

Abstract
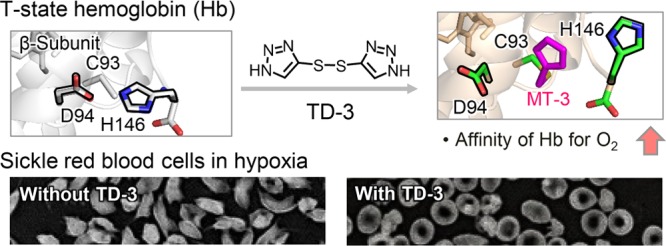
Sickle cell disease is an inherited disorder of hemoglobin (Hb). During a sickle cell crisis, deoxygenated sickle hemoglobin (deoxyHbS) polymerizes to form fibers in red blood cells (RBCs), causing the cells to adopt “sickled” shapes. Using small molecules to increase the affinity of Hb for oxygen is a potential approach to treating sickle cell disease, because oxygenated Hb interferes with the polymerization of deoxyHbS. We have identified a triazole disulfide compound (4,4′-di(1,2,3-triazolyl)disulfide, designated TD-3), which increases the affinity of Hb for oxygen. The crystal structures of carboxy- and deoxy-forms of human adult Hb (HbA), each complexed with TD-3, revealed that one molecule of the monomeric thiol form of TD-3 (5-mercapto-1H-1,2,3-triazole, designated MT-3) forms a disulfide bond with β-Cys93, which inhibits the salt-bridge formation between β-Asp94 and β-His146. This inhibition of salt bridge formation stabilizes the R-state and destabilizes the T-state of Hb, resulting in reduced magnitude of the Bohr effect and increased affinity of Hb for oxygen. Intravenous administration of TD-3 (100 mg/kg) to C57BL/6 mice increased the affinity of murine Hb for oxygen, and the mice did not appear to be adversely affected by the drug. TD-3 reduced in vitro hypoxia-induced sickling of human sickle RBCs. The percentage of sickled RBCs and the P50 of human SS RBCs by TD-3 were inversely correlated with the fraction of Hb modified by TD-3. Our study shows that TD-3, and possibly other triazole disulfide compounds that bind to Hb β-Cys93, may provide new treatment options for patients with sickle cell disease.
Keywords: hemoglobin, red blood cells, sickle cell disease, sickle hemoglobin, oxygen binding affinity, P50, Bohr effect, disulfide compound
Introduction
Sickle cell disease (SCD) is an inherited disorder of hemoglobin (Hb) that affects millions of people worldwide.1,2 During a sickle cell crisis, deoxygenated sickle hemoglobin (deoxyHbS) polymerizes to form fibers of HbS in red blood cells, causing the cells to adopt a “sickled” shape. The formation of sickle red blood cells (SS RBCs) causes hemolysis, microvascular occlusion, thrombosis, and severe pain.1,3 Previous studies identified compounds that can be used to treat SCD. Hydroxyurea increases the concentration of fetal Hb (HbF), which does not form polymers, and improves survival in SCD.4−8 However, the use of hydroxyurea is limited by its lack of effectiveness in some patients,9,10 as well as potential side effects, including neutropenia, infection, and gastrointestinal bleeding.11,12 Recently, l-glutamine was approved in the United States to reduce the acute complications of SCD;13 the mechanism of action of l-glutamine for the treatment of SCD is not yet fully understood.14−16
Hb functions between multiple allosteric structural states that facilitate the efficient uptake and release of oxygen in vivo: the R-state, which exhibits high-affinity for oxygen, and the T-state, which exhibits low-affinity for oxygen. The affinity of Hb for oxygen is dependent upon pH (the Bohr effect), a property that arises from the effects of protons on the equilibrium between the T-state and R-state structures.17,18 Hb β-chain residues β-His146 and β-Asp94 have important roles in the Bohr effect.17,18 At low pH, the β-chain imidazole of β-His146 becomes protonated, resulting in a salt-bridge interaction with the carboxylate of β-Asp94. The salt-bridge stabilizes the T-state of Hb and facilitates oxygen release. At higher pH, the salt-bridge is broken, and the allosteric equilibrium shifts toward the R-state, with a concomitant increase in the affinity of Hb for oxygen.
Oxygenated HbS (oxyHbS) inhibits the polymerization of deoxyHbS, because fully oxygenated HbS does not polymerize.4,7 Therefore, one approach to the treatment of SCD is to use small molecules to increase the affinity of Hb for oxygen4,19,20 and thereby increase the ratio of oxyHbS to deoxyHbS. In a previous study, we screened a library of small molecules and identified a triazole disulfide compound (TD-1, Figure 1A) that increases the affinity of human Hb and intact RBCs for oxygen.21 TD-1 is an oxidized dimer form of mercaptotriazole (MT-1, Figure 1A). The crystal structure of carboxy human adult Hb (COHbA) complexed with TD-1 revealed that MT-1 forms a disulfide bond with the thiol of β-Cys93 and disrupts the T-state salt-bridge interaction between β-Asp94 and β-His146.21 As a result, the allosteric equilibrium of Hb is shifted to the R-state, leading to increased affinity of Hb for oxygen.
