

Abstract
Background
The level of fat deposition in carcass is a crucial factor influencing meat quality. Guangling Large-Tailed (GLT) and Small-Tailed Han (STH) sheep are important local Chinese fat-tailed breeds that show distinct patterns of fat depots. To gain a better understanding of fat deposition, transcriptome profiles were determined by RNA-sequencing of perirenal, subcutaneous, and tail fat tissues from both the sheep breeds. The common highly expressed genes (co-genes) in all the six tissues, and the genes that were differentially expressed (DE genes) between these two breeds in the corresponding tissues were analyzed.
Results
Approximately 47 million clean reads were obtained for each sample, and a total of 17,267 genes were annotated. Of the 47 highly expressed co-genes, FABP4, ADIPOQ, FABP5, and CD36 were the four most highly transcribed genes among all the known genes related to adipose deposition. FHC, FHC-pseudogene, and ZC3H10 were also highly expressed genes and could, thus, have roles in fat deposition. A total of 2091, 4233, and 4131 DE genes were identified in the perirenal, subcutaneous, and tail fat tissues between the GLT and STH breeds, respectively. Gene Ontology (GO) analysis showed that some DE genes were associated with adipose metabolism. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that PPAR signaling pathway and ECM-receptor interaction were specifically enriched. Four genes, namely LOC101102230, PLTP, C1QTNF7, and OLR1 were up-regulated and two genes, SCD and UCP-1, were down-regulated in all the tested tissues of STH. Among the genes involved in ECM–receptor interaction, the genes encoding collagens, laminins, and integrins were quite different depending on the depots or the breeds. In STH, genes such as LAMB3, RELN, TNXB, and ITGA8, were identified to be up regulated and LAMB4 was observed to be down regulated.
Conclusions
This study unravels the complex transcriptome profiles in sheep fat tissues, highlighting the candidate genes involved in fat deposition. Further studies are needed to investigate the roles of the candidate genes in fat deposition and in determining the meat quality of sheep.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4747-1) contains supplementary material, which is available to authorized users.
Keywords: Adipose tissue, Fat deposition, Gene expression, RNA sequencing, Sheep
Background
Sheep is a major livestock resource of meat, milk, wool, and fur around the world. Fat-tailed sheep account for approximately 25% of the world’s sheep population [1]. The “fat tail” trait of sheep is regarded as an adaptive response to harsh environment and the fat stored in such tail is a valuable reserve for sheep during migration and in winter when food is scarce [2]. The tail fat is also used as food by humans. However, because too much fat in the daily diet is considered harmful to human health, the carcass adiposity, especially the fat tail, is not desirable to customers and reduces the value of meat in sheep. Therefore, reduction of fat deposition in the body and tail for production of lean meat is a major goal of the sheep industry. This has been attained by docking the fat tail, slaughtering at an early age, or by crossing of the fat-tailed breed with the lean-tailed breed. A better understanding of the molecular mechanism underlying fat deposition is very important for controlling the fat mass in carcass through sheep breeding.
The tail types in the two Chinese domestic fat-tailed sheep breeds, Guangling Large Tailed (GLT) and Small Tailed Han (STH), show great divergence. Both these breeds originated from the ancient Mongolian sheep. The GLT breed is characterized by large tails and good meat quality but shows low fecundity whereas the STH breed typically has small tails and higher fecundity [3]. The difference in the content of tail fat between these two breeds has stimulated extensive interest in characterizing the expression of key genes associated with fat deposition, namely UCP-1, ANGPTL4, Lpin2, and Lpin3 [3–5]. However, the fat deposition trait is a complex quantitative trait that is controlled by multiple genes. The mechanism responsible for the observed differences in fat deposition between the GLT and STH breed is not yet clear.
The burgeoning developments in the “omics” technologies, such as in trascriptomics, have dramatically changed the approach for investigating the mechanisms underlying metabolism, development, growth, disease resistance in various organisms. In sheep, the transcriptome profiles of tissues including heart, skin, muscle, mammary gland, and adipose tissue, have been determined by deep sequencing [6–11]. Miao et al. [6] analyzed the transcriptome information of subcutaneous adipose tissue between STH and Dorset sheep. Wang et al. [7] investigated the differences in the transcriptome profiles of tail fat tissue between Kazak and Tibetan sheep. It is known that adipose tissues from different anatomical locations are heterogeneous with respect to metabolic activities, functions, and genetic regulation [12–16]. In addition, animal species and breeds can also dictate the characteristics of a given adipose tissue depot [17]. Thus, to better understand the genetic regulation of fat deposition in sheep and to elucidate the differences in fat metabolism between the GLT and STH breed, we used perirenal, subcutaneous, and tail fat tissues from both the breeds to obtain a more comprehensive gene expression profile using RNA sequencing (RNA-seq) technology. We identified some genes that play important roles in fat metabolism. These data provide a valuable theoretical basis for selection of fat deposition trait in sheep breeding.
Methods
Adipose tissue collection
The indigenous Chinese sheep breeds, GLT and STH, were raised as described previously [3]. Four healthy 10-month-old male sheep from each breed were slaughtered. The adipose tissues in the perirenal (PEF), subcutaneous (SUF), and tail fat (TAF) were rapidly sampled after death. All the samples were immediately frozen in liquid nitrogen and stored at − 80 °C until subsequent use.
RNA preparation, cDNA library generation, and sequencing
Total RNA was extracted from the different adipose tissues using Trizol reagent (TaKaRa, USA) according to the manufacturer’s protocol. The concentration and integrity of RNA were evaluated using the 2100 Bioanalyzer (Agilent Technologies, Waldronn, Germany). All the samples had RNA Integrity Number values greater than 7.0 and 28S/18S ratio greater than 1.0. The RNA samples from four independent biological replicates (GLT = 4, STH = 4) for each tissue depot were pooled in equal quantities.
After DNase I treatment, poly (A) mRNA was isolated using oligo (dT) magnetic beads (Invitrogen, Carlsbad, CA, USA) and was fragmented into short fragments in the fragmentation buffer. The mRNA fragments were subsequently used as templates for the synthesis of the first-strand cDNA using random hexamer primers and reverse transcriptase. Following the second-strand cDNA generation, short fragments were added with poly (A) tails and ligated to adaptors. After agarose gel electrophoresis, suitable fragments were selected as templates for PCR amplification. Six paired-end cDNA libraries (three for each breed) were constructed. Finally, sequencing of the libraries was performed using Illumina HiSeq 2000 at the Beijing Genomics Institute (Shenzhen, China).
The raw reads were subjected to quality control through SOAPnuke tool, and low quality reads, including reads with adapters, reads in which unknown bases were more than 10%, and reads in which the percentage of the low quality bases (quality value ≤10) was more than 50%, were filtered. The clean reads, thus obtained, were then aligned against the reference genome and the reference genes of Ovis aries (Oar_v3.1, http://www.ncbi.nlm.nih.gov/assembly/GCF_000298735.1) with SOAPaligner/SOAP2 [18] and were annotated.
Gene expression analysis
The gene coverage and RPKM values were calculated to analyze the gene expression. The gene coverage value is equal to the ratio of the number of bases in a gene covered by the uniquely mapped reads to the total number of bases in that gene. The RPKM means numbers of reads per kilobase of exon model in a gene per million mapped reads [19]. The genes detected in all the six samples were referred to as “co-genes.” A heatmap showing the top recurrently expressed co-genes was generated based on log2 RPKM values using the OmicShare tools, a free online platform for data analysis (www.omicshare.com/tools).
The differentially expressed (DE) genes between the GLT and STH samples in pairwise-comparisons of PEF, SUF, and TAF were determined using the method described by Audic and Claverie [20]. The fold changes (log 2Ratio) were estimated according to the normalized gene expression level in each sample [21, 22]. The false discovery rate (FDR) ≤ 0.001 and the absolute value of log 2Ratio ≥ 1 were used as the threshold to judge the significant differences in gene expression.
Validation of gene expression using quantitative real-time PCR
The expression levels of eight DE genes related to fat deposition were validated using quantitative real-time PCR (qRT-PCR). These genes included FABP4 (fatty acid binding protein 4), SLC27A6 (solute carrier family 27 member 6), SCD (stearoyl-CoA desaturase), THRSP (thyroid hormone responsive protein), ALDH1A1 (aldehyde dehydrogenase 1 family member A1), PPT1 (palmitoyl-protein thioesterase 1), FABP5 (fatty acid binding protein 5), and GPI (glucose-6-phosphate isomerase). The ribosomal protein L 13 gene (RPL13A) was used as an internal control [4] because it showed consistent expression. The gene-specific primers were designed by Primer-BLAST available at the National Center for Biotechnology Information (NCBI) website; the sequences of the primers used are listed in Additional file 1. The first-strand cDNA was synthesized using the PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa, Dalian, China).
