

Abstract
Background
Recent large-scale whole genome sequencing efforts in birds have elucidated broad patterns of avian phylogeny and genome evolution. However, despite the great interest in economically important phasianids like Gallus gallus (Red Junglefowl, the progenitor of the chicken), we know little about the genomes of closely related species. Gallus gallus is highly sexually dichromatic and polygynous, but its sister genus, Bambusicola, is smaller, sexually monomorphic, and monogamous with biparental care. We sequenced the genome of Bambusicola thoracicus (Chinese Bamboo Partridge) using a single insert library to test hypotheses about genome evolution in galliforms. Selection acting at the phenotypic level could result in more evidence of positive selection in the Gallus genome than in Bambusicola. However, the historical range size of Bambusicola was likely smaller than Gallus, and demographic effects could lead to higher rates of nonsynonymous substitution in Bambusicola than in Gallus.
Results
We generated a genome assembly suitable for evolutionary analyses. We examined the impact of selection on coding regions by examining shifts in the average nonsynonymous to synonymous rate ratio (dN/dS) and the proportion of sites subject to episodic positive selection. We observed elevated dN/dS in Bambusicola relative to Gallus, which is consistent with our hypothesis that demographic effects may be important drivers of genome evolution in Bambusicola. We also demonstrated that alignment error can greatly inflate estimates of the number of genes that experienced episodic positive selection and heterogeneity in dN/dS. However, overall patterns of molecular evolution were robust to alignment uncertainty. Bambusicola thoracicus has higher estimates of heterozygosity than Gallus gallus, possibly due to migration events over the past 100,000 years.
Conclusions
Our results emphasized the importance of demographic processes in generating the patterns of variation between Bambusicola and Gallus. We also demonstrated that genome assemblies generated using a single library can provide valuable insights into avian evolutionary history and found that it is important to account for alignment uncertainty in evolutionary inferences from draft genomes.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4711-0) contains supplementary material, which is available to authorized users.
Keywords: Galliforms, Phasianidae, Nonsynonymous to synonymous substitution rate ratio, Selection, Effective population size, Alignment uncertainty
Background
The availability of whole-genome sequences from phylogenetically diverse bird species has provided broad insights into avian evolution [1, 2]. Galliformes, which contains chickens, turkeys, quail, guineafowl, and pheasants, is the most economically important avian order, and their genetics, physiology, development, and behavior have been studied extensively. Indeed, the first published avian genome was from Red Junglefowl (Gallus gallus), the ancestor of domesticated chickens. Several additional galliform genomes have been sequenced [3–6], but none of these genomes are close relatives to the Red Junglefowl. Thus, little is known about genome evolution for the closest relatives of the junglefowl (Gallus species). Of particular interest are genomes of the bamboo partridges (Bambusicola), the sister genus of Gallus [7], with the Chinese Bamboo Partridge (Bambusicola thoracicus) being the best-characterized species. Gallus and Bambusicola diverged from each other in the Miocene [8], approximately 15 million years ago (MYA), while the Gallus/Bambusicola clade diverged from the francolins about 20 MYA [8]. Comparative genomic studies between the Bambusicola and Gallus genera provide an opportunity to study changes in life history traits at the molecular level.
There are a number of phenotypic and behavioral differences between the junglefowl (Gallus) and Bambusicola. Junglefowl exhibit striking sexual dichromatism. Males are more colorful than females, with elaborated tail, hackle, and saddle feathers. In addition, combs and wattles in males are much larger, and typically redder, than in females (with Gallus varius females lacking combs and wattles completely). Males are also larger than females, and frequently possess large spurs used in combat with other males. Although some Gallus may nest monogamously, they are frequently polygynous [9]. Sexual selection is well documented in Gallus gallus, with females exhibiting strong preferences for males with larger, redder combs [10, 11]. In contrast, the much smaller Bambusicola are monochromatic and exhibit little size dimorphism [9]. Although male Bambusicola have spurs, which are weapons used in male-male competition in galliforms, they are much smaller (even relative to body size) than Red Junglefowl spurs [12]. Bamboo partridges appear to be monogamous, as they are frequently seen in pairs that often duet [13]. These differences between junglefowl and bamboo partridges imply that different characteristics have been selected for in each lineage.
Bambusicola thoracicus and Gallus gallus also differ demographically. Although Gallus gallus did not have the nearly worldwide distribution of domestic chickens, the historical range of Gallus gallus was still relatively large, covering much of southeast Asia, where they were typically found in various habitats going up to 2000 m in elevation [13]. Overall, the estimated historical range size for Gallus gallus is 5,100,000 km2 [13]. Bambusicola thoracicus is the most widespread of the three species of Bambusicola, but it still had a much smaller historic range than Gallus gallus. Its native range is restricted to China where it has an estimated range size of 1,280,000 km2 [14]. Although the historic range sizes suggest that the ancestral population size of Gallus gallus was larger than that of Bambusicola thoracicus, Bambusicola are much smaller than Gallus, and small body sizes are associated with larger effective population sizes [15]. Thus, it is not clear which species had a larger ancestral effective population size (Ne).
Here we present a draft genome of Bambusicola thoracicus as a resource for comparative evolutionary analyses within galliforms, especially with respect to Gallus. We explored the impact of demographic and phenotypic change on genome evolution in these two species. Demographically, we considered two mutually exclusive alternative hypotheses (Table 1). First, the larger range size of the Red Junglefowl reflects a larger Ne in that species, and therefore, we should observe a lower nonsynonymous to synonymous substitution rate ratio (dN/dS) in Gallus gallus compared to Bambusicola thoracicus. Alternatively, the smaller body size of Bambusicola thoracicus may have led to a larger historic Ne, leading to a smaller dN/dS compared to Gallus gallus. The expectation of a lower dN/dS in whichever taxon had the larger Ne reflects the greater efficacy of purifying selection in large populations. However, strong natural and sexual selection also could have affected patterns of genome evolution, leading to two additional hypotheses that are not mutually exclusive (Table 1). Bamboo partridges appear more similar to francolins, the sister taxa of the common ancestor of Bambusicola and Gallus. Francolins are largely monogamous with little dichromatism, and they do not exhibit elaborate ornamental traits. If the phenotypic differences between Gallus and Bambusicola reflect strong directional selection (either natural or sexual), we predict an elevated dN/dS due to positive selection in Gallus, at least for a proportion of genes associated with phenotypes under selection. Additionally, if sexual selection has been more intense in the Gallus gallus lineage, we expect Gallus gallus to exhibit lower heterozygosity across the genome than Bambusicola thoracicus.
Table 1.
Predicted hypotheses for rates of molecular evolution in Bambusicola and Gallus
| Hypothesis | Mean dN/dS | dN/dS > 1 | Heterozygosity | |
|---|---|---|---|---|
| Demography (assuming no selection) | Higher Ne in Gallus | Lower in Gallus | Higher in Gallus | Higher in Gallus |
| Lower Ne in Gallus | Higher in Gallus | Lower in Gallus | Lower in Gallus | |
| Selection (assuming equal Ne) | Directional selection on Gallus | Higher in Gallus | Elevated for a proportion of genes in Gallus | Equal between species |
| Sexual Selection on Gallus | Higher in Gallus | Elevated for a proportion of genes in Gallus | Lower in Gallus (due to reduced male population size) |
Predicted patterns of molecular evolution and heterozygosity for Gallus compared to Bambusicola are given for both demographic and selection-based hypotheses of genome evolution. The mean dN/dS refers to results for gene-wide differences in molecular evolution from branch tests while dN/dS > 1 is determined by results from branch-site tests
We assembled a draft Bambusicola thorasicus genome sequence using a single Illumina library with 25× coverage. In addition to testing the proposed biological hypotheses, we addressed technical questions regarding the use of relatively low-coverage genomes generated using short-read technologies. Specifically, we assessed multiple approaches for assembling the Bambusicola genome from such data and evaluated the quality of our assembly using both a standard BUSCO analysis [16] and an assessment of our ability to recover an independently generated set of curated ultraconserved elements (UCEs) from our genome assembly [17]. Given the interest in identifying selection in many comparative genomic studies, we determined whether alignment errors could have an impact on estimates of dN/dS. Our evaluation of the de novo Bambusicola thoracicus genome demonstrates that relatively low-coverage bird genomes can provide valuable evolutionary insights and that they allow the annotation of genes as well as large-scale genomic comparisons.
