

Abstract
An innovative approach for cardiac regeneration following injury is to induce endogenous cardiomyocyte (CM) cell cycle re-entry. In the present study, CMs from adult rat hearts were isolated and transfected with cel-miR-67 (control) and rno-miR-210. A significant increase in CM proliferation and mono-nucleation were observed in miR-210 group, in addition to a reduction in CM size, multi-nucleation, and cell death. When compared to control, β-catenin and Bcl-2 were upregulated while APC (adenomatous polyposis coli), p16, and caspase-3 were downregulated in miR-210 group. In silico analysis predicted cell cycle inhibitor, APC, as a direct target of miR-210 in rodents. Moreover, compared to control, a significant increase in CM survival and proliferation were observed with siRNA-mediated inhibition of APC. Furthermore, miR-210 overexpressing C57BL/6 mice (210-TG) were used for short-term ischemia/reperfusion study, revealing smaller cell size, increased mono-nucleation, decreased multi-nucleation, and increased CM proliferation in 210-TG hearts in contrast to wild-type (NTG). Likewise, myocardial infarction (MI) was created in adult mice, echocardiography was performed, and the hearts were harvested for immunohistochemistry and molecular studies. Compared to NTG, 210-TG hearts showed a significant increase in CM proliferation, reduced apoptosis, upregulated angiogenesis, reduced infarct size, and overall improvement in cardiac function following MI. β-catenin, Bcl-2, and VEGF (vascular endothelial growth factor) were upregulated while APC, p16, and caspase-3 were downregulated in 210-TG hearts. Overall, constitutive overexpression of miR-210 rescues heart function following cardiac injury in adult mice via promoting CM proliferation, cell survival, and angiogenesis.
Keywords: MiR-210, Cardiomyocyte, Adenomatous polyposis coli, Myocardial infarction
Introduction
Cardiovascular diseases (CVD) are the leading cause of death worldwide. According to the recent heart disease and stroke statistics report of American Heart Association, CVD-associated global mortality represents 17.3 million deaths each year. Its medical care costs more than $215 billion per year in the USA alone, that is expected to raise up to $1208 billion per year by 2030 [1]. Ischemic cardiomyopathy is one of the most common cardiac myopathies, originates due to inadequate oxygen and nutrient supply, resulting in myocardial infarction (MI). Following MI, active myocardium degenerates and is replaced by physiologically inactive fibrotic tissue leading to cardiac remodeling and heart failure. Even though several cell-based and non-cellular therapeutic strategies are under trial to treat ischemic heart diseases including stem cell transplantation, tissue engineering approaches, direct reprogramming of host fibroblasts, and mobilization of resident bone marrow cells [2], none of these have transitioned into routine clinical use.
At present, induction of cardiac regeneration following myocardial injury is one of the emerging areas of research with the potential of long-term favorable outcomes. Contrary to the long-held belief that the mammalian heart is a post-mitotic organ without any regeneration potential, recent studies in neonatal mice have shown cardiomyocytes (CMs) to proliferate up to day 7 after birth [3]. However, adult mammalian CMs become fully differentiated and lose their ability to proliferate, thereby are unable to regenerate/repair lost myocardium following ischemic and non-ischemic cardiac damage. Therefore, targeting induction of adult CM mitosis is a key therapeutic approach for cardiac regeneration. One of the novel strategies under extensive research is the use of microRNAs (miRs), to promote CM proliferation. Similar to studies using neonatal CMs [4], miRs also have the potential to induce adult CM cell cycle re-entry [5, 6]. MiRs are evolutionary conserved, small segments of ~ 22 nucleotide long non-coding eukaryotic RNAs, which regulate the expression of numerous genes via mRNA degradation and translational inhibition. So far, several miRs are documented to be involved in various cardiomyopathies leading to cardiac failure [7, 8]. Out of these, miR-210 was reported to be upregulated in animal models of atherosclerosis [9], cardiac hypertrophy, acute MI [10], and heart failure [11, 12]. Interestingly, earlier studies showed that intra-myocardial injection of miR-210 can improve heart function, reduces infarct area, prevents CM apoptosis, and induces angiogenesis post-MI in mice [13], suggesting miR-210 delivery might be cardioprotective during ischemic injury. Moreover, various broadly conserved gene targets of miR-210 have been identified, which are known to be associated with mitochondrial metabolism, DNA repair, cell survival, cell cycle progression, and angiogenesis [14]. These targets could significantly affect the development of major CVD, such as MI, which subsequently leads to heart failure. Notably, miR-210 has been previously implicated in activation of cell proliferation [15–21] revealing its potential for cell cycle induction. However, there is no published report to this date, supporting the role of miR-210 in adult CM cell cycle re-entry. Earlier reports showed that miR-210 is a master hypoxamir, as it is upregulated in various cell types during hypoxia [14] under the influence of hypoxia inducible factor-1-alpha (HIF-1α) [22–24]. Interestingly, a recent study has demonstrated that hypoxia promotes hyper-activation of HIF-1α and induction of endogenous CM proliferation leading to cardiac regeneration, reduction in myocardial fibrosis, and improvement of heart function post-cardiac injury in adult mice [25].
Altogether, these studies support the possible therapeutic potential of miR-210 for induction of CM proliferation, cell survival and angiogenesis leading to cardiac tissue regeneration. Thus, there is an essential need for in-depth analysis of miR-210-mediated signaling pathways that could possibly unfold the basic mechanism behind the induction of CM cell cycle re-entry. This strategy could be a novel step for the development of a promising therapeutic approach to treat ischemic cardiomyopathies. In the present study, for the first time, we report miR-210-mediated CM proliferation under in vitro and in vivo systems, along with cell survival and improved angiogenesis. Thus, our proposed strategy could be used as an effective therapeutic approach following cardiac damage to regenerate lost myocardium leading to restoration of heart function.
Materials and methods
All chemicals and laboratory reagents were purchased from Sigma-Aldrich, MO unless otherwise mentioned. Antibodies used in the current study were obtained from Sigma-Aldrich, MO (Rabbit anti-β-actin, Cat. A1978; rabbit anti-GAPDH, Cat. G9545; rabbit anti-Aurora B, Cat. A5102); Santa Cruz Biotechnology, TX (Rabbit anti-cardiac troponin-I, Cat. sc-15368; rabbit anti-VEGF, Cat. sc-152); Abcam, MA (Rabbit anti-p16, Cat. ab51243); Cell Signaling Technology, MA (Rabbit anti-APC, Cat. 2504S; rabbit anti-caspase-3, Cat. 9661S; rabbit anti-Bcl-2, Cat. 2870S; rabbit anti-β-catenin, Cat. 9562S); BD Pharmingen, CA (Mouse anti-Ki67, Cat. 550,609); GE Healthcare, IL (ECL anti-mouse-HRP, Cat. NA931V; ECL anti-rabbit-HRP, Cat. NA9340V); Life Technologies, CA (Goat anti-mouse Alexa fluor 488, Cat. A11029; goat anti-mouse Alexa fluor 594, Cat. A11005; goat anti-rabbit Alexa fluor 488, Cat. A11008; goat anti-rabbit Alexa fluor 594, Cat. A11037), and Thermo Scientific, MA (Mouse anti-cardiac troponin-T, Cat. MA5-12960).
All experimental animals were housed and maintained at Laboratory Animal Medical Services (LAMS), University of Cincinnati, Ohio, USA. The animals were fed ad libitum standard food and were utilized in accordance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC).
Adult rat cardiomyocyte (ARCM) isolation, transfection, and culture
Primary cultures of ventricular cardiomyocytes were obtained from 12-week-old Fisher rats. For the same, rats were euthanized, hearts were dissected and perfused using solution A (118 mM NaCl, 4.8 mM KCl, 25 mM HEPES, 1.25 mM MgSO4, 1.25 mM K2HPO4, 10 mM glucose, 4.95 mM Taurine, 9.89 mM 2,3-Butanedione monoxime; pH −7.35). The hearts were then adjusted on Langendorff system and digested using digestion buffer [solution A with 0.1% Bovine serum albumin (BSA), 0.05 mM CaCl2, 0.07% collagenase type II, 0.02% hyaluronidase type I]. Then, ventricular tissue was taken and minced in digestion buffer. The cell suspension was filtered using 100 μm cell strainer (BD Biosciences, CA) and the filtrate was centrifuged at 300 rpm for 3 min at room temperature (RT). The cell pellet was resuspended in solution B (solution A with 1% BSA, 0.1 mM CaCl2) and cells were allowed to settle under gravity. Then, the cell pellet was resuspended and seeded in DMEM/High glucose media (GE Healthcare, UK) containing 10% fetal bovine serum (FBS) (Fisher Scientific, MA) and 1% Penicillin/Streptomycin (Fisher scientific, MA). 24 h post-seeding, cells were transfected with rno-miR-210 mimic (50 nM) and negative control, Caenorhabditis elegans-specific miR, cel-miR-67 (50 nM) (Dharmacon, CO), and APC siRNA (siAPC) (50 nM) (Ambion, MA) using Lipofectamine RNAiMAX reagent (Life Technologies, CA) according to the manufacturer’s instructions. 24 h following transfection, media was removed and fresh culture media containing 0.5% EdU (5-ethynyl-2′-deoxyuridine) (Life Technologies, CA) was added. Cells were cultured for 1 week post-transfection and used for further studies. Likewise, in another setup, seeded cells were transfected using siUBC (Ubiquitin-C siRNA) (50 nM) (Ambion, MA) and transfection control mimic tagged with Dy547 (50 nM) (Dharmacon, CO), and transfection efficiency was calculated at day 4 post transfection.
