

Abstract
Photoredox catalysis is a versatile approach for the construction of challenging covalent bonds under mild reaction conditions, commonly using photoredox catalysts (PCs) derived from precious metals. As such, there is need to develop organic analogues as sustainable replacements. Although several organic PCs have been introduced, there remains a lack of strongly reducing visible light organic PCs. Herein, we establish the critical photophysical and electrochemical characteristics of both a dihydrophenazine and a phenoxazine system that enables them success as strongly reducing visible light PCs for trifluoromethylations and dual photoredox/nickel catalyzed C-N and C-S cross-couplings, reactions which have been historically exclusive to precious metal PCs.
Keywords: photocatalysis, photochemistry, organocatalysis, radicals, sustainable chemistry catalysis
COMMUNICATION
The new iridium. The photophysical and electrochemical properties of a strongly reducing phenazine and phenoxazine enable their application as visible light photoredox catalysts to catalyze reactions that were previously restricted to precious metals. These organic visible light photoredox catalysts not only present sustainable alternatives to precious metals, but can possess enhanced properties to their metal counterparts for catalysis.
Visible light photoredox catalysis has gained prominence orchestrating challenging chemical transformations under mild reaction conditions.[1] A large majority of this work has employed precious metal polypyridyl iridium and ruthenium photoredox catalysts (PCs). The rapid establishment of these metal complexes as practical PCs leveraged their well-studied photophysical and photoredox properties, that in turn have enabled their incorporation in a range of applications including photovoltaics,[2] light emitting diodes,[3] imaging and sensing in biological systems,[4] therapeutics,[5] and redox active antibiotics.[6]
In regards to photoredox catalysis, these metal complexes exhibit essential characteristics, including strong absorption of visible light via spin-allowed metal-to-ligand charge transfer (MLCT), efficient conversion to long-lived triplet MLCT excited states (3MLCT),[7] and redox reversibility.[7a, 8] Furthermore, ligand and metal modifications tailor the redox properties of the ground and excited states.[9] For example, fac-Ir(ppy)3 (tris[2-phenylpyridinato-C2,N]iridium(III), 1, ppy = 2-phenylpyridine) is amongst the strongest reducing PCs, while [Ru(bpy)3]2+ (tris(2,2’-bipyridine)ruthenium(II), 2, bpy = 2,2′-bipyridine) possesses redox properties that enable it to function either as a reductant or as an oxidant from the 3MLCT state.
However, iridium and ruthenium are precious metals and amongst the rarest elements on earth, escalating their costs and presenting concerns related to sustainability and scalability, driving the need to realize new PCs incorporating non-precious metals[10] or to develop entirely organic replacements.[11] Several organic molecules have proven successful as visible light PCs for small molecule and polymeric transformations.[12] The majority of these organic PCs, such as Eosin Y,[13] rhodamine dyes,[14] acridinium salts,[15] perylene diimides,[16] and carbazolyls[17] are excited state oxidants and operate through a reductive quenching cycle. Although a few strongly reducing organic PCs exist,[18] many do not absorb visible light. PCs that operate using mild visible light and do not require sacrificial reductants are desired to minimize side reactions.
Our interest in organic PCs[19] initiated with the development of organocatalyzed atom transfer radical polymerization (O-ATRP).[20] ATRP has historically been mediated by transition metal catalysts, most commonly copper or ruthenium complexes, which can contaminate the polymer product and restrict applications.[21] A primary challenge in developing a photoredox mediated O-ATRP is presented by the strong reducing power that is required of the PC to activate a dormant alkyl halide.[22] In general, PCs that possess such strong excited state reducing powers are rare, and this is particularly true for organic systems (vide supra).
To address this challenge, we have introduced visible light organic PCs including perylene,[19a] N,N-diaryl dihydrophenazines,[19b, 23] and N-aryl phenoxazines[24] as organic PCs to mediate O-ATRP via an oxidative quenching pathway.[25] Dihydrophenazine and phenoxazine contain electron rich chromophore motifs that form rather stable radical cations upon oxidation and enable them to be strong excited state reductants.[26] However, a detailed comprehension of the characteristics of these molecules in regards to catalysis or their ability to catalyze other transformations has not been established. Herein, through investigation of their photophysical and electrochemical properties, we report the critical characteristics of N,N-5,10-di(2-naphthalene)-5,10-dihydrophenazine (3) and 3,7-(4-biphenyl)-1-naphthalene-10-phenoxazine (4) that enable them to serve as successful PCs. We further establish 3 and 4 as PCs through their employment in atom transfer radical additions or substitutions with CF3I to alkenes and heterocycles as well as dual photoredox/nickel catalyzed C-N and C-S cross-couplings.
