

Abstract
Background
Accurately predicting the impact of rare non-synonymous variants on disease risk is an important goal in precision medicine. Variants in the cardiac sodium channel SCN5A (Nav1.5) are associated with multiple arrhythmia disorders, including Brugada Syndrome (BrS1) and Long QT Syndrome (LQT3). Rare SCN5A variants also occur in approximately 1% of unaffected individuals. We hypothesized that in vitro electrophysiologic functional parameters explain a statistically significant portion of the variability in disease penetrance.
Methods and Results
From a comprehensive literature review, we quantified the number of carriers presenting with and without disease for 1,712 reported SCN5A variants. For 356 variants, data were also available for five NaV1.5 electrophysiologic parameters: peak current, late/persistent current, steady state V1/2 of activation and inactivation, and recovery from inactivation. We found that peak and late current significantly associate with BrS1 (p < 0.001, rho = −0.44, Spearman’s rank test) and LQT3 disease penetrance (p < 0.001, rho = 0.37). Steady state V1/2 activation and recovery from inactivation associate significantly with BrS1 and LQT3 penetrance, respectively. Continuous estimates of disease penetrance align with the current American College of Medical Genetics classification paradigm.
Conclusions
NaV1.5 in vitro electrophysiologic parameters are correlated with BrS1 and LQT3 disease risk. Our data emphasize the value of in vitro electrophysiologic characterization and incorporating counts of affected and unaffected carriers to aid variant classification. This quantitative analysis of the electrophysiologic literature should aid the interpretation of NaV1.5 variant electrophysiologic abnormalities and help improve Nav1.5 variant classification.
Keywords: SCN5A, Brugada syndrome, long QT syndrome, electrophysiology, ion channel
Journal Subject Terms: Arrhythmias, Electrophysiology, Ion Channels/Membrane Transport, Genetics
Rare variants in SCN5A are implicated in several heart diseases, including type 3 long QT and Brugada syndromes (LQT3 and BrS1).1, 2 Rare SCN5A variants are also collectively present in ~1% of healthy individuals.3 As genetic testing becomes increasingly prevalent, the ability to predict which SCN5A variants are associated with disease will be useful and potentially actionable.4 Computational methods exist to predict variant pathogenicity, and these generally rely on a combination of variant features such as evolutionary conservation, allele frequency, and conservativeness of the amino acid change.5–7 While computational predictive models provide widely accepted supporting evidence for variant classification,8 they often struggle to predict the exact effects of the variants.9 This is due in part to the inability of models to account for the many factors that cause variable penetrance, including other genetic and environmental factors that contribute to risk.10, 11 The classification of variant pathogenicity, broadly defined, “flattens” information related to a carrier’s true risk of presenting with a disease. The insufficient prediction of pathogenicity also ignores the details of disease presentation, which diverges considerably between BrS1 and LQT3, with occasional overlap between the two.12
Patch clamp electrophysiology in heterologous expression systems is often used to characterize the function of ion channel variants. For NaV1.5, several parameters are typically measured, including peak current, late current, voltage shifts in activation and inactivation, and recovery from inactivation. Alterations in these parameters are often assumed to correlate with disease presentation, although the exact relationships between NaV1.5 electrophysiologic parameters and disease risk have not been systematically tested.13, 14
The probability a carrier presents with a disease lies on a continuous range from 0 (impossible) to 1 (certain). We hypothesize that variant pathogenicity can be estimated as a continuous variable and that it has a quantitative relationship to the degree of in vitro perturbation of NaV1.5 function across multiple in vitro electrophysiologic metrics. Here we use the term “penetrance”, analogous to the extent to which a genetic variant associates with a clinical phenotype in a kindred. To address this hypothesis, we curated a large dataset comprised of SCN5A variants characterized by patch clamp electrophysiology in heterologous expression systems. We then compared in vitro electrophysiologic perturbation and homogeneity of presentation to quantitatively establish a relationship between channel function and clinical presentation.
Methods
The data, analytic methods, and study materials have been made available to other researchers for purposes of reproducing the results or replicating the procedure. All analyses were done using the datasets provided in supplemental tables 1 and 2 available in the supplemental material and at [website to be assigned at time of publication].
Collection of the SCN5A variant dataset
On November 15, 2017, we searched Pubmed with the term “SCN5A or Nav1.5” and obtained abstracts from 2123 papers. A comprehensive manual review of the abstracts and papers identified 711 papers describing 1,028 variants, including 356 variants that had been functionally characterized by patch clamp electrophysiology. We supplemented this dataset with all SCN5A variants in the gnomAD database of population variation (http://gnomad.broadinstitute.org/; release 2.0), yielding a total of 1,712 variants. We searched each publication for the number of carriers of each variant, the number of unaffected and affected individuals with BrS1 and LQT3, and the functional electrophysiologic parameters peak current, V1/2 activation and inactivation, recovery from inactivation, and late/persistent current (Tables S1, S2, and S3). All recorded values were normalized to wild-type values reported in the same publications. We excluded papers that reported duplicate patients by searching for papers describing the same variant/phenotype from the same authors or institutions (Table S1). For each variant, we summed all instances for each category of unaffected (including carriers found in gnomAD), BrS1, and LQT3 (Table S2). Since gnomAD has a large population of drawn from several studies of common diseases (much more common than BrS1 or LQT3) we assume the subjects included are at most minimally enriched for BrS1 or LQT3 compared to the broader population and have therefore included them in the unaffected category. We further collected in silico pathogenicity predictions from four commonly used servers: SIFT,5 Polyphen-2,7 CADD,15 and PROVEAN.6 We also include basic local alignment search tool position-specific scoring matrix (BLAST-PSSM) for SCN5A16 and the per residue evolutionary rate,17 previously suggested to have great predictive value for predicting KCNQ1 functional perturbation,18 and point accepted mutation score (PAM).19 For Figure 1, variants with above 20% of carriers presenting with BrS1 or LQT3 were considered disease–associated, whereas variants with above 80% of carriers being unaffected were considered not disease–associated.
