

This protocol describes a semiquantitative glycoproteomics method using sequential treatment with endoglycosidases to create unique mass signatures to determine glycan occupancy and proportion of high-mannose and complex glycans at each glycosite.
Supplementary information
The online version of this article (doi:10.1038/nprot.2018.024) contains supplementary material, which is available to authorized users.
Subject terms: Glycomics, Glycoproteins, Mass spectrometry, Proteomics, Proteolysis
Abstract
N-glycans contribute to the folding, stability and functions of the proteins they decorate. They are produced by transfer of the glycan precursor to the sequon Asn-X-Thr/Ser, followed by enzymatic trimming to a high-mannose-type core and sequential addition of monosaccharides to generate complex-type and hybrid glycans. This process, mediated by the concerted action of multiple enzymes, produces a mixture of related glycoforms at each glycosite, making analysis of glycosylation difficult. To address this analytical challenge, we developed a robust semiquantitative mass spectrometry (MS)-based method that determines the degree of glycan occupancy at each glycosite and the proportion of N-glycans processed from high-mannose type to complex type. It is applicable to virtually any glycoprotein, and a complete analysis can be conducted with 30 μg of protein. Here, we provide a detailed description of the method that includes procedures for (i) proteolytic digestion of glycoprotein(s) with specific and nonspecific proteases; (ii) denaturation of proteases by heating; (iii) sequential treatment of the glycopeptide mixture with two endoglycosidases, Endo H and PNGase F, to create unique mass signatures for the three glycosylation states; (iv) LC-MS/MS analysis; and (v) data analysis for identification and quantitation of peptides for the three glycosylation states. Full coverage of site-specific glycosylation of glycoproteins is achieved, with up to thousands of high-confidence spectra hits for each glycosite. The protocol can be performed by an experienced technician or student/postdoc with basic skills for proteomics experiments and takes ∼7 d to complete.
Supplementary information
The online version of this article (doi:10.1038/nprot.2018.024) contains supplementary material, which is available to authorized users.
Introduction
Asparagine-linked-glycans (N-glycans) are among the most common and important post-translational modifications of proteins. They have critical roles in the folding, conformation and stability of the proteins themselves1,2, and participate as ligands in intra- and intercellular recognition and host–pathogen interactions3,4. Altered biosynthesis of N-glycans has been associated with many diseases, such as cancer5,6, influenza7,8,9, and AIDS10,11,12,13,14. For example, increased levels of underprocessed high-mannose-type glycans have been reported to occur during breast cancer progression in both mice and humans6. Some broadly neutralizing antibodies (bnAbs) of the highly glycosylated HIV envelope glycoprotein include underprocessed high-mannose glycans in their epitopes, whereas others require fully processed complex-type structures containing sialic acid11,15. As the structures of N-glycans affect the activity and pharmacodynamics of glycoprotein biotherapeutics16, careful choice of cell lines used to express proteins10, growth conditions17, and purification methods18,19,20 to control the consistency of N-glycosylation is needed.
Given the importance of N-glycans to the structure and functions of glycoproteins, there is increasing need for robust methods for analysis of N-linked glycosylation that can be integrated with state of the art proteomics. Complicating the analysis is the inherent diversity of N-glycan structures present on any glycoprotein, which is a consequence of the non-template-driven biosynthesis. It begins with the en bloc transfer of Glc3Man9GlcNAc2 from a lipid-linked glycosyl donor to the nascent polypeptide by an oligosaccharyltransferase (OST) to a sequence-defined glycosite, Asn-X-Thr/Ser (where X can be any amino acid residue except proline). Although none of the proteins analyzed in the present study contains an atypical glycosylation site (N-X-Cys/Val), these sites have been verified in previous studies by the presence of glycans on intact glycopeptides21,22. As illustrated in Figure 1, the glycan is then subjected to trimming by removal of the glucose residues to a high-mannose-type glycan, then further trimming to the conserved Man3GlcNAc2 core, followed by addition of sugars by the sequential action of glycosyltransferases that produce a highly related set of complex-type structures (Fig. 1). Moreover, for glycoproteins with more than one glycosite, processing at each site may differ based on the access of the glycans to the processing enzymes. Indeed, well documented examples of proteins that contain high-mannose-type glycans at one or more glycosites, and highly processed complex-type glycans at other sites are IgM23,24, influenza hemagglutinin8,25,26, and the HIV envelope glycoprotein10,27,28,29.
Figure 1. N-linked glycan processing in the endoplasmic reticulum and Golgi apparatus.

N-glycans that are Endo H-sensitive are shown in the red box; these include high-mannose and hybrid glycans that have two terminal mannose residues, which is required for recognition by Endo H (red oval). Asn, asparagine; ER, endoplasmic reticulum; Fuc, fucose; Gal, galactose; Glc, glucose; GlcNAc, N-acetylglucosamine; Man, mannose; Sia, sialic acid.
Analysis of N-glycans of glycoproteins
Over the past two decades, several strategies have emerged to characterize N-glycans of glycoproteins and identify the glycosites that are recognized and used by the cellular glycosylation machinery. Several methods employ endoglycosidases, such as PNGase F and PNGase A, to release the glycans from the protein, followed by analysis of glycoforms using MS30,31,32,33,34,35 or high-performance liquid chromatography (HPLC)36,37 with or without prior derivatization38. The MS-based methods provide a composition for each molecular ion, which is annotated as a high-mannose or complex-type glycan consistent with biosynthetic principles. Furthermore, tandem mass spectra of derivatized N-glycans generated by both matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI) MS have been widely used for characterization of detailed structural information of N-glycans expressed in different biological systems30,34. Freely available software tools such as GRITS Toolbox (http://www.grits-toolbox.org/) are able to automatically process, annotate, and archive glycomics data in a high-throughput manner. The HPLC methods rely on retention times of N-glycan standards for identification of the glycan species. With both methods, supporting experiments using glycosidase digestions and/or permethylation analysis can provide additional support for structure assignments. Such methods find wide utility in characterizing glycosylation of highly studied glycoproteins such as immunoglobulins (e.g., IgG), or detecting individual differences in N-glycans of serum glycoproteins36,37,39. Although these methods provide key information about the nature of the glycoforms present on a glycoprotein and their relative abundance, they do not reveal which glycosites are utilized or the extent to which each glycosite is occupied.
A number of proteomics-based methods focus on identification of the glycosites that are utilized by the OST and the degree to which the site is occupied by a glycan40,41,42,43,44. The basic strategy is to immobilize glycopeptides by using lectins or by coupling periodate-treated glycopeptides to hydrazide-activated beads. Then the peptides are released from the bound glycan with an endoglycosidase (e.g., PNGase F) that, in the process, converts the Asn-X-Thr/Ser sequence to Asp-X-Thr/Ser. When the reaction is performed in H2O18, it creates a mass difference from Asn to Asp of +3. Then MS/MS analysis of the eluted peptides provides positive identification of the sites that are glycosylated. These methods are particularly useful for surveying complex biological systems, such as whole cells or tissues, to identify proteins that are glycosylated.
Recently, there have been major advances in glycoproteomics MS/MS methods that analyze intact glycopeptides and determine which glycoforms are present at each glycosite based on the combined masses of the glycopeptides and N-glycan8,28,29,45,46,47,48. Major limitations of this approach are the relatively low abundance of glycopeptides in the protein digest mixture, as well as the inherent low ionization efficiency of glycopeptides during MS analysis40,41,49,50. For these reasons, glycopeptides are typically enriched from peptide digests prior to MS analysis with different purification methods, such as hydrophilic interaction chromatography (HILIC)29,51,52 and hydrazide chemistry40. Despite the use of different fragmentation methods, including collision-induced dissociation (CID), high-energy collision dissociation (HCD), and electron-transfer dissociation (ETD), the inherent heterogeneity of glycoforms at each glycosylation site, as well as difficulties in obtaining good peptide backbone fragmentations for peptides with large glycans, impedes comprehensive identification of intact glycopeptides28,30. Annotation of LC-MS/MS data from such experiments is possible by using commercial algorithms such as Byonic (http://www.proteinmetrics.com/products/byonic/). However, quantitative assessment of the relative abundance of the glycan structures detected at each glycosite is still a substantial challenge due to unknown ionization efficiencies for peptides with each glycoform, especially for those with sialic acid-containing structures38. Moreover, if the glycopeptides are enriched prior to analysis, peptides with no glycan are lost, and the degree of glycosite occupancy cannot be assessed.
Development of the protocol and applications of the method
The protocol described here arose from the need of the HIV vaccine effort for robust semiquantitative glycoproteomics methods that would: (i) establish the occupancy of N-glycans at each glycosite and (ii) assess the degree to which glycans were processed from high-mannose type to complex type. The need stems from the fact that the primary candidate for a vaccine is the HIV envelope glycoprotein trimer (Env) that contains 75–90 N-glycans, creating a glycan shield to protect against attack by the immune system53,54. Despite the dense cover of N-glycans, bnAbs that bind to HIV Env do occur in 10–30% of HIV-1-infected patients55,56. Importantly, some bnAbs show interactions with high-mannose glycans57,58, whereas others show dependence on complex-type glycans12,15,59,60. Thus, to support the rational design of an HIV Env vaccine, we developed a method that could assess the proportion of high-mannose-type and complex-type N-glycans at each glycosite on the HIV Env10. However, as demonstrated here, the method is broadly applicable to any glycoprotein.
One of the key aspects of our method is the sequential use of two endoglycosidase treatments to introduce unique mass signatures for glycosites that carry no glycan, high-mannose/hybrid-type glycans, and complex-type glycans, as illustrated in Figure 2a. After protease digestion, endoglycosidase H (Endo H) is used to remove all high-mannose- and hybrid-type N-glycans that have at least 5 mannose residues (Fig. 1). This enzyme leaves a GlcNAc-Asn residue that adds +203 to the peptide mass. Subsequently, the remaining complex N-glycans are removed with PNGase F in the presence of H2O18, which both removes the glycan and converts Asn to Asp, with a +3 addition to the peptide mass. Peptides carrying glycosites (Asn-X-Thr/Ser) with Asn (unoccupied), GlcNAc-Asn (Endo H–treated), or Asp (PNGase F–treated), display similar ionization efficiencies during MS analysis10,45, allowing us to use ion intensity peak area to quantify the relative distribution of the three glycosylation states at each glycosite detected10,61. Another distinguishing feature of the method is the use of multiple proteases, which can generate up to thousands of spectra hits for each glycosite, resulting in >95% sequence coverage. This simple strategy effectively converts the glycoproteomics analysis to a proteomics analysis, allowing the use of robust proteomics software to analyze data in a high-throughput manner.
