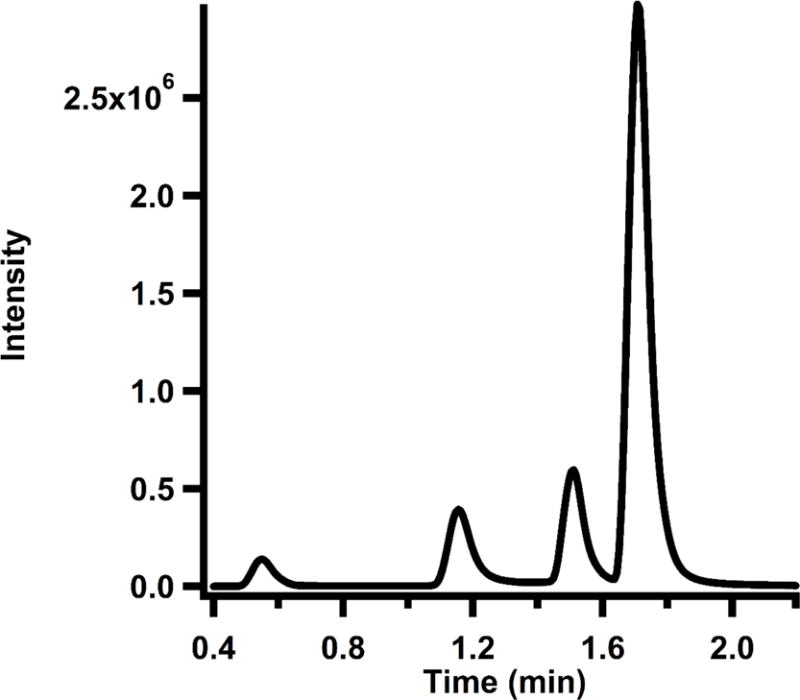
Elution profile of cobalamin standard curve monitored by MS/MS. Retention times for Tyr, OHCbl, AdoCbl, and MeCbl are 0.48 min, 1.17 min, 1.53 min, and 1.70 min, respectively.
5. Quantitative Analysis of Turnover
Some Class B RS methylases generate 5′dA, SAH, and their respective products stoichiometrically. These products—as well as respective substrates—can be quantified by LC-MS/MS as described below for a TsrM-catalyzed reaction, wherein Trp, MeTrp, SAH, SAM, and 5′-dA are monitored.
5.1 Sample preparation protocol for analysis of reaction products by HPLC coupled to electrospray-ionization mass spectrometry
Assays should contain final concentrations of the following in a total volume of 150 μL: 50 mM HEPES, pH 7.5, 100 mM KCl, 5% glycerol, 1 mM SAM, 1 mM substrate, 0.1 – 1 μM TsrM, 100 μM Tyr (internal standard), and reductant. The E. coli flavodoxin/flavodoxin reductase reducing system— which contains 25 μM flavodoxin, 10 μM flavodoxin reductase, and 1 mM NADPH, is typically used as a source of electrons—but can be substituted with other chemical reductants (e.g. dithionite). DTT also can be used as a reductant for the TsrM-catalyzed reaction; however, corresponding reaction rates are about two-fold lower.
Before initiating the reaction by adding substrate, incubate the enzyme with reductant and SAM for 15 min at ambient temperature to allow for methylation of the cobalamin cofactor.
Initiate the reaction by adding substrate, then remove 20 μL aliquots at specified time points and quench the reactions with an equal volume of 100 mM H2SO4.
Centrifuge the samples for 15 min at 14,000 × g before removing supernatants and placing them into MS vials.
5.2 Protocol for quantification of reaction products by HPLC coupled to electrosprayionization mass spectrometry
Prepare standards of SAM, SAH, 5′dA, Trp, and MeTrp using extinction coefficients for each molecule. SAM, SAH, and 5′dA display absorption features at 260 nm with extinction coefficients of 15,400 M−1 cm−1, 16,000 M−1 cm−1, and 15,400 M−1 cm−1, respectively, while Trp and MeTrp display absorption features at 278 nm with an extinction coefficient of 5,560 M−1 cm−1 (Wood, 1987). Using these standards, generate a standard curve containing 0.5 – 250 μM of each molecule and 100 μM Tyr (internal standard).
