

Abstract
Background
Mutations in the SLC26A4 gene are associated with Pendred syndrome and autosomal recessive non-syndromic deafness (DFNB4). Both disorders have similar audiologic characteristics: bilateral hearing loss, often severe or profound, which may be associated with abnormalities of the inner ear, such as dilatation of the vestibular aqueduct or Mondini dysplasia. But, in Pendred syndrome (OMIM #274600), with autosomal recessive inheritance, besides congenital sensorineural deafness, goiter or thyroid dysfunctions are frequently present. The aim of this study was to determine whether mutations in SLC26A4 are a frequent cause of hereditary deafness in Brazilian patients.
Methods
Microsatellite haplotypes linked to SLC26A4 were investigated in 68 families presenting autosomal recessive non-syndromic deafness. In the probands of the 16 families presenting segregation consistent with linkage to SLC26A4, Sanger sequencing of the 20 coding exons was performed. In an additional sample of 15 individuals with suspected Pendred syndrome, because of the presence of hypothyroidism or cochleovestibular malformations, the SLC26A4 gene coding region was also sequenced.
Results
In two of the 16 families with indication of linkage to SLC26A4, the probands were found to be compound heterozygotes for probably pathogenic different mutations: three novel (c.1003 T > G (p. F335 V), c.1553G > A (p.W518X), c.2235 + 2 T > C (IVS19 + 2 T > C), and one already described, c.84C > A (p.S28R). Two of the 15 individuals with suspected Pendred syndrome because of hypothyreoidism or cochleovestibular malformations were monoallelic for likely pathogenic mutations: a splice mutation (IVS7 + 2 T > C) and the previously described c.1246A > C (p.T416P). Pathogenic copy number variations were excluded in the monoallelic cases and in those with normal results after Sanger sequencing. Additional mutations in the SLC26A4 gene or other definite molecular cause for deafness were not identified in the monoallelic patients, after exome sequencing.
Conclusions
Biallelic pathogenic mutations in SLC26A4 explained ~ 3% of cases selected because of autosomal recessive deafness. Monoallelic mutations were present in ~ 13% of isolated cases of deafness with cochleovestibular malformations or suspected Pendred syndrome. These data reinforce the importance of mutation screening of SLC26A4 in Brazilian subjects and highlight the elevated frequency of monoallelic patients.
Keywords: Hereditary deafness, SLC26A4, Pendrin gene, Brazil
Background
Hearing loss may be explained by genetic, environmental or multifactorial causes. Hereditary hearing loss is considered one of the most striking examples of genetic heterogeneity, since it might exhibit all Mendelian inheritance patterns, as well as mitochondrial inheritance. Near 70% of individuals with hereditary hearing loss are referred as non-syndromic and, in remaining 30%, additional clinical features are present, characterizing syndromic hearing loss. Autosomal recessive inheritance contributes to 80% of nonsyndromic hereditary hearing loss, and mutations in the gene encoding the gap junction protein Connexin 26 (GJB2 – DFNB1) are present in about 50% of the recessive cases; autosomal dominant inheritance is observed in 10–20% of the cases, and X-linked inheritance in 2–3%. The frequency of mitochondrial mutations is about 1% [1–3].
Mutations in the SLC26A4 gene (NM_000441) were found to be causative of two autosomal recessive disorders, Pendred syndrome (OMIM # 274600) and one of the forms of non-syndromic autosomal recessive hearing loss (DFNB4; #600791) [4]. Pendred syndrome (PS) is characterized by bilateral sensorineural hearing loss, commonly severe to profound with prelingual onset, vestibular dysfunction, cochleovestibular malformations, such as Mondini dysplasia and euthyroid goiter, and with onset in late childhood to early adulthood. The observed variability of these clinical features is frequently substantial, even within the same family. DFNB4 exhibits similar clinical features, except that the most common temporal bone abnormality is the enlarged vestibular aqueduct (EVA), and thyroid defects are not observed [5]. Because of the variable expressivity and overlap of the clinical features, the two conditions may be considered as subsets of the spectrum of clinical manifestations of one single genetic entity [5].
The SLC26A4 gene encodes pendrin, a protein with 780 amino acids that belongs to the SLC26 anion transporter family [4, 6]. The human pendrin is generally expressed in the inner ear, mainly in endolymphatic sac and hair cells, and in the follicular cells of the thyroid [6–9]. Impaired function of pendrin was associated with endolymph acidification, leading to auditory sensory transduction defects. It is believed that its function in normal inner ear is related to Ca2+ re-absorption inhibition and bicarbonate/chloride exchanging, playing a role in pH homeostasis [8, 9]. In the thyroid, iodide efflux from cells to follicular lumen is allowed by pendrin functioning as an electroneutral iodide/chloride exchanger [6, 7].
Mutations in the SLC26A4 gene are reported to be the most frequent cause of hereditary hearing loss in East Asia, and the second most common cause worldwide, after Connexin 26 (GJB2) gene mutations [10–16]. The purpose of our study was to investigate the contribution of SLC26A4 mutations to hearing loss in Brazilian patients.
Methods
Patients
A total of 31 unrelated patients were selected to the molecular study of the SLC26A4 gene by Sanger sequencing, divided into two groups, based on clinical and genetic findings, as follows: (A) A sample of 68 pedigrees with hearing loss with presumed autosomal recessive inheritance (pedigrees with at least two affected sibs, born to hearing parents, or with at least one affected individual born to consanguineous parents, regardless of clinical data) was considered eligible to haplotype analysis with microsatellite (STR) markers linked to the DFNB4. The haplotype analysis with microsatellite (STR) markers allowed exclusion of segregation with DFNB4 in 52 pedigrees. Thus, 16 probands from the remaining pedigrees with haplotype segregation compatible with DFNB4 were selected to sequencing of the SLC26A4 gene. (B) 15 patients suspected of Pendred syndrome, because of the presence of hearing loss associated with thyroid dysfunction, or because of the presence of hearing loss with EVA or presenting hearing loss associated to other cochlear/vestibular malformation. In this group, 6 had hearing loss and EVA, 5 had hearing loss and Mondini dysplasia, and 4 had hearing loss and hypothyroidism. Most patients in this group were isolated cases.