Immunocytochemistry (IC)
One week post transfection, CM cultures were fixed with 4% paraformaldehyde in PBS (Phosphate buffered saline) (pH 7.4) for 20 min followed by permeabilization using 0.1% Triton X-100 for 15 min. Cells were washed with PBS and blocked with CAS-Block (Life Technologies, CA) for 1 h at RT. Cells were then incubated with primary antibody in CAS-Block overnight at 4 °C. Cells were then washed with PBS followed by incubation with secondary antibody in CAS-Block for 1 h at RT. Cells were again washed with PBS and incubated with nuclear stain DAPI (4′,6-diamidino-2-phenylindole) for 10 min. Imaging was done under a fluorescence microscope (Olympus).
Assessment of CM proliferation and apoptosis
Post one week following transfection, CM number, size, nucleation, EdU and Ki67 labeling were examined. To analyze CM number, CMs were labeled with cardiac-specific Troponin-I (TnI) primary antibody followed by a secondary antibody linked with Alexa fluor, and DAPI. TnI positive (TnI+) CMs and respective nuclei were counted under a fluorescence microscope. For cell size, individual TnI+ CM area was quantified using ImageJ analysis program (NIH, MD). EdU labeling was performed using EdU imaging kit (Invitrogen, CA) according to manufacturer protocol. Similarly, apoptosis in CM culture was analyzed using TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) detection kit (Roche, IN).
MiR-210 overexpressing mice model
MiR-210 overexpressing transgenic (210-TG) mice were generated at the University of Cincinnati Transgenic core using the C57BL/6 mouse strain. Briefly, precursor miR-210 along with H1 promoter (HmiR0167-MR01 vector from GeneCopoeia, MD) was amplified and cloned into pShuttle vector (Stratagene, CA) using restriction enzymes Not1 and Sal1. Amplified vector was digested, purified, and used for microinjection into the pro-nuclei of single-cell embryos derived from superovulated C57BL/6 females. The surviving embryos were implanted into foster female mice. 210-TG and non-transgenic wild-type (NTG) mice were identified by genotyping of the tail genomic DNA via PCR for custom designed primers (Table S1). To examine the possible tumorigenic effect of miR-210 overexpression on major organs, we compared the size of different organs via measuring organ weight to body weight ratios in one-year-old NTG and 210-TG mice (N = 8) (Fig. S3A). Additionally, we evaluated the expression profile of cell proliferation marker, Aurora B via qPCR, in the lung, liver, and kidney in one-year-old NTG and 210-TG mice (Fig. S3B) (N = 4).
Target analysis and confirmation
Using in silico analysis tool, APC was predicted to be a direct conserved target having miR-210 binding sites on its 3′UTR (3′ untranslated region) in mouse and rat. Additionally, to validate the target sequence in rodents, we performed dual luciferase assay in adult rat CMs which is described in detail below. In another parallel experiment, APC was knockdown in ARCMs via siAPC and was examined for EdU, Ki67, and TUNEL labeling.
Luciferase assay
Rat APC 3′UTR with miR-210 target sequence was retrieved from NCBI (https://www.ncbi.nlm.nih.gov/gene/). Target sequence was PCR amplified by using specific primers (Forward; 5′ GTTGTTGAATTCAGGAAAACAAAGTGTGGGCAG; Reverse 5′ GTTGTTACTAGTCGCTGCTTTTCCTAGG GCTT) and cloned into dual luciferase vector (GeneCopoeia) designated as WT 3′UTR-APC. For mutagenesis assay, seed sequence mutation in the miR-210 binding site of APC-3′UTR was created by site directed mutagenesis and vector designated as MT 3′UTR-APC. Afterward, adult Fisher rat CMs were isolated and seeded in 80% confluency in a 24-well plate with 500 μl of culture medium [DMEM/High glucose media (GE Healthcare, UK) containing 10% FBS (Fisher scientific, MA) and 1% Penicillin/Streptomycin (Fisher scientific, MA)]. Post overnight culture, CMs were cotransfected with 300 ng of WT 3′UTR-APC and 50 nM rno-miR-210 mimic using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s recommendations. Likewise, for mutagenesis assay, CMs were co-transfected with 300 ng of MT 3′UTR-APC and 50 nM rno-miR-210 mimic. Cel-miR-67 mimic (50 nM) serves as a control. Luciferase assay was performed after 36 h post-transfection. For the same, cell lysates were prepared by using passive lysis buffer and then assayed for luciferase activity using the Luc-Pair Duo-Luciferase assay kit 2.0 (GeneCopoeia). Luciferase activity of the fusion proteins was measured with a Microplate Luminometer and normalized to the activity of Renilla luciferase.
RNA extraction and real-time PCR
Total RNA was extracted from mouse hearts or cultured cells using Trizol reagent in accordance with the manufacturer protocol (Invitrogen, CA). In short, heart or other organ samples were homogenized in 1 ml Trizol reagent followed by 5-min incubation at RT. 0.2 ml chloroform was added followed by 15-sec shaking and further incubated for 3 min at RT. Samples were centrifuged at 12000×g for 15 min at 4 °C. The aqueous phase was transferred to a new tube and 0.5 ml isopropanol was added. Samples were then incubated at RT for 10 min followed by centrifugation at 1200×g for 10 min at 4 °C. RNA pellet was separated, washed with 1 ml of 75% ethanol and was centrifuged at 7500×g for 5 min at 4 °C. Wash was discarded, RNA pellet was air-dried for 10–30 min at RT and dissolved in 50 μl of RNase-free water. RNA concentration was estimated using Nanodrop and purity was checked by A260/280 ratio > 1.6. Purified RNA was further used for qRT-PCR of miR-210 using miScript Primer assay kit (Qiagen, MD) according to manufacturer recommended procedure. RT-PCR was performed using Bio-Rad Real-Time PCR Detection System. Results were normalized with internal control and analyzed via comparative Ct method. Results were also examined using PCR product via 1.5% agarose gel electrophoresis.
Echocardiography
Indices of left ventricular function and chamber dimensions during systole and diastole were examined via echocardiography (Echo), performed just prior to surgery and then 4 weeks following MI with a Vevo 2100 Imaging digital ultrasound system (VisualSonics) using a 22–55 MHz (MS550D) for adult mice. Prior to echocardiography, mice anesthetized with oxygen mixed with isoflurane (2–2.5%) at a heart rate of ~ 400 beats/min. Mice chest hairs were shaved and the animals were then placed on a warm platform in supine position. Data acquisition was initiated with the parasternal cardiac long axis view followed by a transition to the short axis view, at the level of mid-papillary muscles. Measurements were obtained from short axis M-mode images and graphically represented as % LVEF (left ventricular ejection fraction) and % LVFS (left ventricular fraction shortening).
Ischemia/reperfusion (I/R) injury mice model
The ischemic injury was created in 12-week-old miR-210 overexpressing (210-TG) and wild-type (NTG) mice by 30-min temporary LAD (left anterior descending) artery ligation followed by reperfusion. Post-surgery, EdU (5-ethynyl-2′-deoxyuridine) was administrated in-traperitoneally (i.p.) at a dose of 500 μg/100 μl PBS on alternate days up to day 12. On day 14, echocardiography was performed and animals were euthanized. Finally, CMs were isolated, seeded on a culture dish, and stained for EdU, TnI, and DAPI followed by further analysis.
Myocardial infarction (MI)
Myocardial infarction was created in 12-week-old 210-TG and wild-type (NTG) mice via permanent ligation of left anterior descending (LAD) coronary artery, following which EdU was administrated intraperitoneal at a dose of 500 μg/100 μl PBS on alternate days up to day 12. For long-term studies 4 weeks post LAD, echocardiography was performed, and animals were euthanized to collect hearts. The hearts were fixed, embedded in paraffin blocks for sectioning followed by staining with Masson’s Trichrome or hematoxylin/eosin to assess percent attenuation of infarct size. In addition, heart sections were used for immunohistochemistry (IHC) analysis. Heart samples were also saved for molecular studies.
Histological examination and immunohistochemistry
Prior to isolation, the hearts were arrested in diastole with 1 M KCl and perfused and fixed in 10% buffered formalin solution overnight at 4 °C. Tissue samples were embedded in paraffin blocks followed by sectioning. Formalin fixed tissue slides were de-paraffinized via incubation at 65 °C for 30 min followed by immediately dissolving paraffin in xylene and re-hydrating through sequential 5-min incubations in graded ethanol (100, 90, and 80%) and water. Antigen retrieval was performed via heating the slides in a pressure cooker in 0.1 M citrate buffer (pH 6.0) for 3 min followed by cooling for 45 min. Tissue sections were blocked with CAS-Block (Invitrogen, CA) for 1 h, incubated with primary antibody against analyzing antigen for 1–3 h at 37 °C followed by incubation with appropriate secondary antibody linked with Alexa fluor for 1 h at 37 °C. Slides were subsequently treated with DAPI for 10 min, washed with PBS, and mounted with Fluoromount-G (SouthernBiotech, AL). Slides were examined using a confocal microscope (Olympus). Cell area/size was defined by staining of cardiac tissue with TnI and wheat germ agglutinin (WGA) (Invitrogen, CA). EdU labeling and apoptotic cell death were determined via EdU imaging kit (Invitrogen, CA) and TUNEL kit (Roche, IN) respectively, according to manufacturer’s instructions. CMs undergoing mitosis were examined via anti-Aurora B (cell cytokinesis marker) staining. To analyze angiogenesis, blood vessels were stained using anti-smooth muscle actin (Sigma, MO) and VWF (von Willebrand factor) (Dako, CA) antibodies.