Figure 1.
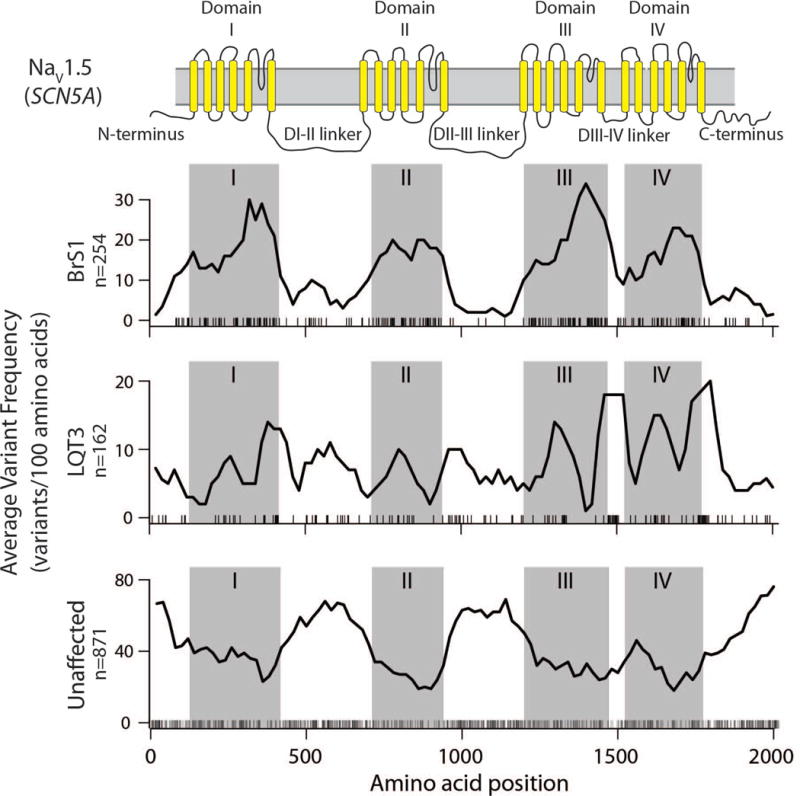
Dataset Variant location in SCN5A coding region (protein Nav1.5). Window count of variants averaged over 100 residues where at least 20% of carriers have been diagnosed with BrS1 (top), LQT3 (middle), or unaffected carriers (bottom). Residue sequence is shown along the x-axis and corresponding domains are highlighted above the figure and throughout as gray boxes. Individual variants used to calculate the sliding average line plot are shown as black ticks at the bottom of each of the three individual plots.
Data collected from functional studies of SCN5A
Most functionally characterized variants were characterized by heterologous expression in Human Embryonic Kidney (HEK) cells (291 of 356 total), so we used only patch clamp data derived in HEK cells when available. For variants where no HEK cell data was available, we used values generated from other cell types including Chinese Hamster Ovary (CHO), Xenopus oocytes, or human induced pluripotent cells, in that order of preference. We averaged the individual parameters in cases where multiple papers reported functional characterization of the same variant in the same cell system. Additionally, when values were reported in the context of multiple splice variants (e.g. with or without Q1077), or common variants such as H558R or R34C, we used functional parameters from the most common isoform, ΔQ1077/H558/R34 (https://www.gtexportal.org/home/), and excluded other measurements.
Calculation of probability distribution
We limited our analyses here to the two most prevalent SCN5A-associated diseases, BrS1 and LQT3. Data for SCN5A variant presence in cases of other diseases/symptoms was curated and available in Table S1. Besides BrS1 and LQT3, the most commonly reported diseases in SCN5A patients were atrial fibrillation, sudden infant death syndrome, sudden unexplained death syndrome, sick sinus syndrome, conduction disease, dilated cardiomyopathy, atrioventricular conduction block, and irritable bowel syndrome. The vast majority of patients in the dataset were either unaffected or presented with a single phenotype. Rare cases of overlap syndrome—individual patients presenting with both BrS1 and LQT3—were classified as either BrS1 or LQT3 according to their more severe phenotype.20 We then aggregated over all variants the number of carriers that present with one disease, yielding the number of carriers presenting with BrS1 or LQT3, the number of unaffected carriers, and the total number of carriers. We define penetrance as the number of affected carriers divided by the total number of carriers of a single variant (Figures S1 and S2). We then used an empirical Bayesian approach to approximate the penetrance of each variant, assuming a binomial distribution as the likelihood function and a beta distributed prior (Figures S3 and S4). Specifically, we calculated a by variant average penetrance using the entire dataset resulting in 24% and 10% penetrance for BrS1 or LQT3, respectively. We then chose beta distribution shape parameters α (defined as “b” in Figure S4) and β (defined as “n” in Figure S4) such that the resulting expectation value of the prior matched 24% and 10%. The resulting beta posterior distribution is slightly biased toward the marginal penetrance. This was done intentionally to offset possible selection bias in the variants that are characterized in the literature, while still allowing carrier count observations to quickly dominate the prior. The resulting penetrance values used in all analyses presented here correspond to penetrance expectation value. Additional information on Bayesian penetrance calculations is presented in Figure S4.