Mutations in GJB2, two deletions near GJB6 gene [Δ (GJB6–D13S1830) and Δ(GJB6–D13S1854)], and the mitochondrial m.1555A > G mutation, had been previously excluded as the genetic cause of hearing loss in all probands, from both groups.
Molecular analysis
Blood samples were collected after written informed consent was obtained from all individuals or their legal guardians (if patients were under 18). The project was approved by the Ethics Committee, Instituto de Biociências, Universidade de São Paulo (Protocol no 109/2010). DNA was extracted by standard procedures using phenol/chloroform or using Autopure LS (Gentra Systems, Minneapolis, MN, USA).
STR genotyping
Microsatellite STR markers on 7q31, linked to the DFNB4 locus, were genotyped and used to perform haplotype analysis (D7S2420, D7S496, D7S2459, D7S2456, D7S799). One of the microsatellites, D7S2459, maps to intron 10 of the SLC26A4 gene. Primer sequences for the STR amplification were obtained from databases (http://genome.ucsc.edu and http://www.ncbi.nlm.nih.gov), except for the D7S799 marker, for which the reverse primer was designed using PRIMER 3 [17] (http://bioinfo.ut.ee/primer3-0.4.0/). The polymorphic fragments were analyzed using the GeneMapper software following capillary electrophoresis in the ABI 3730 DNA Analyzer (Applied Biosystems, Carlsbad, CA, USA).
Sanger sequencing of SLC26A4 coding region
PCR amplification of the 20 coding exons of SLC26A4 (2–21) and their flanking intronic sequences was performed using primers already described in Everett et al. [4], Scott et al. [18] and Lofrano-Porto et al. [19], except for exons 11 and 12, 15, 17 and 21, which had the primers designed using “Primer3” software. The PCR fragments were sequenced using the ABI BigDye Terminator v3.1 Cycle Sequencing Kit and the ABI 3730 DNA Analyzer (Applied Biosystems, Carlsbad, CA, USA).
MLPA analysis
When only one pathogenic variant was detected (two patients) or none possibly pathogenic allele was identified after Sanger sequencing (27 patients), samples were submitted to MLPA analysis, searching for possible copy number variations (CNVs) encompassing the SLC26A4 gene. MLPA was performed using the SALSA MLPA KIT P280-B1 Pendred-SLC26A4 kit (MRC Holland, Amsterdam, Netherlands), following the manufacturer’s instructions. The amplification products were subjected to capillary electrophoresis using the ABI 3730 DNA Analyzer (Applied Biosystems, Carlsbad, CA, USA). The results were analyzed using the Gene Marker software (https://softgenetics.com/GeneMarker.php).
Massive parallel sequencing and bioinformatic analysis
Massive parallel sequencing of the whole exome was performed in two samples, from individuals with only one detected likely pathogenic variant in the SLC26A4 gene (monoallelic). DNA samples were submitted to whole-exome sequencing at Laboratório Central de Tecnologias de Alto Desempenho em Ciências da Vida (LaCTAD) (University of Campinas, SP, BR). Sample libraries were prepared using the ‘TruSeq Custom DNA Library Preparation Kit’ from Illumina (Illumina INC, San Diego, California, USA). Whole exome was captured with Illumina’s ‘TruSeq Expanded Exome Enrichment Kit’ (target regions of 62 Mb). Illumina HiSeq 2500 was used to sequence the samples with paired-end fragments of 100 × 100 and average coverage of 120×. Alignment of fastq files to human reference hg19 was done with Burrows-Wheeler Aligner (BWA) [20], generating SAM files. SAM to BAM conversion, BAM files sorting and PCR duplicates marking were executed with Picard (http://broadinstitute.github.io/picard/). Genome Analysis Tool Kit (GATK) [21] was used in the steps of BAM processing (realignment based upon known local indels and variant quality score recalibration), and variant call (with Unified Genotyper). Variants in the VCF file were annotated with Annovar [22]. Variant frequencies were compared to the 1000 genomes (http://www.1000genomes.org), ESP6500 (http://evs.gs.washington.edu/EVS/), 65000 exomes from Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/) databanks and ABraOM (http://abraom.ib.usp.br/) [23]. The Deafness Variation Database (http://deafnessvariationdatabase.org) [24] and ClinVar were also consulted (https://www.ncbi.nlm.nih.gov/clinvar/). PolyPhen2 [25] SIFT [26] and Mutation Taster [27] were used for in silico pathogenicity prediction of the mutations. To address the effect of the splice site mutations, two softwares were used: NNSPLICE 0.9 version (http://www.fruitfly.org/seq_tools/splice.html) and NetGene 2 Server (http://www.cbs.dtu.dk/services/NetGene2/).
Results
In order to determine whether mutations in the SLC26A4 gene are a frequent cause of hereditary deafness in Brazilian patients, we analyzed 68 families presenting autosomal recessive non-syndromic hearing loss. Microsatellite haplotypes linked to the SLC26A4 gene were investigated, and segregation was consistent with linkage to this gene in 16 families; linkage was excluded in the remaining 52 pedigrees. Sanger sequencing of the 20 coding exons was performed in samples from the 16 probands, as well as in 15 patients with suspected PS and/or presenting EVA or other cochlear-vestibular malformations. Detected variants are listed in Table 1.
Table 1.