SDS gel electrophoresis and Western blot
Cell lysate preparation
Cell cultures were washed with PBS and mechanically detached from the surface using a cell scraper or mild trypsin treatment. Cell suspensions were then collected in a microcentrifuge tube and centrifuged at 5000 rpm for 5 min. Pellets were resuspended in 100 μl of 1X RIPA buffer and incubated at 4 °C for 1 h. 5× SDS sample buffer (60 mM Tris-Cl pH 6.8, 2.5% Glycerol, 2% SDS, 0.1% Bromophenol Blue) was added and boiled for 15 min followed by centrifugation at 13000 rpm for 15 min at 4 °C. The supernatants containing the lysate proteins were collected and stored at −20 °C for further use.
Tissue lysate preparation
Mice hearts were collected, perfused with PBS and homogenized in Laemmli buffer with β-mercaptoethanol followed by immediate boiling in boiling water bath for 20 min. Subsequently, the lysate was centrifuged at 12000×g for 30 min at 4 °C and the supernatant was stored at −80 °C until use.
Protein content was estimated using Pierce 660 nm reagent kit (Thermo Scientific, MA). Linear slab gel electrophoresis under the denaturing condition (in the presence of 0.1% SDS) was performed. Protein samples were stacked and were electrophoresed using 10–12% polyacrylamide gel (Invitrogen, CA) and running buffer (containing 0.025 M Tris-base, 0.192 M glycine pH 8.3, and 0.1% SDS) in Novex apparatus (Life Technologies, CA). Standard molecular weight marker was electrophoresed alongside to observe the subunit size of the analyzed protein. Electrophoresed protein samples were electroblotted onto PVDF membrane by applying 0.8 mA/h current in a wet transfer unit (BioRad, CA) and were detected using anti-rabbit or anti-mouse antibodies conjugated with horseradish peroxidase (HRP). Then, blots were treated with SuperSignal W. Femto max sensitivity (Thermo Fisher, MA) enhanced chemiluminescence (ECL) substrate. Blot was kept in a cassette: a film exposed to it and developed to visualize the protein bands.
Aortic ring assay
To study angiogenesis in mice, aortic ring assay was performed according to published protocol [26] with few changes. In short, 12-week-old 210-TG and NTG mice were euthanized, and aorta, located along with spine, were dissected, cleaned in Opti-MEM media, and cut into rings of 1 mm width. Rings were serum starved overnight in Opti-MEM at 37 °C and 5% CO2 followed by embedding in Matrigel matrix (Sigma, MO). Rings were fed with 199-media (Sigma, MO) containing 2.5% FBS, 1% Penicillin/Streptomycin. In addition, aortic rings from NTG mice were also treated with same media supplemented with purified VEGF (Vascular endothelial growth factor) (30 ng/ml) and considered as a positive control for neovascularization. Post 1 week, rings were fixed with 4% paraformaldehyde in PBS for 20 min followed by washing with PBS. Imaging was done under bright field and numbers of microvessel sprouts were counted in each aortic ring.
Statistical analysis
Statistical analysis was performed using ANOVA and Student’s t test. All values were expressed as mean ± SEM. P < 0.05 was considered statistically significant.
Results
MiR-210 induces proliferation in ARCM
MiR-210 has already been shown to induce proliferation in various cell lines [15–18, 21]; however, there is no report to this date that reflects its effect on adult CMs mitosis. To investigate the proliferative potential of miR-210, 12-week-old rat CMs were isolated and transfected with cel-miR-67 (control) and rno-miR-210 mimic (Fig. 1a). One week post-transfection, cells were fixed, permeabilized, and stained for cardiac-specific TnI, and cell proliferation markers, EdU and Ki67. As compared to control, miR-210 transfected group showed a twofold increase in cell number (Fig. 1b, e) while cell size was significantly reduced (Fig. 1b, f). Moreover, control group CMs showed intact and well-organized myofibrils which were found to be disorganized in miR-210 transfected cells, suggesting dedifferentiation of these CMs while undergoing proliferation (Fig. 1b). Likewise, miR-210-transfected CMs showed a significant increase in mono-nucleation while multi-nucleation was decreased (Fig. 1g). In contrast to control, miR-210 group represented a fourfold increase in EdU+ (Fig. 1c, h) and Ki67+ (Fig. 1d, i) cells, suggesting CM proliferation. Additionally, for validation of our transfection protocol, transfection efficiency was analyzed using cel-miR-67 mimic (as control), siUBC (Ubiquitin-C siRNA), and Dy547 mimic, and we observed ~ 65–73% transfection efficiency in ARCMs (Fig. S1).
Fig. 1.
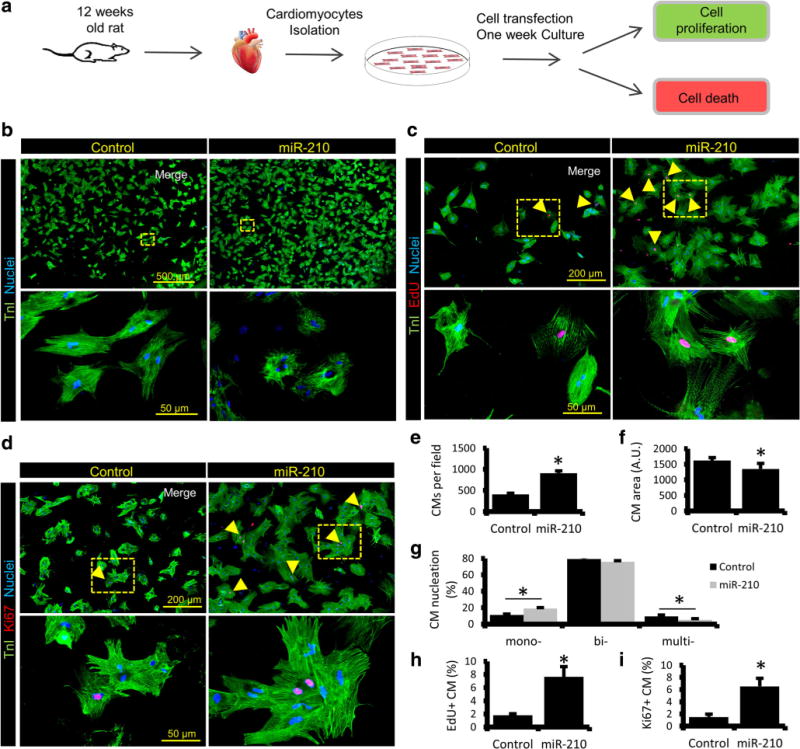
MiR-210 induces proliferation of ARCM. a Experimental design workflow illustrating CM isolation, transfection with cel-miR-67 (control) and rno-miR-210 mimic followed by 1-week culture under controlled conditions. Cells were then fixed, permeabilized, and utilized for further analysis such as cell proliferation and apoptosis. b–d Representative immunostained images of adult rat CMs, post 1 week of cel-miR-67 and miR-210 transfection for cardiac-specific Troponin I (TnI), stained as green, and proliferation markers EdU and Ki67, stained as red (indicated by arrowheads). Cell nuclei were counterstained with DAPI (blue). Panels (yellow) are representing respective enlarged sections. e–g Quantification of the total count of TnI+ CMs, nucleation, and cell size showing a significant increase in CM number and a smaller cell size in miR-210 group. A significant increase was also seen in mono-nucleated CMs while multi-nucleation was reduced in miR-210 group. h–i Quantification of EdU and Ki67 labeled CMs indicating CM proliferation in miR-210 group. N = 4. *P < 0.05 vs Control
MiR-210 reduces cell death in ARCM
MiR-210 is also documented to reduce CM apoptosis in mice post-cardiac injury [13]. Thus, to examine the antiapoptotic effect of miR-210 in adult CMs in vitro, we analyzed the rno-miR-210 transfected ARCM post 1 week of transfection. In contrast to cel-miR-67 transfected control, miR-210 group showed threefold reductions in TUNEL positive cells, suggesting a significant decrease in CM apoptosis (Fig. 2a–b). In addition, a twofold upregulation of cell survival factor Bcl-2 and threefold downregulation of apoptosis marker cleaved caspase-3 to caspase-3 ratio were seen in miR-210 group that further supports the pro-survival role of miR-210 in ARCM (Fig. 2c).
Fig. 2.