The qRT-PCR was carried out on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster, CA, USA) using SYBR Premix Ex Taq II kit (Takara, Dalian, China). The following conditions were used for the amplification: 95 °C for 10 min, followed by 45 cycles of 95 °C for 15 s, 60 °C for 1 min, and a melt curve stage of 95 °C for 45 s, 60 °C for 1 min, and 95 °C for 15 s. The relative expression level of each gene was estimated by the 2-ΔΔCT method [23]. The qRT-PCR analysis was performed in triplicate for each sample.
Analysis of DE genes
Gene Ontology (GO) analysis, which is used to describe the properties of genes in any organism, was employed to analyze the functions of the DE genes. All the DE genes were mapped to the GO terms in the database (http://www.geneontology.org/). The gene numbers for each term were calculated. Thereafter, a hypergeometric test was employed to find the significantly enriched GO terms for the DE genes. The calculated p-value was subjected to Bonferroni Correction [24], taking corrected P value ≤0.05 as the threshold for GO terms for all the DE genes.
The pathway-based analysis helps in the understanding of specific functions of the DE genes in certain biological process. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.kegg.jp/kegg/pathway.html), which is a major public pathway-related database [25], was used to perform pathway enrichment analysis of the DE genes. The method for calculation was the same as that used in the GO analysis. The P and Q values were returned after the pathway analysis. The pathways with Q value ≤0.05 were considered to be significantly enriched for the DE genes.
Results
Sequencing and mapping
We generated six cDNA sequencing libraries using PEF, SUF, and TAF from both the GLT and STH individuals. The libraries were sequenced, and six sets of reads were obtained. All the reads were 90 base pair in length. After filtering the low quality raw reads, a total of 47,445,516, 47,347,456, 47,381,754, 47,344,348, 46,953,510, and 47,047,532 clean reads were obtained from GLTPEF, GLTSUF, GLTTAF, STHPEF, STHSUF, and STHTAF samples, respectively (Table 1). About 76.17–79.43% clean reads were mapped to the sheep genome, and 55.76–63.13% reads were mapped to the known reference genes. Of all the clean reads, 52.46–57.27% reads were mapped perfectly to the sheep reference genome without any mismatch, 64.3–71.43% reads had unique matches, 7–11.86% reads showed multi-position matches, and the total number of unmapped reads was 20.57–23.84%. As for the known sheep reference genes, 42.5–47.45% of the clean reads had 100% match, 51.51–57.22% showed only one match, and the percentage of clean reads with multiple matches was 2.6–5.91%. Unfortunately, 36.87–44.24% reads could not be aligned to any of the reference genes.
Table 1.
Statistics of total reads and mapped reads
| Reads | GLPEF | GLTSUF | GLTTAF | STHPEF | STHSUF | STHTAF | |
|---|---|---|---|---|---|---|---|
| Total | Total reads | 47,445,516 | 47,347,456 | 47,381,754 | 47,344,348 | 46,953,510 | 47,047,532 |
| Total base pairs | 4,270,096,440 | 4,261,271,040 | 4,264,357,860 | 4,260,991,320 | 4,225,815,900 | 4,234,277,880 | |
| Matches to genome | Total mapped reads | 37,675,194 | 36,058,604 | 36,171,524 | 37,605,664 | 36,184,083 | 36,699,831 |
| Perfect match | 26,543,043 | 24,837,018 | 25,023,096 | 27,114,486 | 25,768,571 | 25,233,708 | |
| ≤ 5 bp mismatch | 11,132,151 | 11,221,586 | 11,148,428 | 10,491,178 | 10,415,512 | 11,466,123 | |
| Unique match | 32,990,142 | 30,442,820 | 30,831,328 | 33,818,446 | 32,895,221 | 31,906,341 | |
| Multi-position match | 4,685,052 | 5,615,784 | 5,340,196 | 3,787,218 | 3,288,862 | 4,793,490 | |
| Total unmapped reads | 9,770,322 | 11,288,852 | 11,210,230 | 9,738,684 | 10,769,427 | 10,347,701 | |
| Matches to genes | Total mapped reads | 26,559,610 | 26,402,159 | 27,534,197 | 26,776,917 | 28,230,599 | 29,701,546 |
| Perfect match | 20,476,006 | 20,123,960 | 20,965,259 | 20,683,554 | 21,641,557 | 22,323,002 | |
| ≤ 5 bp mismatch | 6,083,604 | 6,278,199 | 6,568,938 | 6,093,363 | 6,589,042 | 7,378,544 | |
| Unique match | 24,440,852 | 25,169,009 | 26,230,487 | 24,656,176 | 26,501,874 | 26,918,689 | |
| Multi-position match | 2,118,758 | 1,233,150 | 1,303,710 | 2,120,741 | 1,728,725 | 2,782,857 | |
| Total unmapped reads | 20,885,906 | 20,945,297 | 19,847,557 | 20,567,431 | 18,722,911 | 17,345,986 | |
General gene expression and annotation
The gene coverage was calculated as the percentage of a gene covered by the reads. Among the six libraries, approximately 60% of the reference genes had 90–100% coverage, and 10% or 11% of the annotated genes had 80–90% coverage (Additional file 2). Similar gene coverage was observed in all the RNA-seq libraries. A total of 17,267 annotated genes with RPKM > 0 were detected in all the tested tissues (Additional file 3). Interestingly, the number of genes found among all the samples was very similar with respect to the RPKM value (Fig. 1a). In all the six samples, 82.14% (14,183/17267) of these genes were detected, and 92.78% (16,021/17267) genes were common in both the GLT and STH breeds (Additional file 4). In terms of the breeds, 95.52% (16,493/17267) of the total genes were detected in GLT, and a similar percentage, of 96.15% (16,602/17267) of the total genes, was detected in STH. A total of 472 and 581 genes were exclusively detected in GLT and STH, respectively. In GLT sheep, 15,567, 15,527, and 15,498 genes were detected in SUF, PEF, and TAF, respectively, and 14,565 genes were found to be common in all the three tissues. Similarly, 15,833, 15,699, and 15,575 genes were examined in all the corresponding fat tissues of STH, respectively, and 14,817 genes were detected in all of them. A total of 235, 483, and 241 genes were discovered specifically in PEF, SUF, and TAF of GLT sheep, respectively (Fig. 1b). The number of genes that were uniquely expressed in the corresponding tissues of STH was 301, 378, and 235, respectively.
Fig. 1.
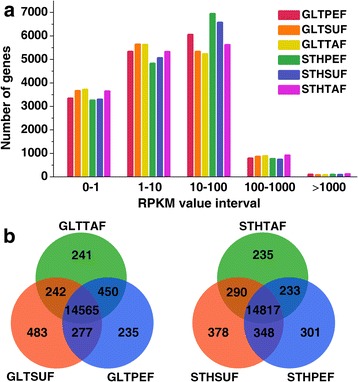
a The numbers of annotated genes with different expression levels against the range of RPKM values. b Venn diagrams of genes among different fat depots in GLT or STH sheep
Validation of RNA-seq results by qRT-PCR
To validate the RNA-seq results, eight DE genes related to fat deposition were selected for qRT-PCR analysis. We found that the expression patterns of these genes were consistent between the two methods. The Spearman’s correlation coefficient suggested that the data obtained from RNA-seq had a highly significant correlation with that obtained from qRT-PCR (Fig. 2). These results suggested that the expression profile determined by RNA-seq was reliable.
Fig. 2.