Heterozygous Variants and their predicted consequences detected after Sanger in both samples (pedigrees with microsatellite segregation compatible with linkage to SLC26A4 and individuals presenting cochlear-vestibular malformations)
| Cohort | Patient | Nucleotide | Protein | Location | Deafness variation database | SIFT (score), PolyPhen2 (score), MutationTaster (score), Splice Prediction Tools | 1000 g | ESP6500 | Conclusion (According to ACMG criteria) | Genotype | Protein locationb |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Families with autosomal recessive inheritance | 6 | c.1003 T > G a | p.F335V a | exon 9 | – | Damaging (0.003), Probably damaging (0.99), Disease causing (0.99) | – | – | likely pathogenic | Compound heterozygosis | External loop |
| c.1553G > A a | p.W518X a | exon 14 | – | -, −, Disease causing (1) | – | – | pathogenic | C-terminal | |||
| 7 | c.15C > A | p.G5G | exon 2 | benign | – | 0.006 | 0.005 | benign | – | – | |
| IVS10 + 35G > T | – | intron 10 | benign | – | 0.003 | 0.003 | benign | – | – | ||
| 24 | c.84C > A | p.S28R | exon 2 | Likely Pathogenic | Damaging (0.003), Possibly damaging (0.92), Polymorphism (0.79) | 0 | 0 | likely pathogenic | Compound heterozygosis | N-terminal | |
| IVS19 + 2 T > C a | SS a | intron 19 | – | -, −, Disease causing (1), splice donor site abolished | – | – | pathogenic | C-terminal | |||
| 44 | c.218A > G | p.E73G | exon 3 | – | Tolerated (0.313), Benign (0.00), Disease causing (0.77) | – | – | benign | – | – | |
| IVS15-18 T > A | – | intron 15 | benign | – | 0.013 | 0.02 | benign | – | – | ||
| 51 | IVS15 + 76G > C | – | intron 15 | benign | – | 0.039 | 0 | benign | – | – | |
| c.1826 T > G | p.V609G | exon 17 | benign | Tolerated (0.54), Benign (0.00), Polymorphism (0.19) | 0.04 | 0.049 | benign | – | – | ||
| c.2130C > T | p.D710D | exon 19 | benign | – | 0.019 | 0.023 | benign | – | – | ||
| c.2218G > A | p.G740S | exon 19 | benign | Tolerated (0,091), Benign (0.00), Polymorphism (0.99) | 0.015 | 0.017 | benign | – | – | ||
| Cases of deafness with suspected PS and/or presenting EVA or other cochleovestibular malformation | 71 | c.1246A > C | p.T416P | exon 10 | Pathogenic | Damaging (0.00), Probably damaging (1.00), Disease causing (0.99) | 0 | 0 | likely pathogenic | Heterozygosis (monoallelic) | TM10/Cytosolic Interface |
| 76 | c.898A > C | p.I300L | exon 7 | benign | Damaging (0.02), Probably damaging (0.97), Disease causing (0.99) | 0.005 | 0.004 | benign | – | – | |
| 78 | IVS8-143 T > C | intron 8 | – | – | – | – | benign | – | – | ||
| 83 | IVS7 + 2 T > C | SS | intron 7 | Pathogenic | -, −, Disease causing (1) | 0 | 0 | pathogenic | Heterozygosis (monoallelic) | TM7 | |
| 85 | IVS15-18 T > A | – | intron 18 | benign | – | 0.013 | 0.022 | benign | – | – |
In bold, variants considered as pathogenic or likely pathogenic
a indicates variants reported for the first time in this study. bAccording to Bassot et al. 2017 [52]
After variant filtering, according to classification in different databases and bioinformatics prediction of pathogenicity, six different variants were found in four probands that could be considered as probably causative of their autosomal recessive hearing loss. These variants were classified according to ACMG guidelines [28]. Half of them were missense variants (3/6, 50%); a novel nonsense mutation [c.1553G > A (p.W518X)] was identified; two splice site variants were detected, one novel (IVS19 + 2 T > C), and the other, previously described as pathogenic (IVS7 + 2 T > C).
Probands 6 and 24 (Table 1) are compound heterozygotes for two different likely pathogenic mutations: c.1003 T > G (p.F335 V) and c.1553G > A (p.W518X) in Patient 6; the variant c.84C > A (p.S28R) and the novel splice site mutation IVS19 + 2 T > C in Patient 24. Figure 1 shows the STR haplotypes and chromatograms of the mutations present in the two families.
Fig. 1.
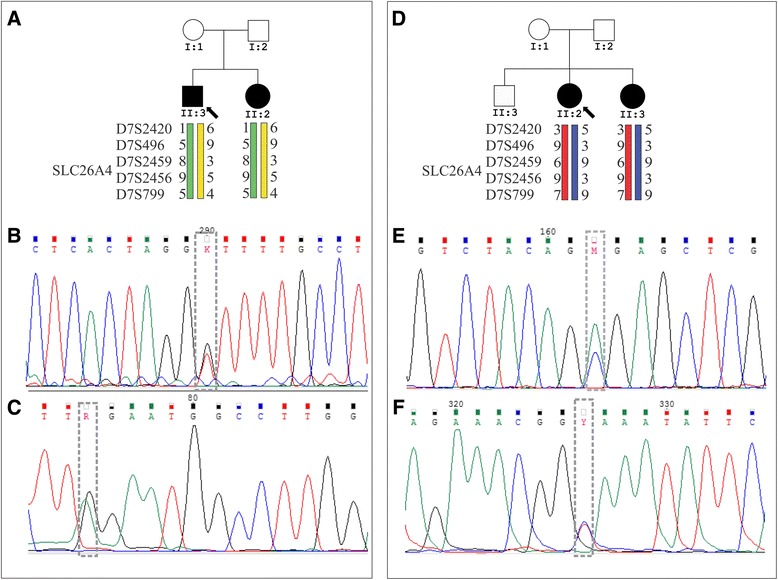
Pedigrees of families 6 (a) and 24 (d) showing the segregation of the pathogenic mutations. Chromatograms showing the probably pathogenic mutations found to segregate with deafness in family 6 (b and c) and family 24 (e and f)
It is noteworthy that in two probands (Patients 71 and 83), only one likely pathogenic mutation was detected and they were thus classified as “monoallelic” (Table 1). MLPA analysis performed in these two patients with only one detected pathogenic variant, and in those 27 patients who had no possibly pathogenic allele detected after Sanger sequencing, revealed no SLC26A4 deletion or duplication.
The DNA samples of the two monoallelic patients were submitted to whole exome sequencing. A summary of the variants filtered from known deafness associated genes is presented in Table 2, along with pathogenicity prediction from different in silico bioinformatics tools.
Table 2.