Figure 2. Schematic overview of the protocol.
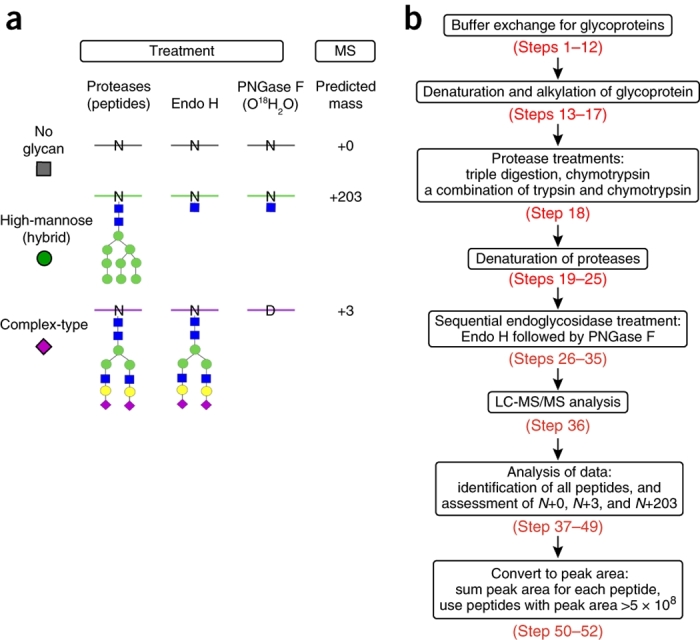
(a) Introduction of novel mass signatures for peptide glycosites that are not occupied, or that are occupied by high-mannose/hybrid or complex-type glycans. (b) The workflow of the protocol. a adapted from ref. 10, Nature Publishing Group.
A major advantage of this method is that it provides a glimpse into the site occupancy and processing of N-glycans at each glycosylation site10. It provides a semiquantitative analysis of the three glycosylation states, and a complete analysis can be conducted on only 30 μg of protein, even for complex glycoproteins such as HIV Env, comprising up to 30 glycosites per monomer. It should be noted that hybrid structures, which can potentially contain sialic acid (see Fig. 1), are included in the high-mannose-type category because they are cleaved by Endo H. However, hybrid structures typically have low abundance29,48.
One major limitation of the method is that glycan structures are removed before LC-MS/MS, and the class of the glycan is inferred by the specificities of the endoglycosidases. MS/MS methods based on analysis of intact glycopeptides provide more information on the spectra of glycans at individual glycosites27,28,29,62, and are complementary to this protocol, which provides semiquantitative information on site occupancy and glycan processing. However, neither method provides the precise structure complete with glycosidic linkages between monosaccharides. Although this protocol is designed for site-specific glycosylation analysis of purified glycoproteins, it is, in principle, applicable to more-complex protein samples, such as membrane-enveloped viruses (e.g., HIV, influenza virus, coronavirus) that comprise only 10–12 proteins. However, as described in the 'Experimental design' section, modification of the protocol would be needed to survey glycosites in more complex samples such as cells or human serum.
Overview of the procedure
The procedure for site-specific analysis of glycoprotein N-glycan processing is summarized in Figure 2b and consists of the following key stages.
In the first stage, buffer exchange for glycoproteins is needed if they are dissolved in the buffers that contain nonvolatile salts (Steps 1–12). Glycoproteins are then denatured and alkylated at pH 6 (Steps 13–17) to minimize non-enzymatic deamidation while retaining protein sequence coverage. Proteins are digested with several different protease treatments, including 'triple digestion', chymotrypsin, and a combination of trypsin and chymotrypsin, in order to maximize sequence coverage and increase the confidence of detecting each glycosite (Step 18).
In the second stage (Steps 19–25), proteases are denatured by heating to prevent incorporation of 18O-water into the C termini of peptides during the subsequent PNGase F treatment.
In the third stage (Steps 26–36), sequential endoglycosidase treatment is employed to create unique mass signatures relative to the predicted amino acid sequence, for peptide glycosites that are not occupied (+0 Da) or that are occupied by high-mannose/hybrid- (+203 Da) or complex-type glycans (+3 Da) (Fig. 2a).
The resulting samples are subjected to LC-MS/MS analysis (Step 37).
Data processing for identification and quantitation of peptides for the three glycosylation states is done using the Integrated Proteomics Pipeline (IP2) software package (stage 5, Steps 41–53). The MS1 and MS2 data are extracted from MS raw files with RawConverter and processed using multiple components from the IP2 software package (Steps 38–53).
Alternative methods
Over the past decade there has been substantial progress in site-specific glycosylation analysis of purified glycoproteins8,27,28,29,45,48,62,63,64. Exemplary, and perhaps the most relevant to this protocol, is the work on HIV-1 Env by the Desaire and Cripsin groups27,28,29,62. The methods developed by the two groups focus on characterizing intact glycopeptides with or without enrichment of glycopeptides prior to MS analysis, and thus can provide complementary information about individual glycoforms at each glycosite. However, milligram quantities of material may be required for their methods, which is attributed in part to the relatively low ionization efficiencies of glycopeptides and in part to the fact that each glycopeptide is actually a mixture of many different glycoforms28,29. Although the complexity is reduced by using only two proteases, chymotrypsin and trypsin, identification of all forms of a glycopeptide with multiple N-glycans is still a challenge, even when using a combination of CID and ETD for fragmentation27,28. Quantitative measurements are also challenging, due to unknown ionization of glycopeptides with different N-glycan species during MS analysis. Moreover, there is limited information about site occupancy, due to markedly different ionization efficiencies between peptides and the corresponding glycopeptides, and/or enrichment of glycopeptides prior to MS analysis.
Other methods that use endoglycosidases to identify glycosites have long been in use40,41,44. However, typically, glycopeptides are selectively captured using treatment with periodate followed by binding to hydrazide-activated beads, or are enriched using lectins. The peptides are then released using PNGase F, in which glycosylated asparagine is converted to aspartic acid (+1 Da mass shift), allowing indirect identification of glycosites in the released peptides40,41. Alternatively, for a purified protein, PNGase F can be applied directly, and site occupancy of glycosylation sites can be determined by measuring the ratio of peptides with glycosites containing asparagine to aspartic acid during ESI-MS analysis28. By comparison with the protocol described here, addition of a second endoglycosidase, Endo H, provides additional information about the extent of glycan processing, in addition to the degree of site occupancy.
Experimental design
Proteolysis with a combination of proteases. Glycoproteins are denatured and alkylated at pH 6 instead of a mildly alkaline pH to minimize the non-enzymatic deamidation of Asn to Asp, which can complicate data analysis65. To maximize sequence coverage, a number of different protease digestions are used, including digestion by chymotrypsin, a combination of trypsin and chymotrypsin, and combinations of Arg-C, trypsin, elastase and subtilisin ('triple digestion')66 (Step 18). Of note, a combination of all proteolytic digestions is essential for detecting all glycosites on heavily glycosylated proteins with large molecular weights, such as the HIV Env trimer and the spike glycoprotein of Middle East respiratory syndrome coronavirus (MERS-CoV S-2P protein). Triple digestion alone is able to identify all glycosites in glycoproteins containing only a few glycosites, whereas for most glycoproteins, a combination of triple digestion and chymotrypsin is enough to generate detectable peptides that contain all the glycosylation sites10. Another benefit of the use of multiple proteases, including trypsin and nonspecific proteases, is that it produces a sequence ladder that contains a series of peptides with variable numbers of amino acid residues on both sides of glycosite, allowing for highly confident identification for a given glycosite (Supplementary Table 1). For single glycoproteins or simple mixtures (e.g., viruses), the use of multiple proteases allows for much higher confident detection and semiquantitative analysis of the three states of glycosylation for each glycosite. However, for more-complex mixtures (e.g., cells, plasma) use of specific proteases such as trypsin will result in reduced complexity and the identification of glycosites while retaining the ability to detect the three different glycosylation states. Another key aspect of the protocol is that only volatile salts, such as ammonium bicarbonate and ammonium acetate, are used, so that no column purification is needed, resulting in high specificity of the protocol (requires ∼30 μg of starting material).
Denaturation of proteases. In the next stage, PNGase F treatment is employed for deglycosylation of glycoproteins, resulting in conversion of Asn to Asp upon removal of the carbohydrate, and a mass change of +3 when carried out in O18-water (Steps 19–25). Residual active trypsin and other proteases used for proteolysis can incorporate O18 into the C termini of the peptides during the deglycosylation step67, resulting in a high false-positive identification of peptides that may have glycosites. To avoid this, we denature all proteases used in the protocol by heating to 100 °C prior to the deglycosylation steps.
Sequential endoglycosidase treatment. Sequential treatment of glycopeptides with Endo H followed by PNGase F is employed to generate different mass signatures for glycosites that contain no glycan (+0), high-mannose/hybrid-type glycan (+203), and complex-type glycan (+3) (Steps 26–36, Fig. 2a). The endoglycosidase digestions are highly efficient and progress rapidly to completion, so the deglycosylation reactions are conducted for 1 h to minimize non-enzymatic deamidation that can occur during this step. Removal of N-glycans increases the ionization efficiencies of peptides and allows us to localize glycosylation sites on peptides with multiple modifications, which is challenging for analysis of intact glycopeptides (Supplementary Fig. 1).
LC-MS/MS analysis. In principle, the deglycosylated peptides can then be analyzed by any type of LC-MS/MS (Step 37). A high-resolution mass spectrometer, such as the Orbitrap Elite, provides satisfactory sequence coverage for most glycoproteins. An instrument with a higher scan speed, such as an Orbitrap Fusion or Orbitrap Lumos, is more sensitive and generates several times more MS/MS spectra for a given sample than an Orbitrap Elite. Thus, they are able to provide site-specific glycan-processing information for those sites that may be missed in the results generated by the Orbitrap Elite as a result of heavy glycosylation and the large molecular weights of the glycoproteins. Although single-dimension separation is sufficient for characterization of site-specific glycosylation of purified proteins, multidimensional protein identification technology (MudPIT), in which peptides are systematically separated based on the charge in the first dimension and hydrophobicity in the second, will accelerate measurement of site-specific N-glycan processing of glycoproteins in these complex protein samples68.