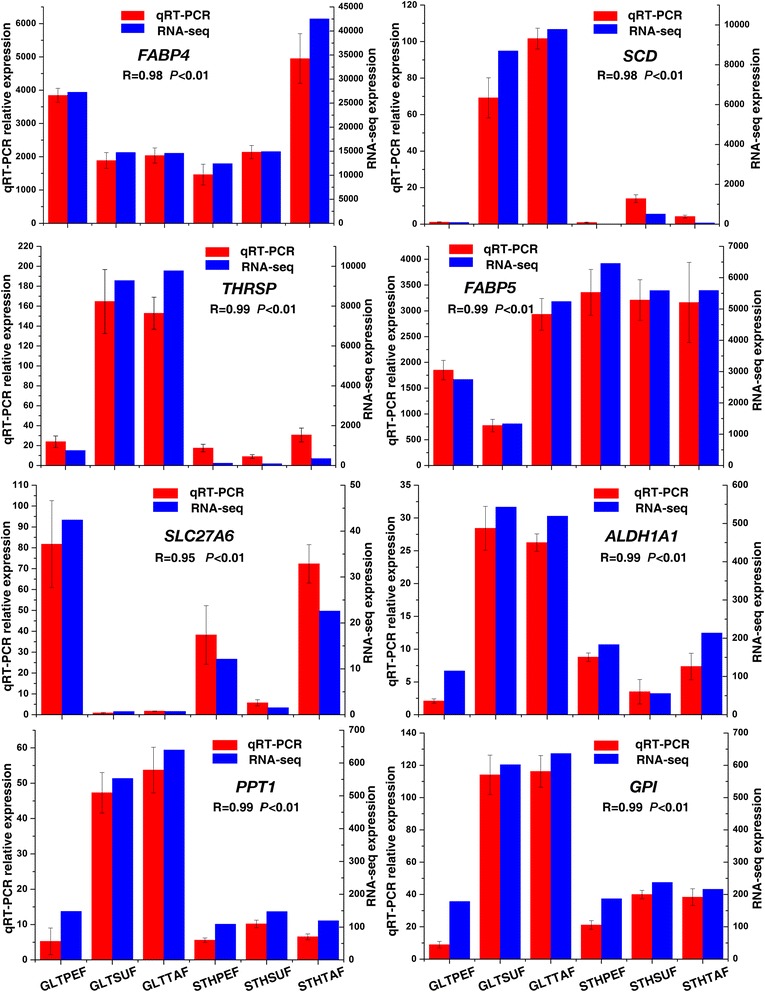
Expression levels of eight genes from qRT-PCR and RNA-seq. The X axis represents the different adipose tissues from both sheep breeds. The Y axis on the left represents the relative gene expression levels of qRT-PCR by columns and bars. The Y axis on the right represents the relative values of RPKM by lines
Analysis of highly expressed co-genes
To further illustrate the specific gene expression patterns in the adipose tissues, we selected 47 genes that recurrently appeared in the top 100 most-highly expressed co-genes in each sample, and grouped them into three major clusters based on the RPKM value (Additional file 5 and Fig. 3). The first group consisted of 28 genes, which encoded ribosomal proteins or ribosomal protein homologue (the FAU gene), as well as MYL, ZC3H10, and UBC. Several genes related to fat deposition, namely ADIPOQ, FABP5, CD36, and FABP4, formed the second group, along with other genes (FHC, FHC-pseudogene, UBA52, and RPLP1). Eight genes (ITM2B, UBB, TMSB10, TMSB, TMSB4X, LGALS1, VIM, and ACTG1) with diverse functions formed another group.
Fig. 3.
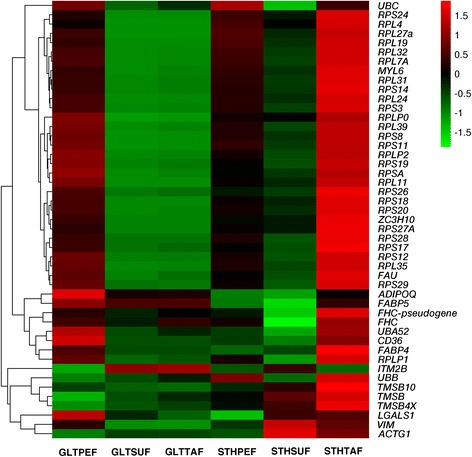
A heat-map showing the top 47 highly expressed co-genes. The cluster analysis of gene expression is based on log2 RPKM data. The red color represents higher expression and the green color represents lower expression
In the heatmap, different colors represent the different expression levels. The red color represents higher expression, and the green color represents lower expression. In the first group, all the genes except UBC showed similar differential expression pattern between the two breeds (GLTPEF vs. STHPEF, GLTSUF vs. STHSUF, and GLTTAF vs. STHTAF). Especially in TAF, the ribosomal protein genes had stronger expression in STH than in GLT. Most of the genes from the other two groups exhibited a higher level of expression in STHTAF than in GLTTAF. FABP4 had the highest mRNA expression level in all the tested fat tissues. There was no obvious difference in the expression in STHTAF vs. GLTTAF for ADIPOQ and FABP5.
Expression of DE genes in GLT and STH sheep
To gain a global perspective on the differences in gene expression in the GLT and STH breeds, we performed the following pairwise comparisons: GLTPEF vs. STHPEF, GLTSUF vs. STHSUF, and GLTTAF vs. STHTAF. The DE genes were screened using the filtering criteria for the FDR value ≤0.001 and the absolute value of log 2Ratio ≥ 1 (Additional file 6). In the comparisons of PEF, SUF, and TAF for both the breeds, we found 1917 up-regulated and 717 down-regulated genes, 3895 up-regulated and 1606 down-regulated genes, and 3160 up-regulated and 2235 down-regulated genes, respectively (Fig. 4).
Fig. 4.
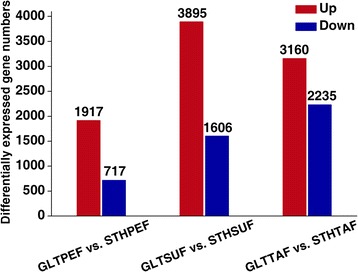
The number of up or down regulated genes in STH as compared to GLT sheep
The DE genes were classified by GO enrichment based on cellular component, molecular function, and biological process. The results of the selected significant (corrected p-value ≤0.05) GO annotation are presented in Additional file 7. Among the variety of cellular component terms, the category of extracellular matrix or extracellular region part was significantly enriched in all the three comparisons. For molecular function, binding related terms were the abundant categories. The term “lipid metabolic process” was also involved in the SUF and TAF comparisons. The top enriched biological process terms are represented in Fig. 5.
Fig. 5.

The significantly enriched GO biological process terms of DE genes
To identify the biological pathways that were involved in fat deposition, the DE genes from the three comparisons were mapped to the KEGG pathway database (Additional file 8). The pathways with the Q value ≤0.05 were thought to be significantly enriched. Several pathways related to lipid metabolism were found. In the PEF comparison, the PPAR signaling pathway (33 genes, Q = 0.002916), glycerolipid metabolism (23 genes, Q = 0.006579), Type II diabetes mellitus (20 genes, Q = 0.01202), and adipocytokine signaling pathway (25 genes, Q = 0.03863) were identified. The terms, ECM–receptor interaction (47 genes, Q = 0.00009957274), cell adhesion molecules (59 genes, Q = 0.000004805), and valine, leucine, and isoleucine degradation (18 genes, Q = 0.004779), were also significantly enriched, suggesting that these processes might contribute to fat deposition or fatty acid metabolism.
In GLTSUF vs. STHSUF, the significantly enriched pathways contained the PPAR signaling pathway (55 genes, Q = 0.003290), valine, leucine, and isoleucine degradation (33 genes, Q = 0.009917), Type II diabetes mellitus (33 genes, Q = 0.01313), lipoic acid metabolism (7 genes, Q = 0.04033), and ECM–receptor interaction (68 genes, Q = 0.01587673) pathways. For TAF comparison, the significant pathways did not include the pathways related to fat metabolism; however, Insulin signaling pathway (74 genes, Q = 0.07256), PPAR signaling pathway (49 genes, Q = 0.08449), and ECM–receptor interaction (62 genes, Q = 0.1917799) pathways were among the top enriched pathways.
Together, the PPAR signaling pathway and ECM–receptor interaction were identified as the common pathways with top rank in all the three comparisons. Therefore, we focused on these two pathways for further characterization. The DE genes enriched in the both pathways are listed in Table 2. In the PPAR signaling pathway, a total of nine DE genes, including four up-regulated genes (LOC101102230, PLTP, C1QTNF7, and OLR1) and two down-regulated genes (SCD and UCP-1) were commonly annotated in all the three comparisons (Table 3).
Table 2.