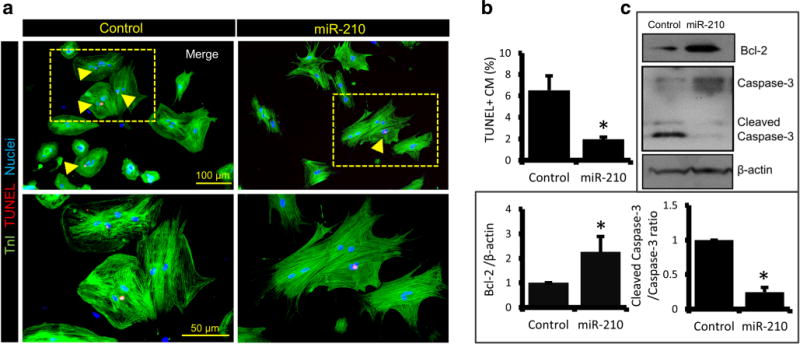
MiR-210 promotes survival of ARCM. a Immunostaining of ARCM, post 1 week of miR-210 and cel-miR-67 transfection, for TnI (green) and cell apoptosis marker TUNEL (red; indicated by arrowheads). Cell nuclei were counterstained with DAPI (blue). b Quantification of apoptotic TUNEL+ nuclei showing a significant decline in CM death in miR-210 group. c Western blot analysis in 12-week-old rat CMs, post 1 week of cel-miR-67 and rno-miR-210 mimic transfection, showing protein expression levels of cell survival factor Bcl-2 and apoptosis marker cleaved caspase-3 to caspase-3 ratio. Densitometry representation of respective blots in fold change expression normalized by β-actin as a loading control, indicating Bcl-2 upregulation and caspase-3 downregulation in miR-210 group. For all studies N = 4. *P < 0.05 vs Control
APC is the direct target of miR-210
APC is a part of canonical Wnt signaling pathway that negatively regulates cell cycle progression via promoting β-catenin degradation. In silico analysis predicted binding sites for miR-210 on 3′UTR region of APC gene of the mouse, and rat (Fig. 3a). To validate this experimentally, dual luciferase assay was performed in ARCMs, and we found a twofold downregulation in luciferase activity in miR-210 + WT 3′UTR-APC group containing a normal 3′UTR target sequence as compared to miR-210 + MT 3′UTR-APC group containing a mutated 3′UTR target site (Fig. 3b), suggesting miR-210-mediated inhibition of APC. In a parallel experiment, compared to cel-miR-67 (control), miR-210 transfected CMs showed a significant downregulation of cell cycle inhibitors like APC and p16, while cell cycle activator β-catenin was upregulated (Fig. 3c). Further, to evaluate the effect of miR-210 mediated inhibition of APC on CM proliferation, ARCMs were isolated and transfected with cel-miR-67 (control), miR-210, and siAPC individually. One week post transfection, cultured cells were fixed, permeabilized, and stained for TnI, EdU, and Ki67. As compared to control, miR-210 and siAPC transfected groups showed threefold and twofold increase in EdU+ and Ki67+ cells respectively (Fig. 3d–e, f–g), suggesting induction of CM proliferation in both the groups when compared to control. Moreover, miR-210 and siAPC transfected CMs also showed a threefold decrease in TUNEL labeling (Fig. 3h–i), indicating a significant reduction in cell death in these groups.
Fig. 3.
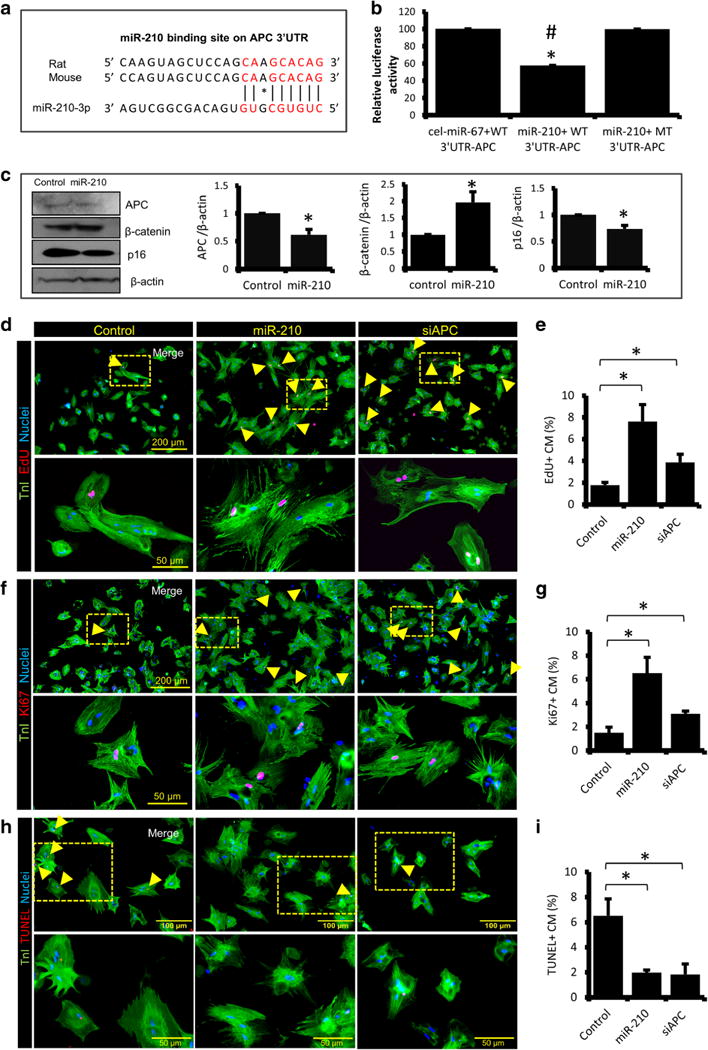
APC is a conserved target of miR-210. a In silico analysis for miR-210 target prediction using Findtar3 and RNAhybrid 2.2 tools showing complementarity of the miR-210 sequence to 3′UTR region of APC gene in rat and mouse. b Dual luciferase assay performed in adult rat CMs, and quantitative analysis showing a significant downregulation in luciferase activity in miR-210 + WT 3′UTR-APC group containing a normal 3′UTR target sequence as compared to miR-210 + MT 3′UTR-APC group containing a mutated 3′UTR target site. Here, cel-miR-67 mimic serves as a control. c Western blot analysis showing fold change expression levels of APC, β-catenin, and p16 in cel-miR-67 (Control) and miR-210 transfected ARCM. Respective densitometry using β-actin as a loading control, showing downregulation of APC and p16, and up-regulation of β-catenin in miR-210 group. d, f, h Immunostaining of ARCMs, post 1 week of cel-miR-67 (Control), miR-210 and siAPC transfection for TnI (green), EdU (red), Ki67 (red), and TUNEL (red). Cell nuclei were counterstained with DAPI (blue). e, g, i Quantification of EdU+, Ki67+, and TUNEL+ CMs, indicating an increase in CM proliferation and a decrease in apoptosis in miR-210 and siAPC transfected groups as compared to control. For all studies N = 4. *P < 0.05 vs Control
MiR-210 overexpression promotes CM proliferation in adult mice post-ischemic injury
To analyze the effect of miR-210 under in vivo system, we generated miR-210 overexpressing transgenic mice (210-TG) using C57BL/6 strain (Fig. 4a). Genotyping confirmed the upregulation of miR-210 in 210-TG mice as compared to NTG (non-transgenic/wild type) (Fig. 4b). In addition, qPCR data of different organs (spleen, heart, lung, skeletal muscle, kidney) and blood samples showed significant upregulation of miR-210 expression in 210-TG as compared to NTG (Fig. 4c–d). Moreover, to study the effect of miR-210 overexpression following cardiac injury, 12-week-old mice were subjected to 30-min LAD ligation followed by permanent reperfusion for 2 weeks [Ischemia/reperfusion (I/R) model] (Fig. 4e). EdU was administered on alternate days following surgery until day 12. The hearts were dissected, and CMs were isolated and seeded for 24 h. Immunocytochemistry and quantification of EdU labeling showed a significant increase in EdU+ CMs in 210-TG hearts post I/R injury (Fig. 4f–g), indicating CM proliferation. However, CMs from baseline hearts were lacking EdU labeling, suggesting the absence of CM proliferation in these mice (Fig. S2). In contrast to NTG, 210-TG hearts showed a significant increase in mono-nucleated CMs while multi-nucleation was decreased following I/R injury (Fig. 4h). Likewise, contrary to NTG, CM size decreased significantly in 210-TG hearts at baseline. In addition, compared to basal groups, CM size further decreased following cardiac injury. However, we observed no significant difference in CM size between NTG and 210-TG hearts post I/R (Fig. 4i). To examine the possible tumorigenic effect of overexpressed miR-210, we compared the body weight as well as size of different organs via measuring organ weight to body weight ratios in 1-year-old NTG and 210-TG mice (N = 8), which were found to be similar for the spleen, lung, liver, kidney, and brain (Fig. S3A). In continuation, we also examined the expression profile of cell proliferation marker, Aurora B via qPCR, that was also non-significant in the lung, liver, and kidney in 1-year-old NTG and 210-TG mice (Fig. S3B). These observations clearly indicate that although miR-210 promotes cell proliferation and angiogenesis in injured heart tissue, it did not show any tumorigenic sign in other major organs. Thus, present data supports the non-carcinogenic behavior of miR-210 therapy in 1-year-old 210-TG mice.
Fig. 4.
