

Key Points
Cofactor-independent aPLs cause PDI- and complement-dependent monocyte TF activation.
C3, but not C5, is required for TF activation and aPL-induced thrombosis.
Abstract
The complement and coagulation cascades interact at multiple levels in thrombosis and inflammatory diseases. In venous thrombosis, complement factor 3 (C3) is crucial for platelet and tissue factor (TF) procoagulant activation dependent on protein disulfide isomerase (PDI). Furthermore, C5 selectively contributes to the exposure of leukocyte procoagulant phosphatidylserine (PS), which is a prerequisite for rapid activation of monocyte TF and fibrin formation in thrombosis. Here, we show that monoclonal cofactor-independent antiphospholipid antibodies (aPLs) rapidly activate TF on myelomonocytic cells. TF activation is blocked by PDI inhibitor and an anti-TF antibody interfering with PDI binding to TF, and requires C3 but unexpectedly not C5. Other prothrombotic, complement-fixing antibodies, for example, antithymocyte globulin, typically induce TF activation dependent on C5b-7–mediated PS exposure on the outer membrane of monocytes. We show that aPLs directly induce procoagulant PS exposure independent of C5. Accordingly, mice deficient in C3, but not mice deficient in C5, are protected from in vivo thrombus formation induced by cofactor-independent aPLs. Only immunoglobulin G (IgG) fractions with cofactor-independent anticardiolipin reactivity from patients with antiphospholipid syndrome (APS) induce complement-independent monocyte PS exposure and PDI-dependent TF activation. Neither a human monoclonal aPL directed against β2-glycoprotein I (β2GPI) nor patient IgG with selective reactivity to β2GPI rapidly activated monocyte TF. These results indicate that inhibitors of PDI and TF, but not necessarily clinically available drugs targeting C5, have therapeutic benefit in preventing thrombosis associated with APS caused by pathogenic aPLs primarily reactive with lipid, independent of β2GPI.
Visual Abstract
Introduction
The complement system not only cooperates with hemostasis in host defense, but plays increasingly recognized roles in thrombosis and the coagulopathy in sepsis.1,2 Complement activation following binding of antithymocyte globulin (ATG) contributes to rapidly converting tissue factor (TF) expressed by myelomonocytic cells to a procoagulant form.3 In this context, the complement-fixing antibodies trigger complement factor 3 (C3)–dependent thiol-disulfide exchange involving cell surface protein disulfide isomerase (PDI) and C5b-C7 membrane insertion leading to procoagulant phosphatidylserine (PS) exposure and TF activation.4 Complement activation also plays a pivotal role in TF-dependent venous thrombus development in the mouse,5 but complement contributions to other settings of thrombosis remain incompletely understood.
The antiphospholipid syndrome (APS) causes thrombosis, but pathogenic effects of antiphospholipid antibodies (aPLs) frequently also involve activation of the complement cascade. Although aPLs are known to induce TF transcription in myelomonocytic cells,6-9 it is an open question whether aPLs, through complement fixation, also rapidly induce cell surface TF activation, as previously elucidated for the thrombosis-promoting ATG applied clinically in stem cell therapy.3 Fetal loss by aPLs is reduced in complement- and C5a receptor–deficient mice, indicating that complement triggers leukocyte activation during pregnancy complications.10,11 The aPL cofactor β2 glycoprotein-I (β2GPI) has complement regulatory activity,12 and prothrombotic effects of β2GPI-reactive aPLs cause endothelial activation and thrombophilia dependent on C5 and C6.13-16 In part, the prothrombotic effects of cofactor-dependent aPLs involve C5a receptors,17 but β2GPI-reactive antibodies also activate platelets18 and require low-density lipoprotein receptor–related protein 8 (LRP8) for thrombosis induction in vivo.19 Although there are anecdotal reports that C5 inhibition can improve thrombotic complications in catastrophic APS in patients with cardiolipin- and β2GPI-reactive aPLs,20,21 the role of complement in thrombosis caused by β2GPI-independent aPLs22 remains unclear.