Sequence location of variants
To examine the location of disease-associated and control variants within SCN5A, we examined a 100 amino acid-wide sliding window across SCN5A from amino acid number 1 to 2016. For each window we calculated the number of missense variants that were BrS1-associated, LQT3-associated, or unaffected. Variants with at least a 20% posterior penetrance estimate for each disease were considered to be disease associated, and variants with less than a 20% combined BrS1 and LQT3 estimated penetrance were considered to be unaffected. Locations of transmembrane domains were obtained from UniProt (www.uniprot.org/uniprot/Q14524).
Variant classification
We used the American College of Medical Genetics (ACMG) guidelines8 to classify variants in our dataset by prevalence in population (estimated using gnomAD), in silico predicted pathogenicity, and enrichment of disease in carriers (Table S4). For each variant, we calculated four ACMG criteria: (1) BS1/PM2: variant rate in the general population is common/rare (based on gnomAD allele frequency), (2) BP4/PP3: sequence–based in silico prediction methods SIFT and Polyphen-2 classified as benign/deleterious, (3) PS4: enriched in affected population, (4) PP2: missense variant in gene with low rate of benign missense variants and where missense variants are a common mechanism of disease (true for all SCN5A variants). Likely pathogenic/pathogenic were classified based on PS4 (with the enrichment set to >20% of carriers presenting with either BrS1 or LQT3), PP2, PP3, and PM2. Variants of uncertain significance were classified based on PM2, BP4, and PP2. Likely benign/benign were classified based on PP2, BP4, or BS1 (Table S4). Next we classified variants according to the ACMG criteria as pathogenic/likely pathogenic, VUS, or benign/likely benign. We marginalized the posterior penetrance probability distribution over variants for each of the three classes. The resulting densities reflect a penetrance probability characteristic of each ACMG category. The University of Maryland annotation server (www.medschool.umaryland.edu/Genetic_Variant_Interpretation_Tool1.html/) was used to aid in these calculations.21
Weighted correlations and rank tests
We used a Spearman’s rank correlation coefficient (ρ) weighted by number of carriers to assess the relationship among functional parameters, in silico predictive models, and posterior penetrance estimates. We also fit linear regression models of BrS1, LQT3, or both BrS1 and LQT3 penetrance using features fit linearly or with restricted cubic spline. We use this approach rather than generalized mixed-effects models because we do not have patient-specific functional perturbation covariates. Fitting the patient level model in the dataset resulted in severe overfitting. For a more direct comparison between electrophysiologic features and in silico predictive models in predicting penetrance, we analyzed the subset of variants where functional data were available (maximum n = 215; Table 1). All calculations were performed in R; weighted Spearman’s rank correlation coefficient (rho) were calculated using the wCorr package. All weighting was calculated from
to ensure that variants with a greater number of carriers, and therefore greater certainty in penetrance, had a greater weight in the analysis.
Table 1.
Spearman rank order correlation by function perturbation and penetrance of BrS1, LQT3 or either BrS1 or LQT3
| Parameter | BrS1 or LQT3 (p-value) | BrS1 (p-value) | LQT3 (p-value) | n | |||
|---|---|---|---|---|---|---|---|
| Peak Current | −0.20/−0.18 | (0.008) | −0.44/−0.41 | (< 0.001) | 0.15/0.09 | (0.049)† | 212 |
| Late Current | 0.33/0.29 | (0.006) | 0.00/0.02 | (0.980) | 0.37/0.31 | (< 0.001) | 91 |
| Late Current (norm)* | 0.28/0.27 | (0.023) | −0.05/−0.04 | (0.674) | 0.33/0.28 | (0.005) | 86 |
| V1/2 Activation | 0.18/0.19 | (0.031) | 0.31/0.30 | (< 0.001) | −0.09/0.01 | (0.331) | 170 |
| V1/2 Inactivation | −0.09/−0.09 | (0.273) | −0.14/−0.13 | (0.082) | 0.03/0.00 | (0.685) | 193 |
| Rec. from Inact. | −0.12/−0.10 | (0.269) | 0.17/0.16 | (0.088) | −0.25/0.20 | (0.010)† | 127 |
| Evolution Rate | −0.46/−0.45 | (< 0.001) | −0.29/−0.30 | (< 0.001) | −0.26/0.31 | (< 0.001) | 224 |
| BLAST-PSSM | −0.24/−0.24 | (0.001) | −0.20/−0.22 | (0.004) | −0.08/−0.10 | (0.293) | 224 |
| PolyPhen-2 | 0.43/0.40 | (< 0.001) | 0.36/0.21 | (< 0.001) | 0.18/0.21 | (0.015) | 221 |
| PROVEAN | −0.46/0.45 | (< 0.001) | −0.34/−0.36 | (< 0.001) | −0.23/−0.25 | (0.007) | 221 |
| SIFT | −0.48/−0.48 | (< 0.001) | −0.35/−0.37 | (< 0.001) | −0.24/0.31 | (< 0.001) | 219 |
| CADD | 0.41/0.39 | (< 0.001) | 0.37/0.37 | (< 0.001) | 0.12/0.17 | (0.156) | 189 |
| PAM Score | −0.05/−0.03 | (0.538) | −0.08/−0.07 | (0.314) | 0.00/0.03 | (0.992) | 224 |
Normalized to peak current relative to WT
Bonferroni nominal p-value at the 0.05 threshold is p = 0.002
Correlations were calculated with two different priors. The Spearman rank order correlations are shown as calculated with “an empirical Bayesian prior”/”an uninformative prior”, to the left and right of the slash, respectively. Twenty-four of the 26 significant correlations with the empirical Bayesian prior were also significant with an uninformative prior. The two exceptions are indicated with †.