The annotated differentially expressed genes enriched in the PPAR signaling pathway or ECM–receptor interaction
| Pathway | Up-down regulation | GLTPEF vs. STHPEF | GLTSUF vs. STHSUF | GLTTAF vs. STHTAF |
|---|---|---|---|---|
| PPAR signaling pathway | Up | LOC101114861, LOC101102230, CRABP1, PLTP, SCD5, FADS2, ACSBG2, OLR1, CPT1C, C1QTNF3, COL8A1, C1QTNF7, LOC101116326, CBLN3, LOC101114586, PDLIM2 | LOC101102230, LOC101113077, SLC27A1, SLC27A5, SLC27A6, PLTP, OLR1, CPT1A, CPT1B, ANGPTL4, COL8A1, C1QTNF3, COL8A2, C1QTNF7, CBLN3, CAPRIN2, C1QTNF5, C1QTNF2, NEBL, PDLIM2, SORBS3 | LOC101102230, LOC101113339, LOC101113077, SLC27A6, SLC27A5, PLTP, SCD5, LOC101102230, OLR1, CPT1B, CPT1C, CPT1A, ANGPTL4, PLIN2, FABP4, C1QTNF7, LOC101116326, CAPRIN2, C1QTNF5, LOC101114586, NEBL, PDLIM2 |
| Down | LOC101113339, FABP4, FABP5, SLC27A6, RXRG, SCD, ANGPTL4, PLIN1, PLIN5, ADIPOQ, UCP1 | FABP5, CRABP2, CRABP1, SLC27A2, RXRA, SCD, FADS2, CYB5A, ACSL6, ACSL5, ACSL1, ACSBG1, PLIN2, PLIN5, PLIN3, ADIPOQ, UCP1 | FABP12, CRABP2, RXRG, RXRA, SCD, FADS2, CYB5A, ACSL6, ACSL1, ACSL5, ACSL3, LOC101117914, SORBS1, UCP1 | |
| ECM-receptor interaction | Up | COL4A6, COL4A5, COL4A3, COL23A1, ARID5A, COL1A1, COL13A1, COL1A2, LAMB3, MEGF9, LAMA5, RELN, THSD1, ISM1, FN1, ITGA5, TNC, ANGPTL1, TNXB, TENM3, ITGA8, ITGA9, ITGA11, ZAN, FCGBP, ITGBL1, ITGB4 | COL6A5, COL6A6, COL6A1, COL6A3, COL6A2, PIMREG, KRBA1, TNKS1BP1, ARID5A, EHBP1L1, AHNAK, LAMB3, LAMA2, LAMB1, RELN, THBS1, COMP, THBS2, THBS3, THBS4, THSD1, FN1, ITGA5, SDK2, ANGPTL6, ANGPTL1, TNXB, TNC, IGDCC4, ITGA8, ITGA9, ITGA11, ITGB3, ITGBL1, ITGA2, ITGA4, SPP1, PRG4, ITGB7, ITGB5 | COL9A3, COL2A1, COL13A1, COL11A2, TNKS1BP1,COL6A5, COL23A1, EHBP1L1, LAMB3, LAMB1, LAMA2, RELN, THBS2, THBS1, SCARF1, TNXB, ITGA8, VWF, ITGA2, SPP1, VTN, TGB7, ITGB5 |
| Down | WFIKKN2, WAP, PIMREG, LAMB4, COMP | COL24A1, GRTP1, COL11A1, LOC101117707, LAMB4, SSPO, C5H19orf70, ITGA3 | COL24A1, PIMREG, GRTP1, COL5A1, COL3A1, COL5A3, COL5A2, COL11A1, COL1A2, COL1A1, LAMC2, LAMB4, LOC101117707, MEGF9, ATRN, SM1, LOC101120408, TENM4, TENM1, ITGA1, ITGA6, TGAV, ITGB8 |
The differentially expressed genes (DE genes) were generated from comparisons, GLTPEF vs. STHPEF, GLTSUF vs. STHSUF, and GLTTAF vs. STHTAF. The PPAR signaling pathway and ECM–receptor interaction were identified through KEGG pathway analysis. “Up-down regulation” means that the genes were up- or down-regulated in STH as compared to their expression levels in GLT sheep
Table 3.
The common differentially expressed genes enriched in PPAR signaling pathway or ECM–receptor interaction
| Pathway | Gene information from NCBI website | Expression ration | ||||
|---|---|---|---|---|---|---|
| Gene ID | Gene name | Gene annotation | STHPEF/GLTPEF | STHSUF/GLTSUF | STHTAF/GLTTAF | |
| PPAR signaling pathway | 101,102,230 | LOC101102230 | platelet glycoprotein 4-like, FATCD36 | Up | Up | Up |
| 101,115,982 | PLTP | phospholipid transfer protein | Up | Up | Up | |
| 101,117,918 | OLR1 | oxidized low density lipoprotein receptor 1 | Up | Up | Up | |
| 101,105,159 | C1QTNF7 | C1q and tumor necrosis factor related protein 7 | Up | Up | Up | |
| 101,104,743 | SLC27A6 | solute carrier family 27 member 6 | Down | Up | Up | |
| 101,104,317 | ANGPTL4 | angiopoietin like 4 | Down | Up | Up | |
| 101,108,615 | FADS2 | fatty acid desaturase 2 | Up | Down | Down | |
| 443,185 | SCD | stearoyl-CoA desaturase | Down | Down | Down | |
| 494,434 | UCP-1 | uncoupling protein 1 | Down | Down | Down | |
| ECM-receptor interaction | 101,105,176 | LAMB3 | laminin subunit beta 3 | UP | UP | UP |
| 101,120,658 | RELN | reelin | UP | UP | UP | |
| 100,820,740 | TNXB | tenascin XB | UP | UP | UP | |
| 101,117,689 | ITGA8 | integrin subunit alpha 8 | UP | UP | UP | |
| 101,106,520 | LAMB4 | laminin subunit beta 4 | Down | Down | Down | |
The differentially expressed (DE) genes listed in this table were common in the comparisons, GLTPEF vs. STHPEF, GLTSUF vs. STHSUF, and GLTTAF vs. STHTAF. The PPAR signaling pathway and ECM–receptor interaction were enriched through KEGG pathway analysis. The gene expression level was evaluated by RPKM. The ratio was defined as the RPKM value of a gene in one tissue of STH to that in the corresponding tissue of GLT
In the ECM–receptor interaction, the collagen genes (for example, COL4A6, COL6A5, and COL9A3), laminin genes (for example, LAMB3, LAMB4, and LAMA2), and Integrin genes (for example, ITGA5, ITGA9, and ITGA1) were differentially expressed between the two breeds (Table 2). The Collagen IV genes (COL4A6, COL4A5, and COL4A3) were up-regulated in GLTPEF. The Collagen VI genes (COL6A5, COL6A6, COL6A1, COL6A3, and COL6A2) were up-regulated in GLTSUF. However, Collagen V genes (COL5A1, COL5A3, and COL5A2) were up-regulated in STHTAF. Among the common genes involved in the ECM–receptor interaction, five genes (LAMB3, RELN, TNXB, and ITGA8) were differentially up-regulated in STH, and LAMB4 was down-regulated (Table 3).
Discussion
The mechanisms of fat metabolism are complex, and manipulation of fat deposition for meat production is very important in sheep breeding. Crossing of sheep breeds with different patterns of fat deposition is one of the agronomical strategies for improving the meat quality. Previous studies have investigated the differences in gene expression patterns or molecular genetic mechanisms between characteristically different sheep breeds [3, 5, 6, 26–28]. To investigate the genetic profiles of fat tissues and to understand the differences in the genetic mechanisms determining fat deposition between distinct breeds, we characterized the transcriptome of PEF, SUF, and TAF from GLT and STH sheep breeds.
The qRT-PCR analysis showed that the transcriptome profiles determined by RNA-seq were reliable. The percentage of reads that could be mapped to the reference genome of sheep was comparable to that reported for pigs [29, 30] and cattle [31, 32], where 60.2–78.3% of the reads could be mapped to the respective reference genomes. The percentage of reads mapped to the reference genes were lower than that mapped to the sheep genome, owing to the GC content, type of cells, and for other reasons, as described earlier [33, 34]. It should be noted that the percentage of total reads not mapped to the reference genome (20.57–23.84%) or to the reference genes (36.87–44.24%) was not low. This could be caused by imperfections in the reference genome, reference errors, sequencing errors, and the defined mapping criteria [35].
The identification of co-genes transcribed in all the tested fat tissues and the genes that were differentially expressed between the two breeds not only reveals the possible mechanism controlling fat deposition in sheep and the possible novel functions of known genes, but also provides valuable information for understanding the phenotypic and functional differences in the deposition of fat in livestock.
Top highly expressed candidate genes for fat deposition in sheep
We found four candidate genes for fat deposition, namely FABP4, CD36, FABP5, and ADIPOQ, which were abundantly expressed in the fat tissues of sheep. FABP4 is thought to play roles in fatty acid transport and fat deposition in animals as well as in human metabolic syndrome [36]. Previous studies have shown that FABP4 is involved in fat accumulation in cattle [37] and in determining the tenderness of meat in sheep [38]. CD36 can bind long chain fatty acids and plays an important role in the absorption and storage of dietary lipids [39, 40]. FABP5 has similar roles as FABP4 and could compensate for the loss of FABP4 in adipocytes [41]. ADIPOQ (Adiponectin), an important adipocytokine that is secreted by adipocytes, modulates the regulation of glucose and fatty acid oxidation [42–44]. Polymorphisms in ADIPOQ have been suggested to be associated with fat deposition and carcass traits in pigs [45], and with meat marbling in cattle [46, 47]. The analysis of the Sheep Quantitative Trait Locus (QTL) Database (Sheep QTLdb: https://www.animalgenome.org/cgi-bin/QTLdb/OA/index) showed that the chromosomal location of ADIPOQ is within the QTL region for “carcass fat percentage.” This suggested that ADIPOQ also might be associated with the sheep fatness trait. Strong transcription of these four genes observed in the present study confirms their extreme importance in adipose deposition in sheep.