Replicates and controls. Ideally, each glycoprotein is digested in at least two technical replicates and analyzed by the same MS instrument. Invertase produced by the yeast Saccharomyces cerevisiae and α-1-acid glycoprotein from bovine serum are known to be occupied by high-mannose-type and complex-type glycans, respectively, and can be used as controls to test completion of endoglycosidase treatment. As a check on the overall success of the protocol, non-enzymatic deamidation of Asn residues and/or O18-incorpation at the C terminus should be seen in <5% of the total peptides.
Materials
REAGENTS
Urea (MilliporeSigma, cat. no. U5128)
Ammonium acetate (CH3COONH4; MilliporeSigma, cat. no. A1542)
DTT (Fisher BioReagents, cat. no. BP172-5)
Iodoacetamide (IAA; MilliporeSigma, cat. no. I1149)
Water (purified by the GenPure Pro water purification system; Thermo Fisher Scientific)
Ammonium bicarbonate (NH4HCO3; MilliporeSigma, cat. no. A6141)
Formic acid (MilliporeSigma, cat. no. F0507)
Acetic acid (Fisher Scientific, cat. no. A38-212)
-
Hydrochloric acid (36.5–38% (vol/vol) HCl; Fisher Scientific, cat. no. A144-212)
Caution
Concentrated hydrochloric acid is a corrosive acid and forms acidic mists. Both the mist and the solution have a corrosive effect on human tissue, with the potential to damage respiratory organs, eyes, skin, and intestines irreversibly. Handle it in a hood while wearing personal protective equipment, including lab coat, gloves, and safety glasses.
Arg-C (Promega, cat. no. V1881)
Trypsin (Promega, cat. no. V5111)
EDTA disodium salt dihydrate (MilliporeSigma, cat. no. E5134)
Ethylene bridged hybrid (BEH) 1.7-μm C18 resin (Waters, cat. no. 186003556)
4-μm Jupiter C18 (Phenomenex, cat. no. 04A-4396)
-
Acetonitrile (ACN; VWR, cat. no. 200004-350)
Caution
ACN has modest toxicity in small doses and should be handled in a hood while wearing gloves.
Tris base (Fisher Scientific, cat. no. BP152-10)
Calcium chloride dihydrate (CaCl2·2H2O; MilliporeSigma, cat. no. C3306)
Elastase (Promega, cat. no. V1891)
Subtilisin (MilliporeSigma, cat. no. P5380)
Chymotrypsin (Promega, cat. no. V1061)
Endo H (New England Biolabs, cat. no. P0702L)
PNGase F (New England Biolabs, cat. no. P0705S)
-
18O-water (97 atom% of 18O; MilliporeSigma, cat. no. 329878)
Critical
Store the reagent in a desiccator at room temperature (20–25 °C) for up to 1 year.
Fetuin (MilliporeSigma, cat. no. F3385)
Invertase (MilliporeSigma, cat. no. I0408)
IgG (MilliporeSigma, cat. no. I4506)
IgM (MilliporeSigma, cat. no. I8260)
α-1-Acid glycoprotein (AGP; MilliporeSigma, cat. no. G3643)
Transferrin (MilliporeSigma, cat. no. T8158)
NaOH pellets (MilliporeSigma, cat. no. 221465)
Glycoprotein of interest: In this protocol, we use three example proteins: BG505 SOSIP.664 trimer (laboratory-made, expressed in HEK 293-F cells as described in a previous study10), MERS-CoV S-2P protein (laboratory-made, expressed in HEK 293-F cells as described in a previous study69), and influenza virus hemagglutinin (HA, laboratory-made, expressed in HEK 293-F cells as described in a previous study10).
EQUIPMENT
Eppendorf Research Plus pipette (Eppendorf)
Biotix microcentrifuge tubes, 1.5 ml (VWR, cat. no. MTL-0150-BC)
Low-binding pipette tips, 10 μl (Corning, cat. no. 4150)
Low-binding pipette tips, 200 μl (Corning, cat. no. 4151)
Centrifugal filter, 10 kDa (MilliporeSigma, cat. no. MRCPRT010)
Centrifugal filter, 30 kDa (MilliporeSigma, cat. no. MRCF0R030)
Incubator set to 37 °C (Fisher Scientific, cat. no. 15-103-0514)
Incubator set to 56 °C (Fisher Scientific, cat. no. 15-103-0514)
Incubator set to 100 °C (Fisher Scientific, cat. no. 05-412-500)
Water bath set to 30 °C (Fisher Scientific, cat. no. 15-462-5Q)
pH meter (MilliporeSigma, cat. no. Z283037)
NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, cat. no. ND-2000)
Eppendorf microcentrifuge 5415R (MilliporeSigma, cat. no. Z605212)
Microcentrifuge (Santa Cruz Biotechnology, cat. no. sc-358765)
Lyophilizer (Labconco)
Desiccator (Hach)
Fisher Vortex Genie 2 (Fisher Scientific, cat. no. 12-812)
Parafilm (VWR, cat. no. 52858-000)
Kimtech Science Kimwipes tissues (Kimberly-Clark)
Freezer set to −80 °C (Forma Scientific)
Mass spectrometer: the protocol below is optimized for either (i) an Orbitrap Elite Hybrid Ion Trap-Orbitrap mass spectrometer equipped with the EASY-nLCII system (Thermo Fisher Scientific) or (ii) an Orbitrap Fusion Tribrid mass spectrometer equipped with the EASY 1000 system (Thermo Fisher Scientific)
Software
RawConverter, v1.1.0.19 (http://fields.scripps.edu/rawconv/)
Notepad++, v7.5.4 (https://notepad-plus-plus.org/download/v7.5.3.html)
glyco_motif_filter, v1.0 (http://fields.scripps.edu/yates/wp/?page_id=687)
IP2 (Integrated Proteomics Pipeline, v5.0.1; http://goldfish.scripps.edu/ip2/login.jsp)
REAGENT SETUP
Ammonium acetate (100 mM, pH 6)
Dissolve 0.077 g of ammonium acetate in 10 ml of water. Adjust the buffer pH to 6 by adding acetic acid. Dispense into aliquots and store the buffer at −20 °C for up to 2 months.
Urea (8 M)
Dissolve 0.048 g of urea in 100 μl of 100 mM ammonium acetate (pH 6). Prepare fresh solution for each experiment.
DTT (500 mM)
Dissolve 0.0077 g of DTT in 100 μl of water. Prepare fresh solution for each experiment.
Iodoacetamide (500 mM)
Dissolve 0.00925 g of iodoacetamide in 100 μl of water. Prepare fresh solution for each experiment.
Ammonium bicarbonate (100 mM, pH 8)
Dissolve 0.079 g of ammonium bicarbonate in 10 ml of water. Measure the buffer pH. Dispense into aliquots and store the buffer at −20 °C for up to 2 months.
100 mM Tris-HCl (pH 7.8)
Dissolve 12.114 g of Tris in 800 ml of water. Adjust the pH to 7.8 with concentrated HCl. Bring the final volume to 1 liter with water. Store the buffer at 4 °C for up to 2 months.
CaCl2 (1 M)
Dissolve 0.147 g of calcium chloride dihydrate in 1 ml of water. Dispense into aliquots and store the buffer at −20 °C for up to 2 months.
EDTA (1 M, pH 8)
Add 186.12 g of EDTA·2H2O to 300 ml of H2O. Stir vigorously on a magnetic stirrer. Adjust the pH to 8.0 with NaOH pellets. The disodium salt of EDTA will not go into solution until the pH of the solution is adjusted to ∼8.0 by the addition of NaOH. Bring the final volume to 500 ml with water. Store the buffer at 4 °C for up to 2 months.
Arg-C (0.5 μg/μl)
Dissolve 10 μg of Arg-C in 20 μl of a buffer that contains 50 mM Tris-HCl (pH 7.8), 5 mM CaCl2, 2 mM EDTA. Dispense into aliquots and store the solution at −80 °C for up to 2 months.
Trypsin (0.5 μg/μl)
Dissolve 20 μg of trypsin in the resuspension buffer provided by manufacturer. Dispense into aliquots and store the solution at −80 °C for up to 2 months.
Elastase (0.5 μg/μl)
Dissolve 0.5 mg of elastase in 1 ml of water. Dispense into aliquots and store the solution at −80 °C for up to 2 months.
Subtilisin (0.5 μg/μl)
Dissolve 0.5 mg of subtilisin in 1 ml of water. Dispense into aliquots and store the solution at −80 °C for up to 2 months.
Chymotrypsin (0.5 μg/μl)
Dissolve 25 μg of chymotrypsin in 50 μl of 1 mM HCl. Dispense into aliquots and store the solution at −80 °C for up to 2 months.
Ammonium acetate (100 mM, pH 5.5)
Dissolve 0.077 g of ammonium acetate in 10 ml of water. Adjust the buffer pH to 5.5 by adding acetic acid. Dispense into aliquots and store the buffer at −20 °C for up to 2 months.
18O-water (97 atom% of 18O)
Dispense the 18O-water into 200-μl aliquots, seal the aliquots, and store them in a desiccator at room temperature for up to 1 year.
Ammonium bicarbonate (100 mM, prepared with 18O-water, pH 8)
Place ammonium bicarbonate into a desiccator for at least 48 h to dry it before use. Dissolve 0.0079 g of ammonium bicarbonate in 1 ml of 18O-water. Place it into a desiccator and store the buffer at room temperature for up to 2 months.
PNGase F (glycerol-free)
Lyophilize the solution that contains PNGase F and redissolve it in an equal volume of 18O-water. Prepare fresh solution for each experiment.
Endo H activity assays
A set of control experiments in which a defined amount of the desired glycoprotein is treated with a series of Endo H concentrations according to the manufacturer's instructions should be done before using this protocol. Gel shifts of high-mannose glycans on SDS-PAGE can be used to determine completion of deglycosylation.