Results
Dataset summary
The collated dataset of SCN5A variants had 1,712 total variants, including 519 where at least one carrier presented with BrS1, 297 where at least one carrier presented with LQT3, and 278 where at least one carrier presented with a disease other than BrS1 or LQT3. Means and interquartile ranges for all functional parameters are listed in table S1. Interestingly, the median value of late current is 182% wild-type (WT) across all studied variants, suggesting a gain-of-function bias within the dataset. Further, the range of late current extends up to almost 4000% WT, with a 3rd quartile of 389% WT. Peak current median is 84% that of WT, suggesting a slight bias towards loss-of-function variants. We next explored the distribution of disease-associated variants within SCN5A. We calculated a 100 amino acid-wide sliding window average of the number of unaffected, BrS1-associated, and LQT3-associated variants to aid the eye in finding regions where disease-associated variants cluster (Figure 1). Interestingly, there was a marked enrichment of BrS1 variants within the four transmembrane domains, and a depletion of unaffected variants in these domains. LQT3-associated variants were prevalent in and near domains III and IV, as well as the DIII-IV linker, consistent with the observation that those segments are largely responsible for channel inactivation. Conversely, we observed a depletion of BrS1 and LQT3-associated variants and enrichment of unaffected variants in the D1-DII and DII-DIII interdomain linkers and the N- and C- terminal regions, suggesting that most variants in these regions are not disease-causing.
Penetrance Uncertainty
Because many variants have little clinical presentation data available, we employed a Bayesian strategy to estimate penetrance. This approach enables us to include prior information about relative background rate of BrS1 and LQT3. A benefit of this approach is that single observations are not interpreted as 100% or 0% penetrant for variants with a single carrier with disease or without phenotype, respectively. Further, with the addition of data (a greater number of carriers) the mean posterior value centers on the true fraction of carriers with disease and probability distributions narrow, reflecting the decrease in uncertainty (Figure 2). As expected, posterior penetrance probability distributions for variants with little patient data available were broad and therefore less certain, whereas for variants with more patient data the probability distributions are more narrow. For example, in Figure 2 D1790G and R1644C each have at least one carrier presenting with LQT3 and BrS1, but since there are more total carriers of D1790G, the distribution of posterior penetrance probability is much narrower. This phenomenon can also be seen when comparing distributions of the posterior penetrance probability for the same variant over time (Figure S5). As more patients presenting with and without disease are described in the literature the probability distributions become more narrow. Estimates of BrS1 and LQT3 penetrance for functionally characterized variants are presented in Figures S1 and S2, respectively.
Figure 2.
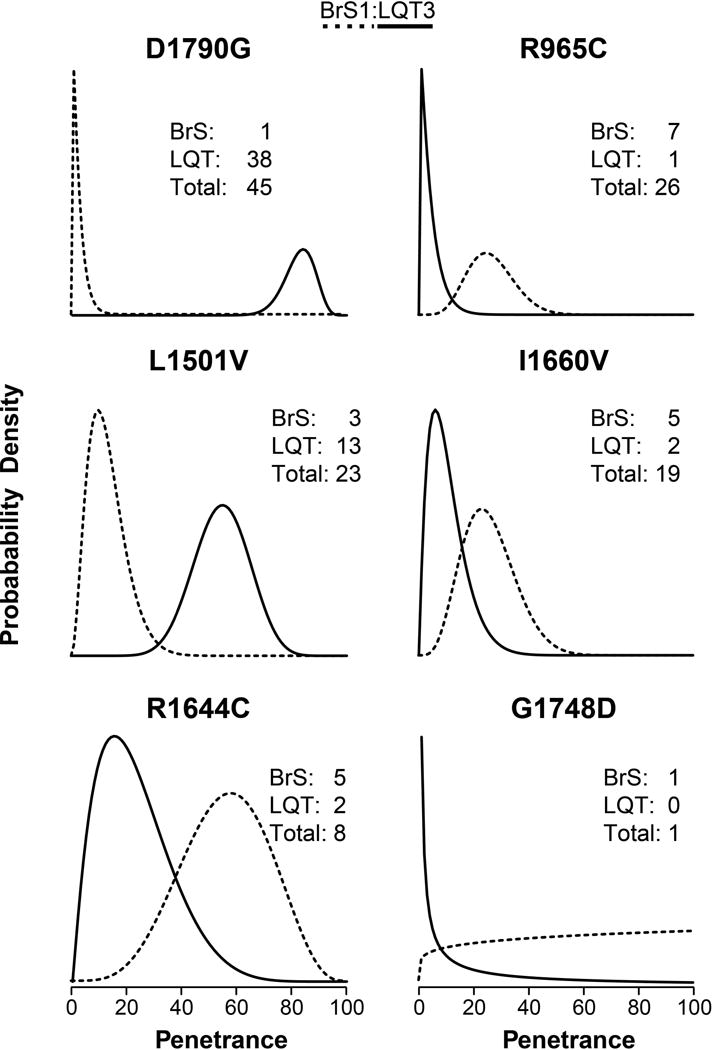
Penetrance probability distribution for six representative variants. The x-axes are penetrance values (the fraction of all carriers that present with either BrS1 or LQT3. The y-axes are probability density normalized such that the integral from x = 0 to x = 1 equals one. This figure is meant to illustrate some variants have enough carriers to be more certain about the risk of presentation, while some have fewer known carriers and thus more uncertainty shown as larger probability distributions. BrS1 penetrance probability density is shown as dashed lines; LQT3 penetrance probability density shown as a solid line.