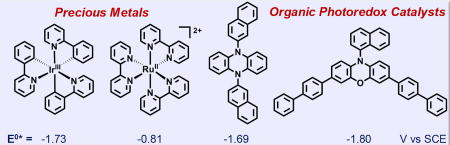
The photophysical properties of 3 and 4 were investigated and compared to that of transition metal complexes 1 and 2 (Figure 1). As photoexcitation is the first step in photoredox catalysis, PCs should be strong light absorbers. In N,N-dimethylacetamide (DMA), the molar absorptivities (ε) for transition metal complexes 1 and 2 are 13,100 and 12,500 M−1cm−1 at their maximum peak wavelengths of absorption (λmax,abs) of 377 nm and 454 nm, respectively (Figure 1A). Dihydrophenazine 3 has a lower molar extinction (εmax,abs = 5,950 M−1cm−1; λmax,abs = 343 nm) in comparison to 1 and 2, while phenoxazine 4 is an excellent light absorber and superior to the other three PCs (εmax,abs = 26,600 M−1cm−1; λmax,abs = 388 nm). In a similar fashion to 1, although the λmax,abs values are < 400 nm, the absorption profiles of organic PCs 3 and 4 extend into the visible region and enable them to function as visible light PCs.
Figure 1.

Photophysical and electrochemical properties of precious metal and organic photoredox catalysts. (A) UV-vis absorption spectra of PCs 1–4 in N,N-dimethylacetamide (DMA). (B) Structures of precious metal and organic PCs. (C) Values enclosed in parentheses are from density functional theory (DFT) calculations. All experimental values for PCs 3 and 4 were measured in DMA at room temperature. [a] Triplet excited state reduction potential, E0*, in units of V vs. SCE. [b] Ground state oxidation potential [c] Triplet energy (Etriplet), estimated from the fluorescence wavelength of the charge transfer singlet state. [d] Maximum absorption wavelength (λmax,abs); molar absorptivity (εmax,abs) at λmax,abs. [e] Emission maximum wavelength (λmax,em). [f] Triplet excited state lifetime (τtriplet). [g] Quantum yield (Φtriplet) of charge transfer triplet excited state (3CT*), and metal-ligand charge transfer triplet state (3MLCT*). [h] λmax,abs and εmax,abs were measured in this work in DMA. All other values were obtained from ref. [8a,9b], and were measured in acetonitrile, except for the λmax,em, which was measured in alcoholic solvent at 77K. [i] λmax,abs and εmax,abs were measured in this work in DMA solvent for [Ru(bpy)3](PF6)2. All other values were obtained from ref. [8b,9a], and were measured in acetonitrile. See Supporting Information for details.
1 is known to be one of the strongest excited state transition metal PC reductants available, with an excited state reduction potential (E0*) of E0(Ir(IV)/3Ir(III)*) = −1.73 V vs. SCE (Figure 1C). Excitingly, organic PCs 3 and 4 are equally reducing with E0* = E0(2PC•+/3PC*) values of −1.69 and −1.80 V vs. SCE, respectively. Although 2 is not as reducing in the excited state [E0* = E0(Ru(III)/3Ru(II)*) = −0.81 V vs. SCE], the Ru(III) generated after participating in a photoreduction event is strongly oxidizing, with an oxidation potential [E0ox = E0(Ru(III)/Ru(II))] of 1.29 V vs. SCE. Notably, 1 and the organic PC 4 have similar E0ox values [E0(Ir(IV)/Ir(III)) = 0.77 and E0(2PC•+/1PC) = 0.65 V vs. SCE, respectively], while 3 forms a rather stable radical cation [E0ox = E0(2PC•+/1PC) = 0.21 V vs. SCE]. In regards to triplet energy (Etriplet), both 1 and 4 have energetic triplets with Etriplet of 2.50 and 2.45 V, respectively. Meanwhile, Etriplet of 2 and 3 are lower, with respective values of 2.10 and 1.90 V.