0.1% (vol/vol) Formic acid in 80% (vol/vol) ACN
Mix 800 ml of 100% (vol/vol) ACN and 200 ml of water. Add 1 ml of formic acid. Store the solution at room temperature for up to 1 month.
0.1% (vol/vol) Formic acid in 5% (vol/vol) ACN
Mix 50 ml of 100% (vol/vol) ACN and 950 ml of water. Add 1 ml of formic acid. Store the solution at room temperature for up to 1 month.
0.1% (vol/vol) Formic acid
Add 1 ml of formic acid to 1 liter of water. Store the solution at room temperature for up to 1 month.
0.1% (vol/vol) Formic acid in ACN
Add 1 ml of formic acid to 1 liter of ACN. Store the solution at room temperature for up to 1 month.
EQUIPMENT SETUP
LC setup
-
The given information is for use with an Easy 1000 or Easy nLCII pump (Thermo); parameters may have to be adjusted for other LC systems. Easy 1000 and Easy nLCII are very similar in nature and only differ in pressure limits and thus type of column that can be used.
Table 2.
Easy 1000 Easy nLCII Column BEH 1.7-μm C18 resin (Waters) pressure-loaded into a 100-μm inner diameter × 25-cm length capillary column 4-μm Jupiter C18 (Phenomenex) pressure-loaded into a 100-μm inner diameter × 15 cm length capillary column Mobile phases A: 0.1% (vol/vol) formic acid in waterB: 0.1% (vol/vol) formic acid in ACNA: 0.1% (vol/vol) formic acid in 5% (vol/vol) ACNB: 0.1% (vol/vol) formic acid in 80% (vol/vol) ACNFlow rate 200 nl per min 400 nl per min Sample injection volume 3–5 μl with a 1- to 2-μg sample 10 μl with a 1- to 2-μg sample Gradient program for Easy 1000:Table 3.
Step Time (min) % Solvent B Gradient 0 5 150 25 200 40 210 100 240 100 The column is re-equilibrated with 20 μl of buffer A prior to the injection of sample.Gradient program for Easy nLCII:Table 4.
Step Time (min) % Solvent B Gradient 0 0 10 0 20 10 105 60 125 100 130 100 135 0 140 0 The column is re-equilibrated with 20 μl of buffer A prior to the injection of sample.
Mass spectrometer setup
- The instrument should be mass-calibrated prior to use. The detailed settings below were used with the Orbitrap Fusion and the Orbitrap Elite.
Table 5.
Parameter Orbitrap fusion Orbitrap elite Ionization mode Positive Positive Electrospray voltage 2.5 kV 2.5 kV MS1 resolution 120 k 120 k MS2 resolution 15 k 15 k MS1 scan range 400–1,500 400–2,000 MS1 maximum injection time 50 ms 200 ms MS2 maximum injection time 250 ms 1,000 ms MS1 AGC target 4 × 105 1 × 106 MS2 AGC target 5 × 104 5 × 104 Isolation 2 m/z with quadrupole 2 m/z with ion trap Fragmentation CID at 35% CID at 35% Dynamic exclusion 5 s 60 s Charge states 2–7 selected +1 and unassigned are rejected Intensity threshold 5e4 500 DDA method 3 s cycle time Top ten AGC, automatic gain control.
Procedure
Buffer exchange for glycoproteins
Timing ∼11 h
Critical Step
To minimize sample loss, low-protein-binding microcentrifuge tubes and pipette tips should be used during sample preparation. Only volatile salts are used in the protocol and these can be removed by lyophilization, resulting in higher sensitivity due to minimal protein loss. High-purity reagents should be used for sample preparation and MS analysis to minimize signals derived from contaminants.
Critical Step
If the protein is a lyophilized powder, then proceed to the 'Denaturation and alkylation of glycoprotein' section (Step 13).
Insert a 10-kDa centrifugal filter into a tube.
Rinse the filter by adding 100 μl of 100 mM ammonium acetate immediately before use. Seal the tube with the attached cap.
Centrifuge the device (the tube and the filter) at 8,000g for 50 min at 4 °C.
-
Pipette the solution containing ∼30 μg of target protein into the filter. Seal the tube with the attached cap. If the volume of the solution is <100 μl, bring the final volume to 100 μl with water.
Critical Step
Protein concentration is estimated with the NanoDrop A280 assay.
Centrifuge the device at 8,000g for 50 min at 4 °C.
Pipette 100 μl of 100 mM ammonium acetate into the filter.
Centrifuge the device at 8,000g for 50 min at 4 °C.
-
Repeat Steps 6 through 7 at least two times.
Critical Step
To complete the buffer exchange, it is important to extensively wash the filter that contains glycoproteins (at least three times) with 100 mM ammonium acetate (pH 6).
Critical Step
To minimize non-enzymatic deamidation, the acidic buffer, 100 mM ammonium acetate (pH 6), should be used instead of mildly alkaline buffers during buffer exchange.
Wash the filter membrane with 100 μl of 100 mM ammonium acetate (pH 6).
Collect the solution and store it in a low-protein-binding microcentrifuge tube.
-
Repeat Steps 9 and 10 at least four times and combine the fractions.
Critical Step
Wash the filter membrane with the buffer at least five times after buffer exchange in order to achieve maximum recovery of proteins.
Pause point
The solution can be stored up to 1 week at −80 °C or in liquid nitrogen.
-
Lyophilize 500 μl of the solution at room temperature for at least 5 h.
Critical Step
If volatile salts such as ammonium acetate are added to the buffer, it is important to completely remove them with the lyophilizer.
Denaturation and alkylation of glycoprotein
Timing ∼7 h
-
13
Dissolve the glycoprotein in 100 μl of 8 M urea in 100 mM ammonium acetate (pH 6) and place the solution at room temperature for 1 h.
Critical Step
The acidic buffer, 100 mM ammonium acetate, should be used for sample preparation instead of mildly alkaline buffers, if possible, in order to keep non-enzymatic deamidation to a minimum.
-
14
Add 2 μl of 500 mM DTT (to a final concentration of ∼10 mM) and incubate the solution at 56 °C for 1 h.
Critical Step
Spin down (1,000g, 25 °C, 1 min) all the solution from the walls of microcentrifuge tube before adding DTT.
-
15
Add 11 μl of freshly prepared 500 mM iodoacetamide (to a final concentration of ∼50 mM) and incubate the solution in the dark at room temperature for 45 min.
Critical Step
To preserve activity of iodoacetamide, prepare iodoacetamide solution immediately before use, as it is unstable. Perform the alkylation step in the dark, as iodoacetamide is light-sensitive.
-
16
Buffer-exchange to 100 mM ammonium bicarbonate (pH 8) by using a centrifugal filter with a membrane nominal molecular weight limit of 10 kDa.
Critical Step
To complete buffer exchange, it is important to wash the filter at least three times with 100 μl of 100 mM ammonium bicarbonate at 8,000 g for 50 min at 4 °C.
-
17
Dispense the sample into five equal aliquots for the following proteolytic digestion.
Critical Step
Do not dry glycoproteins after denaturation, as they may not be fully soluble in the buffer after lyophilization.
Pause point
The samples can be stored at −80 °C or in liquid nitrogen for at least 2 weeks.
Protease treatments
Timing ∼24 h
-
18Five aliquots (containing 6 μg of denatured glycoproteins each) are subjected to treatments with multiple proteases and combinations of proteases involving: Arg-C followed by trypsin (option A), elastase (option B), and subtilisin (option C). Aliquots A–C will later be combined into a 'triple digestion' sample (Step 21). The remaining two aliquots are digested with chymotrypsin (option D) or a combination of trypsin and chymotrypsin (option E).
-
AArg-C followed by trypsin
-
iAdd Arg-C to one aliquot that contains ∼6 μg of denatured glycoproteins at an enzyme/protein ratio of 1:20 (wt/wt). Bring the final volume to 100 μl with 100 mM ammonium bicarbonate (pH 8). Add DTT and EDTA to final concentrations of 5 mM and 0.2 mM, respectively.Critical StepArg-C is able to cleave at the C terminus of arginine residues, including sites next to proline, resulting in increased sequence coverage when combined with trypsin digestion.
-
iiIncubate the solution at 37 °C for 4 h.
-
iiiLyophilize the resulting peptide mixture for at least 3 h to remove water and volatile salt.
-
ivRedissolve the peptide mixture in 500 μl of 100 mM ammonium acetate (pH 6).
-
vAdd sequencing-grade modified trypsin to the solution at a trypsin/protein ratio of 1:10 (wt/wt).Critical StepSequencing-grade modified trypsin is able to digest glycoproteins at pH 6 while preserving adequate digestion efficiency.
-
viIncubate the reaction at 37 °C for 16 h.
-
i
-
BElastase
-
iAdd elastase to the second aliquot of the denatured glycoprotein at an elastase/protein ratio of 1:20 (wt/wt).Critical StepThe utility of triple digestion can generate higher sequence coverage than digestion with any of the single enzymes alone.
-
iiBring the final volume to 500 μl with 100 mM ammonium bicarbonate (pH 8).
-
iiiIncubate the reaction at 37 °C for 16 h.
-
i
-
CSubtilisin
-
iAdd subtilisin to the third aliquot of the denatured glycoprotein at a subtilisin/protein ratio of 1:20 (wt/wt).
-
iiBring the final volume to 500 μl with 100 mM ammonium bicarbonate (pH 8).
-
iiiIncubate the reaction at 37 °C for 4 h.Critical StepDo not incubate the reaction longer than 4 h, in order to obtain appropriate lengths of peptides for MS detection.
-
i
-
DChymotrypsin
-
iAdd chymotrypsin to the fourth aliquot of the denatured glycoprotein at a chymotrypsin/protein ratio of 1:13 (wt/wt).Critical StepDo not add too much chymotrypsin to the solution, as it can be self-digested, which will suppress the ESI signals of analytes.
-
iiBring the final volume to 500 μl with 100 mM ammonium bicarbonate (pH 8).
-
iiiIncubate the reaction in a 30 °C water bath for 10 h.
-
i
-
EA combination of trypsin and chymotrypsin
-
iAdd trypsin and chymotrypsin to the fifth aliquot of the denatured glycoprotein at enzyme/protein ratios of 1:20 (wt/wt) and 1:13 (wt/wt) respectively.