Distributions of penetrance within ACMG classifications
We generated the individual densities for each variant classified using ACMG criteria as either pathogenic/likely pathogenic, VUS (variant of uncertain significance), or benign/likely benign. By marginalizing over each variant in a given class, we obtained a distribution of penetrance for variants within that class. As expected, variants classified as pathogenic were more likely to have a high penetrance than variants classified as benign or of uncertain significance. Variants classified as pathogenic had a 91% probability of penetrance over 20%, whereas variants classified as benign had only a 5% probability of penetrance over 20%. Variants of unknown significance fall between the two, with a 32% probability of penetrance over 20% (Figure 3).
Figure 3.
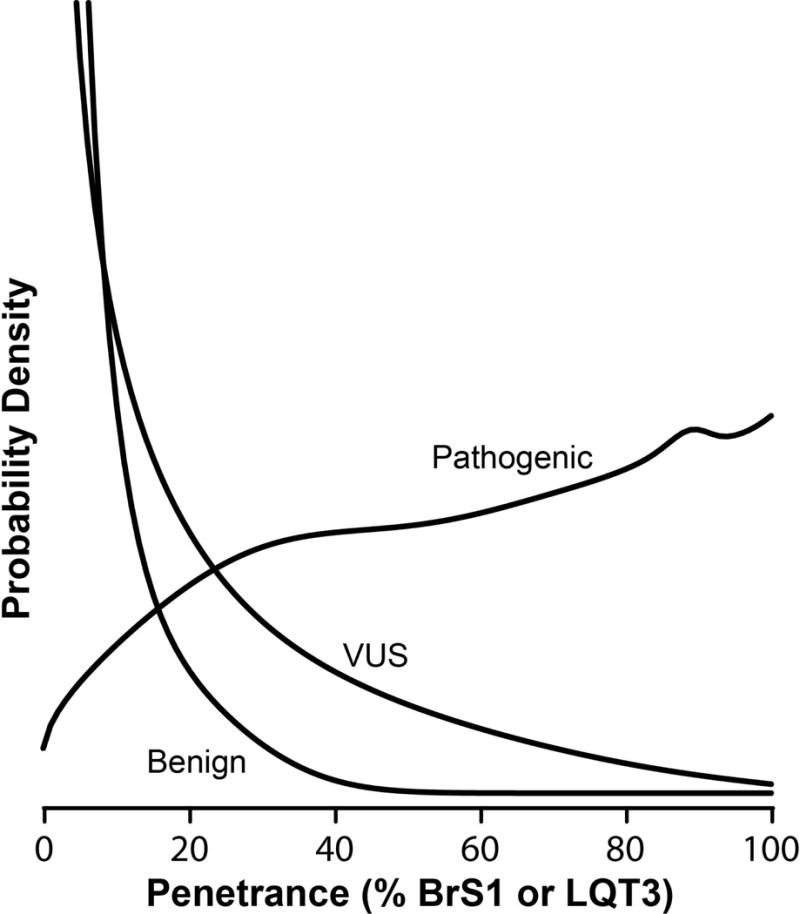
Penetrance distribution for a variant classifiable as likely benign, likely pathogenic, or variant of uncertain significance. 265, 33, and 262 variants are classifiable as likely pathogenic, benign, and VUS, respectively, using the classification criteria explained in the methods section.
Relationship between functional parameters and penetrance
We observed a continuous gradation of penetrance for both BrS1 and LQT3 (Figure S6). We hypothesized that this continuous gradation was due in part to the difference in the severity of the electrophysiological perturbations caused by each variant. To test this, we analyzed the relationship between variants’ functional parameters and their associations with disease (Table 1). Peak current and V1/2 activation are significantly associated with BrS1 penetrance (rho = −0.44 and 0.31, respectively; p < 0.001 for both). Increasing peak current decreases BrS1 penetrance, whereas a positive shift in V1/2 activation increases BrS1 penetrance. This finding supports the commonly held view that BrS1 is linked to a loss of overall NaV1.5 function. The only electrophysiological parameters that were statistically associated with LQT3 penetrance were late current and recovery from inactivation. Late/persistent current, often considered a predictor of LQT3,22, 23 had a significant correlation in the expected direction (rho = 0.37, p < 0.001). Shorter recovery from inactivation times, which would lead to a greater number of channels available to open in a given interval, predict increased LQT3 penetrance (rho = −0.25, p = 0.01). This is consistent with the common view that an increase in overall NaV1.5 activity can cause LQT3. V1/2 inactivation was not predictive of either BrS1 or LQT3. The observed correlations were robust to the choice of prior used to calculate the penetrance values (Table 1). Correlations for penetrance of BrS1 and LQT3 with respect to peak and late current, respectively, persisted in most penetrance subsets (Figure S7).
Relationship between in silico predictive models and penetrance
To compare the prediction accuracy of in silico predictive models and electrophysiological parameters, we analyzed a subset of the variants that could be assessed for each (missense variants that had electrophysiological measurements). Notably, for predicting BrS1 or LQT3 separately, functional parameters outperformed all in silico predictors. However, for predicting both LQT3 and BrS1 penetrance together, SIFT, Polyphen-2, CADD, PROVEAN, and evolutionary rate each outperform any single functional parameter (Table 1). The difference in performance is likely due to the fact that in silico predictive tools predict pathogenicity broadly, but do not account for the mechanisms of pathogenicity, which differ between BrS1 and LQT3.