Analyze samples on an Agilent Technologies Zorbax Extend-C18 RRHD column (4.6 mm × 50 mm, 1.8 μm particle size) equilibrated in 90.5% solvent A (0.1% formic acid, pH 2.6) and 9.5% solvent B (methanol). Apply a gradient of 9.5-9.8% solvent B from 0.2 to 3.3 min, and then a gradient of 9.8-30% solvent B from 3.3 to 4.1 min. Next, increase solvent B to 60% from 4.1 to 4.7 min and hold constant from 4.7 to 5.1 min before returning to 9.5% solvent B from 5.1 to 5.5 min. Allow the column to re-equilibrate for 3.2 min under initial conditions before subsequent sample injections.
Detect products using MS/MS with electrospray ionization in positive mode. All products are detected in their +1 charge state. Table 4 displays the m/z values (parent and daughter), fragmentor voltages, collision energies, and retention times of all molecules that are monitored in a TsrM reaction.
Quantify product formation. Shown in Figure 7 is the time-dependent formation of MeTrp, SAH, and 5′-dA in TsrM catalyzed reaction.
Table 4.
Parameters for LC-MS/MS analysis of reaction products
| Retention Time (min) | Parent Ion | Fragmentor Energy (V) | Product Ion 1 | Collision Energy 1 | Product Ion 2 | Collision Energy 2 | |
|---|---|---|---|---|---|---|---|
| MeTrp | 6.6 | 202.0 | 130 | 160.1 | 10 | 132.0 | 21 |
| Trp | 6.2 | 188 | 130 | 146.1 | 10 | 118 | 21 |
| SAM | 0.8 | 399.1 | 90 | 250.1 | 14 | ||
| SAH | 1.4 | 385.4 | 136.0 | 100 | 24 | 134.0 | 24 |
| 5′-dA | 2.8 | 252.1 | 136.0 | 90 | 13 | 119.0 | 50 |
| Tyr (IS) | 1.7 | 182.0 | 165.0 | 60 | 3 | 123.0 | 15 |
Figure 7.
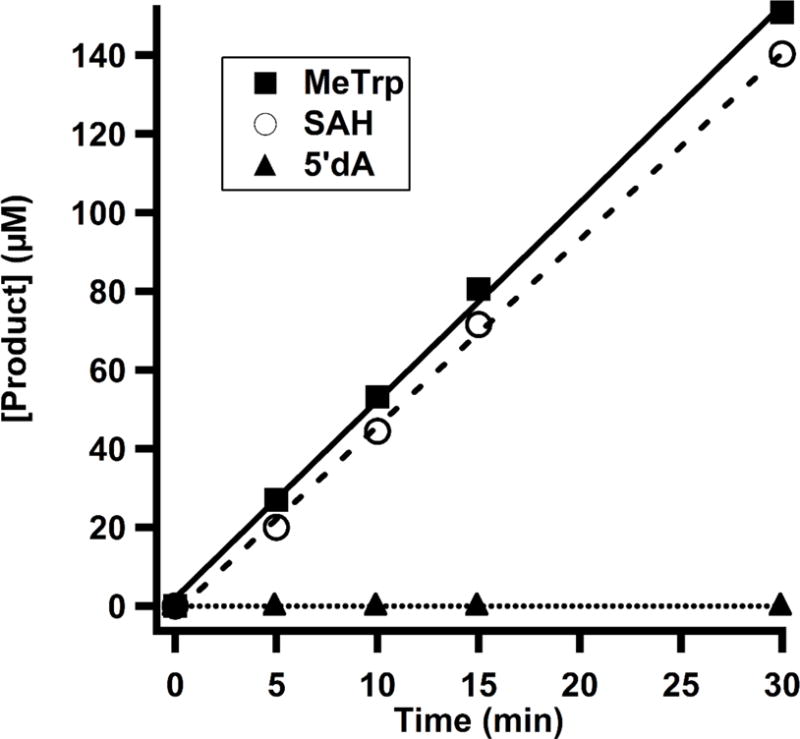
Time-dependent formation of MeTrp (solid line), SAH (dashed line), and 5′-dA (dotted line) monitored by LC-MS/MS in a reaction with TsrM (1 μM), SAM (1 mM), Trp (1 mM), flavodoxin (25 μM), flavodoxin reductase (10 μM), and NADPH (1 mM).