Upon photoexcitation, transition metal complexes 1 and 2 form a MLCT excited state, which is suggested to facilitate electron transfer mechanisms in photocatalytic cycles.[8b, 9a] Recently, we reported that the lowest energy excited state of dihydrophenazine 3 is also CT in nature.[23] Specifically, intramolecular CT occurs from the electron rich phenazine core (donor) to one of the 2-naphthyl N-substituents (acceptor). Here we show that phenoxazine PC 4 similarly undergoes photoinduced intramolecular CT, as evidenced by a significant solvatochromic effect in the emission (Figure 2A). Additionally, the broad and featureless emission peaks are characteristic of emission from a CT state (Figure 2B).[27]
Figure 2.

Photophysical properties of organic PCs. (A) Photograph showing solvatochromic shifts in the emission of 4 when irradiated with 365 nm light in solvents of increasing polarity; from left to right: 1-hexene, benzene, dioxane, ethyl acetate, pyridine, and DMF. (B) Normalized emission spectra of 4 in solvents of varying polarity. Transient absorption spectra of 3 (C) and 4 (D) in DMA at room temperature. Cyclic voltammetry (CV) experiments with various scan rates for (E) 3 and (F) 4. See Supporting Information for details.
The efficient access of a long-lived excited species by the PC enables sufficient time for bimolecular electron transfer with the desired substrate(s). In the case of transition metal complexes 1 and 2, ultrafast intersystem crossing produces the 3MLCT and is useful by way of extending excited state lifetimes (τ). Photoexcitation quantitatively leads to a long-lived 3MLCT which survives for 1.9 and 1.1 µs (in acetonitrile) for 1 and 2, respectively (Figure 1C). Using nanosecond transient-absorption (TA) spectroscopy performed in DMA at room temperature, we identified long-lived excited states for the organic PCs 3 and 4. The τ of 3 was determined to be 4.3 ± 0.5 µs (Figure 2C) whereas for 4, it is a remarkable two orders of magnitude longer than that of 1, 2 or 3, with τ = 480 ± 50 µs (Figure 2D). By use of a triplet-triplet energy transfer method, we have determined the triplet quantum yield (Φtriplet) of 3 and 4 in DMA at ambient temperature: 3’s Φtriplet is relatively low at 2.0 ± 0.7% while 4 has an impressively high Φtriplet of 90% ± 10%.
Another critical characteristic for successful PCs is radical stability following single electron transfer events. Transition metal complexes 1 and 2 exhibit reversible waves in cyclic voltammetry (CV), a property that indicates stability of the redox-altered catalyst.[7a, 8] Similarly, the CVs corresponding to the 2PC•+/1PC couple of 3 and 4 are reversible (Figure 2E and F). In particular, the difference between the anodic and cathodic peak potential (ΔEp) for 3 is 67 mV (compared to theoretical value of 59 mV),[28] while the ratio of the peak anodic current (ipa) to the peak cathodic current (ipc) is 0.97 (compared to theoretical value of 1) (Figure 2E). Redox reversibility of 3 is in part attributed to the stability of 3’s 2PC•+, as indicated by low E0ox value of 0.21 V vs. SCE; this value is even lower than the redox couple producing ferrocenium (E0ox = ~ 0.4 V vs. SCE).[29] Likewise, the CV of 4 reveals ΔEp = 68 mV and ipa/ipc = 1.28.[30] Additionally, a linear relationship between ipa and the square root of the scan rate (ν1/2) reveals that the CV of 3 and 4 are diffusion limited (Figure 2E and F, insets); this supports the idea that electron transfer between the PC and the electrode (for either 3 or 4) is fast and likely facilitated by small structural reorganization[24] between 1PC and 2PC•+.
With the confirmation that 3 and 4 possess key photophysical and electrochemical characteristics critical for photoredox catalysis, we set out to establish their broader catalytic ability and potential to replace precious metal PCs through performing challenging chemical transformations, particularly ones that have been previously directed by polypyridyl iridium and ruthenium PCs such as 1 and 2.