Methods
Antibodies and inhibitors
We used C3 inhibitor compstatin (Tocris); PDI inhibitor 16F16, Acridine Orange, Rhodamine B (Sigma); Annexin V–fluorescein isothiocyanate (FITC) (BD Biosciences); ATG (a gift from Florian Langer, II. Medical Clinic and Polyclinic, University Medical Center Eppendorf, Hamburg, Germany); anti-TF 10H1023; anti-TF 21E1024; and isotype-matched control produced in-house.24 Anti-C5 antibody eculizumab was obtained from Alexion. The human monoclonal aPLs HL5B, RR7F, HL7G, and rJGG9 were characterized previously25-29 and compared with immunoglobulin G (IgG) control antibody isolated from a healthy donor. F(ab′)2 fragments of HL5B were generated with the F(ab′)2 Preparation kit (ThermoFisher). IgG fractions from APS patients were isolated as described.29 Briefly, IgG fractions from 20 APS patients were isolated by protein G affinity chromatography. Cofactor-independent aPLs were detected on enzyme-linked immunosorbent assay (ELISA) plates coated with cardiolipin dissolved in ethanol and blocked with 1% Tween 20. To assess the effect of cofactors in this ELISA format, 1% β2GPI or 10% human serum was added to the incubation buffer. Binding to β2GPI was analyzed in commercially available ELISA plates (AESKU Diagnostics, Wendelsheim, Germany). All antibody preparations had <0.1 endotoxin units per mL.
Mice
Lrp8−/− mice30 on a C57BL/6-129S6 mixed genetic background (The Jackson Laboratory) were backcrossed for 8 generations into the C57BL/6J background and compared with wild-type (wt) mice derived from littermates obtained during the backcrossing. C3- and C5-deficient animals and controls were obtained from The Jackson Laboratory, as previously described.5 All animal procedures were approved by the local committee on legislation on protection of animals (23177-07/G14-1-043; Landesuntersuchungsamt Rheinland-Pfalz, Koblenz, Germany).
Cell isolation and clotting assay
Mouse spleen monocytes were selected with the CD115 MicroBead kit (Miltenyi). Human MonoMac1 (MM1) cells were maintained as described.31 Cells in serum-free medium were exposed to aPLs with indicated inhibitors for 10 to 15 minutes at 37°C followed by 1:1 (volume-to-volume ratio) mixing with normal human or mouse, or C3- or C5-deficient, plasma for 10 minutes at 37°C. The anti-human TF antibody 10H10 was either incubated with cells during stimulation with aPLs or, as a control for inhibition of fully activated TF, added to plasma prior to recalcification. Clotting times after recalcification were recorded in a KC4 coagulation analyzer (Amelung, Lemgo, Germany) and converted into procoagulant activity (PCA) units based on a standard curve of TF (Innovin; Siemens, Marburg, Germany). Cells were similarly activated for 10 minutes in the presence of autologous serum and stained with Annexin V–FITC to quantify cell surface PS exposure.
Phospholipid-free FXa-generation assay with soluble TF
The regulation of TF by PDI was analyzed in a phospholipid-free, solution-phase assay of TF–factor VIIa (FVIIa)-mediated FX activation, as described previously.32 Briefly, FXa generation by soluble TF with a leucine zipper domain (2 nM), FVIIa (2 nM), PDI (20 nM), and FX (100) in the absence or presence of anti-TF 10H10 (100 µg/mL) was determined in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)-buffered saline, 2 mM Ca2+, 1 mM Mg2+ at 37°C, and FXa levels in EDTA-quenched samples were measured with chromogenic substrate Spectrozyme FXa.
Inferior vena cava thrombosis model
aPL-amplified thrombus development was evaluated as described.22 Briefly, aPL HL5B (1 μg) was injected via jugular catheter into 8- to 12-week-old male mice 1 hour before flow reduction in the inferior vena cava induced by ligation over a transiently positioned spacer (0.26 mm). Rhodamine B–labeled platelets and 100 μL of 50 μg/μL Acridine Orange to label leukocytes were infused for imaging of thrombus formation by high-speed fluorescence video microscopy (BX51WI; Olympus).
Statistics
GraphPad Prism 7 software was used for group comparisons with 1-way analysis of variance (ANOVA) and multicomparison correction (Dunnett).