Conclusions
Class B RS methylases catalyze the methylation of sp2- and sp3-hybridized carbon centers and phosphinate phosphorous centers in myriad biomolecules that play critical roles in human health and disease. Their characterization and mechanistic interrogation can lead to new insight into how cobalamin functions within enzyme-catalyzed reactions, and also facilitate the development of biomolecules that have better pharmacological properties. Class B RS methylases, however, have been somewhat resistant to overproduction and purification in a homogenous and soluble form, which has hampered their study. The successful purification of some of these enzymes can be enhanced when they are produced as fusion proteins with SUMO and under conditions that facilitate cobalamin entry into the cell.
Table 2.
Amino Acid Mixturea
| 4 L of 5× Amino Acid | Quantity (g) |
|---|---|
| Alanine | 1.426 |
| Arginine Monohydrochloride | 0.9656 |
| Asparagine | 1.065 |
| Aspartic Acid | 1.057 |
| Cysteine Monohydrochloride Monohydrate | 0.2422 |
| Glutamic Acid | 2.224 |
| Glutamine | 1.755 |
| Glycine | 1.201 |
| Histidine Monohydrochloride Monohydrate | 0.839 |
| Isoleucine | 1.050 |
| Leucine | 2.099 |
| Lysine Monohydrochloride | 1.462 |
| Methionine | 0.5970 |
| Phenylalanine | 1.322 |
| Proline | 0.9210 |
| Serine | 21.02 |
| Threonine | 0.9530 |
| Tryptophan | 0.4090 |
| Tyrosine | 0.7250 |
| Valine | 1.407 |
Amino acids should be added directly to 20× M9 Salt Solution prior to autoclaving the medium.
Table 3.
Parameters for MS/MS analysis of cobalamin
| Transition (m/z) | Collision Energy | |
|---|---|---|
| Hydroxocobalamin | 664.9➔635.8 | 11 |
| 664.9➔147.0 | 41 | |
| Methylcobalamin | 673.0➔665.6 | 17 |
| 673.0➔147.0 | 57 | |
| Adenosylcobalamin | 790.6➔665.6 | 17 |
| 790.6➔147.0 | 75 |
Acknowledgments
This work was supported by the National Institutes of Health (GM-103268, GM-101957, GM-122595, and AI-111419) and Penn State Funds for Undergraduate Research (RVW). SJB is an investigator of the Howard Hughes Medical Institute.
References
- Aasa R, Vänngård T. EPR signal intensity and powder shapes: A reexamination. Journal of Magnetic Resonance (1969) 1975;19(3):308–315. doi: http://dx.doi.org/10.1016/0022-2364(75)90045-1. [Google Scholar]
- Akiva E, Brown S, Almonacid DE, Barber AE, Custer AF, Hicks MA, Babbitt PC. The Structure–Function Linkage Database. Nucleic Acids Res. 2014;42:D521–D530. doi: 10.1093/nar/gkt1130. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KD, Wang SC. Initial characterization of Fom3 from Streptomyces wedmorensis: The methyltransferase in fosfomycin biosynthesis. Arch Biochem Biophys. 2014;543:67–73. doi: 10.1016/j.abb.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KD, Xu H, White RH. Identification of a unique radical S-adenosylmethionine methylase likely involved in methanopterin biosynthesis in Methanocaldococcus jannaschii. J Bacteriol. 2014;196(18):3315–3323. doi: 10.1128/jb.01903-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderle C, Alt S, Gulder T, Bringmann G, Kammerer B, Gust B, Heide L. Biosynthesis of clorobiocin: investigation of the transfer and methylation of the pyrrolyl-2-carboxyl moiety. Arch Microbiol. 2007;187(3):227–237. doi: 10.1007/s00203-006-0190-9. [DOI] [PubMed] [Google Scholar]
- Babior BM, Moss TH, Orme-Johnson WH, Beinert H. The mechanism of action of ethanolamine ammonia-lyase, a B-12-dependent enzyme. The participation of paramagnetic species in the catalytic deamination of 2-aminopropanol. J Biol Chem. 1974;249(14):4537–4544. [PubMed] [Google Scholar]