FHC (Ferritin heavy chain) is one of the subunits of Ferritin, a ubiquitous intracellular protein that stores iron [48]. Previous studies have reported the up-regulation of FHC by adiponectin in skeletal muscle cells [49] and a consistent increase in FHC expression during the differentiation of 3 T3-L1 preadipocytes [50]. Our results suggest that FHC and FHC pseudogene express abundantly in sheep fat tissues, and the two genes were found to cluster together with other genes related to fat deposition. Genes with similar expression pattern could be grouped together through clustering analysis, suggesting that such genes might have similar functions. Therefore, we infer that the two FHC related genes are closely associated with fat cell activity. Another candidate gene for fat deposition, ZC3H10 (Zinc Finger CCCH-Type Containing 10), was reported to be down-regulated in breast cancer cells [51] and is expected to have a tumor suppressor function [52]. The finding on the chromosomal location of ZC3H10 within the QTL region for “internal fat amount” obtained from the Sheep QTLdb indicated its association with fat deposition in sheep. However, its role in adipocytes has not been studied. Further investigation is required to determine whether or not these genes are involved in adipocyte homeostasis.
Several DE genes are responsible for fat deposition in sheep
The PPAR signaling pathway regulates cellular differentiation, energy balance, and lipid metabolism [53]. In the present study, multiple genes enriched in PPAR signaling pathway were found to be differentially regulated in the tissues of the two sheep breeds (Table 3). PLTP, OLR1, C1QTNF7, and LOC101102230 were up-regulated in the STH breed. It is known that PLTP (phospholipid transfer protein) is involved in transferring surface lipids from triglyceride-rich lipoproteins to HDL during lipolysis [54]. OLR1 (Oxidized low-density lipoprotein receptor 1) is expressed in multiple cell types, including adipocytes [55]. It can bind to and degrade oxidized low-density lipoproteins. Recent studies have demonstrated that OLR1 is closely linked to obesity [56], and it was found to be highly expressed in fat pigs compared to lean animals [57]. There are evidences that show that SNPs in PLTP [58] or OLR1 [59, 60] are significantly associated with economic traits, such as marbling score, in beef. C1QTNF7 (C1q and tumor necrosis factor related protein 7), also called CTRP7, shares a high structural similarity with adiponectin. It is assumed that C1QTNF7 mimics the effects of adiponectin and is activated by PPAR. The C1QTNF7 transcripts were found to be expressed predominantly in the adipose tissue [61], and deletion of C1QTNF7 attenuated obesity-linked glucose intolerance, adipose tissue inflammation, and hepatic stress [62]. The mRNA sequence of LOC101102230 (Accession No.: XM_012176565) showed 91% identity with the sequence of sheep CD36 (Accession: XM_012176587), suggesting that LOC101102230 might exert roles similar to those of CD36 in lipid absorption. At present, little is known about LOC101102230, and further investigation should be conducted to explore its possible role in fat tissues. Both SCD and UCP-1 were up-regulated in GLT sheep. SCD (Stearoyl-CoA desaturase) is a key enzyme that catalyzes a rate-limiting step in the synthesis of unsaturated fatty acids [63]. The elevated SCD activity is positively correlated with increased fat accumulation and monounsaturation of saturated fatty acids [64]. Thus, SCD is considered to be a principle gene in animal breeding that can improve the meat quality by modulating the fat deposition and saturated fatty acid content. The association of the SNPs in SCD and meat quality traits in beef [64], pig [65], and sheep [26] has been reported extensively. In this study, much higher mRNA concentrations of SCD were detected in SUF than in TAF for both the sheep breeds. The expression pattern of SCD observed in this study suggests lower content of saturated fatty acids in GLT compared to that in STH. UCP1 (the uncoupling protein 1) is found in brown adipose tissues and plays a pivotal role in thermogenesis and in the regulation of lipolysis [66]. The polymorphisms in UCP1 were found to be associated with milk quality in dairy cows [67] or with carcass traits in sheep [68]. Large tail shape and high mount fat mass present in GLT sheep could partially contribute to the higher expression levels of SCD and UCP1.
The other genes, such as ANGPTL4, FASD2, and SLC27A6, also behave differently between GLT and STH. The ANGPTL4 (angiopoietin like 4) gene showed a dramatic increase in transcription during and after adipocyte differentiation [69]. Recent research suggests that ANGPTL4 reduces the LPL protein in adipocytes by promoting its intracellular degradation [70]. The delta-6 desaturase encoded by FASD2 (fatty acid desaturase 2) is one of the important enzymes in the endogenous formation of long-chain polyunsaturated fatty acids. Genetic variation in human FADS2 was associated with the activity of the desaturation–elongation pathway, whole-body fat oxidation [71], and inflammation of adipose tissue [72]. The SNPs of ANGPTL4 [73] or FADS2 [74] showed significant effects on the quality of pig meat. SLC27A6 (the solute carrier family 27A) is involved in the translocation of long-chain fatty acids across the plasma membrane [75]. A recent study showed that there was significant association of SLC27A6 polymorphisms with milk quality [76]. ANGPTL4, FASD2, and SLC27A6 might play roles in the differences observed in fat metabolism between the two sheep breeds.
Roles of extracellular matrix in adipose tissues
In adipose tissue, adipocytes are embedded in the extracellular matrix (ECM) network, which predominantly consists of collagen [77]. The ECM provides structural support and anchorage for adipocytes and regulates adipogenesis. Some of the genes that were differentially expressed between GLT and STH sheep were enriched in the ECM–receptor interaction pathway. Different collagens make up the different ECM components; for example, collagen 1, 3, and 5 form the fibrils, collagen 6 forms the microfibril, and collagen 4 forms the basal membrane. Collagen 1 and 3 are secreted by preadipocytes during the early stage of adipocyte differentiation. The levels of collagen 4, 5, and 6 peak at the middle stage of adipocyte differentiation [78]. In the present research, the differential expression of COL4 in PEF, COL6 in SUF, and COL5 in TAF suggests structural distinction of ECM or adipogenetic difference based on the position of the fat depots. Some minor collagen genes, including collagen 2, 11, 13, 23, and 24, were also expressed differentially, which also indicates the complicated histological differences in the adipose tissues. The different expression of other ECM-related genes, like LAMB3, LAMB4, ITGA8, RELN, and TNXB, also shows the difference in the ECM structure or adipogenic signaling between GLT and STH breeds.
Conclusions
This study provides a global view of the transcriptome based on three different fat tissue depots from two fat-tailed sheep breeds. The highly transcribed co-genes were identified and the DE genes between these two breeds were analyzed through GO and KEGG database. Our data showed that FABP4, ADIPOQ, FABP5, and CD36 were related to fat deposition, and were transcribed at very high levels. Nine fat deposition related genes (LOC101102230, PLTP, C1QTNF7, OLR1, SCD, UCP-1, ANGPTL4, FASD2, and SLC27A6) and five ECM related genes (LAMB3, RELN, TNXB, ITGA8, and LAMB4) might be responsible for the difference in fat deposition between the GLT and STH sheep breeds. Although the RNA samples used for sequencing were obtained by pooling equal amounts of RNA from four independent biological replicates, the qRT-PCR validation confirmed the reliability of our results. The deep sequencing results might need to be validated using more biological replicates. Further studies are required to investigate the roles of the candidate genes in fat deposition for improvement of sheep breeding programs.
Additional files
Table S1. Primers used in qRT-PCR. (XLS 19 kb)
Figure S1. Gene coverage distribution of the six adipose tissue transcriptomes. Gene coverage is calculated as the percentage of a gene covered by reads. This value is equal to the ratio of the base number in a gene covered by unique mapping reads to the total base number of coding region in that gene. Pies with different colors represent proportions of genes with certain coverage. For example, green pie is indicating proportion of genes with coverage between 90 and 100%. (TIF 7235 kb)
Table S2. All the annotated genes in the six libraries. Unique reads are reads aligned to only one position in the reference sequence. Gene coverage and RPKM values were calculated to analyze gene expression. (XLSX 3230 kb)
Table S3. Summary of the number of genes (RPKM> 0) detected in the six adipose tissues of two breeds. (XLS 20 kb)
Table S4. The most highly expressed 47 genes and their RPKM values in the six samples. (XLS 24 kb)
Table S5. DE genes with the FDR value≤0.001 and the absolute value of log 2 Ratio ≥ 1. Gene Length, length of all exon in gene; Expression, reads number that uniquely mapped to gene; P value, p value for hypothesis testing. (XLS 2504 kb)
Table S6. GO analysis of DE genes with corrected P value ≤0.05. The letter “n” is number of DE genes which are annotated to this ontology. The letter “m” is number of DE genes which are annotated to this term. (XLS 789 kb)
Table S7. KEGG pathway analysis of DE genes with Q value ≤1. Significantly enriched biological pathways were identified in DE genes based on the entire genome background. Q value is a P value that has been adjusted for the FDR value. Pathways with Q value ≤0.05 are thought to be significantly enriched. (XLS 105 kb)
Acknowledgments
The authors thank the staff of the Laboratory of Animal Genetics, Breeding and Reproduction, Shanxi Agricultural University for providing the research laboratories.