Figure 1.

(A) Structure of triazole disulfides (TD-1 and TD-3) and mercaptotriazoles (MT-1 and MT-3). (B) Left-shift of the oxygen dissociation curve (ODC) of purified HbA with increasing molar ratio of TD-3:Hb tetramer. The ODC for TD-3:Hb tetramer molar ratio of 6:1 overlaps with that of the molar ratio of 4:1. (C) The dose-dependent effect of TD-3 on the P50 of HbA. The plots of TD-3 (circle) overlap with the plots of TD-1 (triangle). (D) Left-shift of the ODC of normal human blood with increasing molar ratio of TD-3 to Hb tetramer. (E) The dose-dependent effect of TD-3 on reducing the P50 of normal RBCs. (F) Left-shift of the ODC of SS RBCs with increasing molar ratio of TD-3:Hb tetramer. (G) The dose-dependent effect of TD-3 on reducing the P50 of SS RBCs. The ODCs were measured in triplicate at 37 °C. Each data point in panel C, E, and G represents the mean value of the P50 determined from the ODCs. Error bars represent standard deviation (sd). Most error bars are too short to visualize because the sd was less than 1.2 mmHg.
TD-1 reduced hypoxia-induced sickling of human SS RBCs in vitro and increased the affinity of HbS for oxygen, demonstrating that triazole disulfides have the potential to be new treatments for SCD. Unfortunately, TD-1 is relatively insoluble in water, a feature that limits its use in preclinical investigations.
In this study, we identified a novel triazole disulfide compound (4,4′-di(1,2,3-triazolyl)disulfide, designated “TD-3″, Figure 1A), which has higher aqueous solubility than TD-1, while maintaining the ability to increase the affinity of Hb for oxygen. We investigated the mechanism by which TD-3 increases the affinity of Hb for oxygen using HbA that was chemically modified by N-ethylmaleimide (NEM) at β-Cys93 and a HbA mutant with an alanine replacing β-Cys93 (Hb C93A). We also studied the crystal structures of COHbA and deoxyHbA, each complexed with TD-3, and showed that modification of the thiol of β-Cys93 by TD-3 inhibits salt-bridge formation between β-Asp94 and β-His146, resulting in destabilization of T-state Hb, decreased magnitude of the Bohr effect, and increased affinity of Hb for oxygen. Moreover, we demonstrated that intravenous administration of TD-3 to mice increased the affinity of murine Hb for oxygen, and TD-3 reduced hypoxia-induced sickling of human SS RBCs.
Experimental Section
Materials
Di(5-(2,3-dihydro-1,4-benzodioxin-2-yl)-4H-1,2,4-triazol-3-yl)disulfide (TD-1) was purchased from ChemPartner (Shanghai, China). 4,4′-Di(1,2,3-triazolyl)disulfide hydrate (TD-3) and 5-Mercapto-1H-1,2,3-triazole sodium salt (MT-3) were purchased from TCI America (Portland, OR, USA). N-Ethylmaleimide (NEM) and PEG400 (Catalog No. 202398) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Normal human blood and blood from patients with sickle cell disease (homozygous SS disease) were obtained from volunteers using protocols that were reviewed and approved by Institutional Review Boards (IRB) of the Partners Healthcare Human Research Committee, Boston Medical Center, and the Children’s Hospital of Philadelphia. Outdated donated red blood cells (RBCs) were obtained from the blood bank of the Massachusetts General Hospital and used to obtain HbA. Studies using mice were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital.