First, we investigated if the strongly reducing dihydrophenazine 3 could directly reduce CF3I (peak reduction potential (Ep) of −1.52 V vs. SCE on glassy carbon),[31] thereby generating CF3• for the trifluoromethylation of unsaturated substrates (Figure 3A).[32] Using white LED irradiation of 3 (1 to 5 mol %) in the presence of 1.5 eq. of potassium formate (HCOOK), CF3 was successfully installed onto five-membered heteroarenes (indoles, pyrroles), arenes, and alkenes at moderate to excellent yields (42% to 98%). For alkenes, the presence of HCOOK base affords the substitution product while the absence of HCOOK favors the addition product. The reduction of CF3CF2I was also accomplished, generating CF3CF2• for substitution onto indoles and alkenes. The trifluoromethylation of 3-methylindole was achieved with similar yield using natural sunlight. The substitution reaction between 10-undecene-1-ol and CF3I could be performed using lower catalyst loading (0.25 mol %, 69 % yield) or on a larger 10 mmol scale (1.74 g product, 73% yield) while maintaining good yields.
Figure 3.

Photoredox catalyzed transformations using organic PCs 3 and 4. (A) Radical trifluoromethylations using PC 3 on alkenes, five-membered heteroarenes, arenes, and cross-addition on alkenes. (B) Dual organic photoredox and nickel catalyzed C-N cross-coupling reaction scope. (C) Dual organic photoredox and nickel catalyzed C-S cross-coupling scope. Data reported as isolated yields. Values in parentheses are the ratio of Z : E : β-hydride elimination product. aReaction was also conducted using sunlight for 1 week (67% yield for trifluoromethylation, 83% yield for C-N coupling, 94% yield for C-S coupling). bCF3CF2I was used instead of CF3I. cReaction time 6 hours. dReaction was also conducted on a larger 10 mmol scale (73% yield for trifluoromethylation, 53% yield for C-N coupling, 98% yield for C-S coupling). eReaction was also conducted at reduced catalyst loading of 0.25 mol %, instead of standard 1.0 mol % (69% yield for trifluoromethylation, 53% yield for C-N coupling, 98% yield for C-S coupling after 24 hours). fPerformed without HCOOK. gReaction performed with 10 mol % pyrrolidine as the ligand and reduced nickel loading to 1.0 mol %. hReaction catalyzed by PC 4. iReaction catalyzed by PC 3.
Dual catalytic approaches integrating photoredox catalysis using iridium PCs and nickel-catalyzed cross-coupling reactions have enabled access to C-O,[33] C-S,[34] C-N[35] and various C-C[36] reactions. Incorporating the photoredox cycle introduces redox or energy transfer[37] mechanisms with the nickel complexes to complete otherwise demanding catalytic cycles. Cross-coupling reactions have traditionally been catalyzed by palladium complexes at elevated temperatures to construct such critical bonds.[38] Thus, to entirely remove precious metals out of cross-couplings through dual catalytic reactions, we sought to determine if organic PCs 3 and 4 could also enable such challenging reactions.
Previously, a dual photoredox/nickel catalytic approach employing 0.02 mol % of polypyridyl iridium PC Ir[dF(CF)3ppy]2(dtbbpy)PF6 [dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine; dtbbpy = 4,4′-ditertbutyl-2,2′-bipyridine] in conjunction with NiBr2•glyme could efficiently catalyze C-N bond formation under mild reaction conditions.[35a] At similar reaction conditions, albeit using a higher catalyst loading (0.4 mol %), PC 3 or PC 4 in combination with NiBr2•glyme successfully catalyzed C-N coupling reactions at good to excellent yields (68% to 96%, Figure 3B). The scope of amines included both primary (aniline, furfurylamine, and propylamine) and secondary amines (pyrrolidine and morpholine) and were effectively coupled with electron rich, electron poor, and heterocyclic aryl bromides. For secondary amines, both PC 3 and 4 catalyzed C-N bond formations, although PC 3 generally gave slightly higher yields. Whilst PC 3 was unsuccessful in effecting C-N cross-coupling involving primary amines, PC 4 proved to be effective to couple primary amines in high yields.