Funding
The work was supported by the projects of the Chinese National Natural Science Foundation (31372292) and the Program for the Top Innovative Talents of Shanxi Agricultural University (BJRC201203).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files. The transcriptome data is freely available at the National Center for Biotechnology Information (NCBI) database Short Read Archive (Accession number SRP113440).
Abbreviations
- Co-genes
Common highly expressed genes
- DE genes
Differentially expressed genes
- GLT
Guangling large tailed
- GO
Gene ontology
- KEGG
Kyoto encyclopedia of genes and genomes
- PEF
Perirenal fat
- qRT-PCR
Quantitative real-time PCR
- RNA-seq
RNA sequencing
- RPKM
Reads per kilobase of exon model in a gene per million mapped reads
- STH
Small tailed han
- SUF
Subcutaneous fat
- TAF
Tail fat
Authors’ contributions
WZL and LYQ designed the study. LYQ, WWW and JHL performed sample collection and total RNA preparation. BJL and LXA performed the qRT-PCR validation. BJL analyzed the RNA-Seq data with the assistance of YYP and JJJ. BJL prepared the manuscript with contribution from WZL and YSR. All authors read and approved the final manuscript.
Ethics approval
The animal use procedures were approved by the Institutional Animal Care and Ethics Committee of Shanxi Agricultural University, China, and conducted according to the National (GB13078–2001 and GB/T17237–1998) and the Agricultural Standards (NY 5148–2002) of the People’s Republic of China.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4747-1) contains supplementary material, which is available to authorized users.
Contributor Information
Baojun Li, Email: bjlee0520@aliyun.com.
Liying Qiao, Email: liyingqiao1970@163.com.
Lixia An, Email: an-li-xia@163.com.
Weiwei Wang, Email: sxndweiwei@163.com.
Jianhua Liu, Email: ljhbeth@163.com.
Youshe Ren, Email: rys925@126.com.
Yangyang Pan, Email: pyybiology@163.com.
Jiongjie Jing, Email: 445776281@qq.com.
Wenzhong Liu, Email: tglwzyc@163.com.
References
- 1.Davidson A. The Oxford companion to food. Oxford: Oxford University Press; 1999. pp. 290–3. [Google Scholar]
- 2.Kashan NEJ, Azar GHM, Afzalzadeh A, Salehi A. Growth performance and carcass quality of fattening lambs from fat-tailed and tailed sheep breeds. Small Rumin Res. 2005;60(3):267–271. doi: 10.1016/j.smallrumres.2005.01.001. [DOI] [Google Scholar]
- 3.Yuan YN, Liu WZ, Liu JH, Qiao LY, Wu JL. Cloning and ontogenetic expression of the uncoupling protein 1 gene UCP1 in sheep. J Appl Genet. 2012;53(2):203–212. doi: 10.1007/s13353-012-0086-0. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Jing JJ, Jia XL, Qiao LY, Liu JH, Liang C, Liu WZ. mRNA expression of ovine angiopoietin-like protein 4 gene in adipose tissues. Asian-Australas J Anim Sci. 2016;29(5):615–623. doi: 10.5713/ajas.15.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiao X-L, Jing J-J, Qiao L-Y, Liu J-H, Li L-A, Zhang J, Jia X-L, Liu W-Z. Ontogenetic expression of Lpin2 and Lpin3 genes and their associations with traits in two breeds of Chinese fat-tailed sheep. Asian Australas J Anim Sci. 2016;29(3):333–342. doi: 10.5713/ajas.15.0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miao X, Luo Q, Qin X, Guo Y, Zhao H. Genome-wide mRNA-seq profiling reveals predominant down-regulation of lipid metabolic processes in adipose tissues of small tail Han than Dorset sheep. Biochem Biophys Res Commun. 2015;467(2):413–420. doi: 10.1016/j.bbrc.2015.09.129. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Zhou G, Xu X, Geng R, Zhou J, Yang Y, Yang Z, Chen Y. Transcriptome profile analysis of adipose tissues from fat and short-tailed sheep. Gene. 2014;549(2):252–257. doi: 10.1016/j.gene.2014.07.072. [DOI] [PubMed] [Google Scholar]
- 8.Cox LA, Glenn JP, Spradling KD, Nijland MJ, Garcia R, Nathanielsz PW, Ford SP. A genome resource to address mechanisms of developmental programming: determination of the fetal sheep heart transcriptome. J Physiol. 2012;590(12):2873–2884. doi: 10.1113/jphysiol.2011.222398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu N, He JN, Yu WM, Liu KD, Cheng M, Liu JF, He YH, Zhao JS, Qu XX. Transcriptome analysis of skeletal muscle at prenatal stages in polled Dorset versus small-tailed Han sheep. Genet Mol Res. 2015;14:1085–1095. doi: 10.4238/2015.February.6.12. [DOI] [PubMed] [Google Scholar]
- 10.Yue YJ, Liu JB, Yang M, Han JL, Guo TT, Guo J, Feng RL, Yang BH. De novo assembly and characterization of skin transcriptome using RNAseq in sheep (Ovis aries) Genet Mol Res. 2015;14(1):1371–1384. doi: 10.4238/2015.February.13.16. [DOI] [PubMed] [Google Scholar]
- 11.Suarez-Vega A, Gutierrez-Gil B, Klopp C, Tosser-Klopp G, Arranz JJ. Comprehensive RNA-Seq profiling to evaluate lactating sheep mammary gland transcriptome. Sci Data. 2016;3:160051. doi: 10.1038/sdata.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD, Kirkland JL. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. 2013;17(5):644–656. doi: 10.1016/j.cmet.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samulin J, Lien S, Grindflek E, Berget I, Ruyter B, Sundvold H. Depot specific differences during adipogenesis of porcine stromal-vascular cells. Cell Biol Int. 2008;32(5):525–531. doi: 10.1016/j.cellbi.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Ortiz-Colon G, Grant AC, Doumit ME, Buskirk DD. Bovine intramuscular, subcutaneous, and perirenal stromal-vascular cells express similar glucocorticoid receptor isoforms, but exhibit different adipogenic capacity. J Anim Sci. 2009;87(6):1913–1920. doi: 10.2527/jas.2008-1350. [DOI] [PubMed] [Google Scholar]
- 15.Gondret F, Guitton N, Guillerm-Regost C, Louveau I. Regional differences in porcine adipocytes isolated from skeletal muscle and adipose tissues as identified by a proteomic approach. J Anim Sci. 2008;86(9):2115–2125. doi: 10.2527/jas.2007-0750. [DOI] [PubMed] [Google Scholar]
- 16.Tchoukalova YD, Votruba SB, Tchkonia T, Giorgadze N, Kirkland JL, Jensen MD. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc Natl Acad Sci U S A. 2010;107(42):18226–18231. doi: 10.1073/pnas.1005259107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hausman GJ, Basu U, Wei S, Hausman DB, Dodson MV. Preadipocyte and adipose tissue differentiation in meat animals: influence of species and anatomical location. Annu Rev Anim Biosci. 2014;2:323–351. doi: 10.1146/annurev-animal-022513-114211. [DOI] [PubMed] [Google Scholar]
- 18.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 19.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 20.Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- 21.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Statist. 2001;29(4):1165–1188. doi: 10.1214/aos/1013699998. [DOI] [Google Scholar]
- 22.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Abdi H. Encyclopedia of Measurement and Statistics. Edited by Salkind NJ. Thousand Oaks: Sage; 2007. Bonferroni and Sidak corrections for multiple comparisons; pp. 103–107. [Google Scholar]
- 25.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aali M, Shahrbabak MM, Shahrbabak HM, Sadeghi M. Identifying novel SNPs and allelic sequences of the Stearoyl-CoA desaturase gene (SCD) in fat-tailed and thin-tailed sheep breeds. Biochem Genet. 2014;52(3):153–158. doi: 10.1007/s10528-013-9635-4. [DOI] [PubMed] [Google Scholar]
- 27.Xu XC, Li BB, Wei X, Yang YX, Wang XL, Chen YL. Differential expression of peroxisome proliferator-activated receptor gamma, fatty acid synthase, and hormone-sensitive lipase in fat-tailed and thin-tailed sheep breeds. Genet Mol Res. 2015;14(4):15624–15633. doi: 10.4238/2015.December.1.14. [DOI] [PubMed] [Google Scholar]
- 28.Wu JL, Liu WZ, Liu JH, Qiao LY, Yuan YN. Distribution and quantification of beta-3 adrenergic receptor in tissues of sheep. Animal. 2011;5(1):88–93. doi: 10.1017/S1751731110001564. [DOI] [PubMed] [Google Scholar]