Purification of HbA
RBCs were centrifuged at 3000 rpm for 15 min at 4 °C. The supernatant was discarded, and the remaining RBCs were washed three times with an equal volume of saline (aqueous solution of 0.9 wt % sodium chloride). Two equivalents of distilled water (4 °C) were added to the washed RBCs to lyse the cells, and the mixture was centrifuged at 20 000 g for 1 h at 4 °C. The supernatant was collected and centrifuged again (under the same conditions), and the resulting supernatant was collected and dialyzed against Dulbecco’s phosphate buffered saline (PBS, pH 7.4) at 4 °C for 18 h. The dialyzed Hb was passed through 0.2 μm poly(ether sulfone) membrane (Thermo Scientific, Cat. No. 725–2520) to remove protein aggregates. The absorption spectrum of the obtained Hb was measured from 500–700 nm, fitted to a linear combination of pure oxygenated hemoglobin (oxyHb), deoxygenated hemoglobin (deoxyHb), met hemoglobin (metHb), and a baseline using the Solver program (Excel 2013, Microsoft, Redmond, WA, USA) to determine the total concentration of Hb.21
Measurement of the Oxygen Dissociation Curve (ODC) of Purified Hb
The ODC of purified Hb was measured at 37 °C using a HEMOX analyzer (TCS Scientific Corporation, New Hope, PA, USA). TD-1 and TD-3 were dissolved in DMSO, and MT-3 was dissolved in PBS as stock solutions. HbA (20 μM tetramer) was incubated with each of the compounds, which were diluted in PBS containing DMSO (5 vol %) and antifoaming agent (0.2 vol %, TCS Scientific Corporation). After the incubation, the ODC of the samples were measured at 37 °C. In evaluating the Bohr effect of HbA, sodium phosphate buffer (0.1 M phosphate, pH 6, 6.5, 7, 7.5, or 8.0) was used instead of PBS to measure the ODC of Hb with TD-3 (Hb tetramer/TD-3 = 1/6 mol/mol). The partial pressure of oxygen at which 50% of Hb is oxygenated (P50), was determined from the ODC.
Measurement of the ODC of Human RBCs
After obtaining informed consent, blood was collected from healthy volunteers or a sickle disease patient into tubes containing EDTA. The human sickle cell blood was obtained from a single patient who has homozygous SS disease and was treated with hydroxyurea but not chronic blood transfusions. The concentration of Hb in the collected blood was determined using a blood gas analyzer (ABL 800 FLEX, Radiometer America, Brea, CA, USA).
A PEG solution of TD-3 (267 mM) was mixed with PBS (PEG solution of TD-3/PBS = 3/7 v/v) at 37 °C to obtain a stock solution of TD-3 (80 mM of TD-3 or 16 mg/mL of TD-3 hydrate) in PBS with PEG400 (30 vol %). The obtained mixture was filtrated through 0.2 μM poly(ether sulfone) membrane (Catalog No. 725–2520, Thermo Fisher Scientific, Waltham, MA, USA) to remove aggregates, and the filtrate was kept at 37 °C. PBS with PEG400 (30 vol %) was prepared as a control or vehicle for TD-3-treated samples.
The stock solution of TD-3 in PBS with PEG400 (30 vol %) was added to human whole blood, and the mixtures were incubated at 37 °C for 10 min. The molar ratios of TD-3 to Hb tetramer in the mixtures were 0, 1, 2, and 3, and the concentration of PEG400 was 2 vol %. After the incubation, 20 μL of each mixture was diluted with 3 mL HEMOX solution (TCS Scientific Corporation) and 6 μL of the antifoaming agent. HEMOX solution contains N-[Tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid (TES, 30 mM), sodium chloride (135 mM), and potassium chloride (5 mM) in water (pH 7.4). The ODCs of the diluted blood samples were measured using a HEMOX analyzer with the sample maintained at 37 °C.
Intravenous Administration of TD-3 to C57BL/6 Mice and Blood Collection from Mice
C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). A micro catheter, consisting of a 30 G BD PrecisionGlide needle (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and PE10 tubing was used to intravenously administer TD-3 or vehicle to mice. The metal part of the needle was snapped and the tip partially slipped into the tubing. The mouse was restrained and anesthetized with sevoflurane (3–3.5 vol %) in air. Once the mouse was asleep, breath rate was monitored continuously. The tail was then punctured until a flash of blood was observed in the catheter. Saline (50 μL) was injected to confirm that there was no resistance to the injection. The syringe was connected to a volume controlled infusion pump (Braintree Scientific, MA, USA), and the desired volume of the sample was injected at a rate of 30 μL/min. The mouse was kept anesthetized for the duration of the injection. The dose of PEG400 in the vehicle or TD-3 treatment was 1.96 mL/kg. This dose was similar to the maximum dose of PEG400 (2 mL/kg) that can be given to a mouse without causing injury.24 Murine blood was collected before and after the treatment with TD-3 by a tail knick. Drops of blood were collected using a heparinized microhematocrit capillary tube (Fisher Scientific, Pittsburgh, PA, USA).
Measurement of the ODC of Intact Murine RBCs and Murine Hb (as Hemolysate)
The ODC of murine whole blood was measured as described above for human RBCs. To measure the ODC of murine Hb, 20 μL of whole blood was mixed with 180 μL of distilled water to prepare murine Hb as hemolysate. The obtained hemolysate was mixed with 2.8 mL of HEMOX solution containing 6 μL of antifoaming reagent. The ODC of the mixture was measured using HEMOX analyzer at 37 °C.