- Bandarian V, Matthews RG. Measurement of energetics of conformational change in cobalamin-dependent methionine synthase. Methods Enzymol. 2004;380:152–169. doi: 10.1016/s0076-6879(04)80007-7. [DOI] [PubMed] [Google Scholar]
- Banerjee R, Ragsdale SW. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- Banerjee RV, Frasca V, Ballou DP, Matthews RG. Participation of Cob(I)alamin in the reaction catalyzed by methionine synthase from Escherichia coli: a steady-state and rapid reaction kinetic analysis. Biochemistry. 1990;29(50):11101–11109. doi: 10.1021/bi00502a013. [DOI] [PubMed] [Google Scholar]
- Banerjee RV, Harder SR, Ragsdale SW, Matthews RG. Mechanism of reductive activation of cobalamin-dependent methionine synthase: an electron paramagnetic resonance spectroelectrochemical study. Biochemistry. 1990;29(5):1129–1135. doi: 10.1021/bi00457a005. [DOI] [PubMed] [Google Scholar]
- Bauerle MR, Schwalm EL, Booker SJ. Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. J Biol Chem. 2015;290(7):3995–4002. doi: 10.1074/jbc.R114.607044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann S, Herrmann J, Raju R, Steinmetz H, Mohr KI, Huttel S, Muller R. Cystobactamids: myxobacterial topoisomerase inhibitors exhibiting potent antibacterial activity. Angew Chem Int Ed Engl. 2014;53(52):14605–14609. doi: 10.1002/anie.201409964. [DOI] [PubMed] [Google Scholar]
- Beinert H. Micro methods for the quantitative determination of iron and copper in biological material. Methods Enzymol. 1978;54:435–445. doi: 10.1016/s0076-6879(78)54027-5. [DOI] [PubMed] [Google Scholar]
- Beinert H. Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal Biochem. 1983;131(2):373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- Blackwell CM, Turner JM. Microbial metabolism of amino alcohols. Formation of coenzyme B12-dependent ethanolamine ammonia-lyase and its concerted induction in Escherichia coli. Biochemical Journal. 1978;176(3):751–757. doi: 10.1042/bj1760751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk AJ, Silakov A, Zhang B, Maiocco SJ, Lanz ND, Kelly WL, Booker SJ. Spectroscopic and Electrochemical Characterization of the Iron-Sulfur and Cobalamin Cofactors of TsrM, an Unusual Radical S-Adenosylmethionine Methylase. J Am Chem Soc. 2016;138(10):3416–3426. doi: 10.1021/jacs.5b12592. [DOI] [PubMed] [Google Scholar]
- Bommer M, Kunze C, Fesseler J, Schubert T, Diekert G, Dobbek H. Structural basis for organohalide respiration. Science. 2014;346(6208):455–458. doi: 10.1126/science.1258118. [DOI] [PubMed] [Google Scholar]
- Booker SJ, Grove TL. Mechanistic and functional versatility of radical SAM enzymes. F1000 Biology Reports. 2010;2:52. doi: 10.3410/B2-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Broderick JB, Duffus BR, Duschene KS, Shepard EM. Radical S-Adenosylmethionine Enzymes. Chemical Reviews. 2014;114(8):4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew AG, Bryant DA. Chlorophyll biosynthesis in bacteria: the origins of structural and functional diversity. Annu Rev Microbiol. 2007;61:113–129. doi: 10.1146/annurev.micro.61.080706.093242. [DOI] [PubMed] [Google Scholar]
- Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu-Gogonea C, Souder MG, Booker SJ. Lipoyl synthase requires two equivalents of S-adenosyl-L-methionine to synthesize one equivalent of lipoic acid. Biochemistry. 2004;43(21):6378–6386. doi: 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- Dairi T, Ohta T, Hashimoto E, Hasegawa M. Self cloning in Micromonospora olivasterospora of fms genes for fortimicin A (astromicin) biosynthesis. Mol Gen Genet. 1992;232(2):262–270. doi: 10.1007/BF00280005. [DOI] [PubMed] [Google Scholar]