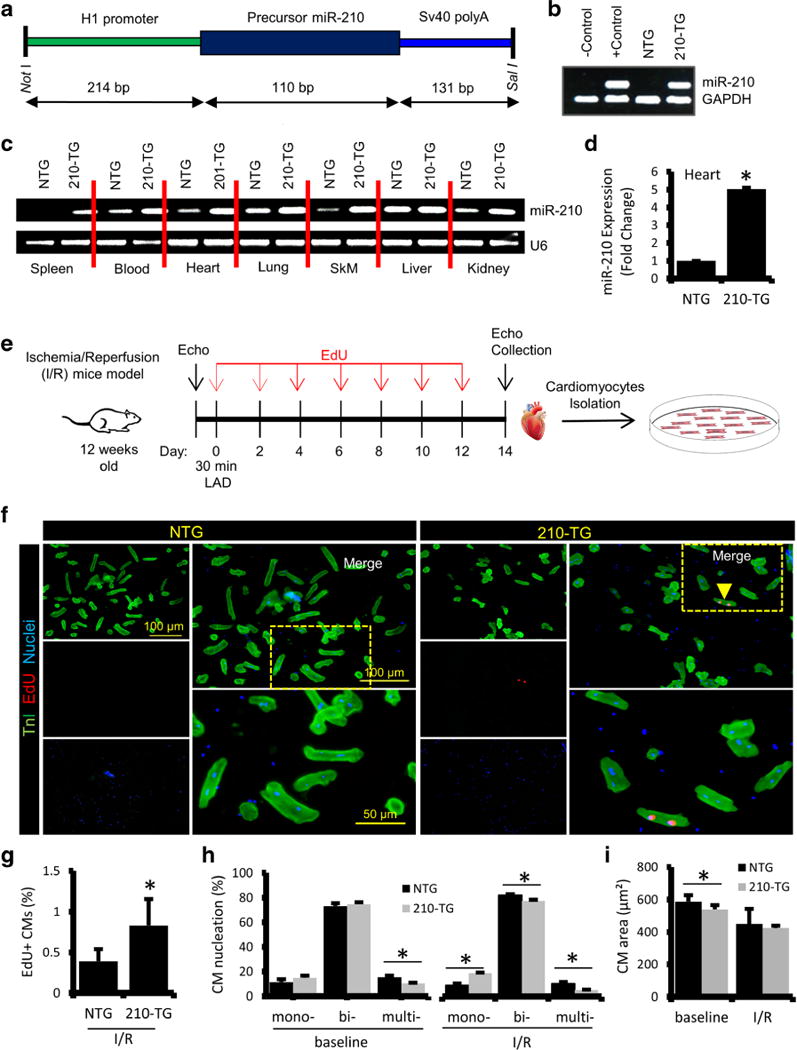
MiR-210 overexpression promotes CM proliferation in I/R injury mice model. a MiR-210 overexpressing transgenic (210-TG) mice were generated using C57BL/6 strain. 210-TG construct was designed to have an H1 promoter, precursor miR-210 cDNA, and SV40 poly A cassette at 3′ end. Precursor miR-210 along with H1 promoter was amplified and cloned into pShuttle vector using restriction enzymes Not1 and Sal1. Amplified vector was digested, purified, and used for microinjection into the pronuclei of single-cell embryos derived from superovulated C57BL/6 females. The surviving embryos were implanted into foster female mice. b Genotyping of mice tail genomic DNA via PCR using custom designed primers for miR-210, showing miR-210 transgenic (210-TG; lane 4) and non-transgenic (NTG; lane 3). GAPDH was used as internal control. c qPCR results showing miR-210 expression in different organs such as the spleen, heart, lung, skeletal muscle (SkM), liver, kidney, and blood of 210-TG and NTG mice. d Quantification ofmiR-210 expression by qPCR showing miR-210 overexpression in 210-TG mice hearts. e Schematic representation of 12-week-old mice subjected to LAD ligation for 30 min followed by reperfusion for 2 weeks (ischemia/reperfusion injury model). EdU (350 μg/100 μl PBS; i.p.) treatments were given at every alternate day till day 12. Post 2 weeks of surgery, echocardiography was performed, and the hearts were collected for CM isolation and culture. f Immunostaining of CMs, against TnI (green) and EdU (Red; indicated by arrowhead). Cell nuclei were counterstained with DAPI (blue). g–i Quantification of EdU labeling, cell nucleation (Mono-, bi-, multi-), and CM size, showing a significant increase in EdU+ CMs and mono-nucleation, while multi-nucleation was decreased in 210-TG hearts following I/R injury. N = 4 for all studies. *P < 0.05 vs NTG
MiR-210 overexpression rescues heart function in adult mice following MI
To evaluate the effect of miR-210 on adult mice cardiac function post injury, myocardial infarction was created by permanent LAD artery ligation in 12-week-old 210-TG and wild-type (NTG) mice. EdU was administered on alternate days up to day 12. Prior to surgery, and 4 weeks after MI, echocardiography was performed. Animals were then euthanized, and hearts were collected for histopathological and molecular analysis (Fig. 5a). Masson’s Trichrome stained tissue sections were used to measure infarct area, showing a significant decrease in infarct size in 210-TG hearts as compared to NTG (Fig. 5b, e). At baseline and post-MI, 210-TG hearts expressed sixfold upregulated levels of miR-210 (Fig. 5d). Heart weight to body weight ratio remains unaffected in both groups at baseline as well as following MI (Fig. 5f). Echocardiographic analysis showed a normal heart function at baseline in both groups. However, post-MI, heart activity, as represented by percent LVEF and LVFS, was significantly restored in 210-TG hearts as compared to NTG (Fig. 5c, g–h).
Fig. 5.
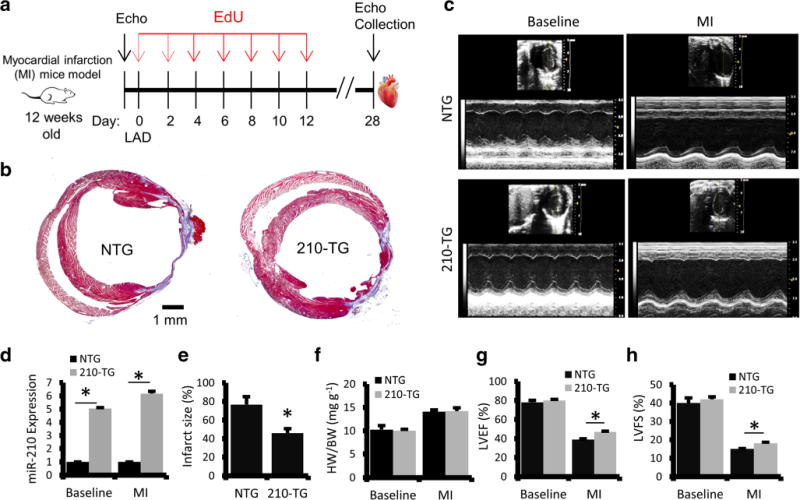
MiR-210 overexpression rescues heart function following MI in adult mice. a Schematic representation showing 12 weeks old mice undergoing permanent LAD ligation followed by EdU (350 μg/100 μl PBS; i.p.) treatments at every alternate day till day 12. Echocardiography (Echo) was performed prior to surgery, and at day 28 following surgery (post-MI). Four weeks post-surgery, mice were euthanized, and the hearts were collected for histopathology and molecular studies. b Representative images of Masson’s Trichrome stained heart sections in NTG and 210-TG following MI, illustrating infarct area (blue). c Representative images for echo analysis at baseline and post-MI. d–f Quantification of miR-210 fold change expression, infarct size, and heart weight to body weight ratio in 210-TG and NTG hearts following MI, indicating miR-210 overexpression, decrease in scar size, and similar HW/BW ratio in 210-TG as compared to NTG. g–h Echocardiographic assessment, and quantification of heart function at baseline and post-MI, showing significant restoration of LVEF and LVFS in 210-TG. For all studies N = 9. *P < 0.05 vs NTG
MiR-210 overexpression promotes CM proliferation in adult mice post-MI
APC is a member of Wnt pathway that forms a complex with other members to degrade β-catenin leading to inhibition of cell mitosis. In other words, APC negatively regulates cell cycle reentry. Thus, to evaluate the proliferative potential of miR-210 under in vivo system, Wnt signaling pathway was explored. Four weeks post-MI, mice were euthanized and hearts were collected for further studies. Immunohistochemistry and quantification of WGA stained peri-infarct zone indicated a significant decrease in CM size in 210-TG hearts as compared to NTG (Fig. 6a, d). Immunostaining and quantification of EdU-stained CMs at peri-infarct zone demonstrated a fourfold increase in EdU+ CMs in 210-TG hearts (Fig. 6b, e), suggesting CM proliferation. Additionally, troponin-T (TnT) and Aurora B specific immunostaining showed a fivefold increase of Aurora B+ CMs at the peri-infarct area in 210-TG hearts, indicating significant CM cell cycle re-entry (Fig. 6c, f). Western blots and densitometry showed nonsignificant changes in APC, β-catenin and p16 at baseline (Fig. 6g). However, following MI, twofold downregulation in APC and p16 expression were observed in 210-TG hearts as compared to NTG (Fig. 6h). In addition, 210-TG mice showed significant upregulation of β-catenin level (Fig. 6h), supporting CM cell cycle progression.
Fig. 6.