Methods
Genome sequencing, assembly, and annotation
The Bambisicola thoracicus sample came from blood preserved in lysis buffer obtained from a female individual from a captive breeding program, and the sample was originally collected for Kimball et al. [18]. This individual was released back to captivity after blood collection. We extracted DNA using the Gentra PureGene DNA Isolation Kit (Qiagen) following manufacturer’s instructions. Mitochondrial regions previously sequenced from this individual [18] showed high identity to published sequences sampled throughout the range [19], indicating it was correctly identified. Library preparation, fragment selection, and sequencing was carried out at the University of Florida Interdisciplinary Center for Biotechnology Research. A single insert library was prepared using Illumina’s NEBNext Ultra DNA Library Prep Kit following the manufacturer’s instructions. A single insert of approximately 500 bp was selected for the NextSeq500 sample preparation protocol. The library was sequenced on a single flow cell using paired-end 150 bp reads on an Illumina NextSeq500.
We assembled a Bambusicola thoracicus draft genome sequence using de novo methods. First, we discarded PCR duplicates from the genome sequencing reads using in-house Perl scripts. Then we removed Illumina barcodes and adapters using Trimmomatic [20]. We only retained sequences with an average Phred score of at least 20, with each four-base sliding window having an average Phred score of at least 15, and with a minimum sequence length of 70. We corrected possible sequencing errors based on the distribution of Kmer frequencies using SOAPec v2.01 with default settings [21]. We then built de novo assemblies from the edited reads using SOAPdenovo2 v2.04 [21] and ABySS v1.9.0 [22]. We also assembled the genome with MaSuRCA v2.3.2 [23], which uses its own raw data quality control tools. For computational feasibility, the three assemblies used Kmer values of 63, 63, and 35 respectively, and we merged scaffolds with Metassembler v1.5 [24]. We estimated proportions of repetitive elements with RepeatMasker v4.0.5 using the “Aves” repeat library [25].
We annotated scaffolds with a length of 1 kb or greater using MAKER v2.31.8 [26] and conducted gene predictions using AUGUSTUS v3.2.1 [27] with a hidden Markov model trained from Gallus gallus RefSeq sequences. In addition to ab initio gene prediction, we used homologous protein evidence for annotated genes from the amino acid sequences of Gallus gallus [28], Meleagris gallopavo [29], and Taeniopygia guttata [30]. We assessed annotation quality with BUSCO v1.1b1 [16], which estimated the proportion of genes missing from our annotations.
To account for possible annotation biases in downstream analyses, in which we compared our Bambusicola data with other galliform genomes, we reannotated the genomes of Coturnix japonica [4, 31] and Colinus virginianus [5, 32] using MAKER [26] as described above. Since the draft genomes of Coturnix japonica and Colinus virginianus also are based solely on computational predictions, this step makes the gene models for the three draft genomes in this study more comparable than if they were generated using different prediction methods. We did not reannotate Gallus or Meleagris because the assembly and annotation for both of those genomes is excellent. In fact, a number of gene models have been validated using RNAseq data.
Finally, we assessed both the assembly quality and our ability to accurately annotate unique sequence features in the Bambusicola thoracicus genome using 3854 ultraconserved elements (UCEs) that were generated by sequence capture from the same individual [17]. The UCE sequences were aligned against the masked Bambusicola assembly using the NUCmer program [33] from MUMmer [34] with default settings. We extracted coverage and mismatch information for each UCE from the pairwise alignments.
Pairwise comparisons between Bambusicola and Gallus
We assessed the completeness of our assembly by comparing it with the reference Gallus gallus genome assembly [35]. We aligned the masked Bambusicola scaffolds to Gallus genomic sequence using the NUCmer [33] with default settings. We calculated the proportion of sequenced bases, pairwise nucleotide diversity, and GC content for non-overlapping 100 kb sliding windows from the alignable sequences using Perl scripts. Results from sliding windows were visualized with CIRCOS [36].
Testing hypotheses of molecular evolution
We circumscribed gene families from the annotated genomes of Gallus gallus, Bambusicola thoracicus, Coturnix japonica, Meleagris gallopavo, and Colinus virginianus using OrthoMCL [37], and we identified those gene clusters that had exactly one sequence from each of the five species. We refer to these as the single-copy orthologous groups. We obtained codon alignments for the single-copy orthologous groups by first aligning amino acids with MUSCLE [38] and then mapping codons onto the multiple sequence alignments with a custom Perl script (https://github.com/gtiley/Alignment_Tools/tree/master/Codon_Alignment). We constructed maximum likelihood gene trees for all single-copy orthologous groups with all five taxa present from the codon alignments using RAxML [39] with the GTR + Γ nucleotide substitution model. We attempted to improve the accuracy of the gene tree topologies using TreeFix [40]. TreeFix uses the species tree, the ML estimate of the gene tree, and the multiple sequence alignment as input, and it searches for the rooted gene tree topology that implies the fewest duplications and losses without a significant decrease in the likelihood compared to the ML tree. For the TreeFix analysis, we used the species relationships from Hosner et al. [41]; however, the relationships among our focal taxa are strongly corroborated by multiple data types [7, 42].
We next attempted to identify and remove anomalous or erroneous gene sequences that might mislead subsequent molecular evolution analyses from the single-copy orthologous groups. We optimized dN/dS on each branch of the gene trees using PAML v. 4.8a [43] and calculated the minimum patristic distance based on dS for each sequence in the single-copy gene trees using the ape package [44] in R [45]. We constructed a Beta distribution from the mean and variance of the minimum patristic dS estimates. We removed any sequence that had a nearest neighbor distance in the 99th percentile of the theoretical Beta distribution from the gene tree and excluded the gene tree from further analyses.
We tested rates of molecular evolution on the remaining single-copy orthologous groups in which the gene tree topology after the TreeFix analysis was identical to the species tree topology (i.e. gene trees in which the sequences appear to be orthologs). We used PAML v. 4.8a [43] to test whether any of these single-copy orthologous groups exhibited shifts in gene-wide dN/dS [46] and to test for episodic positive selection acting on a proportion of sites [47]. For both tests, we designated a single branch of interest as the foreground branch; the remaining branches are the background branches. In the tests for shifts in gene-wide dN/dS (branch tests), the null hypothesis was that all branches of the gene tree had the same dN/dS whereas the alternative hypothesis added one additional free dN/dS parameter on the foreground branch. For branch tests, the p-value can be calculated using the likelihood ratio test (LRT), assuming that the LRT statistic is approximately distributed as χ21. The test for episodic positive selection (branch-site test) similarly used a LRT ~ χ21, but rather than a single dN/dS across all sites in the gene, it modeled molecular evolution with finite mixtures of dN/dS values across sites. For the null hypothesis, all branches in a gene tree had a proportion of sites under purifying selection (i.e. dN/dS < 1) and a proportion of neutrally evolving sites (i.e. dN/dS = 1). The alternative hypothesis allowed a third class where a proportion of sites under positive selection (i.e. dN/dS > 1) are present on the foreground branch.
We tested three branches in the single-copy orthologous groups for differences in the gene-wide dN/dS and for positive selection: 1) the Bambusicola thoracicus terminal branch, 2) the Gallus gallus terminal branch, and 3) the branch leading to the most recent common ancestor (MRCA) of Bambusicola and Gallus. These three branches were treated as unconstrained foreground branches for both branch and branch-site tests. Since our analyses involved hypothesis tests for many gene trees, we controlled for false positives by assuming a false discovery rate of 0.05 and applying the method of Benjamini and Hochberg [48]. Briefly, p-values were ranked from lowest to highest, and the Benjamini-Hochberg correction computed a q-value for each p-value that depended only on the false discovery rate and the number of tests performed. If a p-value was less than its corresponding q-value, we rejected the null hypothesis. This allowed us to reduce the number of false positives while not applying a multiple-testing correction as severe as the Bonferroni [48]. Although branch-site tests can be vulnerable to false discovery rates higher than 0.05, we accepted this correction as a necessary trade-off due to the low power for tests of positive selection using gene trees with only five taxa [49]. We preformed corrections for the Bambusicola, Gallus, and MRCA branch and branch-site tests independently.
Uncertainty in estimates of episodic positive selection
While the branch-site test has high power even when only a small proportion of sites have experienced positive selection [50], it may be susceptible to alignment errors [51]. To account for potential alignment error in the branch-site tests, we reanalyzed the single-copy orthologous groups with significant branch-site tests by integrating the branch-site tests over alignment uncertainty using BAli-Phy [52, 53], a program that implements a Bayesian Markov chain Monte Carlo (MCMC) analysis that jointly estimates posterior distributions of the alignment and dN/dS parameters. For each test, we ran two independent chains of 25,000 MCMC samples and proposed five alignments for each sample, discarding the first 2500 iterations from each chain as burn-in. We applied the Rao-Blackwell estimator for the posterior probability of positive selection. We diagnosed convergence when all parameter posterior distributions had an effective sample size greater than 300 and a potential scale reduction factor of approximately 1 for the 80% credible interval. Complete details of the BAli-Phy analyses are in the supplementary material (Additional file 1).