- Dubendorf JW, Studier FW. Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. Journal of Molecular Biology. 1991;219(1):45–59. doi: 10.1016/0022-2836(91)90856-2. doi: http://dx.doi.org/10.1016/0022-2836(91)90856-2. [DOI] [PubMed] [Google Scholar]
- Frey PA, Booker SJ. Radical mechanisms of S-adenosylmethionine-dependent enzymes. Adv Protein Chem. 2001;58:1–45. doi: 10.1016/s0065-3233(01)58001-8. [DOI] [PubMed] [Google Scholar]
- Frey PA, Hegeman AD, Ruzicka FJ. The Radical SAM Superfamily. Crit Rev Biochem Mol Biol. 2008;43(1):63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- Hidaka T, Goda M, Kuzuyama T, Takei N, Hidaka M, Seto H. Cloning and nucleotide sequence of fosfomycin biosynthetic genes of Streptomyces wedmorensis. Mol Gen Genet. 1995;249(3):274–280. doi: 10.1007/BF00290527. [DOI] [PubMed] [Google Scholar]
- Hidaka T, Hidaka M, Kuzuyama T, Seto H. Sequence of a P-methyltransferase-encoding gene isolated from a bialaphos-producing Streptomyces hygroscopicus. Gene. 1995;158(1):149–150. doi: 10.1016/0378-1119(95)00101-b. [DOI] [PubMed] [Google Scholar]
- Hu Y, Ribbe MW. Maturation of nitrogenase cofactor-the role of a class E radical SAM methyltransferase NifB. Curr Opin Chem Biol. 2016;31:188–194. doi: 10.1016/j.cbpa.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inahashi Y, Zhou S, Bibb MJ, Song L, Al-Bassam MM, Bibb MJ, Challis GL. Watasemycin biosynthesis in Streptomyces venezuelae: thiazoline C-methylation by a type B radical-SAM methylase homologue. Chemical Science. 2017;8(4):2823–2831. doi: 10.1039/C6SC03533G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett JT, Goulding CW, Fluhr K, Huang S, Matthews RG. Purification and assay of cobalamin-dependent methionine synthase from Escherichia coli. Methods Enzymol. 1997;281:196–213. doi: 10.1016/s0076-6879(97)81026-9. [DOI] [PubMed] [Google Scholar]
- Kamigiri K, Hidaka T, Imai S, Murakami T, Seto H. Studies on the biosynthesis of bialaphos (SF-1293) 12. C-P bond formation mechanism of bialaphos: discovery of a P-methylation enzyme. J Antibiot (Tokyo) 1992;45(5):781–787. doi: 10.7164/antibiotics.45.781. [DOI] [PubMed] [Google Scholar]
- Kelly WL, Pan L, Li C. Thiostrepton biosynthesis: prototype for a new family of bacteriocins. J Am Chem Soc. 2009;131(12):4327–4334. doi: 10.1021/ja807890a. [DOI] [PubMed] [Google Scholar]
- Kim HJ, McCarty RM, Ogasawara Y, Liu Y-N, Mansoorabadi SO, LeVieux J, Liu H-W. GenK-Catalyzed C-6′ Methylation in the Biosynthesis of Gentamicin: Isolation and Characterization of a Cobalamin-Dependent Radical SAM Enzyme. J Am Chem Soc. 2013;135(22):8093–8096. doi: 10.1021/ja312641f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Suh JW, Kang SH, Phan TH, Park SH, Kwon HJ. Gene inactivation study of gntE reveals its role in the first step of pseudotrisaccharide modifications in gentamicin biosynthesis. Biochem Biophys Res Commun. 2008;372(4):730–734. doi: 10.1016/j.bbrc.2008.05.133. [DOI] [PubMed] [Google Scholar]
- Kowalak JA, Bruenger E, McCloskey JA. Posttranscriptional modification of the central loop of domain V in Escherichia coli 23 S ribosomal RNA. J Biol Chem. 1995;270(30):17758–17764. doi: 10.1074/jbc.270.30.17758. [DOI] [PubMed] [Google Scholar]