Results
aPLs induce PS exposure and TF activation independent of C5
Monocyte TF procoagulant activity is rapidly induced dependent on PDI and complement by complement-fixing ATG used for preventing allograft rejection.3 We confirmed that ATG also upregulated the TF procoagulant activity of CD115-selected mouse spleen cells (Figure 1A). In addition, TF activity was induced by the cofactor-independent aPLs HL5B and RR7F.31 Consistent with prior data obtained with ATG, TF activation by aPLs was prevented by the PDI inhibitor 16F16 and the C3 inhibitor compstatin (Figure 1A). Experiments with human monocytic MM1 cells confirmed these results (data not shown). ATG activation of human TF requires thiol-disulfide exchange involving PDI and C5-dependent exposure of procoagulant PS mediated by membrane insertion of the C5b-C7 complex.3 As expected from this study, TF activation by ATG was prevented when mouse monocytes were stimulated in the presence of C3- or C5-deficient plasma. However, aPLs induced TF procoagulant activity normally in C5-deficient, but not C3-deficient, plasma (Figure 1B).
Figure 1.

aPLs induce TF activation and PS exposure. (A) CD115-selected spleen cells were stimulated for 15 minutes at 37°C with IgG (500 ng/mL), cofactor-independent aPLs HL5B, HL5B F(ab)2 (100 ng/mL) or RR7F (500 ng/mL), or ATG (100 µg/mL). aPLs were either added alone or together with C3 inhibitor compstatin (50 µM), PDI inhibitor 16F16 (2 µM), anti-TF antibody 21E10, or isotype rat IgG2a control (5 µg/mL). Cell-associated PCA was subsequently measured by single-stage clotting assay. (B) CD115-selected cells from the indicated knockout strains were exposed to antibodies as in panel A in plasma from wt, C3−/−, C5−/−, or Lrp8−/− mice as indicated and clotting times were determined. (C) Murine monocytes were stimulated with antibody and inhibitors in autologous plasma as indicated for 10 minutes. Samples were analyzed for PS exposure (Annexin V–FITC binding) using flow cytometry. (D) Murine monocytes were stimulated with antibody in plasma from wt, C3−/−, C5−/−, or Lrp8−/− as indicated. PS exposure was determined as in panel C. Data in panels A and C are shown as mean ± standard deviation (SD). Data in panels B and D are shown as median, interquartile range, and range; n = 6; *P < .001. One-way ANOVA followed by the Dunnett multiple-comparison test. MFI, mean fluorescence intensity; unst., unstimulated.
To better understand this distinct property of aPLs, we measured cell surface PS exposure by annexin 5 staining. Consistent with prior data, PS exposure by ATG required C3 and C5, but aPLs caused PS externalization independent of PDI and complement activation (Figure 1C-D). Note that the aPL HL5B F(ab′)2 fragment devoid of the complement-fixing Fc portion induced PS exposure, but failed to upregulate TF procoagulant activity. These data indicate that cofactor-independent aPLs can bypass the requirement for C5-dependent membrane perturbation, but nevertheless require C3 for PDI-dependent TF activation.
We next evaluated whether rapid TF activation and PS exposure is a general property of aPLs. HL7G has dual reactivity with cardiolipin and β2GPI, whereas rJGG9 is only reactive with β2GPI.29 HL7G, but not JGG9, induced PS exposure and complement- and PDI-dependent TF activation (Figure 2A-B). Thus, anticardiolipin reactivity appears to be required and sufficient to rapidly induce TF activity on monocytic cells.
Figure 2.