Effects of alignment error on estimates of gene-wide dN/dS
We also examined whether alignment error affected branch tests. For single-copy gene families that had a significant branch test from PAML, we ran BAli-Phy for one chain of 10,000 iterations under a codon model with a single dN/dS across all branches and randomly sampled 100 alignments without burn-in, as the MCMC chain started with the MUSCLE alignment (Additional file 1). We then re-evaluated the branch tests on these 100 alignments using PAML with methods described above.
Enrichment of functional categories in the genes subject to selection
We assigned GO terms to each single-copy orthologous group based on Gallus gallus annotations from AgBase [54]. Enrichment for generic GO slim processes, functions, and components [55] were inferred from genes with significant branch or branch-site tests using a two-sided Fisher exact test implemented in R [45]. We tested for overrepresentation and underrepresentation of GO terms based on the number of significant and non-significant tests for a branch of interest compared to all other significant and non-significant tests. For example, the contingency table when testing for enrichment of a GO category on the Bambusicola branch would have ((Number of significant tests for GO category in Bambusicola, Number of significant tests for GO category not in Bambusicola), (Number of nonsignificant tests for GO category in Bambusicola, Number of nonsignificant tests for GO category not in Bambusicola)) (Additional file 1: Figure S1). For branch tests, we treated orthologous groups that had a dN/dS significantly higher or lower than the background separately. Again, we controlled for a false discovery rate of 0.05 with a Benjamini-Hochberg correction [48].
Estimating long-term effective population size and measuring heterozygosity
In order to explore how demographic differences between Bambusicola thoracicus and Gallus gallus may explain the observed patterns of molecular evolution, we estimated effective population size (Ne) over time for Bambusicola using the Pairwise Sequential Markov Coalescent (PSMC) model [56]. We used SAMtools mpileup on a BWA [57] alignment of Bambusicola thoracicus paired-end reads to their de novo genome assembly to call heterozygous bases, but applied the C50 option to correct mapping quality scores for high quality reads with uncertain mapping positions, such as repetitive regions of the genome. All genomic sites considered for PSMC analyses had a minimum read depth of 10 and a maximum read depth of 50. Because of the reduced recombination on sex chromosomes, we only analyzed the genomic sequence data that mapped to autosomes. Since genome architecture of birds is relatively stable (e.g. [58, 59]), we excluded those Bambusicola scaffolds that mapped to the Z chromosome from the NUCmer alignment with the Gallus reference genome. Scaffolds that did not uniquely align to any Gallus autosome also were excluded from PSMC analysis.
To compare the Ne estimates over time from the Bambusicola genome with those from Gallus gallus, we reanalyzed two Gallus gallus genomes from Wang et al. [60] that had 23× and 35× coverage. We downloaded data from GenBank BioProject accession PRJNA241474 [61] for SRA accessions SRX511214 and SRX511217 respectively. Short reads were trimmed and corrected using methods described above. We generated two PSMC analyses for the 23× Gallus genome: 1) Short reads were aligned to the reference Gallus genome using BWA [57], and 2) a de novo assembly was generated using MaSuRCA [23] and reads were then aligned to the de novo assembly with BWA [57]. We had insufficient computer memory to construct a de novo assembly for the 35× genome. We called heterozygous bases using the same methods as for Bambusicola, except the 35× Gallus genome had a minimum read depth of 15 and a maximum read depth of 70. Again, we used only data that mapped to Gallus gallus autosomes.
We ran all PSMC analyses using 64 atomic time intervals with the options –N 30 –t 15 –r 5 –p “4 + 25*2 + 4 + 6”, such that the recombination rate was relatively constant across each atomic time interval. The PSMC model of Li and Durbin [56] compressed heterozygosity information into bins by examining 100 bp of contiguous sequence and calling a bin heterozygous if there was at least one high quality bi-allelic site in within those 100 bp. We further split the binned sequence data into segments of no more than 10,000 bins and performed 100 bootstrap replicates for each analysis to measure uncertainty in estimates of Ne. We assumed a generation time of 1 year [62, 63] and a mutation rate of 1.91 × 10− 9 per year [64] for both Bambusicola and Gallus to convert from coalescent units to absolute time.
In addition to inferring differences in Ne based on the distribution of heterozygous sites, we wanted to explore differences in heterozygosity across autosomes between Bambusicola and Gallus. We estimated the proportion of heterozygous sites per chromosome using the mpileup calls on our Bambusicola scaffolds that could be aligned to the reference Gallus gallus genome. We only considered bi-allelic sites with a depth of at least 20, bases with phred scores of 20, and a minor allele frequency of at least 0.1. We estimated a false positive rate for our heterozygous base calls using 21 independently Sanger sequenced nuclear introns from the same Bambusicola thoracicus individual [42, 65]. For comparison, we aligned masked scaffolds from the 23× Gallus de novo assembly to the reference Gallus genome and called heterozygous bases as previously described.
Results
Bambusicola assembly and annotation
The paired-end NextSeq run of a single 500 bp insert library resulted in 116 million reads, with approximately 100 million reads retained from the forward and reverse read after quality filtering (Additional file 2: Table S1). The merged assembly resulted in 163,749 scaffolds with an N50 of 13,160 bp. The total length of the assembly was 1.03 Gb, with a minimum scaffold length of 218 bp, a maximum scaffold length of 311 kb, and a median scaffold length of 3145 bp (Table 2). Assembly statistics varied considerably among de novo assembly methods, and the merged assembly most closely resembled the MaSuRCA results (Table 2). The assembly consisted of approximately 11% repetitive content, with the largest component, 6.71%, consisting of CR1 LINES (Table 3). The overall level of repetitive content in Bambusicola is similar to Gallus, and greater than the other galliform genomes we examined (Table 3).
Table 2.
Comparison of Bambusicola assemblies and published genomes utilized in this study
| SOAP denovo2 | ABySS | MsSuRCA | Bambusicola | Colinus | Coturnix | Gallus | Meleagris | |
|---|---|---|---|---|---|---|---|---|
| Total contig length | 1.475 Gb | 1.049 Gb | 1.068 Gb | – | – | – | – | – |
| Total scaffold length | 1.543 Gb | 1.049 Gb | 1.069 Gb | 1.032 Gb | 1.172 Gb | 1.751 Gb | 1.047 Gb | 1.062 Gb |
| # contigs | 3,657,399 | 872,000 | 255,563 | – | – | – | – | – |
| # scaffolds | 2,514,856 | 871,673 | 232,161 | 163,749 | 220,307 | 275,637 | 15,932 | 5884 |
| # Contigs > 1 kb | 188,688 | 194,673 | 145,400 | – | – | – | – | – |
| # Scaffolds > 1 kb | 312,353 | 194,682 | 128,638 | 123,582 | 65,748 | 275,637 | 14,828 | 5866 |
| Med. contig length | 254 | 162 | 1383 | – | – | – | – | – |
| Med. scaf. Length | 282 | 162 | 1296 | 3145 | 559 | 2991 | 1206 | 1561 |
| Max contig length | 97.41 kb | 111.0 kb | 201.7 kb | – | – | – | – | – |
| Max scaf. Length | 400.4 kb | 111.0 kb | 311.0 kb | 311.0 kb | 600.7 kb | 1.501 Mb | 195.3 Mb | 204.1 Mb |
| Contig N50 | 508 | 5668 | 10,589 | – | – | – | – | – |
| Scaffold N50 | 1154 | 5670 | 12,505 | 13,160 | 45,461 | 11,408 | 90.22 Mb | 74.86 Mb |
| Gene Models | – | – | – | 17,772 | 15,984 | 27,554 | 16,354 | 16,494 |
| Comp. BUSCOS | – | – | – | 45% | 54% | 46% | 90% | 81% |
| Frag. BUSCOS | – | – | – | 19% | 18% | 20% | 4% | 7% |
Assembly statistics for individual methods are given as well as the merged Bambusicola assembly. Comparative assembly metrics for other genomes included in this study were taken from primary literature
Table 3.