-
iiBring the final volume to 500 μl with 100 mM ammonium bicarbonate (pH 8).
-
iiiIncubate the reaction at 37 °C for 16 h.Pause pointThe peptide mixtures derived from combination proteolytic digestion can be stored at −80 °C or in liquid nitrogen for at least 2 weeks.
-
i
-
A
Denaturation of proteases
Timing ∼10 h
-
19
Lyophilize the peptide mixtures derived from each of the five protease digestions (Step 18A–E) for at least 5 h.
-
20
Redissolve each sample in 100 μl of water.
-
21
Combine the peptide mixtures derived from Step 18A–C into a 'triple digestion' sample.
-
22
Separately, heat the combined triple digestion sample (Step 21) and the samples generated from digestion with chymotrypsin (Step 18D), and a combination of trypsin and chymotrypsin (Step 18E) at 100 °C for 30 s.
-
23
Cool the samples at room temperature for 30 s.
-
24
Repeat Steps 22 and 23 at least five times.
Critical Step
To completely deactivate the proteases used, Steps 22 and 23 should be repeated at least five times. Any remaining active proteases will accelerate incorporation of 18O-water into the C termini of peptides during the following PNGase F treatment conducted in 18O-water.
-
25
Lyophilize the samples for at least 3 h to remove any remaining volatile salts from the samples.
Critical Step
Complete removal of volatile salts from the samples is important for the following Endo H treatment, which has optimal activity at pH 5.5.
Pause point
The peptide mixtures can be stored at −80 °C or in liquid nitrogen for at least 2 weeks.
Sequential endoglycosidase treatment
Timing ∼7 h
-
26
Separately, redissolve each of the three samples in 20 μl of 100 mM ammonium acetate (pH 5.5).
-
27
Add Endo H to the peptide mixtures at a minimum enzyme/glycoprotein ratio of 250 NEB units per 10 μg.
Critical Step
The quantity of Endo H needed for deglycosylation may vary from protein to protein. A set of control experiments should be done before using this protocol (see Reagent Setup).
-
28
Incubate the reaction at 37 °C for 1 h.
Pause point
The Endo H–treated peptides can be stored up to 1 week at −80 °C or in liquid nitrogen.
-
29
Lyophilize the Endo H–treated samples for at least 3 h immediately before use. In the meantime, proceed to Step 30.
Critical Step
Complete removal of ammonium acetate in the samples ensures that the following PNGase F treatment will proceed to completion.
-
30
Lyophilize the PNGase F solution for at least 1 h immediately before use and redissolve the PNGase F enzyme in the same volume of18O-water (see Reagent Setup).
-
31
Add 20 μl of 100 mM ammonium bicarbonate (pH 8) prepared with 18O-water to the PNGase F solution.
Critical Step
Steps 31 and 32 should be done as quickly as possible to reduce contact of the reaction mixture with air.
-
32
Add PNGase F to the Endo H–treated peptide mixtures at a minimum enzyme/glycoprotein ratio of 500 NEB units per 10 μg.
Critical Step
The quantity of PNGase F that is needed for deglycosylation can be determined by treating a defined amount of the desired glycoprotein with a series of PNGase F concentrations. Gel shifts of N-glycans on SDS-PAGE can be used to determine completion of deglycosylation.
-
33
Seal the microcentrifuge tube that contains the mixture.
-
34
Incubate the reaction at 37 °C for 1 h.
-
35
Dispense the reaction mixture into aliquots that contain ∼2 μg of peptides and store them in liquid nitrogen immediately.
Pause point
The samples can be stored in liquid nitrogen for at least 1 month.
LC-MS/MS analysis
Timing 6–48 h per glycoprotein
-
36
Set up the LC-MS/MS system to characterize the deglycosylated peptides as described in the 'Equipment Setup' section.
Critical Step
Each glycoprotein is digested in two or three technical replicates and analyzed by the same MS instrument.
Data analysis
Timing variable; hours to 1 d per glycoprotein
-
37
Extract the MS1 and MS2 spectra from the MS raw files using the spectrum-converting software RawConverter.
-
38
Add the MS raw files to the 'files to covert' window of RawConverter.
-
39
Set 'Experiment Type' as data dependent. Enable 'Select monoisotopic m/z in DDA'. 'Output formats' should be set to 'MS1, MS2, and MS3', in which both MS1 and MS2 data are extracted from the MS raw files.
-
40
Use a text editor such as Notepad++ (https://notepad-plus-plus.org/) to prepare a file containing the sequences of the target glycoproteins in FASTA format.
-
41
Add the resulting file to a predefined database, such as the European Bioinformatic Institute Bos Taurus protein database, with the 'database' of IP2.
-
42
Set the parameters as: Source: Uniprot; Organism Name: Bos Taurus; Generate reverse (decoy) sequences: yes; Add contaminant proteins: No.
Critical Step
Reverse (decoy) sequences should be generated and included in the final database in order to estimate peptide probabilities and false-discovery rates.
-
43
Upload the resulting file that contains the sequences of the target glycoproteins to the 'database' window of IP2.
-
44
Upload the database to IP2.
-
45
Upload the MS1 and MS2 files to IP2.
-
46
Start a ProLuCID search in the IP2 (v5.0.1) software package.
-
47
Set the mass tolerance at 50 p.p.m. for precursor ions and 20 p.p.m. for fragment ions (MS2 spectra are detected in the Orbitrap instrument). No enzyme specificity is considered for searching. Set carboxyamidomethylation (+57.02146 C) as a fixed modification. Set oxidation (+15.9994 M), deamidation (+2.988261 N), GlcNAc (+203.079373 N), and pyroglutamate formation from N-terminal glutamine residue (−17.026549 Q) as variable modifications.
-
48
Filter the results generated by the ProLuCID search by using DTASelect (v2.0, another component of the IP2 software kit). The parameters are set as: minimum number of peptides per protein: ≥2, spectrum false-positive rate: ≤0.05, and precursor delta mass cutoff: ≤10 p.p.m.
Critical Step
Sequence coverage of target proteins should be >95%.
-
49
Filter the results generated by DTASelect with the software 'Glyco_motif_filter' to remove those peptides with N+3 and/or N+203 modifications that are not located at the motif (N-X-S/T, X can be any amino acid residue except proline).
Critical Step
All peptides with N+203 modifications that are not located at the consensus motif should be manually checked before they are removed. Asparagine residues that are not located at the motif should be considered as potential glycosylation sites when multiple spectra hits with N+203 modifications that do not contain the motif are consistently detected. Further verification is needed for these potential glycosylation sites.
-
50
Start a label-free analysis using Census (another component of the IP2 software package). The parameters are set as: 'find missing peptide': enabled, mass tolerance: ≤10 p.p.m., retention time tolerance: ≤0.1 min.
Critical Step
Ion injection time is used to further normalize the resulting peak area.
-
51
Determine the abundance of each peptide from each raw file by the sum of the ion intensity peak area over all identified charge states.
Critical Step
To improve the accuracy of the method, a set of peptides with N+0, N+3, and N+203 modifications is considered only when at least one of the three has a peak area of at least 5 × 108. Of note, ∼2 μm of purified glycoproteins are loaded onto the column in the present study. This value was empirically determined as optimal to distinguish information from spectral noise and will vary from instrument to instrument. A control experiment should be done by using well-characterized model glycoproteins such as invertase (occupied by high-mannose glycans) and α-1-acid glycoprotein (occupied by complex-type glycans) before setting the peak area threshold. Peak area values will be dependent on the type of LC system and mass spectrometer used, and the appropriate threshold will need to be determined for other instrument types.
-
52
Combine the data derived from two or three technical replicates after analysis of each MS run separately.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 36 | No peptides detectable | The LC-MS instrument is not performing properly | Make sure that the LC-MS instrument is well-calibrated and working properly according to specifications |
| 48 | Low sequence coverage of target glycoproteins | Glycoproteins may not have been washed out completely after buffer exchange (Step 16) | Wash the filter membrane at least five times after buffer exchange to maximize recovery of target glycoproteins |
| No peptides detected after combination proteolytic digestion | Proteases stored in the buffer at −80 °C for too long may not be active (Step 18) | Make fresh protease solution | |
| Too many peaks that belong to non-enzymatic deamidation are detected | The microcentrifuge tube that contains 18O-water is not completely sealed or was exposed to air for too long (Steps 30–33) | Seal the microcentrifuge tube completely and place it into a desiccator. Perform Steps 30–33 as quickly as possible to reduce contact of the reaction mixture with air | |
| 51 | The output file generated by Census is empty or cannot be downloaded | The MS1 file was not extracted correctly by RawConverter or the names of the target glycoproteins are not recognized by Census | Extract the MS1 spectra from raw files again using RawConverter. Delete spaces that are not recognized by Census from the names of the target glycoproteins |
Timing
Steps 1–12, buffer exchange for glycoproteins: ∼11 h
Steps 13–17, denaturation and alkylation of glycoprotein: ∼7 h
Step 18, protease treatments: ∼24 h
Steps 19–25, denaturation of proteases: ∼10 h
Steps 26–35, sequential endoglycosidase treatment: ∼7 h
Step 36, LC-MS/MS analysis: 6–48 h per glycoprotein
Steps 37–52, data analysis: variable; hours to 1 d per glycoprotein, depending on complexity
Anticipated results
This protocol is used to determine site-specific N-glycan processing of glycoproteins. In the most widely used strategy, glycoproteins are digested with specific proteases such as trypsin, resulting in (glyco)peptides that are suitable for LC-MS/MS analysis. Glycosylated peptides, however, have much lower ionization efficiency during MS analysis relative to peptides, and thus milligram quantities of materials are generally used for typical glycoproteomics methods29,61. Characterization of glycopeptides with multiple glycosites by MS/MS is still challenging, even with combination of different types of fragmentation techniques28. Quantitative measurement of glycopeptides is complicated by the fact that ionization efficiencies of glycopeptides differ with variable glycoforms38. This protocol describes an alternative way to overcome these problems by the use of combination proteolytic digestion followed by sequential endoglycosidase treatment.