Prevention of PDI-dependent TF activation by anti-TF 10H10. (A) CD115-selected spleen cells were stimulated for 15 minutes at 37°C with IgG control (1 µg/mL), anti-β2GPI antibody rJGG9 (1 µg/mL), or HL7G (100 ng/mL) either alone or together with indicated inhibitors (using concentrations given in Figure 1). Cell-associated PCA was subsequently measured by single-stage clotting assay. (B) Murine monocytes were stimulated with antibody and inhibitors in autologous plasma, as indicated, for 10 minutes. Samples were analyzed for PS exposure (Annexin V–FITC binding) by flow cytometry. n = 6; *P < .002; 1-way ANOVA followed by the Dunnett multiple-comparison test. (C) Effect of 10H10 (100 µg/mL) on PDI enhanced FXa generation by soluble TF (sTF)-FVIIa. Data are shown as mean ± SD; *P = .004; **P = .006; 1-way ANOVA followed by the Dunnett multiple-comparison test. (D) Single-stage clotting assay of human MM1 cells. 10H10 was added 15 minutes after aPL stimulation, that is, directly before recalcification. Data are shown as mean ± SD; n = 6; *P ≤ .001; 1-way ANOVA followed by the Dunnett multiple-comparison test. (E) Single-stage clotting assay of human MM1 cells stimulated with monoclonal aPLs or ATG for 15 minutes at 37°C either with or without preincubation of 50 µg/mL 10H10. (F) MM1 cells were stimulated as indicated and PS exposure (Annexin V–FITC binding) was measured using flow cytometry. (G) Single-stage clotting assay of human MM1 cells cultured in 10% human serum. Anti-C5 antibody eculizumab (100 µg/mL) was added 5 minutes before HL5B (100 ng/mL) or ATG (100 µg/mL). Data are shown as mean ± SD; n = 6; *P ≤ .001; 1-way ANOVA followed by the Dunnett multiple-comparison test.
We have recently shown that PS exposure on extracellular vesicles released from macrophages is not sufficient for TF prothrombotic activity and that only PDI+ vesicles promote fibrin strand formation in a flow system.33 PDI is involved in a thiol-disulfide exchange that switches TF between coagulation and cell signaling.34 In addition, PDI through its chaperone function enhances TF activity involving TF residues Lys149 and Asp150, which are located in a flexible region of the TF C-terminal domain.32 This region of TF is not directly involved in the interaction with FVIIa or FX,35-37 but Lys149 and Asp150 are in the center of the epitope recognized by anti-TF 10H10.38 Although this antibody typically shows no appreciable inhibition of TF procoagulant activity, it inhibits the prothrombotic activity of TF on PDI+ extracellular vesicles in flowing blood.33 As shown in Figure 2C, anti-TF 10H10 had no effect on FXa generation in the established soluble TF assay,32 but it reduced PDI enhanced TF-FVIIa activity, as previously observed with TF+ and PDI+ extracellular vesicles.33
We hypothesized that 10H10 might interfere with PDI interaction with TF during activation by aPLs on monocytes. Note that addition of anti-TF 10H10 after aPL stimulation had no effect on TF procoagulant activity (Figure 2D), consistent with the previously documented inhibitory properties of the antibody toward fully active TF. We therefore pretreated cells with anti-TF 10H10 prior to aPL exposure. The antibody prevented cardiolipin-reactive aPLs from rapidly inducing TF activity on monocytic MM1 cells (Figure 2E), but did not influence PS exposure (Figure 2F). We next addressed whether inhibition of complement C5 activation with eculizumab interfered with TF activation. Although the inhibitory anti-C5 blocked TF activation by ATG as previously shown,3 it did not prevent TF activation by aPLs (Figure 2G). Thus, PDI and the thiol-disulfide exchange associated with C3 conversion downstream of aPLs contribute to rapid induction of full monocyte TF activity independent of C5b-7–induced PS exposure.
Thrombosis induction by cofactor-independent aPLs requires C3, but not C5
Prior studies showed that aPL affinity isolated on β2GPI induces thrombosis,18 but the cross-reactivity of these antibodies with cardiolipin was not assessed. In addition, LRP8 is a crucial cellular receptor for the pathogenic effects of cofactor-dependent aPLs.19 Rapid activation of monocyte TF by cofactor-independent aPL HL5B was unaltered in Lrp8−/− mice compared with controls (Figure 1B). We therefore tested the response of aPL-amplified venous thrombus development and showed that cofactor-independent aPLs induced thrombosis efficiently in Lrp8-deficient mice (Figure 3), confirming that LRP8 is not involved in signaling induced by these aPLs.29 We next analyzed the contributions of C3 and C5 in this TF-dependent flow restriction model of venous thrombus development.22 Accelerated thrombus formation following infusion of aPL HL5B was prevented in C3-deficient, but not in C5-deficient, mice (Figure 3). We have previously shown that myeloid cell TF-dependent fibrin formation following flow restriction of the vena cava requires both C3 and C5.5 The present findings show that the pathogenic effects of aPLs can also bypass in vivo the crucial role of C5 in myeloid cell TF activation.