Relative abundances of interspersed repeats across genomes
| Bambusicola | Colinus | Coturnix | Gallus | Meleagris | |
|---|---|---|---|---|---|
| Total | 9.51% | 5.69% | 5.17% | 9.64% | 7.67% |
| Retroelements | 8.44% | 4.92% | 4.37% | 8.58% | 6.74% |
| SINEs | 0.08% | 0.06% | 0.07% | 0.08% | 0.07% |
| CR1 LINEs | 6.71% | 4.06% | 3.76% | 6.79% | 5.67% |
| ERV LTRs | 1.64% | 0.80% | 0.53% | 1.71% | 1.00% |
| DNA transposons | 1.02% | 0.73% | 0.75% | 1.01% | 0.88% |
| hobo/Activator | 0.53% | 0.38% | 0.36% | 0.53% | 0.45% |
| Tc1/Mariner | 0.29% | 0.21% | 0.21% | 0.30% | 0.26% |
| Tourist/Harbinger | 0.04% | 0.03% | 0.03% | 0.03% | 0.03% |
| Unclassified | 0.05% | 0.04% | 0.05% | 0.05% | 0.05% |
Percentages are notable repetitive element content for genomes included in this study. All estimates were generated with RepeatMasker v4.0.5
We assembled approximately 90% of the Bambusicola genome based on scaffolds aligned to the reference Gallus gallus genome (Additional file 2: Table S2). While the whole-genome alignment to the Gallus gallus genome suggests we assembled 85%–95% of most chromosomes, our scaffolds could not be aligned to 47% of chromosome 16, 32% of chromosome 25, and 29% of the Z chromosome (Additional file 2: Table S2). Pairwise comparison between the Bambusicola and Gallus NUCmer alignment also revealed local assembly gaps in the Bambusicola genome (Fig. 1). We expected these portions of the genome to be poorly assembled, especially chromosome 16, which contains the rapidly evolving MHC loci, many members of the large olfactory receptor gene family, and repetitive regions such as LTRs, LINEs, and ribosomal DNA repeats (e.g. [66]).
Fig. 1.
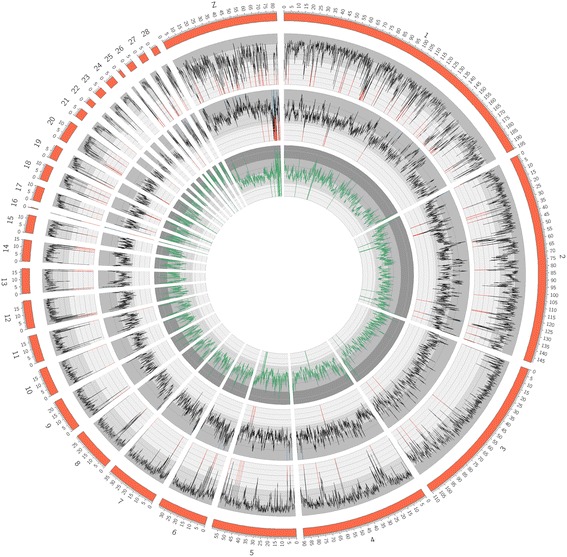
Circular plot of the Gallus gallus genome sequence assembled at the chromosome level compared with our Bambusicola thoracicus genome. Analyses were across non-overlapping 100 kb sliding windows. The outermost line plot represents breadth of coverage of Bambusicola with respect to Gallus. Bases aligned by NUCmer were considered sequenced while all gaps were considered missing data. Only values between 50% and 100% are shown. All areas where the breadth of coverage drops below 50% are drawn in red. The middle line plot shows pairwise nucleotide divergence. Missing data and gaps were excluded from this estimate. Only values between 0.02 and 0.08 are shown. Values below 0.02 are shown in red while values above 0.08 are shown in blue. The innermost ring is variation in GC content. The purple line is Gallus GC content and the green line is Bambusicola GC content. Only values between 20% and 60% are shown. Chromosomes 8 through 28 are magnified in Additional File 1 (Figure S3)
Among the assembled portion of the Bambusicola genome, we achieved an average of 25× coverage, given the distribution of kmer frequencies as well as the depth of short read alignment to the Bambusicola de novo assembly (Additional file 2: Tables S1 and S2). We observed an average pairwise nucleotide divergence of 5.4% between Bambusicola and Gallus; however, pairwise nucleotide divergence varied across the genome and appeared to be especially high in some regions with low coverage (Fig. 1; Additional file 1: Figures. S2 and S3). These regions of the Bambusicola genome were likely difficult to align to the Gallus genome. GC content across the genome appeared relatively consistent in the 100 kb windows between Bambusicola and Gallus, but the GC content was generally higher on the microchromosomes (Fig. 1; Additional file 1: Figure S3), as has been noted in other studies (e.g. [67]).
MAKER annotations produced 17,772 gene models in the Bambusicola genome, with 44% coverage of core vertebrate orthologs from BUSCO. Our new annotations of the Coturnix japonica and Colinus virginianus genomes found 27,544 and 15,948 gene models, with 46% and 54% coverage of core vertebrate orthologs respectively. For comparison, Gallus gallus and Meleagris gallopavo cover 90% and 80% of vertebrate single-copy orthologs from BUSCO respectively (Table 3).
Out of the 3854 reference UCE sequences from the same Bambusicola individual [17], 3842 were present in the masked assembly. The average assembly coverage of a UCE was 98.7%, and the average frequency of mismatches per UCE was 0.09%. The mismatches could represent sequencing errors, assembly errors, or missed heterozygous base calls. 2390 of the UCEs were represented in the Bambusicola assembly at full length with no mismatches between our genome assembly and the previously sequenced UCEs. If we allowed mismatches between the Bambusicola thoracicus UCE sequences and our genome assembly, there were 3172 full-length UCEs represented in the Bambusicola thoracicus genome. Overall, most previously sequenced UCEs from the same Bambusicola thoracicus individual were represented in our assembly with high similarity (Additional file 1: Figure S4).
When we clustered the amino acid sequences from all five genomes, we identified 15,864 orthologous groups, 3285 of which had a single-copy from all five species after removing potentially erroneous sequences. Among these single-copy, putatively orthologous groups, 2822 had gene tree topologies that were identical to the species topology following the TreeFix analysis. From these 2822 gene trees, we identified a subset of 374 orthologous groups with high sequencing accuracy, such that all sites in the Bambusicola sequence had at least 20× depth with all phred scores at least 14. This subset of 374 orthologous groups were analyzed with the total set of 2822 gene trees and separately to observe if sequencing depth and base qualities might influence overall patterns of molecular evolution in our study (Additional file 1).
Variation in dN/dS – Branch tests
Branch tests revealed many genes suggesting relaxed selective pressures (i.e. significantly higher dN/dS than background branches) in both the Bambusicola and Gallus lineages, and strong purifying selection (i.e. significantly lower dN/dS than background branches) in the branch leading to their MRCA (Fig. 2a; Table 4). There were more significant branch tests, indicating a different dN/dS, in Bambusicola (11.1% of the genes) than in Gallus (5.4% of the genes). 75% and 64% of significant branch tests indicated an increased dN/dS in Bambusicola and Gallus respectively (Fig. 2b; Table 4; Additional file 2: Table S3). Only 1.6% of gene trees indicated a different dN/dS on the MRCA branch, with 61% evolving at a lower dN/dS compared to the Bambusicola and Gallus branches. The median foreground dN/dS for significant branch tests in Bambusicola (0.463) was higher than that in Gallus (0.310; Fig. 3a-3c). We also found a similar pattern of more gene trees with elevated dN/dS in Bambusicola in our subset of 374 orthologous groups (Additional file 1).
Fig. 2.

Significant branch and branch-site tests on branches leading to Gallus, Bambusicola, and their most recent common ancestor. Colors correspond to the foreground branches in the branch and branch site tests. a) Numbers above foreground branches are the median foreground dN/dS for significant branch tests; numbers below branches are median background dN/dS for significant branch tests (i.e. the dN/dS on the other branches in the tree). b) The number of significant branch tests on the three foreground branches. Branch tests that were significant in only one foreground branch were classified into genes that are evolving with a higher dN/dS than the background rate / genes that were evolving at a lower dN/dS than the background rate. c) The number of significant branch-site tests across foreground branches, suggesting potential positive selection
Table 4.
Summary of branch and branch-site tests of molecular evolution
| Bambusicola | # trees | Gallus | # trees | MRCA | # trees | |
|---|---|---|---|---|---|---|
| Branch | 264(199) | 2822 | 95(61) | 2822 | 46(18) | 2822 |
| resampled q | 13(9) | 264 | 9(5) | 95 | 2(2) | 46 |
| resampled p | 46(22) | 264 | 24(10) | 95 | 16(6) | 46 |
| Branch-Site | 531 | 2822 | 191 | 2822 | 94 | 2822 |
| BAli-Phy | 3 | 531 | 2 | 191 | 1 | 94 |
Results from ML estimates are shown in bold while subsequent methods are shown in plain text below. Resampled q refers to the use of q-values while resampled p refers to the use of p-values for distributions of alignments for branch tests. The number of trees tested for each species is given to the right of each respective column. Numbers in parentheses for branch tests are the number of gene trees where the foreground dN/dS was greater than the background dN/dS. Numbers for branch-site tests represent the number of genes that showed evidence of episodic positive selection
Fig. 3.