Predictive models of BrS1 and LQT3 penetrance
We observed high pairwise correlations (50-70%, Pearson) between different in silico predictors (Figure S8). However there was much lower correlation between the in silico predictors and the functional parameters, suggesting the in silico predictors and functional parameters might contain complementary information about variant function. To test this, we built a series of models to predict BrS1 or LQT3 penetrance from functional parameters (peak current, recovery from inactivation, and late current) and an in silico predictor (PROVEAN), allowing restrictive cubic spline fitting to account for non-linear features. A chunk test on the resulting models suggest a statistically significant portion of the variable penetrance of BrS1 and LQT3 is explained by peak current and late current, respectively (Table S5). In contrast, PROVEAN alone has a weaker association with BrS1 and LQT3 penetrance, respectively, but improves on models that already include peak current or late current (Table S5).
Discussion
Estimating penetrance from patient data
The growth of genetic sequencing has led to a wealth of data in the literature describing patients with SCN5A variants who present either with specific diseases or as unaffected. For some SCN5A variants, there is clear evidence the variant is benign or pathogenic. For example, several common variants (e.g. H558R and R34C) exist at too high a frequency in the population to cause a highly penetrant form of a rare arrhythmia disorder, and are thus considered benign.24 Several rare variants (e.g. D1790G and E1784K) have been observed in dozens of patients with BrS1 or LQT3 and thus can be confidently annotated as pathogenic. However, the majority of variants fall in between these extremes: rare enough they could conceivably cause a rare arrhythmia disorder, but with only a small number of reported patients and no large families to generate statistically significant linkage to use to assess disease risk. Furthermore, the classification as pathogenic or not does not capture subtype of disease well. Incomplete penetrance of SCN5A variants further complicates the problem of inferring disease risk from sparse patient data.
To address the problem of the variants in our dataset with sparse patient data, we used a Bayesian method to calculate a posterior mean penetrance with a corresponding penetrance probability distribution. Penetrance is a useful quantitative metric to assess the risk a carrier of a rare variant in SCN5A will eventually present with BrS1 or LQT3. As expected, our penetrance predictions for variants with little patient data available were broad and less certain, whereas the penetrance predictions were tighter and more certain as more patient data were available (Figure S6). Variants in our dataset that can be classified as likely benign or benign have a much lower probability of having high penetrance, whereas variants classifiable as likely pathogenic or pathogenic are more likely to have high penetrance (Figure 3). The probability of high penetrance for the middle category—variants of uncertain significance—falls between the two. These findings suggest that penetrance captures the essential elements of the current classification scheme (benign, likely benign, VUS, likely pathogenic, and pathogenic)8 while providing more quantitative and precise information.
Significant correlations between NaV1.5 functional parameters and penetrance of BrS1/LQT3
Penetrance depends on many genetic and environmental factors, many of which are presently unknown.10, 11 In contrast to the binary pathogenic/benign classification scheme commonly in use, we observe that several variants are consistently associated with variable levels of penetrance. For example, in our dataset, patients with SCN5A haploinsufficiency (heterozygotes for a variant with 0% peak current) have a 70% likelihood of presenting with BrS1. Variants with partial loss of function are correlated with a lower penetrance of BrS1 (e.g. L657Q and R367H; Figures 4 and S6). The continuous relationship between the functional perturbations of peak current and late current in NaV1.5 and penetrance of BrS1 and LQT3, respectively, suggest carriers are more susceptible to presenting with these diseases in proportion to the degree of functional perturbation of the channel (Figures 4 and S9 and Tables S6 and 7).
Figure 4.
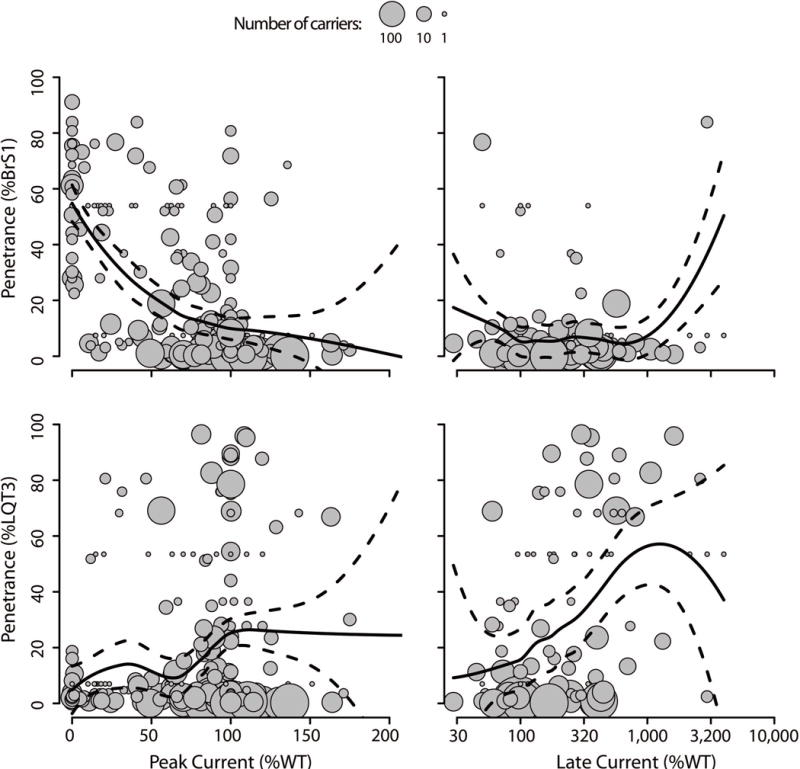
Degree of perturbation in peak current and late current versus BrS1 or LQT penetrance. Each grey circle is an SCN5A variant where the radius is equal to log10(number of carriers). The solid trend line is the locally weighted average of penetrance from functional perturbation; the dashed lines are 95% confidence intervals for the weighted average. Median absolute error for BrS1 penetrance by peak current and late current are 14 and 10%, respectively; Median absolute error for LQT3 penetrance by peak current and late current are 14 and 32%, respectively.”