Figure 3.

aPL-induced thrombosis requires C3 but not C5. (A) Representative images of thrombi developing in the flow-restricted vessels of the indicated mouse strains and (B) quantification of thrombus size 3 hours after aPL injection. Data are median, interquartile range, and range. *P = .0346; **P = .0042; ***P = .0009; 1-way ANOVA followed by the Dunnett multiple-comparison test. In panel A, platelets are depicted in red and nucleated cells in green. Scale bars = 100 μm.
To evaluate the relevance of these findings in patients, we analyzed previously characterized IgG fractions from 20 patients with APS.29 Isolated IgG from 11 patients had properties similar to HL5B and bound to cardiolipin in a cofactor-independent manner, but not to β2GPI.29 These aPLs induced rapid activation of TF (Figure 4A). Whereas TF activation was prevented by the C3 inhibitor compstatin and a PDI inhibitor, PS exposure was induced by patient IgG independent of complement activation and PDI (Figure 4B). IgG fractions from 2 patients showed reactivity similar to the monoclonal antibody rJGG9 and bound only to β2GPI, but not to cardiolipin in the absence of β2GPI. These IgG fractions induced neither TF activity nor PS exposure (Figure 4). The remaining 7 patient IgG fractions had dual reactivity with cardiolipin and β2GPI and, as seen with monoclonal antibody HL7G, efficiently induced both PS surface exposure and TF activity, which required C3 and PDI.
Figure 4.

Cardiolipin reactivity of polyclonal aPLs is required and sufficient for rapid TF activation. (A) Human MM1 cells were stimulated in the presence or absence of C3 inhibitor compstatin (50 µM) or PDI inhibitor 16F16 (2 µM) for 15 minutes at 37°C with IgG fractions isolated from 20 APS patients or 20 healthy controls. APS patient IgG samples can be divided into 3 groups: patient IgG that bind only to cardiolipin (CL) in a cofactor-independent manner similar to HL5B (CL; n = 11); patient IgG that bind to both antigens similar to HL7G (CL + β2GPI; n = 7); and patient IgG that bind only to β2GPI similar to rJGG9 (β2GPI; n = 2). Cell-associated PCA was subsequently measured by single-stage clotting assay; *P < .0001; 1-way ANOVA followed by the Dunnett multiple-comparison test. (B) MM1 cells were stimulated for 10 minutes with IgG fractions as described in panel A and PS exposure was quantified with Annexin V–FITC by flow cytometry.
Discussion
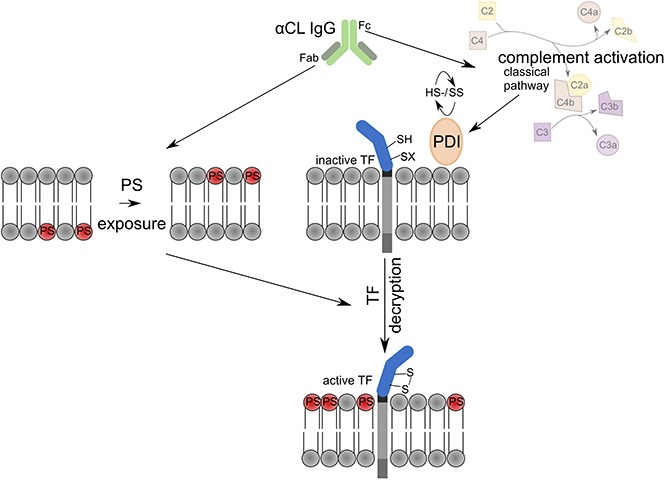
Understanding the pathomechanisms of aPL-induced thrombosis is essential for tailored antithrombotic therapy of APS patients. To date, aPLs directed against β2GPI are considered most important for APS pathogenesis. Several mechanisms including LRP8-dependent platelet activation and inhibition of endothelial nitric oxide synthase39-41 have been implicated in anti-β2GPI–mediated pathology. We have shown that cofactor-independent anticardiolipin aPLs, which have long been regarded as nonpathogenic, induce thrombosis in a mouse model.20 These aPLs can directly induce TF transcription and protein expression in myelomonocytic cells dependent on endosomal reactive oxygen species production.6,22,31 We here show that these antibodies also promote rapid activation of preexisting TF on monocytes and delineate that this pathway is dependent on C3 activation and PDI (Figure 5).