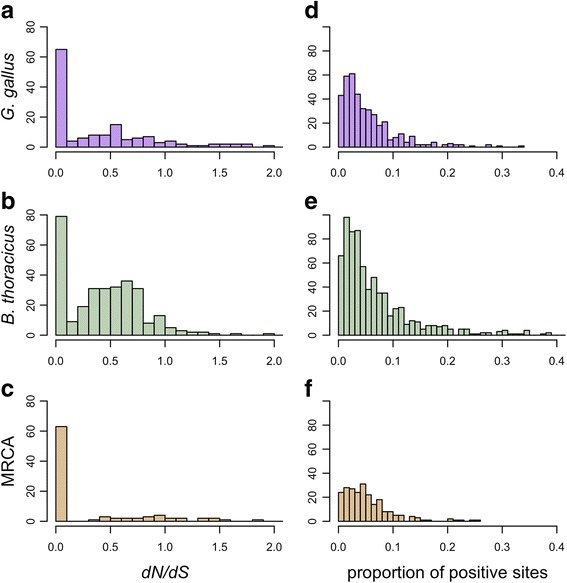
Distributions of foreground gene-wide dN/dS and the ML estimates of the proportions of sites under positive selection for significant branch and branch-site tests. Results are after multiple-testing corrections. dN/dS distributions were truncated at two for ease of visualization and because excessively high dN/dS estimates likely are unreliable. Panels a-c correspond to gene-wide dN/dS from branch tests while panels d-f correspond to the proportion of sites under positive selection from branch-site tests
We re-evaluated the branch tests that were significant after multiple-testing corrections across the distribution of alignments created by BAli-Phy [53]. Comparing LRT statistics to each test’s original q value, only 0.5%, 0.3%, and 0.001% of the original number of gene trees were still significant on the Bambusicola, Gallus, and MRCA branches respectively. If we relaxed the multiple testing requirement and considered genes with a p value < 0.05 for at least 95% of alignments, we found more significant tests, but there was more evidence of relaxed selection on the Bambusicola branch than the other branches in all analyses (Table 4).
Variation in dN/dS – Branch-site tests
We found 31.6%, 14.1%, and 8.1% of the genes in Bambusicola, Gallus, and their MRCA respectively had some proportion of sites with dN/dS > 1 based on branch-site tests (Additional file 2: Table S4). If we only consider genes that were significant in only one of these three branches, the proportion of positive tests was 18.8%, 6.8%, and 3.3% for the Bambusicola, Gallus, and MRCA branches, respectively (Fig. 2c; Table 4). The distribution of the proportion of sites under positive selection was similar across the Bambusicola, Gallus, and MRCA branches (Fig. 3d-3f). Our subsampled data also revealed elevated numbers of genes in Bambusicola that appear to have experienced episodic positive selection (which we define as those orthologous groups for which including a proportion of sites with dN/dS > 1 significantly improves model fit) compared to Gallus or the MRCA of Bambusicola and Gallus (Additional file 1).
Alignment error likely contributed to many significant branch-site tests. Posterior probabilities of positive selection from BAli-Phy only supported 3 genes with sites under positive selection on the Gallus branch, 2 on the Bambusicola branch, and 1 gene on the MRCA branch (Fig. 4; Table 4). There was a weak association between the proportion of sites under positive selection from both the ML and Bayesian estimators for the Gallus branch (Kendall’s τ = 0.125; p < 0.001) and the Bambusicola branch (Kendall’s τ = 0.201, p < 0.001), but not the MRCA branch (Kendall’s τ = 0.07 p = 0.112). Thus, while there is some agreement between the two estimators on the proportion of sites with dN/dS > 1, there is much uncertainty in the inference of episodic positive selection (Fig. 4). In most of our analyses using MUSCLE alignments for which MLEs imply positive selection, there also are possible alignments that do not imply positive selection.
Fig. 4.
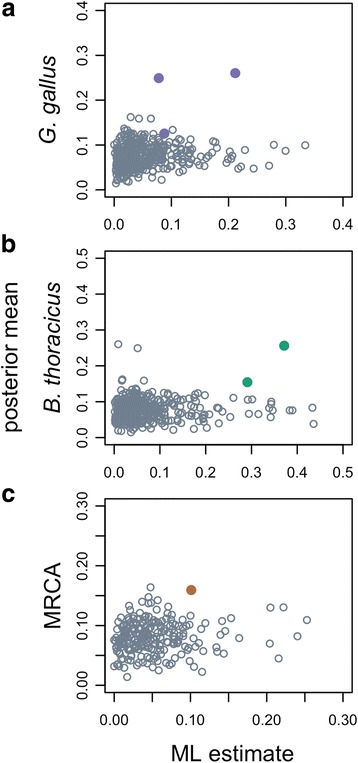
The estimated proportion of sites under positive selection for significant branch-site tests. Significant results are indicated by the LRT after multiple testing corrections. The original ML estimate is from PAML while the mean of the posterior distribution is from post-burnin BAli-Phy estimates. Genes with a posterior probability of positive selection > 0.95 are shown in color while genes with a posterior probability ≤0.95 are shown as empty grey circles. Panels a-c show the respective distributions from the Gallus gallus, Bambusicola thoracicus, and their MRCA foreground branches
For the MUSCLE alignment results, there was some overlap between the significant branch and branch-site tests. For the Bambusicola branch, 224 genes were significant for both tests, with 19 of these having a gene-wide dN/dS > 1 (Additional file 2: Tables S3 and S4). On the Gallus branch, 81 genes had significant branch and branch site tests (Additional file 2: Tables S3 and S4), including 23 genes with a global dN/dS > 1. The MRCA branch had an additional 28 genes with significant branch and branch site tests, with 10 genes having a global dN/dS > 1.
Enrichment of GO categories
There were seven cellular components and two molecular functions significantly overrepresented with higher dN/dS in the Bambusicola branch, while 6 components were underrepresented for increased dN/dS in the MRCA branch (Fig. 5; Additional file 2: Table S5). For five components, there was both an overrepresentation of increased dN/dS in Bambusicola and a simultaneous underrepresentation of increased dN/dS in the MRCA. Overall, we tested 127 GO categories, and none were over or underrepresented in Gallus. Many GO categories were not relevant due to small numbers represented in our sample of orthologous genes, but the results supported an increase in dN/dS in Bambusicola across many different GO categories that was not observed in the MRCA or Gallus branches.
Fig. 5.
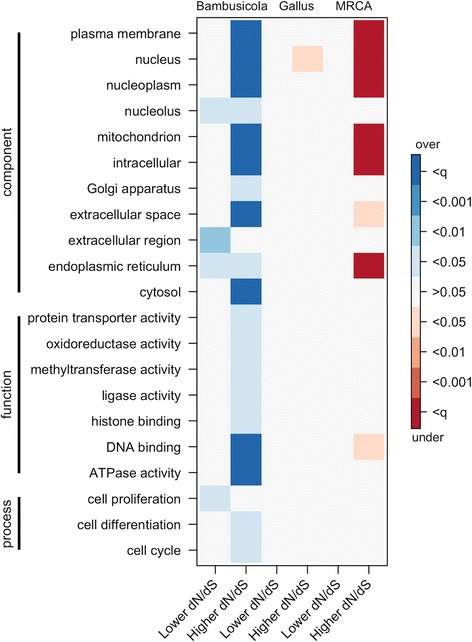
Overrepresentation or underrepresentation of GO slim categories. Tests were conducted for a proportion of sites under positive selection, lower gene-wide dN/dS, and higher gene-wide dN/dS, on the Bambusicola, Gallus, and MRCA branches. Overrepresented terms are blue, and underrepresented terms are red. Levels of significance are shown for p < 0.05, p < 0.01, p < 0.001, and p < q, where q is the Benjamini-Hochberg q-value corrected for a FDR of 0.05
Estimating heterozygosity and Ne
We identified 2,968,298 heterozygous bases across 781,975,390 total bases of Bambusicola genomic data that were alignable to Gallus autosomal sequence, representing 3.80 heterozygous bases per 1 kb. The Gallus gallus reference alignments of 23× and 35× had 3.80 and 6.36 heterozygous bases per 1 kb respectively; however, there were 1,814,172 heterozygous bases across 806,203,193 bases of autosomal scaffolds in the 23× de novo Gallus assembly, representing 2.25 heterozygous bases per 1 kb. Thus, the de novo assemblies estimated lower heterozygosity compared to aligning reads directly to the Galgal4 reference genome, but comparison of heterozygosity for the Bambusicola and Gallus de novo assemblies alone implied higher heterozygosity in Bambusicola (Fig. 6; Additional file 2: Table S6). Evaluation of Sanger sequenced intron data for Bambusicola suggested a false positive rate for heterozygous bases of 0.23%, which was consistent with previous benchmarking datasets for Illumina sequencing technology with SAMTOOLS variant calling methods [68, 69].
Fig. 6.