- 29.Wang T, Jiang A, Guo Y, Tan Y, Tang G, Mai M, Liu H, Xiao J, Li M, Li X. Deep sequencing of the transcriptome reveals inflammatory features of porcine visceral adipose tissue. Int J Biol Sci. 2013;9(6):550–556. doi: 10.7150/ijbs.6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li B, Dong C, Li P, Ren Z, Wang H, Yu F, Ning C, Liu K, Wei W, Huang R, et al. Identification of candidate genes associated with porcine meat color traits by genome-wide transcriptome analysis. Sci Rep. 2016;6:35224. doi: 10.1038/srep35224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang W, Nadeem A, Zhang B, Babar M, Soller M, Khatib H. Characterization and comparison of the leukocyte transcriptomes of three cattle breeds. PLoS One. 2012;7(1):e30244. doi: 10.1371/journal.pone.0030244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee EJ, Malik A, Pokharel S, Ahmad S, Mir BA, Cho KH, Kim J, Kong JC, Lee DM, Chung KY, et al. Identification of genes differentially expressed in myogenin knock-down bovine muscle satellite cells during differentiation through RNA sequencing analysis. PLoS One. 2014;9(3):e92447. doi: 10.1371/journal.pone.0092447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McIntyre LM, Lopiano KK, Morse AM, Amin V, Oberg AL, Young LJ, Nuzhdin SV. RNA-seq: technical variability and sampling. BMC Genomics. 2011;12:293. doi: 10.1186/1471-2164-12-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sendler E, Johnson GD, Krawetz SA. Local and global factors affecting RNA sequencing analysis. Anal Biochem. 2011;419(2):317–322. doi: 10.1016/j.ab.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 35.Chen C, Ai H, Ren J, Li W, Li P, Qiao R, Ouyang J, Yang M, Ma J, Huang L. A global view of porcine transcriptome in three tissues from a full-sib pair with extreme phenotypes in growth and fat deposition by paired-end RNA sequencing. BMC Genomics. 2011;12:448. doi: 10.1186/1471-2164-12-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei S, Zan LS, Wang HB, Cheng G, Du M, Jiang Z, Hausman GJ, DC MF, Dodson MV. Adenovirus-mediated interference of FABP4 regulates mRNA expression of ADIPOQ, LEP and LEPR in bovine adipocytes. Genet Mol Res. 2013;12(1):494–505. doi: 10.4238/2013.January.4.21. [DOI] [PubMed] [Google Scholar]
- 37.Aviles C, Polvillo O, Pena F, Juarez M, Martinez AL, Molina A. Associations between DGAT1, FABP4, LEP, RORC, and SCD1 gene polymorphisms and fat deposition in Spanish commercial beef. J Anim Sci. 2013;91(10):4571–4577. doi: 10.2527/jas.2013-6402. [DOI] [PubMed] [Google Scholar]
- 38.Xu QL, Tang GW, Zhang QL, Huang YK, Liu YX, Quan K, Zhu KY, Zhang CX. The FABP4 gene polymorphism is associated with meat tenderness in three Chinese native sheep breeds. Czech Journal of Animal Science. 2011;56(1):1–6. doi: 10.17221/231/2009-CJAS. [DOI] [Google Scholar]
- 39.Bokor S, Legry V, Meirhaeghe A, Ruiz JR, Mauro B, Widhalm K, Manios Y, Amouyel P, Moreno LA, Molnar D, et al. Single-nucleotide polymorphism of CD36 locus and obesity in European adolescents. Obesity (Silver Spring) 2010;18(7):1398–1403. doi: 10.1038/oby.2009.412. [DOI] [PubMed] [Google Scholar]
- 40.Zhang XJ, Zhou LH, Ban X, Liu DX, Jiang W, Liu XM. Decreased expression of CD36 in circumvallate taste buds of high-fat diet induced obese rats. Acta Histochem. 2011;113(6):663–667. doi: 10.1016/j.acthis.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 41.Hotamisligil GS, Johnson RS, Distel RJ, Ellis R, Papaioannou VE, Spiegelman BM. Uncoupling of obesity from insulin resistance through a targeted mutation in <em>aP2</em>, the adipocyte fatty acid binding protein. Science. 1996;274(5291):1377. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- 42.Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes. 2003;52(7):1779–1785. doi: 10.2337/diabetes.52.7.1779. [DOI] [PubMed] [Google Scholar]
- 43.Tschritter O, Fritsche A, Thamer C, Haap M, Shirkavand F, Rahe S, Staiger H, Maerker E, Häring H, Stumvoll M. Plasma adiponectin concentrations predict insulin sensitivity of both glucose and lipid metabolism. Diabetes. 2003;52(2):239. doi: 10.2337/diabetes.52.2.239. [DOI] [PubMed] [Google Scholar]
- 44.Abbasi F, Chu JW, Lamendola C, McLaughlin T, Hayden J, Reaven GM, Reaven PD. Discrimination between obesity and insulin resistance in the relationship with adiponectin. Diabetes. 2004;53(3):585. doi: 10.2337/diabetes.53.3.585. [DOI] [PubMed] [Google Scholar]
- 45.Dall'Olio S, Davoli R, Buttazzoni L, Zambonelli P, Russo V. Study of porcine adiponectin (ADIPOQ) gene and association of a missense mutation with EBVs for production and carcass traits in Italian Duroc heavy pigs. Livest Sci. 2009;125(1):101–104. doi: 10.1016/j.livsci.2009.03.003. [DOI] [Google Scholar]
- 46.Zhang L, Yang M, Li C, Xu Y, Sun J, Lei C, Lan X, Zhang C, Chen H. Identification and genetic effect of a variable duplication in the promoter region of the cattle ADIPOQ gene. Anim Genet. 2014;45(2):171–179. doi: 10.1111/age.12112. [DOI] [PubMed] [Google Scholar]
- 47.Choi Y, Davis ME, Chung H. Effects of genetic variants in the promoter region of the bovine adiponectin (ADIPOQ) gene on marbling of Hanwoo beef cattle. Meat Sci. 2015;105:57–62. doi: 10.1016/j.meatsci.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 48.Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood. 2002;99(10):3505–3516. doi: 10.1182/blood.V99.10.3505. [DOI] [PubMed] [Google Scholar]
- 49.Ikegami Y, Inukai K, Imai K, Sakamoto Y, Katagiri H, Kurihara S, Awata T, Katayama S. Adiponectin upregulates ferritin heavy chain in skeletal muscle cells. Diabetes. 2009;58(1):61–70. doi: 10.2337/db07-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Festa M, Ricciardelli G, Mele G, Pietropaolo C, Ruffo A, Colonna A. Overexpression of H ferritin and up-regulation of iron regulatory protein genes during differentiation of 3T3-L1 pre-adipocytes. J Biol Chem. 2000;275(47):36708–36712. doi: 10.1074/jbc.M004988200. [DOI] [PubMed] [Google Scholar]
- 51.van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 52.Guardiola-Serrano F, Haendeler J, Lukosz M, Sturm K, Melchner H, Altschmied J. Gene trapping identifies a putative tumor suppressor and a new inducer of cell migration. Biochem Biophys Res Commun. 2008;376(4):748–752. doi: 10.1016/j.bbrc.2008.09.070. [DOI] [PubMed] [Google Scholar]
- 53.Gross B, Pawlak M, Lefebvre P, Staels B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat Rev Endocrinol. 2017;13(1):36–49. doi: 10.1038/nrendo.2016.135. [DOI] [PubMed] [Google Scholar]
- 54.Moerland M, Anghelescu N, Samyn H, van Haperen R, van Gent T, Strouboulis J, van Tol A, Grosveld F, de Crom R. Inducible expression of phospholipid transfer protein (PLTP) in transgenic mice: acute effects of PLTP on lipoprotein metabolism. Transgenic Res. 2007;16(4):503–513. doi: 10.1007/s11248-007-9094-y. [DOI] [PubMed] [Google Scholar]
- 55.Rasouli N, Yao-Borengasser A, Varma V, Spencer HJ, McGehee RE, Jr, Peterson CA, Mehta JL, Kern PA. Association of scavenger receptors in adipose tissue with insulin resistance in nondiabetic humans. Arterioscler Thromb Vasc Biol. 2009;29(9):1328–1335. doi: 10.1161/ATVBAHA.109.186957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nomata Y, Kume N, Sasai H, Katayama Y, Nakata Y, Okura T, Tanaka K. Weight reduction can decrease circulating soluble lectin-like oxidized low-density lipoprotein receptor-1 levels in overweight middle-aged men. Metabolism. 2009;58(9):1209–1214. doi: 10.1016/j.metabol.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 57.Zambonelli P, Gaffo E, Zappaterra M, Bortoluzzi S, Davoli R. Transcriptional profiling of subcutaneous adipose tissue in Italian large white pigs divergent for backfat thickness. Anim Genet. 2016;47(3):306–323. doi: 10.1111/age.12413. [DOI] [PubMed] [Google Scholar]