Figure 5.

Schematic overview of aPL-mediated rapid activation of monocyte TF.
In vivo, we show that cofactor-independent aPLs indeed promote thrombus formation independent of LRP8, which is crucial for thrombosis induction by β2GPI-reactive aPLs.39,40 In addition, we show that the diagnostic reactivity with cardiolipin is sufficient to predict rapid induction of TF activity and that aPL reactivity with β2GPI alone is not sufficient to induce this prothrombotic pathway. Although these conclusions are based primarily on well-characterized monoclonal aPLs,25 we confirmed with polyclonal patient IgG fractions that cardiolipin binding is required for PDI- and complement-dependent TF activation.
The role of PDI in regulating TF prothrombotic activity has been documented by in vitro and in vivo studies.3,5,24,33,34,42,43 Complement activation is known to produce thiol-disulfide exchange on cell surfaces,3 but PDI also regulates TF through chaperone activity that enhances TF-FVIIa affinity and FX turnover.32 With a monoclonal antibody that inhibits PDI chaperone effects on TF, we provide evidence that PDI is required for completing the activation of TF in response to aPL-induced complement activation. This finding suggests a novel therapeutic modality specifically to block TF activation on monocytes without interfering with the hemostatic activities of TF. Of note, this antibody has previously been shown to prevent aPL-induced pregnancy loss in a humanized TF mouse model.44
These data provide new insights into underlying mechanisms by which cofactor-independent aPLs trigger thrombosis. We demonstrate that aPLs uniquely induce PS exposure independent of complement action on myelomonocytic cells in vitro and that thrombosis development is independent of C5 in vivo. The presented data also show that antibody blockade of C5 is ineffective in blocking aPL-mediated TF activation, whereas this approved therapeutic strategy effectively prevented the effect of other complement-fixing antibodies. Thus, targeting C5 directly may not be uniformly beneficial in all APS patients, but one may expect a potential therapeutic benefit of targeting TF or PDI, including its chaperone activity, for thrombosis prophylaxis in this disease.
Acknowledgments
The authors thank Jennifer Royce for excellent technical assistance and Florian Langer for providing ATG.
This work was supported by Federal Ministry of Education and Research of Germany (Bundesministerium für Bildung und Forschung) grants 01EO1003 and 01EO1503 (N.M.-C., K.J.L., and W.R.), the Alexander von Humboldt Foundation (W.R.), and National Institutes of Health, National Heart, Lung, and Blood Institute grant HL60742 (W.R.).
Authorship
Contribution: N.M.-C. designed, performed, and analyzed experiments and wrote the paper; S.R., A.H., and T.F. performed experiments; K.J.L. provided critical reagents, data analysis, and interpretation; and W.R. designed the study and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wolfram Ruf, Center for Thrombosis and Hemostasis, Johannes Gutenberg University Medical Center, 55131 Mainz, Germany; e-mail: ruf@uni-mainz.de; and Karl J. Lackner, Institute for Clinical Chemistry and Laboratory Medicine, Johannes Gutenberg University Medical Center, 55131 Mainz, Germany; e-mail: karl.lackner@unimedizin-mainz.de.