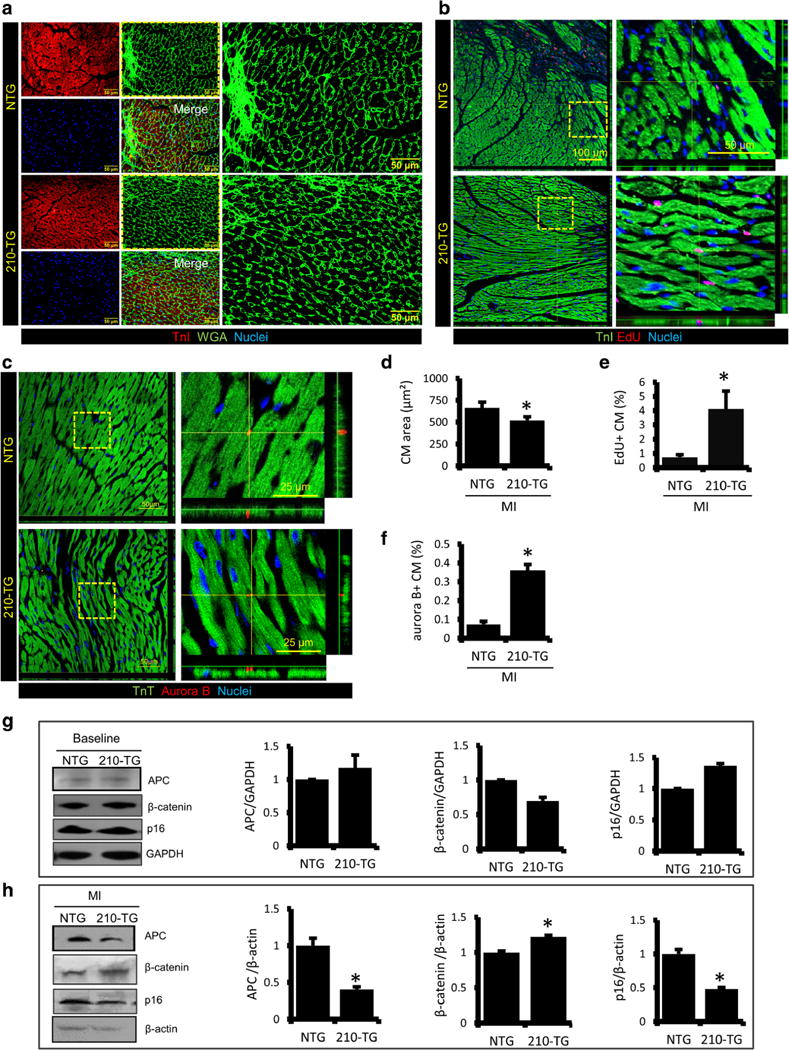
MiR-210 overexpression promotes CM proliferation in adultmice post MI. a Representative immunostained heart sections at peri-infarct zone in 12-week-old mice, post 4 weeks of LAD ligation, illustrating TnI (red) and WGA (green) stained CMs. Cell nuclei were counterstained with DAPI (blue). b Confocal microscope images of heart sections at the peri-infarct region, showing immunostaining for TnI (green) and EdU (red). c Representative confocal images of the peri-infarct zone, indicating cardiac specific TnT (green) and cell cytokinesis marker Aurora B (red). d–f Quantification of CM area, and EdU and Aurora B labeled CMs at the peri-infarct zone, showing a significant decrease in CM size, and a robust increase in EdU and Aurora B+ CMs in 210-TG hearts. g–h Western blots representing fold change expression levels of APC, β-catenin, and p16 in NTG and 210-TG heart at baseline and post-MI. GAPDH and β-actin were used as loading controls for densitometry analysis, indicating downregulation of APC and p16, while β-catenin was upregulated in 210-TG hearts. For all studies N = 4. *P < 0.05 vs NTG
MiR-210 overexpression reduces cell death and promotes angiogenesis in adult mice following MI
Heart sections from 4-week post-MI animals were examined for apoptosis and angiogenesis. Immunohistochemistry and quantification of TUNEL+ CMs at peri-infarct zone showed a twofold decrease in apoptotic CMs in 210-TG hearts (Fig. 7a–b). Western blots and densitometric analysis illustrated the non-significant change in Bcl-2 and caspase-3 expression at baseline (Fig. 7c). However, significant upregulation in Bcl-2 and twofold downregulation in caspase-3 was observed in 210-TG hearts post-MI (Fig. 7d), suggesting reduced cell death. Moreover, 210-TG hearts showed a twofold increase in blood vessel density at the peri-infarct zone, indicating upregulation of angiogenesis (Fig. 8a–b). Likewise, immunoblotting showed unaltered levels of angiogenic factor VEGF at baseline. Though, following MI, twofold upregulation in VEGF expression was observed in 210-TG hearts as compared to NTG (Fig. 8c). In a parallel experiment, angiogenic potential was further assessed in different mice groups via aortic ring assay, showing fourfold increment in aortic microvessel sprout formation, representing upregulated neovascularization, in 210-TG and NTGVEGF (VEGF-treated NTG) groups as compared to NTG (Fig. 8d–e). These observations clearly suggest the higher angiogenic potential of miR-210. In addition, HIF-1α expression was slightly increased in the 210-TG hearts at baseline and following MI (Fig. S4).
Fig. 7.
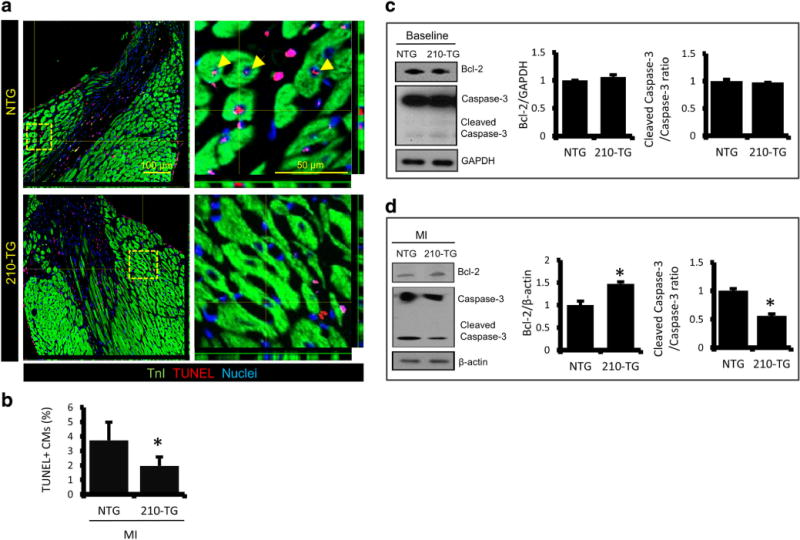
MiR-210 overexpression reduces apoptosis and promotes CM survival in adult mice following MI. a Representative confocal images of NTG and 210-TG mice hearts following MI, illustrating TnI (green) and TUNEL+ CM (red) at the peri-infarct zone. Cell nuclei were counterstained with DAPI (blue). b Quantitative analysis showing a significant decrease in apoptotic TUNEL+ CMs in 210-TG hearts. c–d Western blots indicating fold change expression levels of Bcl-2 and cleaved caspase-3 to caspase-3 ratio in NTG and 210-TG mice heart at baseline and post-MI. GAPDH and β-actin were used as loading controls for densitometry, showing upregulation of Bcl-2 and downregulation of caspase-3 level in 210-TG hearts following MI. For all studies N = 4. *P < 0.05 vs NTG
Fig. 8.
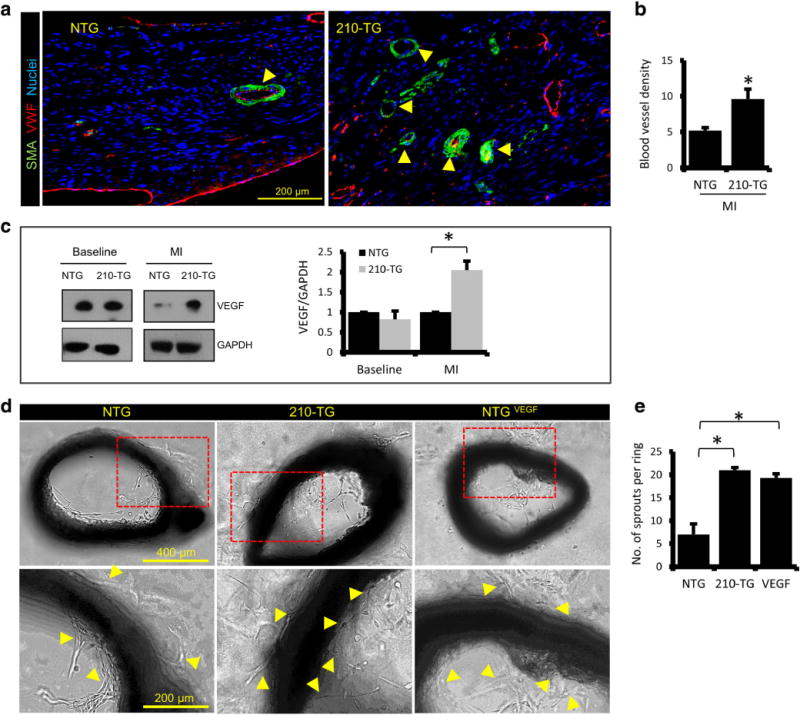
MiR-210 overexpression promotes angiogenesis in adult mice post-cardiac injury. a Adult mice heart sections at the peri-infarct zone, showing blood vessels (indicated by arrowheads) illustrated via outer multilayered smooth muscle actin (SMA) positive cells (green), and inner single layered VWF stained endothelial cells (red). b Quantitative analysis of blood vessel density at the peri-infarct zone, indicating upregulated angiogenesis in the 210-TG heart. c Western blots showing fold change expression of angiogenic factor VEGF in NTG and 210-TG hearts at baseline and post-MI (LAD). β-actin was used as loading control for densitometry, showing upregulation of VEGF in 210-TG hearts following MI. d Representative bright field images for aortic ring assay performed in 12 weeks old NTG and 210-TG mice, illustrating aortic neovascularization, indicated by microvessel sprouting (indicated by arrowheads). NTG mice aorta treated with VEGF (NTGVEGF) was used as positive control. e Quantification of aortic outgrowths/sprouts, showing a significant increase in neovascularization in 210-TG and NTGVEGF groups as compared to NTG. For all studies N = 4. *P < 0.05 vs NTG
Discussion
For a long time, the mammalian heart was presumed to be a post-mitotic organ as CMs undergo cell cycle arrest soon after birth [27–29]. However, recent studies have shown that adult mammalian heart has a small fraction of cycling CMs with limited proliferative capacity, though these are insufficient to rescue heart function following cardiac injury [30]. Therefore, there is a pressing need to develop novel therapeutic approaches to promote cardiac regeneration. In contrast to adult mammals, newts, zebrafish, and neonate mice (before day 7 after birth) can successfully regenerate their hearts post injury via endogenous CM mitosis [31–33]. Based on this, it is a promising strategy to induce cell cycle re-entry of preexisting CMs, surrounding the site of injury. Following this approach, there are few studies that have attempted to induce CM proliferation to regenerate injured myocardial tissue in adult animal models [34, 35]. Likewise, some recent reports have also documented the inherent potential of certain miRNAs that can induce proliferation in terminally differentiated adult CMs [5, 34]. Similarly, earlier studies indicate that master hypoxamiR-210 (miR-210) has cell proliferative potential [15, 17–19, 21]. On the contrary, miR-210 knockdown using siRNA could lead to cell cycle arrest [16, 21]. However, to this date, there is no report on miR-210 mediated induction of adult CM mitosis. Hence, to explore miR-210-associated effect and underlying mechanism on CM proliferation, we proposed the current study, which has a potential therapeutic application for ischemic heart injuries like MI. According to our present study, miR-210 is one of the unique candidate miRNAs which can promote adult CM proliferation, cell survival, and cardiac angiogenesis simultaneously, following myocardial injury. Altogether, these favorable key characteristics of miR-210 can reduce pathological remodeling of the scarred tissue and regenerate new functional myocardium leading to the significant recovery of impaired heart function.