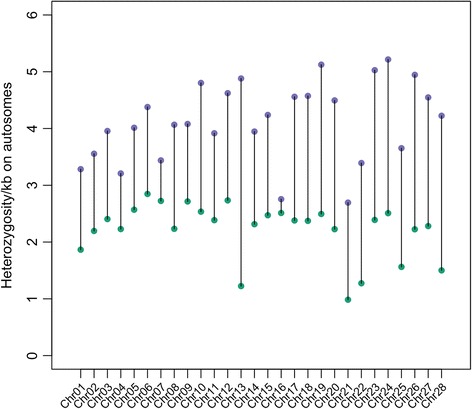
Heterozygosity measured as the number of heterozygous bases per one kilobase. Purple circles represent Bambusicola while green circles represent Gallus. Base calls were performed on assembled contigs that were alignable to the Gallus reference genome, and we only consider the length of these alignable contigs in our heterozygosity estimates. Heterozygosity was higher in Bambusicola than Gallus for all chromosomes
PSMC analysis suggested that Bambusicola reached its maximum Ne between 100,000 and 500,000 years ago (Fig. 7). Both the Gallus 23× and 35× referenced aligned data suggested a maximum Ne in Gallus closer to 100,000 years ago (Fig. 7). However, using heterozygosity data inferred from a reference genome or a de novo assembly affected the PSMC results. The Gallus 35× referenced data indicated the highest maximum Ne. In contrast, the 23× Gallus de novo assembly inferred a much older and smaller maximum Ne than that inferred when the data were aligned to the Galgal4 reference assembly (Fig. 7). Although comparisons of the Gallus 35× referenced data with the de novo Bambusicola genome indicated Gallus had a higher maximum Ne than Bambusicola, comparison of the de novo Bambusicola genome with the de novo 23× Gallus genome implied a larger maximum Ne in Bambusicola.
Fig. 7.
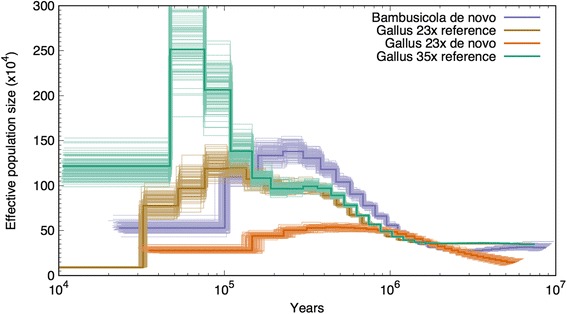
Estimation of Ne over time, inferred from heterozygous bases of individual genome sequences with the PSMC model. Results are shown for the observed data (bold lines) and bootstrap replicates for the Bambusicola genome sequence as well as two previously published Gallus gallus genome sequences. Estimates are included for the 23× and 35× Gallus genomes for short reads aligned directly to the Gallus reference genome (reference) as well as short reads aligned to a de novo assembly (de novo) for the 23× Gallus genome
Discussion
Our results demonstrated it is possible to generate an informative draft avian genome from a single library with only moderate coverage for relatively little expense. Complete avian genome sequences are becoming increasingly common [1, 4–6], and the technical issues explored here are not unique to our genome assembly. We attempted to reduce assembly errors by merging multiple de novo assemblies, and pairwise comparisons with the closely related Gallus gallus reference genome suggested that we assembled at least 85% of the nuclear genome. Often the most variable regions of the Gallus genome, such as chromosome 16, did not align with our Bambusicola assembly. However, this is common for bird genomes [60], and it is not unique to low-coverage genome assemblies. Although we had limited success assembling long scaffolds, the size of most Bambusicola contigs was comparable to other avian genome sequencing projects using two or more insert libraries with similar (or even higher) coverage [e.g. 1]. We identified and recovered unique sequence features such as protein-coding genes, UCEs, transposable elements, and heterozygous sites with considerable completeness and accuracy. Notably our estimates of repetitive content were consistent with other galliforms, including those sequenced using Sanger sequencing (Table 3). A de novo assembly using short-read next generation sequencing might be expected to underestimate repetitive content, which suggests we have excellent capabilities to assemble contigs of genomic sequence data using single insert libraries.
Our goal was to leverage the Bambusicola genome to test molecular evolution hypotheses relevant to the divergent life histories of Bambusicola and Gallus. Our genome-level analyses of molecular evolution, even when controlling for sources of error, supported elevated dN/dS in Bambusicola (Fig. 2; Fig. 3). This result was consistent with the hypothesis that the reduced range of Bambusicola relative to Gallus has led to a lower Ne in Bambusicola, and thus relaxed purifying selection over time. We also found increased levels of episodic positive selection in Bambusicola with respect to Gallus, which was, in contrast to the elevated dN/dS results, consistent with predictions for a lower Ne in Gallus. However, the latter result could also be driven by alignment uncertainty, which likely inflated the error rate for our branch-site tests (Fig. 4). In our case, the use of BAli-Phy greatly reduced the perception of rampant positive selection in the Bambusicola and Gallus lineages relative to our original branch-site tests that used fixed alignments.
To further test the hypothesis that Bambusicola has a lower Ne than Gallus, we used PSMC analyses to estimate changes in Ne for both Bambusicola and Gallus. Similar to PSMC analyses of other avian genomes [70], our results suggested that Ne for Bambusicola and Gallus peaked around 100,000 years ago, followed by a reduction near the last glacial maximum (Fig. 7). While it may be difficult to obtain reliable estimates of recent Ne from PSMC analyses, our results indicate that Ne was higher in Gallus 50,000–100,000 years ago, which may reflect the larger ancestral range of Gallus compared to Bambusicola and would be consistent with our observation that Bambusicola exhibited globally increased dN/dS [13, 14]. However, the PSMC model estimates parameters from contiguous sequence data, and the Bambusicola genome assembly consisted of many fragmented scaffolds. Differences in assembly quality can lead to disparities in Ne estimates; this was shown in a recent analysis of flycatcher genomes [71], and it was consistent with our comparison of the 23× Gallus de novo and reference aligned genomes (Fig. 7). Although these technical issues make it challenging to interpret the results of PSMC analyses, the fact that two very different lines of evidence (the globally increased dN/dS and the PSMC results) similarly indicate a smaller Ne for Bambusicola increases our confidence in our general conclusions.
Although our results suggested a lower Ne in Bambusicola than Gallus, heterozygosity was higher in Bambusicola, at least when comparing de novo assemblies of similar sequencing depth (Fig. 6; Additional file 2: Table S6). Heterozygosity likely reflects recent events. Thus, the increased heterozygosity in Bambusicola may be due to migration events within the last 100,000 years between Pleistocene glacial refugia [72]. Even if Gallus populations experienced similar range shifts and migration following Pleistocene glaciations, a higher growth rate and carrying capacity in Bambusicola could explain a more rapid increase in heterozygosity [73, 74]; Bambusicola populations could increase more quickly than Gallus due to both their smaller mass and more males participating in reproduction. It is plausible that if both Bambusicola and Gallus both experienced reductions in Ne, the Bambusicola population could recover faster than Gallus, which would experience more inbreeding. Thus, there are a number of scenarios that could explain both globally increased dN/dS and heterozygosity in Bambusicola.
We also attempted to infer if phenotypic differences between Bambusicola and Gallus were associated with elevated dN/dS in specific genes by testing for enrichment of GO terms. Although interpretation of GO enrichment analyses can be difficult given the large numbers of uncharacterized genes and protein products (Additional file 2: Table S7), we found an overrepresentation of a few specific GO terms within the Bambusicola lineage, with little to no bias in the Gallus lineage (Fig. 5; Additional file 2: Table S5). The overrepresentation of GO terms in Bambusicola was largely driven by transcription factors involved in DNA repair and cell cycle pathways, but structural protein-coding genes under the terms such as cytoplasm (GO:0005737), extracellular space (GO:0005615), and plasma membrane (GO:0005886) suggest some additional potentially biologically important changes. For example, we found both a proportion of sites under positive selection and globally elevated dN/dS for a serpin peptidase inhibitor ortholog (Entrez Gene: SERPINB5). Given the developmental importance of ovalbumin for birds (e.g. [75]), further investigations of serpin proteins in Bambusicola may be warranted. Despite the enrichment of a few specific terms, there is no obvious biological pattern to the enrichment patterns, further supporting that our observations of elevated dN/dS in Bambusicola are mostly likely due to demographic effects.
Conclusions
Our analyses highlight that a draft avian genome assembled using a single library can produce high quality data and evolutionary insights. We revealed globally relaxed selective pressures acting throughout the Bambusicola genome, which are likely due to demographic effects, such as a lower Ne and smaller range in Bambusicola compared to Gallus. Bambusicola also exhibits high heterozygosity with respect to Gallus, which may be due to a combination of post-glaciation migration events and mating system differences. The Bambusicola genome can serve as a resource for testing the effects of sexual selection and mating systems on molecular sequence evolution in future studies.