References
- 1.Silasi-Mansat R, Zhu H, Popescu NI, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. 2010;116(6):1002-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res. 2016;118(9):1392-1408. [DOI] [PubMed] [Google Scholar]
- 3.Langer F, Spath B, Fischer C, et al. Rapid activation of monocyte tissue factor by antithymocyte globulin is dependent on complement and protein disulfide isomerase. Blood. 2013;121(12):2324-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langer F, Ruf W. Synergies of phosphatidylserine and protein disulfide isomerase in tissue factor activation. Thromb Haemost. 2014;111(4):590-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subramaniam S, Jurk K, Hobohm L, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. 2017;129(16):2291-2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prinz N, Clemens N, Canisius A, Lackner KJ. Endosomal NADPH-oxidase is critical for induction of the tissue factor gene in monocytes and endothelial cells. Lessons from the antiphospholipid syndrome. Thromb Haemost. 2013;109(3):525-531. [DOI] [PubMed] [Google Scholar]
- 7.Reverter JC, Tàssies D, Font J, et al. Effects of human monoclonal anticardiolipin antibodies on platelet function and on tissue factor expression on monocytes. Arthritis Rheum. 1998;41(8):1420-1427. [DOI] [PubMed] [Google Scholar]
- 8.Willemze R, Bradford RL, Mooberry MJ, Roubey RA, Key NS. Plasma microparticle tissue factor activity in patients with antiphospholipid antibodies with and without clinical complications. Thromb Res. 2014;133(2):187-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou H, Wolberg AS, Roubey RA. Characterization of monocyte tissue factor activity induced by IgG antiphospholipid antibodies and inhibition by dilazep. Blood. 2004;104(8):2353-2358. [DOI] [PubMed] [Google Scholar]
- 10.Girardi G, Berman J, Redecha P, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. 2003;112(11):1644-1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Redecha P, Tilley R, Tencati M, et al. Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood. 2007;110(7):2423-2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gropp K, Weber N, Reuter M, et al. β2-glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood. 2011;118(10):2774-2783. [DOI] [PubMed] [Google Scholar]
- 13.Fischetti F, Durigutto P, Pellis V, et al. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood. 2005;106(7):2340-2346. [DOI] [PubMed] [Google Scholar]
- 14.Agostinis C, Durigutto P, Sblattero D, et al. A non-complement-fixing antibody to β2 glycoprotein I as a novel therapy for antiphospholipid syndrome. Blood. 2014;123(22):3478-3487. [DOI] [PubMed] [Google Scholar]
- 15.Pierangeli SS, Girardi G, Vega-Ostertag M, Liu X, Espinola RG, Salmon J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthritis Rheum. 2005;52(7):2120-2124. [DOI] [PubMed] [Google Scholar]
- 16.Carrera-Marín A, Romay-Penabad Z, Papalardo E, et al. C6 knock-out mice are protected from thrombophilia mediated by antiphospholipid antibodies. Lupus. 2012;21(14):1497-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romay-Penabad Z, Liu XX, Montiel-Manzano G, Papalardo De Martínez E, Pierangeli SS. C5a receptor-deficient mice are protected from thrombophilia and endothelial cell activation induced by some antiphospholipid antibodies. Ann N Y Acad Sci. 2007;1108(1):554-566. [DOI] [PubMed] [Google Scholar]
- 18.Proulle V, Furie RA, Merrill-Skoloff G, Furie BC, Furie B. Platelets are required for enhanced activation of the endothelium and fibrinogen in a mouse thrombosis model of APS. Blood. 2014;124(4):611-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romay-Penabad Z, Aguilar-Valenzuela R, Urbanus RT, et al. Apolipoprotein E receptor 2 is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood. 2011;117(4):1408-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shapira I, Andrade D, Allen SL, Salmon JE. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64(8):2719-2723. [DOI] [PubMed] [Google Scholar]
- 21.Erkan D, Salmon JE. The role of complement inhibition in thrombotic angiopathies and antiphospholipid syndrome. Turk J Haematol. 2016;33(1):1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manukyan D, Müller-Calleja N, Jäckel S, et al. Cofactor-independent human antiphospholipid antibodies induce venous thrombosis in mice. J Thromb Haemost. 2016;14(5):1011-1020. [DOI] [PubMed] [Google Scholar]