In regards to C-S cross-coupling, the dual photoredox/nickel catalysis with 2 mol % Ir[dF(CF)3ppy]2(dtbbpy)PF6 and NiCl2•glyme produced C-S coupled products under mild conditions.[34] At analogous reaction conditions, phenoxazine PC 4 achieved C-S cross-couplings at good to excellent yields (64% to 98%, Figure 3C) but proved efficient at a much lower PC loading of 0.2 mol %. Aryl thiol (thiophenol), alkyl thiol (4-Methoxybenzyl mercaptan, 1-octanethiol and cyclohexanethiol) and cysteine (N-(tert-Butoxycarbonyl)-L-cysteine methyl ester) successfully coupled with a variety of aryl bromides. It is important to note that aryl bromide coupling partners were successfully incorporated with organic PC 4, which were shown to be inactive when using Ir[dF(CF)3ppy]2(dtbbpy)PF6.[34] PC 3 was unsuccessful in C-S coupling reactions, presumably due to its stable radical cation (E0ox = 0.21 V vs. SCE) being unable to generate a thiol radical involved in the coupling reaction.[34]
These photoredox/nickel C-N and C-S cross-coupling reactions could be driven by natural sunlight to obtain similarly high yield. Furthermore, both the C-N and C-S couplings could be performed on a larger 10 mmol scale reaction for C-N (1.22g, 53% yield) and C-S (2.92g, 98% yield) couplings. In these scaled reactions, C-S coupling maintained the high yield while C-N coupling suffered a 30% drop in yield. This lower yield was attributed to limited light penetration owing to the opaque solution mixture compounded by the lower molar absorptivity of PC 3.
In sum, photophysical and electrochemical characterizations on dihydrophenazine and phenoxazine PCs 3 and 4 reveal that these molecules possess the key attributes vital to successful photoredox catalysis, including redox reversibility and strong visible light absorption to efficiently access a highly reducing triplet state through formation of CT excited state. The triplet excited state of these organic PCs are long-lived and accessed in 90% ± 10% quantum yield by PC 4. Highlighting that 4 is an organic analogue of the iridium complex 1, both PCs have almost identical E0*, E0ox and Etriplet values. The potential for replacement of polypyridyl iridium and ruthenium complexes by organic PCs 3 and 4 is demonstrated by the ability of these organic analogues to catalyze trifluoromethylations and dual photoredox/nickel C-N and C-S cross-coupling reactions using visible light, including natural sunlight.
Supplementary Material
Acknowledgments
This work was supported by the University of Colorado Boulder and the Advanced Research Projects Agency-Energy (DE-AR0000683). Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM119702. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Acknowledgement is made to the donors of The American Chemical Society Petroleum Research Fund for partial support of this research (56501-DNI7). S.M.S and M.D.R. acknowledge support from a DOE Graduate Assistance in Areas of National Need Fellowship. We gratefully acknowledge the use of XSEDE supercomputing resources (NSF ACI-1053575).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Tucker JW, Stephenson CRJ. J Org. Chem. 2012;77:1617–1622. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]; b) Prier CK, Rankic DA, MacMillan DWC. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schultz DM, Yoon TP. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) You Y, Nam W. Chem. Soc. Rev. 2012;41:7061–7084. doi: 10.1039/c2cs35171d. [DOI] [PubMed] [Google Scholar]; b) O'Regan B, Gratzel M. Nature. 1991;353:737–740. [Google Scholar]; c) Wang P, Zakeeruddin SM, Moser JE, Nazeeruddin MK, Sekiguchi T, Gratzel M. Nat. Mater. 2003;2:402–407. doi: 10.1038/nmat904. [DOI] [PubMed] [Google Scholar]