Validation of sequential endoglycosidase treatment
One of the distinguishing features of this protocol is the use of sequential treatment of glycopeptides with endoglycosidases (Endo H followed by PNGase F) to create unique mass signatures for glycosites that have no N-glycan, high-mannose-type glycan, or complex-type glycan. This strategy converts the glycoproteomics analysis to a proteomics analysis, resulting in higher sensitivity of the protocol (only 30 μg of sample is needed for a complete analysis). The key to success is that the sequential endoglycosidase treatments proceed to completion, avoiding mis-assignment of N-glycan processing status. To this end, we applied the protocol to assess site-specific N-glycan processing of two well-characterized model glycoproteins, invertase produced by the yeast S. cerevisiae and α-1-acid glycoprotein from bovine serum70. Glycosites on invertase are occupied by underprocessed oligomannose, and those on α-1-acid glycoprotein are fully processed complex-type glycosylation (Fig. 3). All N-glycosites on both glycoproteins were identified with multiple MS/MS spectra ranging from 54 to >10,000 per site (Supplementary Tables 2 and 3). Low percentages of spectra hits that contain non-enzymatic deamidation or 18O-incorporation into the C termini of peptides were found among all spectra hits identified, which is attributed to the complete denaturation of proteases used (Supplementary Tables 4 and 5). As described in the procedure section, as well as the previous study10, a set of peptides with N+0, N+3, and N+203 modifications was considered only when at least one of the three had a peak area of at least 5 × 108. As expected, the 14 N-glycosites of invertase were identified as entirely high-mannose-type glycosylation, and site occupancy was >90% for all glycosites except the sites N64 and N275 (Fig. 3a,b). By contrast, the five N-glycosites of α-1-acid glycoprotein were completely complex-type glycosylation, and site occupancy was >98% for all five sites (Fig. 3c,d). These results indicated that sequential endoglycosidase treatment reached completion.
Figure 3. Validation of sequential endoglycosidase treatment.

(a) Scatter plot of the site-specific N-glycan processing of invertase produced by the yeast Saccharomyces cerevisiae. (b) Color-coded bar graph of the site-specific N-glycan processing of invertase. (c) Scatter plot of the site-specific N-glycan processing of α-1-acid glycoprotein. (d) Color-coded bar graph of the site-specific N-glycan processing of α-1-acid glycoprotein. A set of peptides with N+0, N+3, and N+203 modifications was displayed only when at least one of the three had a peak area of at least >5 × 108. Data were obtained from six independent experiments. Mean ± s.e.m. were plotted. a,b adapted from ref. 10, Nature Publishing Group.
Validation of MS detection of glycotypes
Another major assumption is that the endoglycosidase-treated peptide glycosites that are unoccupied or occupied by high-mannose glycans or complex-type glycans are detected equally during MS analysis. To test this assumption, the HIV-1 Env trimer BG505 SOSIP.664, expressed in the presence of kifunensine (high-mannose only), was selected as a model protein (hereafter referred to as Kif_BG505). N-glycans on Kif_BG505 were first removed by sequential endoglycosidase treatment, and as expected, glycosites comprised >95% high-mannose-type glycosylation (green bars), indicating that the kifunensine treatment was effective and Kif_BG505 had high-mannose glycans (Fig. 4a). On the other hand, PNGase F treatment only was also applied to release N-glycans on Kif_BG505, resulting in peptides with homogeneous N+3 modification (>98% of purple bars, Fig. 4b). The resulting two samples were then mixed at a molar ratio of 1:1 in order to assess MS detection of glycotypes (Fig. 4c). Peptides with N+3 and N+203 modifications are both detectable for each glycosite, with a ratio of 1.0 to 1.2, suggesting slightly increased sensitivity for peptides with the N+3 modification. Synthetic peptides that carry asparagine (unoccupied), aspartic acid (PNGase F–treated), and N-acetylglucosamine-linked asparagine residues at the glycosylation site, display similar ionization efficiencies during ESI-MS analysis, further indicating that the protocol is able to semiquantitatively assess site-specific N-glycan processing for glycoproteins61.
Figure 4. Validation of MS detection of glycotypes.

(a) Site-specific N-glycan processing of Kif_BG505 that was treated with Endo H followed by PNGase F. (b) Site-specific glycosylation of Kif_BG505 that was treated with PNGase F only. (c) MS detection of peptides that contain N+3 and N+203 modifications at a molar ratio of 1:1. Peptides that had potential glycosites but were not glycosylated were not included. The proportions of high-mannose and complex-type glycans at the glycosites highlighted in yellow were assigned based on the proportion of spectra hits because peak area did not reach the threshold of 5 × 108. Data were obtained from nine independent experiments. Mean ± s.e.m. were plotted. Image adapted from ref. 10, Nature Publishing Group.
Examples of the protocol
Although the protocol was initially developed for analysis of site-specific N-glycan processing of the HIV Env trimer10, it is applicable to analysis of site-specific N-glycan processing of recombinant glycoprotein therapeutics (Fig. 5a,b), serum glycoproteins (Fig. 5c,d), and soluble or membrane-bound envelope glycoproteins from viruses (Fig. 6). It is also likely to be useful in characterization of glycoprotein processing in more complex systems such as whole cells due to its high sensitivity.
Figure 5. Application of the protocol for characterization of site-specific N-glycan processing of recombinant glycoprotein therapeutics and serum glycoproteins.

(a–d) Site-specific N-glycan processing of the recombinant glycoprotein therapeutics IgG (a) and IgM (b), as well as the serum glycoproteins transferrin (c) and fetuin (d). A set of peptides with N+0, N+3, and N+203 modifications was displayed only when at least one of the three had a peak area of at least >5 × 108. Data were obtained from at least six independent experiments. Mean ± s.e.m. were plotted. d adapted from ref. 10, Nature Publishing Group.
Figure 6. Application of the protocol for characterization of site-specific N-glycan processing of virus envelope glycoproteins.

(a–c) Site-specific N-glycan processing of recombinant envelope glycoproteins derived from viruses, including the prefusion-stabilized spike glycoprotein ectodomain of Middle East respiratory syndrome coronavirus (MERS-CoV S-2P protein, a), influenza virus hemagglutinin from H3N2 strain A/Victoria/361/2011 (b), and HIV-1 envelope glycoprotein (c). The proportions of high-mannose and complex-type glycans at the glycosites highlighted in yellow were assigned based on the proportion of spectra hits because peak area did not reach the threshold of 5 × 108. Data were obtained from at least six independent experiments. Mean ± s.e.m. were plotted. b,c adapted from ref. 10, Nature Publishing Group.
Site-specific N-glycan processing of recombinantly produced therapeutic glycoproteins, including IgG and IgM, was determined (Fig. 5a,b). Human serum IgG, which contains IgG1-4 with IgG1 and IgG2 as the major isotypes, was found to be entirely complex-type glycosylation, consistent with previous studies (Fig. 5a)32,49,52. Of the five N-glycosites on IgM, which is the major antibody produced in the primary immune response, three sites were shown to be completely occupied by complex-type structures (N171, N332, and N395), whereas the other two sites, N402 and N563, were primarily high-mannose-type glycosylation (Fig. 5b). The glycosite N563 that is proximal to the CH4 domain and thus is a poor substrate for OST was found to be partially glycosylated, in agreement with previous studies24,71.
Abnormal glycosylation of serum glycoproteins is a common feature in various human diseases, such as cancers and congenital disorders of glycosylation (CDGs). In particular, serum transferrin was first used to diagnose abnormal glycosylation in CDG patients in the 1990s72. Normal transferrin has two N-glycosylation sites, each of which is fully occupied73,74, whereas in type I CDGs, an increase of mono-glycosylated transferrin was found due to defects of oligosaccharide assembly and transfer to glycoproteins in those patients75. Analysis of site-specific N-glycan processing of commercially available human serum transferrin revealed that the two glycosites of this protein were entirely complex-type glycosylation (Fig. 5c), in line with the previous studies73,74. Another serum glycoprotein, fetuin, was also found to be entirely complex-type glycosylation, with full occupancy of two sites (N99 and N156) and partial glycosylation of the third glycosite (N176, 89% of site occupancy, Fig. 5d).
Membrane-bound envelope glycoproteins of various viruses, such as MERS-CoV S-2P protein and HIV Env trimer, are the target of neutralizing antibodies, and thus are the focus of vaccine development55,69. N-linked glycans on those envelope glycoproteins serve as a shield to protect the underlying protein from immune surveillance and thus confound development of effective vaccines for those viruses54. Importantly, some neutralizing antibodies to those viruses have glycan-dependent epitopes15,57,69, suggesting that vaccine design efforts would benefit greatly from understanding the N-glycan processing status at each glycosylation site. MERS-CoV S-2P protein is a large trimer (∼600 kDa) with ∼25 N-linked glycans per monomer, each of which comprises two noncovalently associated subunits, S1 and S269. Characterization of site-specific N-glycan processing of a prefusion-stabilized MERS-CoV S-2P protein ectodomain (MERS S-2P) revealed that all 23 glycosites on the protein were fully occupied, except for the N104 site (Fig. 6a). High-mannose glycans were predominantly found in the S1-NTD (residues 18–353), whereas other regions of the S protein, including the RBD (367–588), the two subdomains (589–751), and S2 (752–1,291), contained glycosites occupied largely by complex-type glycans (Fig. 6a). The glycan N1176, which is in the epitope for antibody G4 and was reported to mask antibody recognition69, was found to have a complex-type structure. Of note, we did not observe that the proteases used had biases on specific glycotypes (Supplementary Fig. 2). Of the 12 glycosites on the recombinant influenza haemagglutinin (HA) of A/Victoria/361/2011, three were >85% high-mannose (N45, N165, and N285), four were fully complex-type glycosylation (N22, N38, N63, and N483), and the rest were occupied by a mixture of high-mannose and complex-type glycans (Fig. 6b). It is striking to observe that the site N122 was not occupied and another site N144 was only 32% occupied on this HA glycoprotein. We also applied the protocol to the benchmark HIV Env trimer BG505 SOSIP.664, resulting in identification of all 28 glycosites with up to 2,000 spectra hits per glycosite (Fig. 6c). All 28 glycosites were >90% occupied, except the sites N185e, N197, N618, and N625. Of those that were largely occupied, 14 were >75% high-mannose, four were >75% complex-type glycosylation, and six other sites had a mixture of high-mannose and complex-type glycans. In particular, the glycosites, N295, N332, N339, N386, and N392, in the high-mannose patch region, were found to be occupied predominantly by underprocessed oligomannose, consistent with previous studies14,76. The N160 glycan, which is critical for binding of the bnAbs PG9 and PG16 to HIV Env, was composed predominantly of high-mannose structures, confirming the glycan composition at this site described in previous structural studies58,77,78. Interestingly, the high-mannose and complex-type glycans identified at each glycosylation site of BG505 SOSIP.664 matched the pathway of N-glycan processing, in which high-mannose structures are first trimmed from Man9 to Man5 before addition of the terminal monosaccharides that define complex-type/hybrid glycans, as compared to the results of the same protein obtained on intact glycopeptide level29. Thus, the sites were predominantly occupied by Man9 if they were 100% high-mannose glycosylation, whereas other sites were occupied by mixtures of processed high-mannose structures (Man8 to Man5) and simple complex-type structures if they were occupied by a mixture of high-mannose and complex-type glycans10.