In order to examine the effect of miR-210 on adult CMs, we transfected ARCMs with cel-miR-67 (negative control) and rno-miR-210 mimics. One week post transfection, cells were analyzed for proliferation and apoptosis. Compared to control group, miR-210 overexpression induced adult CM proliferation as indicated by significant increase in cell number, smaller cell size, and higher EdU+ and Ki67+ cells. Moreover, contrary to control, miR-210 transfected CMs illustrated disorganized myofibrils, supporting their de-differentiated morphology that is another key characteristic of proliferating CMs. Interestingly, a significant increase in smaller mono-nucleated CMs was seen in miR-210 group, while the number of multi-nucleated CMs was reduced. This observation supports CM cell cycle re-entry resulting in proliferation, and reduced mitotic catastrophe-induced cell death as reported by earlier studies [36]. In continuation, we observed fourfold increment in EdU and Ki67 labeling in miR-210 transfected CMs as compared to control, while the cell number was almost double in miR-210 group. One probable rationale behind such robust increase in cell number in miR-210-transfected population could be due to a higher rate of mitosis and reduced cell death. Thus, to examine this hypothesis, TUNEL assay was performed, and we observed a threefold decrease in TUNEL+ CMs, favoring a significant increase in CM survival in miR-210 transfected group. In addition, upregulation of survival factor Bcl-2 and downregulation of apoptosis marker caspase-3 in miR-210 group provided further support for an anti-apoptotic effect of miR-210. The elevated caspase-3 expression is known for activation of signaling pathways leading to cell death [37], and in addition, is also associated with CM hypertrophy [38]. Similarly, our results also show downregulation of caspase-3 and smaller CM size in miR-210 group. Furthermore, we identified APC, an antiproliferative gene, as a direct target of miR-210, via in-silico tools. In addition, the miR-210 binding site was found to be conserved on 3′UTR of APC gene in the rat and mouse. Similar to our rodent experiments, recent studies have identified regulation of APC by hsa-miR-210 in human [39]. Furthermore, miR-210-mediated downregulation of APC was validated via dual luciferase assay performed in adult rat CMs. APC is a member of canonical Wnt signaling pathway that negatively regulates cell cycle progression via promoting degradation of cell cycle activator, β-catenin. Interestingly, β-catenin has been directly implicated in CM proliferation and normal ventricular maturation [40]. Conversely, in our study, miR-210 promotes APC inhibition to induce CM cell cycle reentry. Additionally, APC knockdown using siAPC also resulted in a significant increase in CM proliferation. Similar to miR-210 group, siAPC also showed an anti-apoptotic effect on CMs, suggesting shared downstream signaling in both the groups. Moreover, cell cycle inhibitor p16 was downregulated in miR-210 group that further favors CM mitosis, as p16 is known to inhibit cyclin-dependent kinases (CDK2/4/6) thereby blocking cell cycle progression from G1 to S phase [41].
Furthermore, we validated our in vitro observations in an in vivo system, by generating the first miR-210 overexpressing transgenic mice model (210-TG). We created myocardial injury via LAD ligation in wildtype and 210-TG animals, to evaluate the impact of constitutive overexpression of miR-210 on infarct size, preservation of cardiac function, endogenous CM proliferation, and survival. To test these objectives, foremost, ischemia/reperfusion (I/R) mice model was used as a short-term cardiac damage (acute injury) prototype, and resident CM proliferation was analyzed post 2 weeks of surgery. Contrary to NTG, 210-TG hearts showed an increase in CM proliferation as reflected by an upregulation in EdU labeling, smaller cell size, and a higher number of mono-nucleated CMs. A significant decrease in CM multi-nucleation in miR-210 hearts at baseline as well as post-I/R injury supports a reduction in mitotic catastrophe promoted cell death [36]. Comparable results were seen in permanent LAD ligation (MI) mice model that was utilized as a long-term cardiac damage (chronic injury) prototype. In these mice, miR-210 overexpression in 210-TG hearts resulted in an induction of CM proliferation as revealed by smaller cell size, and significant elevation of EdU+ and Aurora B+ CMs at border zone of injury. Additionally, similar to in vitro studies, 210-TG hearts showed downregulation of APC, supporting the role of Wnt signaling pathway that directly controls cell cycle progression. Here in these animals, inhibition of APC-induced upregulation of β-catenin could force resident CM cell cycle re-entry. Similar supporting results were also reported by earlier studies stating APC deletion promotes CMs proliferation in mice heart [40]. Moreover, downregulation of cell cycle inhibitor p16 in 210-TG hearts further favors CM mitosis [41]. Thus, both our in vitro and in vivo studies clearly point towards the in-built potential of miR-210 to induce CM proliferation. Furthermore, decrease in the number of TUNEL+ CMs, upregulation of Bcl-2, and downregulation of caspase-3 in 210-TG hearts endorse miR-210-mediated CM survival. Interestingly, following MI, miR-210 overexpression also promotes angiogenesis in 210-TG hearts, as indicated by higher blood vessel density and upregulated angiogenic marker VEGF. Together with endogenous CM proliferation, infarcted myocardial revascularization is also crucial for cardiac repair, as VEGF-induced angiogenesis plays a key role in the healing of ischemic scar [42]. In a supporting experiment, the elevated angiogenic potential of miR-210 was further confirmed by aortic ring assay, showing upregulation of aortic microvessel outgrowths, indicating upregulated neovascularization in aortic ring culture of 210-TG mice. Above stated anti-apoptotic and proangiogenic properties of miR-210 are in accordance with other documented studies in adult mice post-cardiac injury [13]. In addition, HIF-1α expression was slightly increased in the 210-TG heart following an injury along with an increase in CM proliferation which is also supported by earlier reports [25].
Finally, following cardiac injury, miR-210-mediated effects like endogenous CM proliferation, cell survival, and angiogenesis resulted in myocardial tissue regeneration. This idea is evidently demonstrated by our results showing a substantial decline in infarct size in 210-TG hearts post-MI. Eventually, all these events lead to a noteworthy restoration of heart function post-MI in 210-TG mice as evidently reflected by a significant improvement in LVEF and LVFS. Moreover, contrary to other reports showing miR-210 involvement in cancer progression [43], we did not observe any skin outgrowth, change in body weight, and size of major organs in 210-TG mice. In addition, there was no abnormal cell cycle activity, as suggested by similar expression of cytokinesis marker, Aurora B in the lung, liver, and kidney in 210-TG mice. These observations in one-year-old 210-TG mice, that are comparable to ~ 40 year age of its human counterpart [44], clearly suggest the non-tumorigenic effect of miR-210 overexpression in these mice at least up until one year. However, we did not carry out any follow-up studies post-one year to determine if there is any difference in long-term survival or the potential to form tumors during later stages in life.
Altogether, these results clearly define the cardioprotective role of miR-210. Thus, constitutive overexpression of miR-210 rescues cardiac function post-MI via promoting CM proliferation, cell survival, and angiogenesis. Thereby, the current study suggests the inherent potential of miR-210 as an excellent candidate for rescuing heart function post-cardiac injury, which can be translated into the development of a novel therapeutic regime for ischemic heart disease.
Supplementary Material
Key messages.
MiRNA-210 transfected adult rat CMs show proliferation and reduced cell death in vitro.
Cell cycle inhibitor APC is a target of miR-210.
MiR-210 overexpressing (210-TG) mouse hearts show CMs cell cycle re-entry and survival post myocardial injury.
210-TG mice show significant neovascularization and angiogenic potential post myocardial infarction.
210-TG hearts show reduced infarct size following ischemic injury.
Acknowledgments
This work was supported by the grant from National Institutes of Health (NIH) HL106190-01 to Dr. Rafeeq Habeebahmed and NIH GM103638 to Dr. Arghya Paul.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00109-017-1591-8) contains supplementary material, which is available to authorized users.
Compliance with ethical standards All animal have been utilized in accordance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC), and the studies have been performed in accordance with the ethical standards. The manuscript does not contain any clinical studies or patient data.