- Kudo F, Kasama Y, Hirayama T, Eguchi T. Cloning of the pactamycin biosynthetic gene cluster and characterization of a crucial glycosyltransferase prior to a unique cyclopentane ring formation. J Antibiot (Tokyo) 2007;60(8):492–503. doi: 10.1038/ja.2007.63. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T, Hidaka T, Kamigiri K, Imai S, Seto H. Studies on the biosynthesis of fosfomycin. 4. The biosynthetic origin of the methyl group of fosfomycin. J Antibiot (Tokyo) 1992;45(11):1812–1814. doi: 10.7164/antibiotics.45.1812. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T, Seki T, Dairi T, Hidaka T, Seto H. Nucleotide sequence of fortimicin KL1 methyltransferase gene isolated from Micromonospora olivasterospora, and comparison of its deduced amino acid sequence with those of methyltransferases involved in the biosynthesis of bialaphos and fosfomycin. J Antibiot (Tokyo) 1995;48(10):1191–1193. doi: 10.7164/antibiotics.48.1191. [DOI] [PubMed] [Google Scholar]
- Landgraf BJ, McCarthy EL, Booker SJ. Radical S-Adenosylmethionine Enzymes in Human Health and Disease. Annu Rev Biochem. 2016;85:485–514. doi: 10.1146/annurev-biochem-060713-035504. [DOI] [PubMed] [Google Scholar]
- Lanz ND, Grove TL, Gogonea CB, Lee KH, Krebs C, Booker SJ. RlmN and AtsB as models for the overproduction and characterization of radical SAM proteins. Methods Enzymol. 2012;516:125–152. doi: 10.1016/b978-0-12-394291-3.00030-7. [DOI] [PubMed] [Google Scholar]
- Lexa D, Saveant JM. The electrochemistry of vitamin B12. Accounts of Chemical Research. 1983;16(7):235–243. doi: 10.1021/ar00091a001. [DOI] [Google Scholar]
- Li C, Kelly WL. Recent advances in thiopeptide antibiotic biosynthesis. Nat Prod Rep. 2010;27(2):153–164. doi: 10.1039/b922434c. [DOI] [PubMed] [Google Scholar]
- Ljungdahl LG, LeGall J, Lee JP. Isolation of a protein containing tightly bound 5-methoxybenzimidazolylcobamide (factor 3m) from Clostridium thermoaceticum. Biochemistry. 1973;12(9):1802–1808. doi: 10.1021/bi00733a022. [DOI] [PubMed] [Google Scholar]
- Mao Y, Varoglu M, Sherman DH. Molecular characterization and analysis of the biosynthetic gene cluster for the antitumor antibiotic mitomycin C from Streptomyces lavendulae NRRL 2564. Chem Biol. 1999;6(4):251–263. doi: 10.1016/s1074-5521(99)80040-4. [DOI] [PubMed] [Google Scholar]
- Marblestone JG, Edavettal SC, Lim Y, Lim P, Zuo X, Butt TR. Comparison of SUMO fusion technology with traditional gene fusion systems: Enhanced expression and solubility with SUMO. Protein Science : A Publication of the Protein Society. 2006;15(1):182–189. doi: 10.1110/ps.051812706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marblestone JG, Edavettal SC, Lim Y, Lim P, Zuo X, Butt TR. Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci. 2006;15(1):182–189. doi: 10.1110/ps.051812706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marous DR, Lloyd EP, Buller AR, Moshos KA, Grove TL, Blaszczyk AJ, Townsend CA. Consecutive radical S-adenosylmethionine methylations form the ethyl side chain in thienamycin biosynthesis. Proc Natl Acad Sci U S A. 2015;112(33):10354–10358. doi: 10.1073/pnas.1508615112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Ragsdale SW. Role of the [4Fe-4S] cluster in reductive activation of the cobalt center of the corrinoid iron-sulfur protein from Clostridium thermoaceticum during acetate biosynthesis. Biochemistry. 1998;37(16):5689–5698. doi: 10.1021/bi9727996. [DOI] [PubMed] [Google Scholar]
- Nunez LE, Mendez C, Brana AF, Blanco G, Salas JA. The biosynthetic gene cluster for the beta-lactam carbapenem thienamycin in Streptomyces cattleya. Chem Biol. 2003;10(4):301–311. doi: 10.1016/s1074-5521(03)00069-3. [DOI] [PubMed] [Google Scholar]