- 23.Versteeg HH, Schaffner F, Kerver M, et al. Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008;111(1):190-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furlan-Freguia C, Marchese P, Gruber A, Ruggeri ZM, Ruf W. P2X7 receptor signaling contributes to tissue factor-dependent thrombosis in mice. J Clin Invest. 2011;121(7):2932-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Landenberg C, Lackner KJ, von Landenberg P, Lang B, Schmitz G. Isolation and characterization of two human monoclonal anti-phospholipid IgG from patients with autoimmune disease. J Autoimmun. 1999;13(2):215-223. [DOI] [PubMed] [Google Scholar]
- 26.Prinz N, Häuser F, Lorenz M, Lackner KJ, von Landenberg P. Structural and functional characterization of a human IgG monoclonal antiphospholipid antibody. Immunobiology. 2011;216(1-2):145-151. [DOI] [PubMed] [Google Scholar]
- 27.Buschmann C, Fischer C, Ochsenhirt V, Neukirch C, Lackner KJ, von Landenberg P. Generation and characterization of three monoclonal IgM antiphospholipid antibodies recognizing different phospholipid antigens. Ann N Y Acad Sci. 2005;1051(1):240-254. [DOI] [PubMed] [Google Scholar]
- 28.Lackner KJ, von Landenberg C, Barlage S, Schmitz G. Analysis of prothrombotic effects of two human monoclonal IgG antiphospholipid antibodies of apparently similar specificity. Thromb Haemost. 2000;83(4):583-588. [PubMed] [Google Scholar]
- 29.Müller-Calleja N, Hollerbach A, Häuser F, Canisius A, Orning C, Lackner KJ. Antiphospholipid antibody-induced cellular responses depend on epitope specificity: implications for treatment of antiphospholipid syndrome. J Thromb Haemost. 2017;15(12):2367-2376. [DOI] [PubMed] [Google Scholar]
- 30.Trommsdorff M, Gotthardt M, Hiesberger T, et al. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97(6):689-701. [DOI] [PubMed] [Google Scholar]
- 31.Prinz N, Clemens N, Strand D, et al. Antiphospholipid antibodies induce translocation of TLR7 and TLR8 to the endosome in human monocytes and plasmacytoid dendritic cells. Blood. 2011;118(8):2322-2332. [DOI] [PubMed] [Google Scholar]
- 32.Versteeg HH, Ruf W. Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity. J Biol Chem. 2007;282(35):25416-25424. [DOI] [PubMed] [Google Scholar]
- 33.Rothmeier AS, Marchese P, Langer F, et al. Tissue factor prothrombotic activity is regulated by integrin-arf6 trafficking. Arterioscler Thromb Vasc Biol. 2017;37(7):1323-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahamed J, Versteeg HH, Kerver M, et al. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA. 2006;103(38):13932-13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruf W, Kelly CR, Schullek JR, et al. Energetic contributions and topographical organization of ligand binding residues of tissue factor. Biochemistry. 1995;34(19):6310-6315. [DOI] [PubMed] [Google Scholar]
- 36.Ruf W, Miles DJ, Rehemtulla A, Edgington TS. Tissue factor residues 157-167 are required for efficient proteolytic activation of factor X and factor VII. J Biol Chem. 1992;267(31):22206-22210. [PubMed] [Google Scholar]
- 37.Ruf W, Schullek JR, Stone MJ, Edgington TS. Mutational mapping of functional residues in tissue factor: identification of factor VII recognition determinants in both structural modules of the predicted cytokine receptor homology domain. Biochemistry. 1994;33(6):1565-1572. [DOI] [PubMed] [Google Scholar]
- 38.Teplyakov A, Obmolova G, Malia TJ, et al. Crystal structure of tissue factor in complex with antibody 10H10 reveals the signaling epitope. Cell Signal. 2017;36:139-144. [DOI] [PubMed] [Google Scholar]
- 39.de Groot PG, Derksen RH, Urbanus RT. The role of LRP8 (ApoER2′) in the pathophysiology of the antiphospholipid syndrome. Lupus. 2010;19(4):389-393. [DOI] [PubMed] [Google Scholar]
- 40.de Groot PG. Platelets as pivot in the antiphospholipid syndrome. Blood. 2014;124(4):475-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramesh S, Morrell CN, Tarango C, et al. Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via β2GPI and apoER2. J Clin Invest. 2011;121(1):120-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. 2008;118(3):1123-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reinhardt C, von Brühl ML, Manukyan D, et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118(3):1110-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Redecha P, Franzke CW, Ruf W, Mackman N, Girardi G. Neutrophil activation by the tissue factor/factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome. J Clin Invest. 2008;118(10):3453-3461. [DOI] [PMC free article] [PubMed] [Google Scholar]