- 3.Adachi C, Baldo MA, Thompson ME, Forrest SR. J Appl. Phys. 2001;90:5048–5051. [Google Scholar]
- 4.a) Gill MR, Garcia-Lara J, Foster SJ, Smythe C, Battaglia G, Thomas JA. Nat. Chem. 2009;1:662–667. doi: 10.1038/nchem.406. [DOI] [PubMed] [Google Scholar]; b) Lo KK-W. Acc. Chem. Res. 2015;48:2985–2995. doi: 10.1021/acs.accounts.5b00211. [DOI] [PubMed] [Google Scholar]
- 5.Allardyce CS, Dyson PJ. Platin. Met. Rev. 2001;45:62–69. [Google Scholar]
- 6.Li F, Collins JG, Keene FR. Chem. Soc. Rev. 2015;44:2529–2542. doi: 10.1039/c4cs00343h. [DOI] [PubMed] [Google Scholar]
- 7.a) Arias-Rotondo DM, McCusker JK. Chem. Soc. Rev. 2016;45:5803–5820. doi: 10.1039/c6cs00526h. [DOI] [PubMed] [Google Scholar]; b) Lamansky S, Djurovich P, Murphy D, Abdel-Razzaq F, Kwong R, Tsyba I, Bortz M, Mui B, Bau R, Thompson ME. Inorg. Chem. 2001;40:1704–1711. doi: 10.1021/ic0008969. [DOI] [PubMed] [Google Scholar]
- 8.a) Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. In: Photochemistry and Photophysics of Coordination Compounds II. Balzani V, Campagna S, editors. Springer Berlin Heidelberg, Berlin, Heidelberg: 2007. pp. 143–203. [Google Scholar]; b) Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, von Zelewsky A. Coord. Chem. Rev. 1988;84:85–277. [Google Scholar]
- 9.a) Bock CR, Connor JA, Gutierrez AR, Meyer TJ, Whitten DG, Sullivan BP, Nagle JK. J Am. Chem. Soc. 1979;101:4815–4824. [Google Scholar]; b) King KA, Spellane PJ, Watts RJ. J Am. Chem. Soc. 1985;107:1431–1432. [Google Scholar]
- 10.a) Higgins RF, Fatur SM, Shepard SG, Stevenson SM, Boston DJ, Ferreira EM, Damrauer NH, Rappé AK, Shores MP. J Am. Chem. Soc. 2016;138:5451–5464. doi: 10.1021/jacs.6b02723. [DOI] [PubMed] [Google Scholar]; b) Kainz QM, Matier CD, Bartoszewicz A, Zultanski SL, Peters JC, Fu GC. Science. 2016;351:681–684. doi: 10.1126/science.aad8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Romero NA, Nicewicz DA. Chem. Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; b) Neumann M, Füldner S, König B, Zeitler K. Angew. Chem. Int. Ed. 2011;50:951–954. doi: 10.1002/anie.201002992. [DOI] [PubMed] [Google Scholar]
- 12.a) Corrigan N, Shanmugam S, Xu J, Boyer C. Chem. Soc. Rev. 2016;45:6165–6212. doi: 10.1039/c6cs00185h. [DOI] [PubMed] [Google Scholar]; b) Chen M, Zhong M, Johnson JA. Chem. Rev. 2016;116:10167–10211. doi: 10.1021/acs.chemrev.5b00671. [DOI] [PubMed] [Google Scholar]; c) Shaw MH, Twilton J, MacMillan DWC. J Org. Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Hari DP, Konig B. Chem. Comm. 2014;50:6688–6699. doi: 10.1039/c4cc00751d. [DOI] [PubMed] [Google Scholar]; b) Rohokale RS, Koenig B, Dhavale DD. J Org. Chem. 2016;81:7121–7126. doi: 10.1021/acs.joc.6b00979. [DOI] [PubMed] [Google Scholar]; c) Meyer AU, Straková K, Slanina T, König B. Chem. Eur. J. 2016;22:8694–8699. doi: 10.1002/chem.201601000. [DOI] [PubMed] [Google Scholar]
- 14.Yoshioka E, Kohtani S, Jichu T, Fukazawa T, Nagai T, Kawashima A, Takemoto Y, Miyabe H. J Org. Chem. 2016;81:7217–7229. doi: 10.1021/acs.joc.6b01102. [DOI] [PubMed] [Google Scholar]
- 15.a) Matsui JK, Molander GA. Org. Lett. 2017;19:950–953. doi: 10.1021/acs.orglett.7b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Joshi-Pangu A, Lévesque F, Roth HG, Oliver SF, Campeau L-C, Nicewicz D, DiRocco DA. J Org. Chem. 2016;81:7244–7249. doi: 10.1021/acs.joc.6b01240. [DOI] [PubMed] [Google Scholar]; c) Alfonzo E, Alfonso FS, Beeler AB. Org. Lett. 2017;19:2989–2992. doi: 10.1021/acs.orglett.7b01222. [DOI] [PubMed] [Google Scholar]; d) Margrey KA, Nicewicz DA. Acc. Chem. Res. 2016;49:1997–2006. doi: 10.1021/acs.accounts.6b00304. [DOI] [PubMed] [Google Scholar]; e) Kotani H, Ohkubo K, Fukuzumi S. J Am. Chem. Soc. 2004;126:15999–16006. doi: 10.1021/ja048353b. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh I, Ghosh T, Bardagi JI, König B. Science. 2014;346:725–728. doi: 10.1126/science.1258232. [DOI] [PubMed] [Google Scholar]