Summary of results of the massively parallel exome sequencing of two patients with monoallelic mutations in SLC26A4. All variants were found in heterozygous state
| Patient | Gene | Nucleotide | Protein | dbSNP | Deafness Variation Database | SIFT (score), PolyPhen2 (score), MutationTaster (score), Splice Prediction Tools | 1000g | ESP6500 | Abraom |
|---|---|---|---|---|---|---|---|---|---|
| 71 | SLC26A4 | c.1246A>C | T416P | rs28939086 | Pathogenic | Damaging (0), Probably Damaging (0.995), Disease causing (0.971541) | 0.0000 | 0.0000 | 0.0000 |
| GJB2 | c.457G>A | V153I | rs111033186 | benign | Tolerated (1), Benign (0.007), Disease causing (0.8156) | 0.0013 | 0.00223 | 0.0032 | |
| TMIE | c.218C>T | T73M | rs770957465 | unknown significance | Tolerated (0.156), Probably Damaging (1), Disease causing (0.999) | 0.000 | 0.000 | 0.000 | |
| USH1C | c.1823C>G | P608R | rs41282932 | Pathogenic | Damaging (0.002), Probably Damaging (0.984), Disease causing ( 0.9815) | 0.000 | 0.001 | 0.000 | |
| MIR96 | 129414574A>G | rs41274239 | benign | - | 0.0010 | 0.0033 | 0.0032 | ||
| PCDH15 | c.4109_4110insGCCGCC | p.P1370delinsPPP | - | not described | -, -. Polymorphism (0.999) | 0.0018 | 0.000 | 0.0016 | |
| PCDH15 | c.5134_5136del | p.1712_1712del | rs397517462 | not described | -, -. Polymorphism (0.999) | 0.000 | 0.031 | 0.0032 | |
| 83 | SLC26A4 | c.918+2T>C | - | - | Pathogenic | -, -, Disease causing (1), splice donor site abolished | 0.000 | 0.000 | 0.0008 |
| MYO7A | c.2463G>C | Q821H | - | not described | Damaging (0), Probably Damaging (1), Disease causing (0.9999) | 0.000 | 0.000 | 0.000 | |
| GJB6 | c.460T>A | F154I | - | not described | Damaging (0.022), Damaging (0.986), Disease causing (0.9987) | 0.000 | 0.000 | 0.000 | |
| P2RX2 | c.A275G | p.Q92R | rs142844880 | unknown significance | Tolerable (0.116), Benign (0.098), Disease causing (1) | 0.0004 | 0.0004 | 0.000 | |
| POLD1 | c.C211T | p.P71S | not described | Tolerable (0.879), Benign (0), non-disease causing (1) | 0.000 | 0.000 | 0.000 | ||
| TSPEAR | c.1185G>T | E395D | rs143303485 | not described | Tolerated (0.420), Benign (0.02), Polymorphism | 0.0002 | 0.0002 | 0.000 |
In bold, variants that all evidence indicate that they are pathogenic
Discussion
Pedigrees with autosomal recessive hearing loss
According to the Human Gene Mutation Database [http://www.hgmd.cf.ac.uk/ac/index.php], more than 360 mutations in the SLC26A4 gene have been identified to date, including splice site aberrations, frameshift, missense and nonsense mutations, as well as large deletions (rare cases) [29]. The mutation spectrum of SLC26A4 varies widely among ethnic groups, with certain mutations demonstrating a higher prevalence in specific populations [10, 14, 29, 30].
In our study, probably causative mutations in SLC26A4 were found in 3% (2/68) of the pedigrees presenting autosomal recessive non-syndromic hearing loss. In two of the probands (Patients 6 and 24; Table 1), four different likely pathogenic mutations were found in compound heterozygosis. Three of them were never reported - c.1003 T > G (p. F335 V), c.1553G > A (p.W518X), c.2235 + 2 T > C (IVS19 + 2 T > C), and a fourth had already been described, c.84C > A (p.S28R) [31].
The variant c.1003 T > G is not reported the Deafness Variation Database [24], neither in 1000 Genomes or 6500 Exomes. Computational predictions (Polyphen 2 and Mutation Taster) indicated it as probably disease causing (Table 1). A substitution in the same position, c.1003 T > C (p.F335 L) was reported as causative of Pendred syndrome (Deafness Variation Database) [24], being found with a 0.1% frequency in 1000 Genomes and 6500 exomes, thus, very rare. In addition to the c. 1003 T > G, Patient 6 also carried the c.1553G > A (p.W518X) mutation (compound heterozygosis), unreported in databases, but considered as pathogenic, since it leads to a premature stop codon. A variant affecting the same codon, (c.1554G > A) also leading to a premature stop codon, was described by Pourová et al. [32] as pathogenic. Patient 6 (Table 1) did not show the clinical picture of Pendred syndrome. The same combination of mutations was detected in his affected sister, reinforcing their role as causative of hearing loss.
Patient 24 (Table 1) showed the combination of mutations c.2235 + 2 T > C (IVS19 + 2 T > C) and c.84C > A (p.S28R); c.84C > A (p.S28R), which was already reported in ClinVar as pathogenic, was first described by Fugazzola et al. [33]. It is predicted to be damaging by SIFT, possibly damaging by Polyphen 2, but indicated as a polymorphism by Mutation Taster (Table 1). It is not present in 1000 Genomes and 6500 Exomes. The IVS19 + 2 T > C is a splice site mutation, never reported before. NNSsplice 0.9 version (Lawrence Berkeley National Laboratory, Genomic Informatic Groups, 2012 link) and Netgene2 Server (Center for Biological sequence analysis, 2012 link) predict that the mutation abolishes the splicing donor site of exon 19. After detection of variants c.84C > A and (IVS19 + 2 T > C) in Patient 24, mutation analysis revealed the same mutations in the proband’s affected sister, and that the parents were heterozygous, thus documenting that the mutations were in trans. The family was contacted for genetic counseling and recent clinical reassessment indicated that both sisters presented EVA. Given clinical data and segregation data, we concluded that the combination of both mutations explains their phenotype.
Park et al. [10] investigated a collection of Indian and Pakistanese families and selected 15 probands after linkage studies for SLC26A4 sequencing. Adding two previously reported families [34] to their estimates, the authors came to the conclusion that 17/318 (5%) autosomal recessive cases were attributed to pathogenic variants in the SLC26A4 gene. Following a strategy similar to ours, Pera et al. [35] estimated that 3.5% of pedigrees showing autosomal recessive hearing loss were explained by mutations in SLC26A4. The authors investigated 115 pedigrees and selected 20 for mutation screening, after segregation studies with STRs linked to SLC26A4.
Albert et al. [31] found that 40% of selected probands for SLC26A4 screening showed biallelic mutations. However, the selection criteria for analysis included, besides evidence of autosome recessive hearing loss, EVA or other inner ear abnormalities, such as Mondini dysplasia.
Summing up, the finding of 3% of biallelic mutations in our sample, selected on the basis of autosomal recessive hearing loss, is comparable to the studies performed in other populations, with similar criteria of patient selection.