- Ostash B, Saghatelian A, Walker S. A streamlined metabolic pathway for the biosynthesis of moenomycin A. Chem Biol. 2007;14(3):257–267. doi: 10.1016/j.chembiol.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panavas T, Sanders C, Butt TR. SUMO Fusion Technology for Enhanced Protein Production in Prokaryotic and Eukaryotic Expression Systems. In: Ulrich HD, editor. SUMO Protocols. Totowa, NJ: Humana Press; 2009. pp. 303–317. [DOI] [PubMed] [Google Scholar]
- Pandelia ME, Lanz ND, Booker SJ, Krebs C. Mossbauer spectroscopy of Fe/S proteins. Biochim Biophys Acta. 2015;1853(6):1395–1405. doi: 10.1016/j.bbamcr.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Parent A, Guillot A, Benjdia A, Chartier G, Leprince J, Berteau O. The B12-Radical SAM Enzyme PoyC Catalyzes Valine Cbeta-Methylation during Polytheonamide Biosynthesis. J Am Chem Soc. 2016;138(48):15515–15518. doi: 10.1021/jacs.6b06697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne KA, Quezada CP, Fisher K, Dunstan MS, Collins FA, Sjuts H, Leys D. Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation. Nature. 2015;517(7535):513–516. doi: 10.1038/nature13901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre S, Guillot A, Benjdia A, Sandstrom C, Langella P, Berteau O. Thiostrepton tryptophan methyltransferase expands the chemistry of radical SAM enzymes. Nat Chem Biol. 2012;8(12):957–959. doi: 10.1038/nchembio.1091. [DOI] [PubMed] [Google Scholar]
- Rachid S, Scharfe M, Blocker H, Weissman KJ, Muller R. Unusual chemistry in the biosynthesis of the antibiotic chondrochlorens. Chem Biol. 2009;16(1):70–81. doi: 10.1016/j.chembiol.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29(5):1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh SM, Xiong L, Bae T, Mankin AS. The methyltransferase YfgB/RlmN is responsible for modification of adenosine 2503 in 23S rRNA. RNA. 2008;14(1):98–106. doi: 10.1261/rna.814408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Schnell B, Baumann S, Muller R, Begley TP. Biosynthesis of Branched Alkoxy Groups: Iterative Methyl Group Alkylation by a Cobalamin-Dependent Radical SAM Enzyme. J Am Chem Soc. 2017;139(5):1742–1745. doi: 10.1021/jacs.6b10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner WJ, Allen KD, Hu K, Helms GL, Chen BS, Wang SC. In vitro phosphinate methylation by PhpK from Kitasatospora phosalacinea. Biochemistry. 2011;50(42):8986–8988. doi: 10.1021/bi201220r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood EJ. Data for biochemical research (third edition) by R M C Dawson, D C Elliott, W H Elliott and K M Jones, pp 580. Oxford Science Publications, OUP, Oxford, 1986. £35/$59. ISBN 0-19-855358-7. Biochemical Education. 1987;15(2):97–97. doi: 10.1016/0307-4412(87)90110-5. [DOI] [Google Scholar]
- Woodyer RD, Shao Z, Thomas PM, Kelleher NL, Blodgett JA, Metcalf WW, Zhao H. Heterologous production of fosfomycin and identification of the minimal biosynthetic gene cluster. Chem Biol. 2006;13(11):1171–1182. doi: 10.1016/j.chembiol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Zhang Q, van der Donk WA, Liu W. Radical-mediated enzymatic methylation: a tale of two SAMS. Acc Chem Res. 2012;45(4):555–564. doi: 10.1021/ar200202c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Cash VL, Flint DH, Dean DR. Assembly of iron-sulfur clusters. Identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. J Biol Chem. 1998;273(21):13264–13272. doi: 10.1074/jbc.273.21.13264. [DOI] [PubMed] [Google Scholar]