We believe this protocol will be of wide interest to the proteomics and glycomics fields, and will be used by many outside those fields who want to gain high-level information about the glycoproteins they investigate.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Further information on experimental design is available in the Life Sciences Reporting Summary.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Integrated supplementary information
{kind=link}
MS/MS spectra and fragment assignment of the (a) di-glycosylated, (b) tri-glycosylated, and (c) tetra-glycosylated peptides derived from BG505 SOSIP.664.
{kind=link}
(a) Triple digestion, (b) the combination of trypsin and chymotrypsin, (c) chymotrypsin. The proportions of high mannose and complex type glycans at those glycosites highlighted in yellow were assigned based on the proportion of spectra hits since peak area did not reach the threshold. Mean ± SEM were plotted.
Supplementary information
Supplementary Figures 1 and 2, and Supplementary Tables 1–5. (PDF 503 kb)
Life Science Reporting Summary (PDF 129 kb)
Acknowledgements
This work was supported by National Institutes of Health (NIH) grants R01AI113867 (J.C.P., J.R.Y. and W.R. Schief), UM1 AI100663 (D.R.B. and J.C.P.), R01AI127521 (J.S.M.), and P41 GM103533 (J.R.Y.).
Author Contributions
L.C., J.R.Y., and J.C.P. designed the research. L.C. prepared samples for MS analysis. J.K.D., L.C., C.M.D., and Y.M. performed the MS analysis. L.C. and S.-K.R.P. analyzed the data. N.W. and M.P. expressed and purified Env proteins. J.S.M., D.R.B., J.R.Y., and J.C.P. supervised the project. L.C. and J.C.P. wrote the manuscript.
Competing interests
The authors declare no competing financial interests.
References
- 1.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 2.Mathys L, Francois KO, Quandte M, Braakman I, Balzarini J. Deletion of the highly conserved N-glycan at Asn260 of HIV-1 gp120 affects folding and lysosomal degradation of gp120, and results in loss of viral infectivity. PLoS ONE. 2014;9:e101181. doi: 10.1371/journal.pone.0101181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science. 2001;291:2370–2376. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- 4.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 5.Balog CIA, et al. N-glycosylation of colorectal cancer tissues. Mol. Cell Proteomics. 2012;11:571–585. doi: 10.1074/mcp.M111.011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Leoz, M.L.A. et al. High-mannose glycans are elevated during breast cancer progression. Mol. Cell Proteomics10http://www.grits-toolbox.org/ (2011). [DOI] [PMC free article] [PubMed]
- 7.de Vries RP, et al. The influenza A virus hemagglutinin glycosylation state affects receptor-binding specificity. Virology. 2010;403:17–25. doi: 10.1016/j.virol.2010.03.047. [DOI] [PubMed] [Google Scholar]
- 8.Khatri K, et al. Integrated omics and computational glycobiology reveal structural basis for influenza A virus glycan microheterogeneity and host interactions. Mol. Cell. Proteomics. 2016;15:1895–1912. doi: 10.1074/mcp.M116.058016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wanzeck K, Boyd KL, McCullers JA. Glycan shielding of the influenza virus hemagglutinin contributes to immunopathology in mice. Am. J. Resp. Crit. Care. 2011;183:767–773. doi: 10.1164/rccm.201007-1184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao LW, et al. Global site-specific N-glycosylation analysis of HIV envelope glycoprotein. Nat. Commun. 2017;8:14954. doi: 10.1038/ncomms14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falkowska E, et al. Broadly neutralizing HIV antibodies define a glycan-dependent epitope on the prefusion conformation of gp41 on cleaved envelope trimers. Immunity. 2014;40:657–668. doi: 10.1016/j.immuni.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Julien JP, et al. Broadly neutralizing antibody PGT121 allosterically modulates CD4 binding via recognition of the HIV-1 gp120 V3 base and multiple surrounding glycans. PLoS Pathog. 2013;9:e1003342. doi: 10.1371/journal.ppat.1003342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garces F, et al. Structural evolution of glycan recognition by a family of potent HIV antibodies. Cell. 2014;159:69–79. doi: 10.1016/j.cell.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sok D, et al. Promiscuous glycan site recognition by antibodies to the high-mannose patch of gp120 broadens neutralization of HIV. Sci. Transl. Med. 2014;6:236ra63. doi: 10.1126/scitranslmed.3008104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blattner C, et al. Structural delineation of a quaternary, cleavage-dependent epitope at the gp41-gp120 interface on intact HIV-1 Env trimers. Immunity. 2014;40:669–680. doi: 10.1016/j.immuni.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morell AG, Gregoriadis G, Scheinberg IH, Hickman J, Ashwell G. The role of sialic acid in determining the survival of glycoproteins in the circulation. J. Biol. Chem. 1971;246:1461–1467. [PubMed] [Google Scholar]
- 17.Doores KJ, et al. Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc. Natl. Acad. Sci. USA. 2010;107:13800–13805. doi: 10.1073/pnas.1006498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritchard LK, et al. Structural constraints determine the glycosylation of HIV-1 envelope trimers. Cell Rep. 2015;11:1604–1613. doi: 10.1016/j.celrep.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raska M, et al. Glycosylation patterns of HIV-1 gp120 depend on the type of expressing cells and affect antibody recognition. J. Biol. Chem. 2010;285:20860–20869. doi: 10.1074/jbc.M109.085472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong L, et al. Expression-system-dependent modulation of HIV-1 envelope glycoprotein antigenicity and immunogenicity. J. Mol. Biol. 2010;403:131–147. doi: 10.1016/j.jmb.2010.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vance BA, Wu W, Ribaudo RK, Segal DM, Kearse KP. Multiple dimeric forms of human CD69 result from differential addition of N-glycans to typical (Asn-X-Ser/Thr) and atypical (Asn-X-cys) glycosylation motifs. J. Biol. Chem. 1997;272:23117–23122. doi: 10.1074/jbc.272.37.23117. [DOI] [PubMed] [Google Scholar]
- 22.Sun SS, Zhang H. Identification and validation of atypical N-glycosylation sites. Anal. Chem. 2015;87:11948–11951. doi: 10.1021/acs.analchem.5b03886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson DR, Atkinson PH, Grimes WJ. Major carbohydrate structures at five glycosylation sites on murine IgM determined by high resolution 1H-NMR spectroscopy. Arch. Biochem. Biophys. 1985;243:605–618. doi: 10.1016/0003-9861(85)90538-7. [DOI] [PubMed] [Google Scholar]
- 24.Arnold JN, et al. Human serum IgM glycosylation - Identification of glycoforms that can bind to mannan-binding lectin. J. Biol. Chem. 2005;280:29080–29087. doi: 10.1074/jbc.M504528200. [DOI] [PubMed] [Google Scholar]
- 25.An Y, McCullers JA, Alymova I, Parsons LM, Cipollo JF. Glycosylation analysis of engineered H3N2 influenza A virus hemagglutinins with sequentially added historically relevant glycosylation sites. J. Proteome Res. 2015;14:3957–3969. doi: 10.1021/acs.jproteome.5b00416. [DOI] [PubMed] [Google Scholar]
- 26.Parsons LM, An Y, de Vries RP, de Haan CA, Cipollo JF. Glycosylation characterization of an influenza H5N7 hemagglutinin series with engineered glycosylation patterns: implications for structure-function relationships. J. Proteome Res. 2017;16:398–412. doi: 10.1021/acs.jproteome.6b00175. [DOI] [PubMed] [Google Scholar]
- 27.Go EP, et al. Glycosylation benchmark profile for HIV-1 envelope glycoprotein production based on eleven Env trimers. J. Virol. 2017;91:e02428-16. doi: 10.1128/JVI.02428-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Go EP, et al. Comparative analysis of the glycosylation profiles of membrane-anchored HIV-1 envelope glycoprotein trimers and soluble gp140. J. Virol. 2015;89:8245–8257. doi: 10.1128/JVI.00628-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Behrens AJ, et al. Composition and antigenic effects of individual glycan sites of a trimeric HIV-1 envelope glycoprotein. Cell Rep. 2016;14:2695–2706. doi: 10.1016/j.celrep.2016.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaia J. Mass spectrometry and the emerging field of glycomics. Chem. Biol. 2008;15:881–892. doi: 10.1016/j.chembiol.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aldredge D, An HJ, Tang N, Waddell K, Lebrilla CB. Annotation of a serum N-glycan library for rapid identification of structures. J. Proteome Res. 2012;11:1958–1968. doi: 10.1021/pr2011439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao LW, et al. Sample preparation for mass spectrometric analysis of human serum N-glycans using hydrophilic interaction chromatography-based solid phase extraction. Analyst. 2014;139:4538–4546. doi: 10.1039/c4an00660g. [DOI] [PubMed] [Google Scholar]
- 33.Pang PC, et al. Human sperm binding is mediated by the sialyl-Lewis(x) oligosaccharide on the zona pellucida. Science. 2011;333:1761–1764. doi: 10.1126/science.1207438. [DOI] [PubMed] [Google Scholar]
- 34.Ashline D, Singh S, Hanneman A, Reinhold V. Congruent strategies for carbohydrate sequencing. 1. Mining structural details by MSn. Anal. Chem. 2005;77:6250–6262. doi: 10.1021/ac050724z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang HT, Wu SL, Karger BL, Hancock WS. Characterization of the glycosylation occupancy and the active site in the follow-on protein therapeutic: TNK-tissue plasminogen activator. Anal. Chem. 2010;82:6154–6162. doi: 10.1021/ac100956x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bones J, Mittermayr S, O′Donoghue N, Guttman A, Rudd PM. Ultra performance liquid chromatographic profiling of serum N-Glycans for fast and efficient identification of cancer associated alterations in glycosylation. Anal. Chem. 2010;82:10208–10215. doi: 10.1021/ac102860w. [DOI] [PubMed] [Google Scholar]