Patients with EVA or other temporal abnormalities (including Mondini dysplasia) or thyroid malfunctioning
Candidate mutations were found in three out of the 15 patients, (Table 1), but in none of them a second mutation in the same gene was found. In Patient 76, an isolated case born to nonconsanguineous parents, the missense variant c.898C > A (p.I300L) was found in heterozygosis. The variant was previously associated to increased susceptibility to Graves disease (autoimmune hyperthyroidism) [36]. All in silico pathogenicity predictors indicated it as Damaging (Polyphen2–0.02), probably damaging (0.97) and Disease causing (0.99). However, Deafness Variation Database claims it is benign, and databases list this variant, but only in heterozygosis, with a high frequency in healthy African descendent populations (10–18%). There is no phenotype information related to the occurrence of this mutation in homozygosis. Thus, we concluded it is more likely not to be related to the clinical phenotype.
A monoallelic mutation in SLC26A4, c.1246A > C, was found in Patient 71, an isolated case of hearing loss, born to non-consanguineous parents. It was already reported as causing Pendred syndrome, and is frequent in Northern European patients with hearing loss [37]. The patient presented bilateral prelingual and progressive hearing loss and computed tomography revealed Mondini dysplasia. After exome sequencing, some variants that could be potentially related to hearing loss were found, but none could individually explain the clinical findings (Table 2).
A monoallelic splice site mutation IVS7 + 2 T > C (c.918 + 2 T > C), listed in the Deafness Variation Database [24] as pathogenic as the cause of non-syndromic hearing loss, and predicted as probably pathogenic by splicing bioinformatic tools, was found in Patient 83, an isolated case, born to non-consanguineous parents. Postlingual progressive hearing loss was observed, but mixed hearing loss was detected in the right ear. MRI indicated EVA. Exome sequencing revealed a set of potentially pathogenic variants in deafness-associated genes, but none could individually explain the hearing loss phenotype (Table 2).
Thus, in 3/15 probands with hearing loss, selected because of EVA or other temporal abnormalities (including Mondini dysplasia) or thyroid malfunctioning, monoallelic mutations were found, but they could not individually explain the clinical findings. In two of them, evidence for pathogenicity of the variants was convincing.
The mutation detection ratio in this sample is smaller than other studies in the literature, in which patients were selected because of clinical signs of Pendred syndrome. However, in most of these studies, patients were selected because they presented hearing loss and EVA and goiter/hypotiroidism. For instance, in the study of Rendtorff et al. [37], 71% of the probands fulfilled at least three diagnostic criteria of Pendred syndrome, which explains the detection rate of 61% of biallelic mutations and 10% of monoallelic mutations in their series. In our sample, only one clinical sign was present in each patient, in addition to hearing loss. The probability of finding SLC26A4 mutations in this sample is lower, because our criteria of selection were less stringent than most reports in the literature.
De Moraes et al. [38] reported on a SLC26A4 mutation screening in a selected sample of 23 Brazilian individuals with severe-to-profound non-syndromic hearing loss and EVA. They found 13 different mutations in nine individuals. Five individuals had two mutations (21.7%) and four were found to be monoallelic (17.3%). Although a conclusive molecular diagnosis was possible in many cases, the puzzling high proportion of monoallelic mutations was also seen in their sample.
MLPA studies
No copy number variation was detected among the patients with only one pathogenic variant (two patients) or among those with no pathogenic allele, detected after Sanger sequencing (27 patients). There are few reports of MLPA technique results regarding the SLC26A4 gene. Pourová et al. [32] used MLPA to screen for SLC26A4 deletions and duplications in 18 probands with only one detected mutation, and no copy variation was found. In the study of Zhao et al. [39], in a group of 68 patients with monoallelic mutations and in another group of 39 patients without mutation, no alteration was found. Pique et al. [40], in the investigation of 107 probands with monoallelic mutations in SLC26A4, found only one deletion, spanning exons 4–6, which accounted for about 1% of the missing mutations. Pang et al. [41] described a 7666-bp genomic deletion in homozygosis in one patient and in compound heterozygosis in four patients previously classified as monoallelic; this genomic deletion was detected in 18% of the Chinese Han EVA probands with monoallelic SLC26A4 mutations.
In conclusion, duplications and deletions are rare and do not explain a substantial amount of cases with one mutation or without mutations, after sequencing the SLC26A4 gene.
The puzzle of monoallelic mutations
In spite of the well-known recessive nature of mutations in the SLC26A4 gene, it is striking that, in many reports, a high frequency of individuals presenting only one potentially pathogenic variant is found [37, 42–44].
Pique et al. [40] reviewed six studies [11, 30, 45–48] in which EVA was a selection criteria and came to an estimate that 20% of patients have monoallelic mutations. Two genes, KCNJ10 [49] and FOXI1 [48] have been investigated for their role in PDS/DFNB4 disease spectrum and digenic inheritance has been proposed. However, according to Landa et al. [50] and Vona et al. [51], FOXI1 and KCNJ10 mutations are rare. In our study, mutations in these two genes were excluded in two of our monoallelic samples, which were submitted to exome sequencing.
Another reasonable explanation for the finding of monoallelic mutations would be the second mutations being deletions or duplications in the SLC26A4 gene, which, as reasoned above, also seem to be rare in ours and in other studies.
As already stated, we performed exome sequencing in two of the patients with monoallelic mutations (Patients 71 and 83). Although a second mutation was not found in SLC26A4, other potentially pathogenic mutations were revealed in other deafness related genes (Table 2), but none could explain deafness from the viewpoint of monogenic inheritance. Of course a quantitative approach is not possible with such a small sample; however, it is tempting to speculate that hearing impairment and related ear malformations in these cases could be due to a multifactorial mechanism. In such a mechanism, a monoallelic SLC26A4 variant would represent one of the genetic hits needed to phenotype expression. Vona et al. [51] used targeted massive parallel sequencing of a panel of deafness genes in a sample of 30 individuals with hearing loss. About 50% of probands were diagnosed with monogenic forms of nonsyndromic hearing loss, but they found a significant enrichment of potentially pathogenic variants in the undiagnosed affected individuals, when compared to nine hearing controls. Their results, associated with the findings of frequent monoallelic SLC26A4 mutations in individuals presenting hearing loss and/or EVA, strongly suggest that part of the molecularly undiagnosed cases are due to multifactorial mechanism. Certainly, further studies are needed in the field. This hypothesis could only be verified in larger samples of affected and unaffected individuals submitted to massive parallel sequencing of deafness related genes.