Our Bambusicola genome assembly also addressed a number of technical questions. Although the Bambusicola genome is fragmented, especially when compared to some recent avian draft genomes based on multiple libraries [1], we recovered similar numbers of gene models and contig N50 statistics as other avian genomes. Our results also highlighted some uncertainty in results from the popular branch-site test due to sensitivity to data and alignment quality. This has broad implications for other similar studies, as the branch-site test is frequently used in genome-level scans for positive selection (e.g. [76–78]). Moreover, the limitations of the branch-site test that we noted are not necessarily specific to low-coverage genome assemblies because they reflect intrinsic features of protein sequence evolution. Single library genome assemblies may sacrifice assembly quality, but the low cost of these assemblies could permit the collection of genome sequences for a larger number of taxa. If the goals of a study do not require large scaffolds or other large-scale structural information, the approach we used could have benefits. Overall, the insights from our Bambusicola draft genome outweigh limitations from a fragmented genome assembly, and additional single library genome sequences may prove a valuable and cost effective resource for comparative genomics, molecular evolution, and phylogenetics of birds.
Additional files
Figures S1 – S4. Details for the sensitivity of dN/dS analyses to sequence quality and a number of additional analyses are contained within a single pdf. (PDF 5463 kb)
Tables S1 – S7. Additional data from analyses that were too large to fit on a single page are given as Excel speadsheets. (XLS 4168 kb)
Alignments from Analyses. All alignments and topologies of one-to-one orthologs used for analyses of molecular evolution are given as a single tarball. (TGZ 3336 kb)
Acknowledgements
The authors would like to thank Pete Hosner for discussions of this research.
Funding
This work was supported in part by funding from the National Science Foundation, DEB-1208428, awarded to JGB and DEB-1118823 to RTK and ELB.
Availability of data and materials
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession PPHD00000000. The version described in this paper is version PPHD01000000.
The raw sequence data generated for this study are available in the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA), referenced by SRP128936. All relevant metadata can be found under Bioproject PRJNA396570. Alignments and topologies used for molecular evolution analyses are available as a supplementary file (Additional file 3).
Abbreviations
- dN/dS
Nonsynonymous to synonymous substitution rate ratio
- FDR
False discovery rate
- GO
Gene Ontology
- LRT
Likelihood ratio test
- ML
Maximum likelihood
- MRCA
Most recent common ancestor
- MYA
Million years ago
- Ne
Effective population size
- PSMC
Pairwise sequential Markov coalescent
- UCE
Ultraconserved Element
Authors’ contributions
GPT, RTK, ELB, and JGB designed the study. GPT performed analyses. GPT, RTK, ELB, and JGB interpreted results and contributed to writing the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
No permissions were required for this study as blood was collected by a breeder, E. Kempf, and given to R. Kimball for previous research and not for the purpose of this study.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4711-0) contains supplementary material, which is available to authorized users.
Contributor Information
G. P. Tiley, Email: george.tiley@duke.edu
R. T. Kimball, Email: rkimball@ufl.edu
E. L. Braun, Email: ebraun68@ufl.edu
J. G. Burleigh, Email: gburleigh@ufl.edu
References
- 1.Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science. 2014;346:1311–1320. doi: 10.1126/science.1251385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jarvis D, Mirarab S, Aberer AJ, Li B, Houde P, Li C, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346:1320–1331. doi: 10.1126/science.1253451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalloul RA, Long JA, Zimin AV, Aslam L, Beal K, Blomberg LA, et al. Multi-platform next-generation sequencing of the domestic Turkey (Meleagris gallopavo): genome assembly and analysis. PLoS Biol. 2010;8:e1000475. doi: 10.1371/journal.pbio.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawahara-Miki R, Sano S, Nunome M, Shimmura T, Kuwayama T, Takahashi S, et al. Next-generation sequencing revels genomic features in the Japanese quail. Genomics. 2013;101:345–353. doi: 10.1016/j.ygeno.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 5.Halley YA, Dowd SE, Decker JE, Seabury PM, Bhattarai E, Johnson CD, et al. A draft de novo genome assembly for the northern bobwhite (Colinus virginianus) reveals evidence for a rapid decline in effective population size beginning in the late Pleistocene. PLoS One. 2014;9:e90240. doi: 10.1371/journal.pone.0090240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang B, Ekblom R, Bunikis I, Siitari H, Höglund J. Whole genome sequencing of the black grouse (Tetrao tetrix): reference guided assembly suggests faster-Z and MHC evolution. BMC Genomics. 2014;15:180. doi: 10.1186/1471-2164-15-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang N, Kimball RT, Braun EL, Liang B, Zhang Z. Assessing phylogenetic relationships among Galliformes: a multigene phylogeny with expanded taxon sampling in Phasianidae. PLoS One. 2013;8:e64312. doi: 10.1371/journal.pone.0064312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang N, Kimball RT, Braun EL, Liang B, Zhang Z. Ancestral range reconstruction of Galliformes: the effects of topology and taxon sampling. J Biogeogr. 2017;44:122–135. doi: 10.1111/jbi.12782. [DOI] [Google Scholar]
- 9.Madge S, McGowan P. Pheasants, partridges and grouse, including buttonquails, sandgrouse and allies. London: Christopher Helm; 2002. [Google Scholar]
- 10.Zuk M, Johnsen TS, Maclarty T. Endocrine–immune interactions, ornaments and mate choice in red jungle fowl. Proc R Soc London Biol. 1995;260:205–210. doi: 10.1098/rspb.1995.0081. [DOI] [Google Scholar]
- 11.Ligon JD, Kimball R, Merola-Zwartjes M. Mate choice by female red junglefowl: the issues of multiple ornaments and fluctuating asymmetry. Anim Behav. 1998;55:41–50. doi: 10.1006/anbe.1997.0582. [DOI] [PubMed] [Google Scholar]
- 12.Davison GWH. Avian spurs. J Zool. 1985;206:353–366. doi: 10.1111/j.1469-7998.1985.tb05664.x. [DOI] [Google Scholar]
- 13.McGowan PJK, Kirwan GM. Christie DA. Chinese Bamboo-partridge (Bambusicola thoracicus). In: del Hoyo J, Elliott A, Sargatal J, Christie DA, de Juana E. (eds.). Handbook of the Birds of the World Alive. Lynx Edicions, Barcelona 2017. http://www.hbw.com/node/53471. Accessed 30 January 2017.
- 14.McGowan PJK, Kirwan GM, Christie DA. Red Junglefowl (Gallus gallus). In: del Hoyo J, Elliott A, Sargatal J, Christie DA, de Juana E, editors. Handbook of the Birds of the World Alive. Barcelona: Lynx Edicions; 2017. https://www.hbw.com/node/53485. Accessed 30 Jan 2017.
- 15.Frankham R. Relationship of genetic variation to population size in wildlife. Conserv Biol. 1996;10:1500–8.
- 16.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 17.Hosner PA, Faircloth BC, Glenn TC, Braun EL, Kimball RT. Avoiding missing data biases in assembling the landfowl tree of life (Aves: Galliformes) Mol Biol Evol. 2016;33:1110–1125. doi: 10.1093/molbev/msv347. [DOI] [PubMed] [Google Scholar]
- 18.Kimball RT, Braun EL, Zwartjes PW, Crowe TM, Ligon JD. A molecular phylogeny of the pheasants and partridges suggests that these lineages are not monophyletic. Mol Phylogenet Evol. 1999;11:38–54. doi: 10.1006/mpev.1998.0562. [DOI] [PubMed] [Google Scholar]
- 19.Hung C-M, Hung H-Y, Yeh C-F, Fu Y-Q, Chen D, Lei F, et al. Species delimitation in the Chinese bamboo partridge Bambusicola thoracica (Phasianidae; Aves) Zool Scr. 2014;43:562–575. doi: 10.1111/zsc.12071. [DOI] [Google Scholar]
- 20.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimin AV, Marcais G, Puiu D, Roberts M, Salzberg SL, Yorke JA. The MaSuRCA genome assembler. Bioinformatics. 2013;29:2669–2677. doi: 10.1093/bioinformatics/btt476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wences AH, Metassembler SMC. Merging and optimizing de novo genome assemblies. Genome Biol. 2015;16:207. doi: 10.1186/s13059-015-0764-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smit AFA, Hubley R, Green P. RepeatMasker Open-4.0. http://www.repeatmasker.org. Accessed 8 Dec 2015.
- 26.Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;7:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.ftp://ftp.ensembl.org/pub/release-84/fasta/gallus_gallus/pep/. Accessed 21 December 2015.
- 29.ftp://ftp.ensembl.org/pub/release-83/fasta/meleagris_gallopavo/pep/. Accessed 21 December 2015.