Overlap variants associate with both BrS1 and LQT3
Most disease-associated variants in our dataset were linked to only BrS1 or LQT3. However several variants were associated with risk of both BrS1 and LQT3, typically in separate individuals (Figure S7). Many of these variants (e.g. E1784K, R1644C, and R689H) had a mix of the two canonical BrS1 and LQT3 electrocardiographic features, reduced peak current and increased late current.
Curiously weak correlations between NaV1.5 functional parameters and penetrance of BrS1/LQT3
Late current underlies the LQT3 presentation in many cases.25, 26 Several papers were published on variants found in LQT3, SIDS, or SUDS cases where, once functionally characterized, the late current was considered anomalously high (e.g. R1193Q27, 28 and F2004L29). Subsequent sequencing efforts, including the amalgamation of sequencing in gnomAD, revealed that some of these variants were too common in to be canonically pathogenic, leading to several variants with high late current, large carrier count, and very low LQT3 penetrance. There are several potential explanations for this unintuitive low correlation. Perhaps HEK cells, the most commonly used heterologous expression system, do not recapitulate the late current in the native human cardiomyocyte. Another possible explanation is that there are common compensatory mechanisms present in many carriers of these variants that blunt the effect of excessive sodium current.
In silico methods provide additional information. In silico predictive models perform much worse than peak current at correctly ranking BrS1 penetrance, but are competitive with late current at correctly predicting LQT3 penetrance. In silico predictive models, however, are not intended to distinguish between subclasses of diseases associated with “pathogenic” variants, which can explain the improved performance in predicting BrS1 or LQT3 simultaneously (Table 1; Figures S10 and S11). In contrast, peak current (or V1/2 activation) and late current or recovery from inactivation only correlate with BrS1 and LQT3, respectively, with no overlap between the two (Figure 4). This highlights the unique utility of functional parameters to provide greater information than broad pathogenicity prediction; however, in silico-based predictive models do provide additional information, as determined by lower AIC and p-values when PROVEAN is included in modeling BrS1/LQT3 penetrance (Table S5). One potential source for the additional information is that in silico-based models are implicating another mechanism modulating penetrance. A major component of PROVEAN, and many in silico-based predictive models, is the divergence of residue identity across species and similar proteins. This evolutionary pressure could impose a constraint on residue identity through mechanisms other than peak current, V1/2 activation, late current or recovery from inactivation, such as modulating binding partners, regulating phosphorylation or interacting with other cellular components.
Variant classification
Currently, ACMG criteria use functional defects as a binary measure to help classify variants. Since electrophysiological defects are multidimensional and continuous, there is a difficulty in understanding how severe an electrophyiological defect should be supporting evidence that a variant is pathogenic. In addition there is a growing recognition that variants can have a range of disease risk, from nearly 100% penetrant to a statistical risk factor.30 The literature curation approach and analyses presented in this work provide a framework for a quantitative understanding of how to translate continuous electrophysiological functional defects into disease risk. It is our hope this approach will help more accurately determine variant-specific disease risk for SCN5A and for other Mendelian disease genes.
Limitations in analysis
We were unable to collect age or sex information for many individuals in our dataset, and therefore have ignored these influencing factors. Further, we assume that the functional expression of variants in the most common isoform in a heterologous system is largely representative of the function of the variant in the human heart. BrS1 is known to affect males more than females and is more common later in life, so it is possible our BrS1 dataset is enriched in either of those categories. Another potential limitation is the restriction of a single predominant clinical presentation, here as BrS1 or LQT3, while not allowing for overlap. There are many instances where variants result in multiple, concomitant clinical presentations.31–33 Several publications did not describe in detail the clinical phenotypes and only presented disease classification. The likely consequence is that our results reflect a bias towards lower rate of BrS1 penetrance for the subset of variants strongly associated with LQT3.
Conclusion
The examination of penetrance, as defined here, in a set of over 1,700 variants from the SCN5A literature allows us to assert that there is a range of tolerated NaV1.5 perturbation. At one extreme, dramatic perturbations are not well tolerated and clinical presentations are largely predictable, more so for BrS1 than LQT3. However, modest perturbations result in varied clinical presentations including incomplete penetrance. This trend holds when comparing different variants, and within a population of carriers of a single variant. We suggest modeling probability of clinical presentation, here called penetrance, for carriers of SCN5A variants is feasible using Nav1.5 channel electrophysiologic features. We further suggest this approach will be especially useful for determining the risk a patient carrying a rare SCN5A variant will present with a disease.
Collected data are available in Table S1 and S2 and at the following website: (to be finalized with publication)
Supplementary Material
Clinical Perspective.
Electrophysiological defects in cardiac ion channels are multidimensional and continuous. At one extreme, dramatic perturbations are not well tolerated and clinical presentations are largely predictable; however, modest perturbations result in varied clinical presentations including incomplete penetrance. This leads to difficulty in determining how severe an electrophyiological defect should be to consider as supporting evidence that a variant is pathogenic. In addition, there is a growing recognition that variants impose a range of disease risk, from nearly 100% penetrant to a statistical risk factor. The literature curation approach and analyses presented in this work provide a framework for a quantitative understanding of how to translate continuous electrophysiological functional defects of the cardiac sodium ion channel SCN5A into disease risk. It is our hope this approach will help more accurately determine variant-specific disease risk for SCN5A and for other Mendelian disease genes.
Acknowledgments
We thank Joe-Elie Salem and Sarah Dobson for careful reading of the manuscript.