Conclusions
Our strategy of molecular study of the SLC26A4 gene allowed the conclusion that biallelic pathogenic mutations in SLC26A4 explained ~ 3% of cases selected because of autosomal recessive deafness, and that monoallelic mutations were present in ~ 13% of cases of deafness with cochleovestibular malformations or suspected Pendred syndrome. These findings highlight the importance of mutation screening of SLC26A4 in Brazilian subjects with hearing loss and reinforce the puzzling finding of a high proportion of monoallelic patients, particularly among those presenting cochleovestibular malformations.
Acknowledgments
The authors thank all family members for their participation in the study. We thank all professionals from DERDIC and from Department of Otolaryngology of the University of São Paulo School of Medicine Clinics Hospital for their referral of patients and clinical evaluation. We also thank Humberto Cezar Vicente Marcolino for technical support and Dr. Angela M. Vianna-Morgante for critical reading of the manuscript.
Web resources
ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/
Deafness Variation Database: http://deafnessvariationdatabase.org.
ESP6500:http://evs.gs.washington.edu/EVS/
Gene Marker: https://softgenetics.com/GeneMarker.php
Human Gene Mutation Database: http://www.hgmd.cf.ac.uk/ac/index.php
Mutation Taster: http://www.mutationtaster.org.
NCBI: http://www.ncbi.nlm.nih.gov
NetGene 2 Server: http://www.cbs.dtu.dk/services/NetGene2
NNSPLICE: http://www.fruitfly.org/seq_tools/splice.html
OMIM: https://www.omim.org/entry/274600
Picard: http://broadinstitute.github.io/picard/
PolyPhen2: http://genetics.bwh.harvard.edu/pph2/
PRIMER 3: http://bioinfo.ut.ee/primer3-0.4.0/.
SIFT: http://sift.jcvi.org/.
UCSC: http://genome.ucsc.edu/ and.
1000 genomes: http://www.1000genomes.org
65000 exomes: http://exac.broadinstitute.org/.
Funding
This work was financially supported by FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo, Grant number 2013/08028-1), CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and CAPES (Coordenação de Aperfeiçoamento Profissional de Nível Superior).
Availability of data and materials
The dataset during and/or analyzed during the current study is available from the corresponding author on reasonable request.
Abbreviations
- BWA
Burrows-Wheeler Aligner
- DFNB4
Autosomal Recessive Deafness Locus number4
- EVA
enlarged vestibular aqueduct
- ExAC
Exome Aggregation Consortium
- GATK
Genome Analysis Tool Kit
- MLPA
Multiplex Ligand Probe Analysis
- OMIM
Online Mendelian Inheritance In Man
- PS
Pendred syndrome
- STR
Short Tandem Repeats
Authors’ contributions
RWN performed all linkage analysis, Sanger sequencing and MLPA. KL helped in all linkage analysis, Sanger sequencing, manuscript preparation, figures and tables formatting. MRBMA provided technical support in lab experiments and performed exclusion of frequent deafness related mutations. ACB helped in exclusion of frequent deafness related mutations, Sanger sequencing, manuscript preparation, figures and tables formatting. GLY performed the bioinformatic analysis of exome data. RCMN designed the study, and was responsible for mentoring and manuscript preparation. All authors have read and approved the manuscript.
Ethics approval and consent to participate
Blood samples were collected after written informed consent was obtained from all individuals or their legal guardians (if patients were under 18). The project was approved by the Ethics Committee, Instituto de Biociências, Universidade de São Paulo (Protocol no 109/2010).
Consent for publication
All subjects have approved the submission of the manuscript by written informed consent.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Renata Watanabe Nonose, Email: rewanose@gmail.com.
Karina Lezirovitz, Email: lezi.karina@gmail.com.
Maria Teresa Balester de Mello Auricchio, Email: mtbmello@yahoo.com.br.
Ana Carla Batissoco, Email: anacarlabatissoco@gmail.com.
Guilherme Lopes Yamamoto, Email: glyamamoto@gmail.com.
Regina Célia Mingroni-Netto, Email: renetto@ib.usp.br.
References
- 1.Ballana E, Ventayol M, Rabionet R, Gasparini P, Estivill X. Connexins and deafness Homepage. 2017. http://davinci.crg.es/deafness. Accessed 10 Apr 2017.
- 2.Smith RJH, Jones MN. Nonsyndromic Hearing Loss and Deafness, DFNB1 GeneReviews - NCBI Bookshelf. U.S. National Library of Medicine. 2017. [Google Scholar]
- 3.Van Camp G, Smith RJH. Hereditary hearing loss homepage. 2017. [Google Scholar]
- 4.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nature Genet. 1997;17:411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- 5.Alasti F, Van Camp G, and Smith RJH. Pendred Syndrome/DFNB4” GeneReviews - NCBI Bookshelf. U.S National Library of Medicine 2017. http://www.ncbi.nlm.nih.gov/books/NBK1467/. Accessed 20 Apr 2017.