- 17.a) Luo J, Zhang J. ACS Catal. 2016;6:873–877. [Google Scholar]; b) Uoyama H, Goushi K, Shizu K, Nomura H, Adachi C. Nature. 2012;492:234–238. doi: 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]
- 18.a) Poelma SO, Burnett GL, Discekici EH, Mattson KM, Treat NJ, Luo Y, Hudson ZM, Shankel SL, Clark PG, Kramer JW, Hawker CJ, Read de Alaniz J. J Org. Chem. 2016;81:7155–7160. doi: 10.1021/acs.joc.6b01034. [DOI] [PubMed] [Google Scholar]; b) Treat NJ, Sprafke H, Kramer JW, Clark PG, Barton BE, Read de Alaniz J, Fors BP, Hawker CJ. J Am. Chem. Soc. 2014;136:16096–16101. doi: 10.1021/ja510389m. [DOI] [PubMed] [Google Scholar]; c) Discekici EH, Treat NJ, Poelma SO, Mattson KM, Hudson ZM, Luo Y, Hawker CJ, de Alaniz JR. Chem. Commun. 2015;51:11705–11708. doi: 10.1039/c5cc04677g. [DOI] [PubMed] [Google Scholar]
- 19.a) Miyake GM, Theriot JC. Macromolecules. 2014;47:8255–8261. [Google Scholar]; b) Theriot JC, Lim C-H, Yang H, Ryan MD, Musgrave CB, Miyake GM. Science. 2016;352:1082–1086. doi: 10.1126/science.aaf3935. [DOI] [PubMed] [Google Scholar]
- 20.a) Matyjaszewski K, Xia J. Chem. Rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]; b) Ouchi M, Terashima T, Sawamoto M. Chem. Rev. 2009;109:4963–5050. doi: 10.1021/cr900234b. [DOI] [PubMed] [Google Scholar]
- 21.Shanmugam S, Boyer C. Science. 2016;352:1053–1054. doi: 10.1126/science.aaf7465. [DOI] [PubMed] [Google Scholar]
- 22.Isse AA, Lin CY, Coote ML, Gennaro A. J Phys. Chem. B. 2011;115:678–684. doi: 10.1021/jp109613t. [DOI] [PubMed] [Google Scholar]
- 23.Lim C-H, Ryan MD, McCarthy BG, Theriot JC, Sartor SM, Damrauer NH, Musgrave CB, Miyake GM. J Am. Chem. Soc. 2017;139:348–355. doi: 10.1021/jacs.6b11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Pearson RM, Lim C-H, McCarthy BG, Musgrave CB, Miyake GM. J Am. Chem. Soc. 2016;138:11399–11407. doi: 10.1021/jacs.6b08068. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ramsey BL, Pearson RM, Beck LR, Miyake GM. Macromolecules. 2017;50:2668–2674. doi: 10.1021/acs.macromol.6b02791. [DOI] [PMC free article] [PubMed] [Google Scholar]; C) Ryan MD, Pearson RM, French TA, Miyake GM. Macromolecules. 2017 doi: 10.1021/acs.macromol.7b00502. 10/1021/acs.macromol.7b00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theriot JC, McCarthy BG, Lim C-H, Miyake GM. Macromol. Rapid Commun. 2017 doi: 10.1002/marc.201700040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Hiraoka S, Okamoto T, Kozaki M, Shiomi D, Sato K, Takui T, Okada K. J Am. Chem. Soc. 2004;126:58–59. doi: 10.1021/ja0367748. [DOI] [PubMed] [Google Scholar]; b) Zhu Y, Kulkarni AP, Wu P-T, Jenekhe SA. Chem. Mater. 2008;20:4200–4211. [Google Scholar]
- 27.a) Ryan MD, Theriot JC, Lim C-H, Yang H, Lockwood AG, Garrison NG, Lincoln SR, Musgrave CB, Miyake GM. J Polym. Sci. A Polym. Chem. 2017 doi: 10.1002/pola.28574. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Nat. Methods. 2008;5:763–775. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 28.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. Second. Wiley; 2001. [Google Scholar]
- 29.Connelly NG, Geiger WE. Chem. Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 30.The oxidation of 4 occurs near the oxidation window of DMA, which causes the ipa/ipc to deviate from 1; 4’s ipa/ipc values improve to 1.11 in DMF and 1.01 in THF with increasingly wider solvent oxidation window (see supporting information).