- 30.ftp://ftp.ensembl.org/pub/release-83/fasta/taeniopygia_guttata/pep/. Accessed 21 December 2015.
- 31.http://viewer.shigen.info/uzura/download.php. Accessed 22 Dec 2015.
- 32.ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/599/465/GCA_000599465.1_NB1.1/. Accessed 21 Dec 2015.
- 33.Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30:2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.ftp://ftp.ensembl.org/pub/release-84/fasta/gallus_gallus/dna/. Accessed 21 December 2015.
- 36.Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Stoeckert J, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Y-C, Rasmussen MD, Bansal MS, Kellis M. Treefix: statistically informed gene tree error correction using species trees. Syst Biol. 2013;62:110–120. doi: 10.1093/sysbio/sys076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hosner PA, Tobias JA, Braun EL, Kimball RT. How do seemingly non-vagile clades accomplish trans-marine dispersal? Trait and dispersal evolution in the landfowl. Proc Biol Sci. 2017;284:20170210. [DOI] [PMC free article] [PubMed]
- 42.Kimball RT, Braun EL. Does more sequence data improve estimates of galliform phylogeny? Analyses of a rapid radiation using a complete data matrix. PeerJ. 2014;2:e361. doi: 10.7717/peerj.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang ZPAML. 4: a program package for phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 44.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 45.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2015. http://www.R-project.org/.
- 46.Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 48.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995:289–300.
- 49.Anismova M, Yang Z. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol. 2007:1219–28. [DOI] [PubMed]
- 50.Yang Z, dos Reis M. Statistical properties of the branch-site test of positive selection. Mol Biol Evol. 2011;28:1217–1228. doi: 10.1093/molbev/msq303. [DOI] [PubMed] [Google Scholar]
- 51.Fletcher W, Yang Z. The effect of insertions, deletions, and alignment errors on the branch-site test of positive selection. Mol Biol Evol. 2010;27:2257–2267. doi: 10.1093/molbev/msq115. [DOI] [PubMed] [Google Scholar]
- 52.Redelings BD, Suchard MA. Bali-Phy: simultaneous Bayesian inference of alignment and phylogeny. Bioinformatics. 2006;22:2047–2048. doi: 10.1093/bioinformatics/btl175. [DOI] [PubMed] [Google Scholar]
- 53.Redelings BD. Erasing errors due to alignment ambiguity when estimating positive selection. Mol Biol Evol. 2014;31:1979–1993. doi: 10.1093/molbev/msu174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.http://www.agbase.msstate.edu/cgi-bin/download.pl. Accessed 2 April 2016.
- 55.http://geneontology.org/page/go-slim-and-subset-guide. Accessed 15 May 2016.
- 56.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singhal S, Leffler EM, Sannareddy K, Turner I, Venn O, Hooper DM, et al. Stable recombination hotspots in birds. Science. 2015;350:928–932. doi: 10.1126/science.aad0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawakami T, Smeds L, Backström N, Husby A, Qvarnstöm A, Mugal CF, et al. A high-density linkage map enables a second-generation collared flycatcher genome assembly and reveals the patterns of avian recombination rate variation and chromosomal evolution. Mol Ecol. 2014;23:4035–4058. doi: 10.1111/mec.12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang M-S, Li Y, Peng M-S, Zhong L, Wang Z-J, Li Q-Y, et al. Genomic analyses reveal potential independent adaptation to high altitude in Tibetan chickens. Mol Biol Evol. 2015;32:1880–1889. doi: 10.1093/molbev/msv071. [DOI] [PubMed] [Google Scholar]
- 61.http://www.ncbi.nlm.nih.gov/bioproject/PRJNA241474. Accessed 16 June 2016.
- 62.Sawai H, Kim HL, Kuno K, Suzuki S, Gotoh H, Takada M, et al. The origin and genetic variation of domestic chickens with special reference to junglefowls Gallus g. gallus and G. varius. PLoS One. 2010;5:e10639. doi: 10.1371/journal.pone.0010639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mooers AO, Harvey PH. Metabolic rate, generation time, and the rate of molecular evolution in birds. Mol Phylogenet Evol. 1994;3:344–350. doi: 10.1006/mpev.1994.1040. [DOI] [PubMed] [Google Scholar]
- 64.Nam K, Mugal C, Nabholz B, Schielzeth H, Wolf JB, Backström N, et al. Molecular evolution of genes in avian genomes. Genome Biol. 2010;11:R68. doi: 10.1186/gb-2010-11-6-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shen YY, Dai K, Cao X, Murphy RW, Shen XJ, Zhang YP. The updated phylogenies of the phasianidae based on combined data of nuclear and mitochondrial DNA. PLoS One. 2014;9:e95786. doi: 10.1371/journal.pone.0095786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller MM, Robinson CM, Abernathy J, Goto RM, Hamilton MK, Zhou H, Delany ME. Mapping genes to chicken microchromosome 16 and discovery of olfactory and scavenger receptor genes near the major histocompatibility complex. J Hered. 2014;105:203–215. doi: 10.1093/jhered/est091. [DOI] [PubMed] [Google Scholar]
- 67.Axelsson E, Webster MT, Smith NGC, Burt DW, Ellegren H. Comparison of the chicken and Turkey genomes reveals a higher rate of nucleotide divergence on microchromosomes than macrochromosomes. Genome Res. 2005;15:120–125. doi: 10.1101/gr.3021305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hwang S, Kim E, Lee I, Marcotte EM. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep. 2015;5:17875. doi: 10.1038/srep17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Farrer RA, Henk DA, MacLean D, Studholme DJ, Fisher MC. Using false discovery rates to benchmark SNP-callers in next-generation sequencing projects. Sci Rep. 2013;3:1512. doi: 10.1038/srep01512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nadachowska-Brzyska K, Li C, Smeds L, Zhang G, Ellegen H. Temporal dynamics of avian populations during Pleistocene revealed by whole-genome sequences. Curr Biol. 2015;25:1375–1380. doi: 10.1016/j.cub.2015.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nadachowska-Brzyska K, Burri R, Smeds L, Ellegren H. PSMC analysis of effective population sizes in molecular ecology and its application to black-and-white Ficedula flycatchers. Mol Ecol. 2016;25:1058–1072. doi: 10.1111/mec.13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang Z, Liu N, Liang W, Zhang Y, Liao X, Ruan L, Yang Z. Phylogeography of Chinese bamboo partridge, Bambusicola thoracica thoracica (Aves: Galliformes) in South China: inference from mitochondrial DNA control-region sequences. Mol Phylogenet Evol. 2010;56:273–280. doi: 10.1016/j.ympev.2010.01.028. [DOI] [PubMed] [Google Scholar]
- 73.Arenas M, Ray N, Currat M, Excoffier L. Consequences of the range contractions and range shifts on molecular diversity. Mol Biol Evol. 2012;29:207–218. doi: 10.1093/molbev/msr187. [DOI] [PubMed] [Google Scholar]
- 74.Excoffier L. Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol. 2003;13:853–864. doi: 10.1046/j.1365-294X.2003.02004.x. [DOI] [PubMed] [Google Scholar]
- 75.Benarafa C, Remold-O’Donnell E. The ovalbumin serpins revisited: perspective from the chicken genome of clade B serpin evolution in vertebrates. Proc Natl Acad Sci U S A. 2005;102:11367–11372. doi: 10.1073/pnas.0502934102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qiu Q, Zhang G, Ma T, Qian W, Wang J, Ye Z, et al. The yak genome and adaptation to life at high altitude. Nat Genet. 2012;44:946–949. doi: 10.1038/ng.2343. [DOI] [PubMed] [Google Scholar]
- 77.Shaffer HB, Minx P, Warren DE, Shedlock AM, Thomson RC, Valenzuela N, et al. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol. 2013;14:R28. doi: 10.1186/gb-2013-14-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seim I, Fang X, Xiong Z, Lobanov AV, Huang Z, Ma S, et al. Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nat Commun. 2013;4:2212. doi: 10.1038/ncomms3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1 – S4. Details for the sensitivity of dN/dS analyses to sequence quality and a number of additional analyses are contained within a single pdf. (PDF 5463 kb)
Tables S1 – S7. Additional data from analyses that were too large to fit on a single page are given as Excel speadsheets. (XLS 4168 kb)
Alignments from Analyses. All alignments and topologies of one-to-one orthologs used for analyses of molecular evolution are given as a single tarball. (TGZ 3336 kb)
Data Availability Statement
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession PPHD00000000. The version described in this paper is version PPHD01000000.
The raw sequence data generated for this study are available in the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA), referenced by SRP128936. All relevant metadata can be found under Bioproject PRJNA396570. Alignments and topologies used for molecular evolution analyses are available as a supplementary file (Additional file 3).