Sources of Funding: This research was funded by K99 HL135442 (BMK), F32 HL137385 (AMG) and HL118952 (DMR)
Footnotes
Disclosures: None
References
- 1.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337–48. doi: 10.1038/nrcardio.2009.44. [DOI] [PubMed] [Google Scholar]
- 2.Veerman CC, Wilde AA, Lodder EM. The cardiac sodium channel gene SCN5A and its gene product NaV1.5: Role in physiology and pathophysiology. Gene. 2015;573:177–87. doi: 10.1016/j.gene.2015.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, et al. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet. 2013;93:631–40. doi: 10.1016/j.ajhg.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 6.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PloS one. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gray VE, Kukurba KR, Kumar S. Performance of computational tools in evaluating the functional impact of laboratory-induced amino acid mutations. Bioinformatics. 2012;28:2093–6. doi: 10.1093/bioinformatics/bts336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tester DJ, Ackerman MJ. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation. 2011;123:1021–37. doi: 10.1161/CIRCULATIONAHA.109.914838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vink AS, Clur SB, Wilde AAM, Blom NA. Effect of age and gender on the QTc-interval in healthy individuals and patients with long-QT syndrome. Trends Cardiovasc Med. 2018;28:64–75. doi: 10.1016/j.tcm.2017.07.012. [DOI] [PubMed] [Google Scholar]
- 12.Nakaya H. SCN5A mutations associated with overlap phenotype of long QT syndrome type 3 and Brugada syndrome. Circ J. 2014;78:1061–2. doi: 10.1253/circj.cj-14-0319. [DOI] [PubMed] [Google Scholar]
- 13.Antzelevitch C, Belardinelli L. The role of sodium channel current in modulating transmural dispersion of repolarization and arrhythmogenesis. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S79–S85. doi: 10.1111/j.1540-8167.2006.00388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagatomo T, January CT, Makielski JC. Preferential block of late sodium current in the LQT3 DeltaKPQ mutant by the class I(C) antiarrhythmic flecainide. Mol Pharmacol. 2000;57:101–7. [PubMed] [Google Scholar]
- 15.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pupko T, Bell RE, Mayrose I, Glaser F, Ben-Tal N. Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics. 2002;18(Suppl 1):S71–7. doi: 10.1093/bioinformatics/18.suppl_1.s71. [DOI] [PubMed] [Google Scholar]
- 18.Li B, Mendenhall JL, Kroncke BM, Taylor KC, Huang H, Smith DK, et al. Predicting the Functional Impact of KCNQ1 Variants of Unknown Significance. Circ Cardiovasc Genet. 2017;10 doi: 10.1161/CIRCGENETICS.117.001754. pii: e001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dayhoff MO, Schwartz RM, Orcutt BC. A model of evolutionary change in proteins. Atlas of protein sequence and structure. 1978;5:345–351. [Google Scholar]
- 20.Veltmann C, Barajas-Martinez H, Wolpert C, Borggrefe M, Schimpf R, Pfeiffer R, et al. Further Insights in the Most Common SCN5A Mutation Causing Overlapping Phenotype of Long QT Syndrome, Brugada Syndrome, and Conduction Defect. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.116.003379. pii: e003379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleinberger J, Maloney KA, Pollin TI, Jeng LJ. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet Med. 2016;18:1165. doi: 10.1038/gim.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang H, Millat G, Rodriguez-Lafrasse C, Rousson R, Kugener B, Chevalier P, et al. Biophysical characterization of a new SCN5A mutation S1333Y in a SIDS infant linked to long QT syndrome. FEBS Lett. 2009;583:890–6. doi: 10.1016/j.febslet.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Dumaine R, Wang Q, Keating MT, Hartmann HA, Schwartz PJ, Brown AM, et al. Multiple mechanisms of Na+ channel–linked long-QT syndrome. Circ Res. 1996;78:916–24. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- 24.Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19:1151–1158. doi: 10.1038/gim.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baroudi G, Chahine M. Biophysical phenotypes of SCN5A mutations causing long QT and Brugada syndromes. FEBS Lett. 2000;487:224–8. doi: 10.1016/s0014-5793(00)02360-7. [DOI] [PubMed] [Google Scholar]
- 26.Wei J, Wang DW, Alings M, Fish F, Wathen M, Roden DM, et al. Congenital long-QT syndrome caused by a novel mutation in a conserved acidic domain of the cardiac Na+ channel. Circulation. 1999;99:3165–71. doi: 10.1161/01.cir.99.24.3165. [DOI] [PubMed] [Google Scholar]
- 27.Wang Q, Chen S, Chen Q, Wan X, Shen J, Hoeltge GA, et al. The common SCN5A mutation R1193Q causes LQTS-type electrophysiological alterations of the cardiac sodium channel. J Med Genet. 2004;41:e66. doi: 10.1136/jmg.2003.013300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang H, Zhao J, Barrane FZ, Champagne J, Chahine M. Nav1.5/R1193Q polymorphism is associated with both long QT and Brugada syndromes. Can J Cardiol. 2006;22:309–13. doi: 10.1016/s0828-282x(06)70915-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, et al. Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 2007;115:368–76. doi: 10.1161/CIRCULATIONAHA.106.646513. [DOI] [PubMed] [Google Scholar]
- 30.Kaab S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, et al. A large candidate gene survey identifies the KCNE1 D85N polymorphism as a possible modulator of drug-induced torsades de pointes. Circ Cardiovasc Genet. 2012;5:91–9. doi: 10.1161/CIRCGENETICS.111.960930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118:2219–29. doi: 10.1172/JCI34057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–13. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 33.Grant AO, Carboni MP, Neplioueva V, Starmer CF, Memmi M, Napolitano C, et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest. 2002;110:1201–9. doi: 10.1172/JCI15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.