- 37.Zauner G, Deelder AM, Wuhrer M. Recent advances in hydrophilic interaction liquid chromatography (HILIC) for structural glycomics. Electrophoresis. 2011;32:3456–3466. doi: 10.1002/elps.201100247. [DOI] [PubMed] [Google Scholar]
- 38.Kolarich D, Jensen PH, Altmann F, Packer NH. Determination of site-specific glycan heterogeneity on glycoproteins. Nat. Protoc. 2012;7:1285–1298. doi: 10.1038/nprot.2012.062. [DOI] [PubMed] [Google Scholar]
- 39.Huffman JE, et al. Comparative performance of four methods for high-throughput glycosylation analysis of immunoglobulin G in genetic and epidemiological research. Mol. Cell Proteomics. 2014;13:1598–1610. doi: 10.1074/mcp.M113.037465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 41.Kaji H, et al. Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat. Biotechnol. 2003;21:667–672. doi: 10.1038/nbt829. [DOI] [PubMed] [Google Scholar]
- 42.Wada Y, Tajiri M, Yoshida S. Hydrophilic affinity isolation and MALDI multiple-stage tandem mass spectrometry of glycopeptides for glycoproteomics. Anal. Chem. 2004;76:6560–6565. doi: 10.1021/ac049062o. [DOI] [PubMed] [Google Scholar]
- 43.Deeb SJ, Cox J, Schmidt-Supprian M, Mann M. N-linked glycosylation enrichment for in-depth cell surface proteomics of diffuse large B-cell lymphoma subtypes. Mol. Cell Proteomics. 2014;13:240–251. doi: 10.1074/mcp.M113.033977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 45.Sun SS, et al. Comprehensive analysis of protein glycosylation by solid-phase extraction of N-linked glycans and glycosite-containing peptides. Nat. Biotechnol. 2016;34:84–88. doi: 10.1038/nbt.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thaysen-Andersen M, Packer NH, Schulz BL. Maturing glycoproteomics technologies provide unique structural insights into the N-glycoproteome and its regulation in health and disease. Mol. Cell Proteomics. 2016;15:1773–1790. doi: 10.1074/mcp.O115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang N, et al. Quantitation of site-specific glycosylation in manufactured recombinant monoclonal antibody drugs. Anal. Chem. 2016;88:7091–7100. doi: 10.1021/acs.analchem.6b00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Panico M, et al. Mapping the complete glycoproteome of virion-derived HIV-1 gp120 provides insights into broadly neutralizing antibody binding. Sci. Rep. 2016;6:32956. doi: 10.1038/srep32956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao LW, et al. Application of a strong anion exchange material in electrostatic repulsion-hydrophilic interaction chromatography for selective enrichment of glycopeptides. J. Chromatogr. A. 2013;1299:18–24. doi: 10.1016/j.chroma.2013.05.037. [DOI] [PubMed] [Google Scholar]
- 50.Sun BY, et al. Shotgun glycopeptide capture approach coupled with mass spectrometry for comprehensive glycoproteomics. Mol. Cell Proteomics. 2007;6:141–149. doi: 10.1074/mcp.T600046-MCP200. [DOI] [PubMed] [Google Scholar]
- 51.Thaysen-Andersen M, et al. Human neutrophils secrete bioactive paucimannosidic proteins from azurophilic granules into pathogen-infected sputum. J. Biol. Chem. 2015;290:8789–8802. doi: 10.1074/jbc.M114.631622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao LW, et al. N-glycosylation site analysis of proteins from Saccharomyces cerevisiae by using hydrophilic interaction liquid chromatography-based enrichment, parallel deglycosylation, and mass spectrometry. J. Proteome Res. 2014;13:1485–1493. doi: 10.1021/pr401049e. [DOI] [PubMed] [Google Scholar]
- 53.Wei XP, et al. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 54.Sok D, et al. A prominent site of antibody vulnerability on HIV envelope incorporates a motif associated with CCR5 binding and its camouflaging glycans. Immunity. 2016;45:31–45. doi: 10.1016/j.immuni.2016.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burton DR, Hangartner L. Broadly neutralizing antibodies to HIV and their role in vaccine design. Annu. Rev. Immunol. 2016;34:635–659. doi: 10.1146/annurev-immunol-041015-055515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCoy LE, McKnight A. Lessons learned from humoral responses of HIV patients. Curr. Opin. HIV AIDS. 2017;12:195–202. doi: 10.1097/COH.0000000000000361. [DOI] [PubMed] [Google Scholar]
- 57.Julien JP, et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kong L, et al. Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nat. Struct. Mol. Biol. 2013;20:796–803. doi: 10.1038/nsmb.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amin MN, et al. Synthetic glycopeptides reveal the glycan specificity of HIV-neutralizing antibodies. Nat. Chem. Biol. 2013;9:521–526. doi: 10.1038/nchembio.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mouquet H, et al. Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc. Natl. Acad. Sci. USA. 2012;109:E3268–E3277. doi: 10.1073/pnas.1217207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stavenhagen K, et al. Quantitative mapping of glycoprotein micro-heterogeneity and macro-heterogeneity: an evaluation of mass spectrometry signal strengths using synthetic peptides and glycopeptides. J. Mass Spectrom. 2013;48:627–639. doi: 10.1002/jms.3210. [DOI] [PubMed] [Google Scholar]
- 62.Behrens AJ, et al. Molecular architecture of the cleavage-dependent mannose patch on a soluble HIV-1 envelope glycoprotein trimer. J. Virol. 2017;91:e01894-16. doi: 10.1128/JVI.01894-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eshghi ST, Shah P, Yang WM, Li XD, Zhang H. GPQuest: a spectral library matching algorithm for site-specific assignment of tandem mass spectra to intact N-glycopeptides. Anal. Chem. 2015;87:5181–5188. doi: 10.1021/acs.analchem.5b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khatri K, Klein JA, Zaia J. Use of an informed search space maximizes confidence of site-specific assignment of glycoprotein glycosylation. Anal. Bioanal. Chem. 2017;409:607–618. doi: 10.1007/s00216-016-9970-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hao PL, Ren Y, Alpert AJ, Sze SK. Detection, evaluation and minimization of nonenzymatic deamidation in proteomic sample preparation. Mol. Cell. Proteomics. 2011;10:O111.00938. doi: 10.1074/mcp.O111.009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.MacCoss MJ, et al. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc. Natl. Acad. Sci. USA. 2002;99:7900–7905. doi: 10.1073/pnas.122231399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Angel PM, Lim JM, Wells L, Bergmann C, Orlando R. A potential pitfall in 18O-based N-linked glycosylation site mapping. Rapid Comm. Mass Spectrom. 2007;21:674–682. doi: 10.1002/rcm.2874. [DOI] [PubMed] [Google Scholar]
- 68.Wolters DA, Washburn MP, Yates JR. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001;73:5683–5690. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 69.Pallesen J, et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA. 2017;114:E7348–E7357. doi: 10.1073/pnas.1707304114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Imre T, et al. Glycosylation site analysis of human alpha-1-acid glycoprotein (AGP) by capillary liquid chromatography-electrospray mass spectrometry. J. Mass Spectrom. 2005;40:1472–1483. doi: 10.1002/jms.938. [DOI] [PubMed] [Google Scholar]
- 71.Pabst M, et al. A microarray-matrix-assisted laser desorption/ionization-mass spectrometry approach for site-specific protein N-glycosylation analysis, as demonstrated for human serum immunoglobulin M (IgM) Mol. Cell Proteomics. 2015;14:1645–1656. doi: 10.1074/mcp.O114.046748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang NLS. Chemical diagnostics from bench to bedside preface. Top. Curr. Chem. 2014;336:V–VI. [Google Scholar]
- 73.Satomi Y, Shimonishi Y, Hase T, Takao T. Site-specific carbohydrate profiling of human transferrin by nano-flow liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2004;18:2983–2988. doi: 10.1002/rcm.1718. [DOI] [PubMed] [Google Scholar]
- 74.Nilsson J, et al. Enrichment of glycopeptides for glycan structure and attachment site identification. Nat. Methods. 2009;6:809–811. doi: 10.1038/nmeth.1392. [DOI] [PubMed] [Google Scholar]
- 75.Hulsmeier AJ, Tobler M, Burda P, Hennet T. Glycosylation site occupancy in health, congenital disorder of glycosylation and fatty liver disease. Sci. Rep. 2016;6:33927. doi: 10.1038/srep33927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guttman M, et al. CD4-induced activation in a soluble HIV-1 Env trimer. Structure. 2014;22:974–984. doi: 10.1016/j.str.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pancera M, et al. Structural basis for diverse N-glycan recognition by HIV-1-neutralizing V1-V2-directed antibody PG16. Nat. Struct. Mol. Biol. 2013;20:804–813. doi: 10.1038/nsmb.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McLellan JS, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480:336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MS/MS spectra and fragment assignment of the (a) di-glycosylated, (b) tri-glycosylated, and (c) tetra-glycosylated peptides derived from BG505 SOSIP.664.
(a) Triple digestion, (b) the combination of trypsin and chymotrypsin, (c) chymotrypsin. The proportions of high mannose and complex type glycans at those glycosites highlighted in yellow were assigned based on the proportion of spectra hits since peak area did not reach the threshold. Mean ± SEM were plotted.
Supplementary Figures 1 and 2, and Supplementary Tables 1–5. (PDF 503 kb)
Life Science Reporting Summary (PDF 129 kb)
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Further information on experimental design is available in the Life Sciences Reporting Summary.