- 31.Andrieux CP, Gelis L, Medebielle M, Pinson J, Saveant JM. J Am. Chem. Soc. 1990;112:3509–3520. [Google Scholar]
- 32.a) Iqbal N, Choi S, Ko E, Cho EJ. Tetrahedron Lett. 2012;53:2005–2008. [Google Scholar]; b) Nguyen JD, Tucker JW, Konieczynska MD, Stephenson CRJ. J Am. Chem. Soc. 2011;133:4160–4163. doi: 10.1021/ja108560e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pham PV, Nagib DA, MacMillan DWC. Angew. Chem. Int. Ed. 2011;50:6119–6122. doi: 10.1002/anie.201101861. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wallentin C-J, Nguyen JD, Finkbeiner P, Stephenson CRJ. J Am. Chem. Soc. 2012;134:8875–8884. doi: 10.1021/ja300798k. [DOI] [PubMed] [Google Scholar]; e) Koike T, Akita M. Top. Catal. 2014;57:967–974. [Google Scholar]
- 33.Terrett JA, Cuthbertson JD, Shurtleff VW, MacMillan DWC. Nature. 2015;524:330–334. doi: 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oderinde MS, Frenette M, Robbins DW, Aquila B, Johannes JW. J Am. Chem. Soc. 2016;138:1760–1763. doi: 10.1021/jacs.5b11244. [DOI] [PubMed] [Google Scholar]
- 35.a) Corcoran EB, Pirnot MT, Lin S, Dreher SD, DiRocco DA, Davies IW, Buchwald SL, MacMillan DWC. Science. 2016;353:279–283. doi: 10.1126/science.aag0209. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Oderinde MS, Jones NH, Juneau A, Frenette M, Aquila B, Tentarelli S, Robbins DW, Johannes JW. Angew. Chem. Int. Ed. 2016;55:13219–13223. doi: 10.1002/anie.201604429. [DOI] [PubMed] [Google Scholar]
- 36.a) Shields BJ, Doyle AG. J Am. Chem. Soc. 2016;138:12719–12722. doi: 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Le CC, MacMillan DWC. J Am. Chem. Soc. 2015;137:11938–11941. doi: 10.1021/jacs.5b08304. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Noble A, McCarver SJ, MacMillan DWC. J Am. Chem. Soc. 2015;137:624–627. doi: 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jouffroy M, Primer DN, Molander GA. J Am. Chem. Soc. 2016;138:475–478. doi: 10.1021/jacs.5b10963. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tellis JC, Primer DN, Molander GA. Science. 2014;345:433–436. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437–440. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC. Science. 2016;352:1304–1308. doi: 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welin ER, Le C, Arias-Rotondo DM, McCusker JK, MacMillan DWC. Science. 2017;355:380–385. doi: 10.1126/science.aal2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.a) Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457–2483. [Google Scholar]; b) Ruiz-Castillo P, Buchwald SL. Chem. Rev. 2016;116:12564–12649. doi: 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Molnár Á. Chem. Rev. 2011;111:2251–2320. doi: 10.1021/cr100355b. [DOI] [PubMed] [Google Scholar]; d) Jana R, Pathak TP, Sigman MS. Chem. Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.