- 6.Everett LA, Morsli H, Wu DK, Green ED. Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci U S A. 1999;96:9727–9732. doi: 10.1073/pnas.96.17.9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida A, Taniguchi S, Hisatome I, Royaux IE, Green ED, Kohn LD, Suzuki K. Pendrin is an iodide-specific apical porter responsible for iodide efflux from thyroid cells. J Clin Endocrinol Metab. 2002;87:3356–3361. doi: 10.1210/jcem.87.7.8679. [DOI] [PubMed] [Google Scholar]
- 8.Wangemann P, Nakaya K, Wu T, Maganti RJ, Itza EM, Sanneman JD, Harbidge DG, Billings S, Marcus DC. Loss of cochlear HCO3- secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model. Am J Physiol Renal Physiol. 2007;292:F1345–F1353. doi: 10.1152/ajprenal.00487.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi BY, Kim HM, Ito T, Lee KY, Li X, Monahan K, Wen Y, Wilson E, Kurima K, Saunders TL, et al. Mouse model of enlarged vestibular aqueducts defines temporal requirement of Slc26a4 expression for hearing acquisition. J Clin Invest. 2011;121:4516–4525. doi: 10.1172/JCI59353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, Kim HN, Moon SK, Abe S, Tukamoto K, Riazuddin S, Kabra M, Erdenetungalag R, Radnaabazar J, Khan S, Pandya A, Usami SI, Nance WE, Wilcox ER, Riazuddin S, Griffith AJ. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. 2003;40:242–248. doi: 10.1136/jmg.40.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsukamoto K, Suzuki H, Harada D, Namba A, Abe S, Usami S. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: a unique spectrum of mutations in Japanese. Eur J Hum Genet. 2003;11:916–922. doi: 10.1038/sj.ejhg.5201073. [DOI] [PubMed] [Google Scholar]
- 12.Park HJ, Lee SJ, Jin HS, Lee JO, Go SH, Jang HS, Moon SK, Lee SC, Chun YM, Lee HK, Choi JY, Jung SC, Griffith AJ, Koo SK. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin Genet. 2005;67:160–165. doi: 10.1111/j.1399-0004.2004.00386.x. [DOI] [PubMed] [Google Scholar]
- 13.Wu CC, Yeh TH, Chen PJ, Hsu CJ. Prevalent SLC26A4 mutations in patients with enlarged vestibular aqueduct and/or Mondini dysplasia: a unique spectrum of mutations in Taiwan, including a frequent founder mutation. Laryngoscope. 2005;115:1060–1064. doi: 10.1097/01.MLG.0000163339.61909.D0. [DOI] [PubMed] [Google Scholar]
- 14.Wang QJ, Zhao YL, Rao SQ, Guo YF, Yuan H, Zong L, Guan J, Xu BC, Wang DY, Han MK, Lan L, Zhai SQ, Shen Y. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin Genet. 2007;72:245–254. doi: 10.1111/j.1399-0004.2007.00862.x. [DOI] [PubMed] [Google Scholar]
- 15.Dai P, Yuan Y, Huang D, Zhu X, Yu F, Kang D, Yuan H, Wu B, Han D, Wong LJ. Molecular etiology of hearing impairment in Inner Mongolia: mutations in SLC26A4 gene and relevant phenotype analysis. J Transl Med. 2008;6:74. doi: 10.1186/1479-5876-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu C, Zheng H, Zhang S, Chen Y, Su J, Wang J, Xie B, Hu X, Fan X, Luo J, Li C, Chen R, Shen Y, Chen S. Mutation screening of the SLC26A4 gene in a sample of 192 Chinese patients with congenital hypothyroidism. Arch Endocrinol Metab. 2016;60(4):323–327. doi: 10.1590/2359-3997000000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott DA, Wang R, Kreman TM, Andrews M, McDonald JM, Bishop JR, Smith RJH, Karniski LP, Sheffield VC. Functional differences of the PDS gene product are associated with phenotypic variation in patients with Pendred syndrome and non-syndromic hearing loss (DFNB4) Hum Mol Genet. 2000;9:1709–1715. doi: 10.1093/hmg/9.11.1709. [DOI] [PubMed] [Google Scholar]
- 19.Lofrano-Porto A, Barra GB, Nascimento PP, Costa PG, Garcia EC, Vaz RF, Batista AR, Freitas AC, Cherulli BL, Bahmad F, Jr, Figueiredo LG, Neves FA, Casulari LA. Pendred syndrome in a large consanguineous Brazilian family caused by a homozygous mutation in the SLC26A4 gene. Arq Bras Endocrinol Metabol. 2008;52(8):1296–1303. doi: 10.1590/S0004-27302008000800015. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010:1297–303. [DOI] [PMC free article] [PubMed]
- 22.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naslavsky MS, Yamamoto GL, de Almeida TF, Ezquina SAM, Sunaga DY, Pho N, Bozoklian D, Sandberg TOM, Brito LA, Lazar M, Bernardo DV, Jr AE, Duarte YAO, Lebrão ML, Passos-Bueno MR, Zatz M. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum Mutat. 2017;38(7):751–763. doi: 10.1002/humu.23220. [DOI] [PubMed] [Google Scholar]
- 24.Shearer AE, Eppsteiner RW, Booth KT, Ephraim SS, Gurrola J, Simpson A, Black-Ziegelbein EA, Joshi S, Ravi H, Giuffre AC, et al. Utilizing ethnic-specific differences in minor allele frequency to re-categorize reported pathogenic deafness variants. Am J Hum Genet. 2014;95(4):445–453. doi: 10.1016/j.ajhg.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 27.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 28.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The human gene mutation database (HGMD). 2017. http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SLC26A4. Accessed 21 Apr 2017.