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, et al. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131:29. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Doppler SA, Deutsch MA, Lange R, Krane M. Cardiac regeneration: current therapies-future concepts. J Thorac Dis. 2013;5:683–697. doi: 10.3978/j.issn.2072-1439.2013.08.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 5.Liang D, Li J, Wu Y, Zhen L, Li C, Qi M, Wang L, Deng F, Huang J, Lv F, et al. miRNA-204 drives cardiomyocyte proliferation via targeting Jarid2. Int J Cardiol. 2015;201:38–48. doi: 10.1016/j.ijcard.2015.06.163. [DOI] [PubMed] [Google Scholar]
- 6.Pandey R, Ahmed RP. MicroRNAs inducing proliferation of quiescent adult cardiomyocytes. Cardiovasc Regen Med. 2015;2:e519. doi: 10.14800/crm.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nair N, Gongora E. MicroRNAs as therapeutic targets in cardiomyopathies: myth or reality? Biomol Concepts. 2014;5:439–448. doi: 10.1515/bmc-2014-0026. [DOI] [PubMed] [Google Scholar]
- 8.Palacin M, Reguero JR, Martin M, Diaz Molina B, Moris C, Alvarez V, Coto E. Profile of microRNAs differentially produced in hearts from patients with hypertrophic cardiomyopathy and sarcomeric mutations. Clin Chem. 2011;57:1614–1616. doi: 10.1373/clinchem.2011.168005. [DOI] [PubMed] [Google Scholar]
- 9.Li T, Cao H, Zhuang J, Wan J, Guan M, Yu B, Li X, Zhang W. Identification of miR-130a, miR-27b and miR-210 as serum biomarkers for atherosclerosis obliterans. Clin Chim Acta. 2011;412:66–70. doi: 10.1016/j.cca.2010.09.029. [DOI] [PubMed] [Google Scholar]
- 10.Bostjancic E, Zidar N, Glavac D. MicroRNA microarray expression profiling in human myocardial infarction. Dis Markers. 2009;27:255–268. doi: 10.3233/DMA-2009-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 12.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC, Wu JC. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010;122:S124–S131. doi: 10.1161/CIRCULATIONAHA.109.928424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devlin C, Greco S, Martelli F, Ivan M. miR-210: more than a silent player in hypoxia. IUBMB Life. 2011;63:94–100. doi: 10.1002/iub.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giannakakis A, Sandaltzopoulos R, Greshock J, Liang S, Huang J, Hasegawa K, Li C, O’Brien-Jenkins A, Katsaros D, Weber BL, et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol Ther. 2008;7:255–264. doi: 10.4161/cbt.7.2.5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hwang HW, Baxter LL, Loftus SK, Cronin JC, Trivedi NS, Borate B, Pavan WJ. Distinct microRNA expression signatures are associated with melanoma subtypes and are regulated by HIF1A. Pigment Cell Melanoma Res. 2014;27:777–787. doi: 10.1111/pcmr.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masliah-Planchon J, Pasmant E, Luscan A, Laurendeau I, Ortonne N, Hivelin M, Varin J, Valeyrie-Allanore L, Dumaine V, Lantieri L, et al. MicroRNAome profiling in benign and malignant neurofibromatosis type 1-associated nerve sheath tumors: evidences of PTEN pathway alterations in early NF1 tumorigenesis. BMC Genomics. 2013;14:473. doi: 10.1186/1471-2164-14-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothe F, Ignatiadis M, Chaboteaux C, Haibe-Kains B, Kheddoumi N, Majjaj S, Badran B, Fayyad-Kazan H, Desmedt C, Harris AL, et al. Global microRNA expression profiling identifies MiR-210 associated with tumor proliferation, invasion and poor clinical outcome in breast cancer. PLoS One. 2011;6:e20980. doi: 10.1371/journal.pone.0020980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsuchiya S, Fujiwara T, Sato F, Shimada Y, Tanaka E, Sakai Y, Shimizu K, Tsujimoto G. MicroRNA-210 regulates cancer cell proliferation through targeting fibroblast growth factor receptor-like 1 (FGFRL1) J Biol Chem. 2011;286:420–428. doi: 10.1074/jbc.M110.170852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Zhao J, Shi M, Ding Y, Sun H, Yuan F, Zou Z. Elevated expression of miR-210 predicts poor survival of cancer patients: a systematic review and meta-analysis. PLoS One. 2014;9:e89223. doi: 10.1371/journal.pone.0089223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, Sun H, Dai H, Walsh RM, Imakura M, Schelter J, Burchard J, Dai X, Chang AN, Diaz RL, et al. MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle. 2009;8:2756–2768. doi: 10.4161/cc.8.17.9387. [DOI] [PubMed] [Google Scholar]
- 22.Camps C, Buffa FM, Colella S, Moore J, Sotiriou C, Sheldon H, Harris AL, Gleadle JM, Ragoussis J. hsa-miR-210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res. 2008;14:1340–1348. doi: 10.1158/1078-0432.CCR-07-1755. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Ding L, Bennewith KL, Tong RT, Welford SM, Ang KK, Story M, Le QT, Giaccia AJ. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell. 2009;35:856–867. doi: 10.1016/j.molcel.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang X, Le QT, Giaccia AJ. MiR-210—micromanager of the hypoxia pathway. Trends Mol Med. 2010;16:230–237. doi: 10.1016/j.molmed.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, et al. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541:222–227. doi: 10.1038/nature20173. [DOI] [PubMed] [Google Scholar]
- 26.Baker M, Robinson SD, Lechertier T, Barber PR, Tavora B, D’Amico G, Jones DT, Vojnovic B, Hodivala-Dilke K. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc. 2011;7:89–104. doi: 10.1038/nprot.2011.435. [DOI] [PubMed] [Google Scholar]
- 27.Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87:521–544. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bicknell KA, Coxon CH, Brooks G. Can the cardiomyocyte cell cycle be reprogrammed? J Mol Cell Cardiol. 2007;42:706–721. doi: 10.1016/j.yjmcc.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 29.van Amerongen MJ, Engel FB. Features of cardiomyocyte proliferation and its potential for cardiac regeneration. J Cell Mol Med. 2008;12:2233–2244. doi: 10.1111/j.1582-4934.2008.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ali SR, Hippenmeyer S, Saadat LV, Luo L, Weissman IL, Ardehali R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci U S A. 2014;111:8850–8855. doi: 10.1073/pnas.1408233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borchardt T, Braun T. Cardiovascular regeneration in non-mammalian model systems: what are the differences between newts and man? Thromb Haemost. 2007;98:311–318. [PubMed] [Google Scholar]
- 32.Gamba L, Harrison M, Lien CL. Cardiac regeneration in model organisms. Curr Treat Options Cardiovasc Med. 2014;16:288. doi: 10.1007/s11936-013-0288-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lien CL, Harrison MR, Tuan TL, Starnes VA. Heart repair and regeneration: recent insights from zebrafish studies. Wound Repair Regen. 2012;20:638–646. doi: 10.1111/j.1524-475X.2012.00814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, et al. Mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res. 2013;112:1557–1566. doi: 10.1161/CIRCRESAHA.112.300658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S, Keating MT. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med. 2007;13:962–969. doi: 10.1038/nm1619. [DOI] [PubMed] [Google Scholar]
- 36.Zhou J, Ahmad F, Parikh S, Hoffman NE, Rajan S, Verma VK, Song J, Yuan A, Shanmughapriya S, Guo Y, et al. Loss of adult cardiac myocyte GSK-3 leads to mitotic catastrophe resulting in fatal dilated cardiomyopathy. Circ Res. 2016;118:1208–1222. doi: 10.1161/CIRCRESAHA.116.308544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu HL, Xu WH, Cai Q, Feng M, Long J, Zheng W, Xiang YB, Shu XO. Polymorphisms and haplotypes in the caspase-3, caspase-7, and caspase-8 genes and risk for endometrial cancer: a population-based, case-control study in a Chinese population. Cancer Epidemiol Biomark Prev. 2009;18:2114–2122. doi: 10.1158/1055-9965.EPI-09-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Putinski C, Abdul-Ghani M, Stiles R, Brunette S, Dick SA, Fernando P, Megeney LA. Intrinsic-mediated caspase activation is essential for cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2013;110:E4079–E4087. doi: 10.1073/pnas.1315587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eken SM, Jin H, Chernogubova E, Li Y, Simon N, Sun C, Korzunowicz G, Busch A, Bäcklund A, Österholm C, et al. MicroRNA-210 enhances fibrous cap stability in advanced atherosclerotic lesions. Circ Res. 2017;120:633–644. doi: 10.1161/CIRCRESAHA.116.309318. [DOI] [PubMed] [Google Scholar]
- 40.Ye B, Hou N, Xiao L, Xu Y, Boyer J, Xu H, Li F. APC controls asymmetric Wnt/beta-catenin signaling and cardiomyocyte proliferation gradient in the heart. J Mol Cell Cardiol. 2015;89:287–296. doi: 10.1016/j.yjmcc.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McConnell BB, Gregory FJ, Stott FJ, Hara E, Peters G. Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol Cell Biol. 1999;19:1981–1989. doi: 10.1128/mcb.19.3.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang JM, Wang JN, Zhang L, Zheng F, Yang JY, Kong X, Guo LY, Chen L, Huang YZ, Wan Y, et al. VEGF/SDF-1 promotes cardiac stem cell mobilization and myocardial repair in the infarcted heart. Cardiovasc Res. 2011;91:402–411. doi: 10.1093/cvr/cvr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dang K, Myers KA. The role of hypoxia-induced miR-210 in cancer progression. Int J Mol Sci. 2015;16:6353–6372. doi: 10.3390/ijms16036353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dutta S, Sengupta P. Men and mice: relating their ages. Life Sci. 2015;152:244–248. doi: 10.1016/j.lfs.2015.10.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.