- 58.Seong J, Oh JD, Lee HK, Jeon GJ, Kong HS. Identification and analysis of phospholipid transfer protein polymorphisms and their association with marbling score in Hanwoo (Korean cattle) Genet Mol Res. 2013;12(1):731–737. doi: 10.4238/2013.March.13.1. [DOI] [PubMed] [Google Scholar]
- 59.Fonseca PD, de Souza FR, de Camargo GM, Gil FM, Cardoso DF, Zetouni L, Braz CU, Boligon AA, Branco RH, de Albuquerque LG, et al. Association of ADIPOQ, OLR1 and PPARGC1A gene polymorphisms with growth and carcass traits in Nelore cattle. Meta Gene. 2015;4:1–7. doi: 10.1016/j.mgene.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaneda M, Lin BZ, Sasazaki S, Oyama K, Mannen H. Allele frequencies of gene polymorphisms related to economic traits in Bos taurus and Bos indicus cattle breeds. Anim Sci J. 2011;82(6):717–721. doi: 10.1111/j.1740-0929.2011.00910.x. [DOI] [PubMed] [Google Scholar]
- 61.Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Revett T, Gimeno R, Lodish HF. Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR-gamma agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem J. 2008;416(2):161–177. doi: 10.1042/BJ20081240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Petersen PS, Lei X, Wolf RM, Rodriguez S, Tan SY, Little HC, Schweitzer MA, Magnuson TH, Steele KE, Wong GW. CTRP7 deletion attenuates obesity-linked glucose intolerance, adipose tissue inflammation, and hepatic stress. Am J Physiol Endocrinol Metab. 2017;312(4):E309–E325. doi: 10.1152/ajpendo.00344.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ntambi JM. Regulation of stearoyl-CoA desaturase by polyunsaturated fatty acids and cholesterol. J Lipid Res. 1999;40(9):1549–1558. [PubMed] [Google Scholar]
- 64.Jiang Z, Michal JJ, Tobey DJ, Daniels TF, Rule DC, Macneil MD. Significant associations of stearoyl-CoA desaturase (SCD1) gene with fat deposition and composition in skeletal muscle. Int J Biol Sci. 2008;4(6):345–351. doi: 10.7150/ijbs.4.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henriquez-Rodriguez E, Tor M, Pena RN, Estany J. A polymorphism in the stearoyl-CoA desaturase gene promoter increases monounsaturated fatty acid content in dry-cured ham. Meat Sci. 2015;106:38–43. doi: 10.1016/j.meatsci.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 66.Azzu V, Brand MD. The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem Sci. 2010;35(5):298–307. doi: 10.1016/j.tibs.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou H, Cheng L, Byun SO, Aizimu W, Hodge MC, Edwards GR, Hickford JGH. Haplotypic variation in the UCP1 gene is associated with milk traits in dairy cows. J Dairy Res. 2016;84(1):68–75. doi: 10.1017/S0022029916000522. [DOI] [PubMed] [Google Scholar]
- 68.Yang G, Forrest R, Zhou H, Hodge S, Hickford J. Genetic variation in the ovine uncoupling protein 1 gene: association with carcass traits in New Zealand (NZ) Romney sheep, but no association with growth traits in either NZ Romney or NZ Suffolk sheep. J Anim Breed Genet. 2014;131(6):437–444. doi: 10.1111/jbg.12097. [DOI] [PubMed] [Google Scholar]
- 69.Dutton S, Trayhurn P. Regulation of angiopoietin-like protein 4/fasting-induced adipose factor (Angptl4/FIAF) expression in mouse white adipose tissue and 3T3-L1 adipocytes. Br J Nutr. 2008;100(1):18–26. doi: 10.1017/S0007114507882961. [DOI] [PubMed] [Google Scholar]
- 70.Dijk W, Beigneux AP, Larsson M, Bensadoun A, Young SG, Kersten S. Angiopoietin-like 4 promotes intracellular degradation of lipoprotein lipase in adipocytes. J Lipid Res. 2016;57(9):1670–1683. doi: 10.1194/jlr.M067363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roke K, Jannas-Vela S, Spriet LL, Mutch DM. FADS2 genotype influences whole-body resting fat oxidation in young adult men. Appl Physiol Nutr Metab. 2016;41(7):791–794. doi: 10.1139/apnm-2016-0043. [DOI] [PubMed] [Google Scholar]
- 72.Vaittinen M, Walle P, Kuosmanen E, Mannisto V, Kakela P, Agren J, Schwab U, Pihlajamaki J. FADS2 genotype regulates delta-6 desaturase activity and inflammation in human adipose tissue. J Lipid Res. 2016;57(1):56–65. doi: 10.1194/jlr.M059113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ren ZQ, Wu WJ, Liu WH, Zheng R, Li JL, Zuo B, Xu DQ, Li FE, Lei MG, Ni DB, et al. Differential expression and effect of the porcine ANGPTL4 gene on intramuscular fat. Genet Mol Res. 2014;13(2):2949–2958. doi: 10.4238/2014.April.16.3. [DOI] [PubMed] [Google Scholar]
- 74.Renaville B, Prandi A, Fan B, Sepulcri A, Rothschild MF, Piasentier E. Candidate gene marker associations with fatty acid profiles in heavy pigs. Meat Sci. 2013;93(3):495–500. doi: 10.1016/j.meatsci.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 75.Gimeno RE, Ortegon AM, Patel S, Punreddy S, Ge P, Sun Y, Lodish HF, Stahl A. Characterization of a heart-specific fatty acid transport protein. J Biol Chem. 2003;278(18):16039–16044. doi: 10.1074/jbc.M211412200. [DOI] [PubMed] [Google Scholar]
- 76.Nafikov RA, Schoonmaker JP, Korn KT, Noack K, Garrick DJ, Koehler KJ, Minick-Bormann J, Reecy JM, Spurlock DE, Beitz DC. Association of polymorphisms in solute carrier family 27, isoform A6 (SLC27A6) and fatty acid-binding protein-3 and fatty acid-binding protein-4 (FABP3 and FABP4) with fatty acid composition of bovine milk. J Dairy Sci. 2013;96(9):6007–6021. doi: 10.3168/jds.2013-6703. [DOI] [PubMed] [Google Scholar]
- 77.Divoux A, Clement K. Architecture and the extracellular matrix: the still unappreciated components of the adipose tissue. Obes Rev. 2011;12(5):e494–e503. doi: 10.1111/j.1467-789X.2010.00811.x. [DOI] [PubMed] [Google Scholar]
- 78.Ojima K, Oe M, Nakajima I, Muroya S, Nishimura T. Dynamics of protein secretion during adipocyte differentiation. FEBS Open Bio. 2016;6(8):816–826. doi: 10.1002/2211-5463.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used in qRT-PCR. (XLS 19 kb)
Figure S1. Gene coverage distribution of the six adipose tissue transcriptomes. Gene coverage is calculated as the percentage of a gene covered by reads. This value is equal to the ratio of the base number in a gene covered by unique mapping reads to the total base number of coding region in that gene. Pies with different colors represent proportions of genes with certain coverage. For example, green pie is indicating proportion of genes with coverage between 90 and 100%. (TIF 7235 kb)
Table S2. All the annotated genes in the six libraries. Unique reads are reads aligned to only one position in the reference sequence. Gene coverage and RPKM values were calculated to analyze gene expression. (XLSX 3230 kb)
Table S3. Summary of the number of genes (RPKM> 0) detected in the six adipose tissues of two breeds. (XLS 20 kb)
Table S4. The most highly expressed 47 genes and their RPKM values in the six samples. (XLS 24 kb)
Table S5. DE genes with the FDR value≤0.001 and the absolute value of log 2 Ratio ≥ 1. Gene Length, length of all exon in gene; Expression, reads number that uniquely mapped to gene; P value, p value for hypothesis testing. (XLS 2504 kb)
Table S6. GO analysis of DE genes with corrected P value ≤0.05. The letter “n” is number of DE genes which are annotated to this ontology. The letter “m” is number of DE genes which are annotated to this term. (XLS 789 kb)
Table S7. KEGG pathway analysis of DE genes with Q value ≤1. Significantly enriched biological pathways were identified in DE genes based on the entire genome background. Q value is a P value that has been adjusted for the FDR value. Pathways with Q value ≤0.05 are thought to be significantly enriched. (XLS 105 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. The transcriptome data is freely available at the National Center for Biotechnology Information (NCBI) database Short Read Archive (Accession number SRP113440).