- 30.Blons H, Feldmann D, Duval V, Messaz O, Denoyelle F, Loundon N, Sergout-Allaoui A, Houang M, Duriez F, Lacombe D, Delobel B, et al. Screening of SLC26A4 (PDS) gene in Pendred’s syndrome: a large spectrum of mutations in France and phenotypic heterogeneity. Clin Genet. 2004;66:333–340. doi: 10.1111/j.1399-0004.2004.00296.x. [DOI] [PubMed] [Google Scholar]
- 31.Albert S, Blons H, Jonard L, Feldmann D, Chauvin P, Loundon N, Sergent-Allaoui A, Houang M, Joannard A, Schmerber S, et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet. 2006;14:773–779. doi: 10.1038/sj.ejhg.5201611. [DOI] [PubMed] [Google Scholar]
- 32.Pourová R, Janousek P, Jurovcík M, Dvoráková M, Malíková M, Rasková D, Bendová O, Leonardi E, Murgia A, Kabelka Z, Astl J, Seeman P. Spectrum and frequency of SLC26A4 mutations among Czech patients with early hearing loss with and without enlarged vestibular aqueduct (EVA) Ann Hum Genet. 2010;74(4):299–307. doi: 10.1111/j.1469-1809.2010.00581.x. [DOI] [PubMed] [Google Scholar]
- 33.Fugazzola L, Cerutti N, Mannavola D, Crino A, Cassio A, Gasparoni P, Vannucchi G, Beck-Peccoz P. Differential diagnosis between Pendred and pseudo-Pendred syndromes: clinical, radiologic, and molecular studies. Pediatr Res. 2002;51(4):479–484. doi: 10.1203/00006450-200204000-00013. [DOI] [PubMed] [Google Scholar]
- 34.Li XC, Everett LA, Lalwani AK, Desmukh D, Friedman TB, Green ED, Wilcox ER. A mutation in PDS causes non-syndromic recessive deafness. Nat Genet. 1998;18(3):215–217. doi: 10.1038/ng0398-215. [DOI] [PubMed] [Google Scholar]
- 35.Pera A, Villamar M, Viñuela A, Gandía M, Medà C, Moreno F, Hernández-Chico C. A mutational analysis of the SLC26A4 gene in Spanish hearing-impaired families provides new insights into the genetic causes of Pendred syndrome and DFNB4 hearing loss. Eur J Hum Genet. 2008;16(8):888–896. doi: 10.1038/ejhg.2008.30. [DOI] [PubMed] [Google Scholar]
- 36.Hadj-Kacem H, Kallel R, Belguith-Maalej S, Mnif M, Charfeddine I, Ghorbel A, Abid M, Ayadi H, Masmoudi S. SLC26A4 variations among Graves' hyper-functioning thyroid gland. Dis Markers. 2010;29(2):63–69. doi: 10.1155/2010/327518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rendtorff ND, Schrijver I, Lodahl M, Rodriguez-Paris J, Johnsen T, Hansén EC, Nickelsen LA, Tümer Z, Fagerheim T, Wetke R, Tranebjaerg L. SLC26A4 mutation frequency and spectrum in 109 Danish Pendred syndrome/DFNB4 probands and a report of nine novel mutations. Clin Genet. 2013;84(4):388–391. doi: 10.1111/cge.12074. [DOI] [PubMed] [Google Scholar]
- 38.De Moraes VC, Dos Santos NZ, Ramos PZ, Svidnicki MC, Castilho AM, Sartorato EL. Molecular analysis of SLC26A4 gene in patients with nonsyndromic hearing loss and EVA: identification of two novel mutations in Brazilian patients. Int J Pediatr Otorhinolaryngol. 2013;77(3):410–413. doi: 10.1016/j.ijporl.2012.11.042. [DOI] [PubMed] [Google Scholar]
- 39.Zhao J, Yuan Y, Chen J, Huang S, Wang G, Han D, Dai P. SLC26A4 gene copy number variations in Chinese patients with non-syndromic enlarged vestibular aqueduct. J Transl Med. 2012;10:82. doi: 10.1186/1479-5876-10-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pique LM, Brennan ML, Davidson CJ, Schaefer F, Greinwald J, Jr, Schrijver I. Mutation analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. Peer J. 2014;2:384. doi: 10.7717/peerj.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pang X, Chai Y, He L, Chen P, Wang X, Li L, Jia H, Wu H, Yang T. A 7666-bp genomic deletion is frequent in Chinese Han deaf patients with non-syndromic enlarged vestibular aqueduct but without bi-allelic SLC26A4 mutations. Int J Pediatr Otorhinolaryngol. 2015;79(12):2248–2252. doi: 10.1016/j.ijporl.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 42.Choi BY, Stewart AK, Nishimura KK, Cha WJ, Seong MW, Park SS, Kim SW, Chun YS, Chung JW, Park SN, Chang SO, Kim CS, Alper SL, Griffith AJ, Oh SH. Efficient molecular genetic diagnosis of enlarged vestibular aqueducts in east Asians. Genet Test Mol Biomarkers. 2009;13(5):679–687. doi: 10.1089/gtmb.2009.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito T, Noguchi Y, Yashima T, Ohno K, Kitamura K. Hereditary hearing loss and deafness genes in Japan. J Med Dent Sci. 2010;57(1):1–10. [PubMed] [Google Scholar]
- 44.Reyes S, Wang G, Ouyang X, Han B, Du LL YHJ, Yan D, Dai P, Liu XZ. Mutation analysis of SLC26A4 in mainland Chinese patients with enlarged vestibular aqueduct. Otolaryngol Head Neck Surg. 2009;141(4):502–508. doi: 10.1016/j.otohns.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campbell C, Cucci RA, Prasad S, Green GE, Edeal JB, Galer CE, Karniski LP, Sheffield VC, Smith RJ. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mutat. 2001;17(5):403–411. doi: 10.1002/humu.1116. [DOI] [PubMed] [Google Scholar]
- 46.Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS, Nance WE, Yang Y, Zalewski CK, Brewer CC, Butman JA, Griffith AJ. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet. 2005;42(2):159–165. doi: 10.1136/jmg.2004.024208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerbäck S, Smith RJ. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4) Am J Hum Genet. 2007;80(6):1055–1063. doi: 10.1086/518314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu CC, Lu YC, Chen PJ, Yeh PL, Su YN, Hwu WL, Hsu CJ. Phenotypic analyses and mutation screening of the SLC26A4 and FOXI1 genes in 101 Taiwanese families with bilateral nonsyndromic enlarged vestibular aqueduct (DFNB4) or Pendred syndrome. Audiol Neurootol. 2010;15(1):57–66. doi: 10.1159/000231567. [DOI] [PubMed] [Google Scholar]
- 49.Yang T, Gurrola JG, 2nd, Wu H, Chiu SM, Wangemann P, Snyder PM, Smith RJ. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84(5):651–657. doi: 10.1016/j.ajhg.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Landa P, Differ AM, Rajput K, Jenkins L, Bitner-Glindzicz M. Lack of significant association between mutations of KCNJ10 or FOXI1 and SLC26A4 mutations in Pendred syndrome/enlarged vestibular aqueducts. BMC Med Genet. 2013;14:85. doi: 10.1186/1471-2350-14-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vona B, Müller T, Nanda I, Neuner C, Hofrichter MA, Schröder J, Bartsch O, Läßig A, Keilmann A, Schraven S, Kraus F, Shehata-Dieler W, Haaf T. Targeted next-generation sequencing of deafness genes in hearing-impaired individuals uncovers informative mutations. Genet Med. 2014;16(12):945–953. doi: 10.1038/gim.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bassot C, Minervini G, Leonardi E, Tosatto SC. Mapping pathogenic mutations suggests an innovative structural model for the pendrin (SLC26A4) transmembrane domain. Biochimie. 2017;132:109–120. doi: 10.1016/j.biochi.2016.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset during and/or analyzed during the current study is available from the corresponding author on reasonable request.