HPLC Analyses of TD-3-Treated Murine Hb
Murine blood was collected before and after intravenous administration of TD-3. The collected blood (20 μL) was mixed with distilled water (180 μL) to lyse RBCs. The lysed cells were centrifuged at 13 000 g for 10 min to remove precipitates, including RBC ghosts. The supernatant was snap frozen in liquid nitrogen and stored at −80 °C until the HPLC measurement. Clarified hemolysates were analyzed for levels of endogenous (unmodified) or modified adduct forms of Hb by HPLC using the Hitachi D-7000 HPLC System (Hitachi Instruments, San Jose, CA, USA) and a weak cation-exchange column (Poly CAT A: 35 mm × 4.6 mm, Poly LC, Columbia, MD, USA). The samples were eluted with a linear gradient of phase B from 0 to 80% (Mobile Phase A: 20 mM Bis-Tris, 2 mM potassium cyanide, pH 6.95; Phase B: 20 mM Bis-Tris, 2 mM potassium cyanide, 0.2 M sodium chloride, pH 6.55), and the eluted samples were detected by absorbance at 410 nm (A410). The percentage of modified Hb was determined by dividing the area of the peak of modified Hb by the area of the peaks produced by both unmodified and modified Hb.
Preparation of NEM-Treated HbA
NEM-treated HbA was prepared according to a previously reported procedure22 with slight modification. HbA (0.6 mM as a tetramer) was mixed with NEM (12 mM) in PBS and incubated at room temperature in air for 1 h. After the incubation, free NEM was removed by passing the mixture through a Sephadex G25 PD-10 column (GE Healthcare Lifesciences, Pittsburgh, PA, USA). NEM-bound HbA, which appeared as a red chromatographic band, was collected, and the concentration of NEM-HbA was determined as described above for HbA.
Purification of Hemoglobin from Knock-in Mice
Mice expressing human Hb with a β chain C93A mutation (HbC93A mice23) and mice expressing wild-type human Hb (HbC93 mice23) were kindly provided by Dr. Tim Townes (University of Alabama, Birmingham). An animal lancet (Goldenrod, Medipoint, Mineola, NY, USA) was used to collect blood from the facial vein into a heparinized glass capillary. Hb C93A and wt Hb were purified, and the concentration of Hb was determined as described for HbA above.
Crystallization, Data Collection, and Structure Determination of CarboxyHb (COHb) and Deoxygenated Hb (deoxyHb) Complexed with TD-3
To prepare a crystal containing COHbA complexed with TD-3, oxyHbA was vacuumed for 1 h (to remove oxygen), and then carbon monoxide gas was bubbled into the solution. A freshly prepared solution of TD-3 in DMSO was added to COHbA (50 mg/mL) at a one-to-ten molar ratio of Hb tetramer to TD-3 and incubated for 1 h to obtain COHbA complexed with TD-3. To prepare a crystal containing deoxyHbA complexed with TD-3, a freshly prepared solution of TD-3 dissolved in DMSO was added to oxyHbA (50 mg/mL) at a one-to-ten molar ratio of Hb tetramer to TD-3 and incubated for 1 h. The solution of oxyHbA with TD-3 was subjected to a vacuum for at least 1 h to obtain deoxyHbA complexed with TD-3. The resulting COHbA and deoxyHbA were crystallized in solution containing a high concentration of phosphate and/or sulfate salt (3.2–3.4 M) as precipitant using the “batch method,” which was previously described for obtaining COHbA and deoxyHbA crystals.25
X-ray diffraction data were collected at 100 K using Rigaku MicroMax-007HF X-ray Generator and Eiger R 4 M Detector. The crystals were washed using a cryoprotectant solution containing mother liquor (50 μL) and glycerol (8–15 μL) prior to data collection. The data sets were processed with CrysAlisPro (Rigaku, Spring, TX) and the CCP4 suite of programs.26 The COHb crystal is in the space group P41212 with typical cell constant of a = 53, b = 53, c = 192, and isomorphous with the classical R-state COHbA crystal (PDB ID: 1LJW(27)). The deoxyHb was crystallized with the space group P21 and typical cell constant of a = 63, b = 82, c = 54, β = 100, and is isomorphous to the T-state deoxyHbA crystal (PDB ID: 2DN2(28)).
The native human COHbA structure (PDB ID: 1LJW) and native deoxyHbA structure (PDB ID: 2DN2) coordinates were used as starting models to phase and refine the COHbA and deoxyHbA complexed with TD-3, respectively, using the Phenix program.29 The two structures showed MT-3 bound covalently to β-Cys93. It is notable that the use of sodium dithionite as previously described to make deoxygenated Hb25 resulted in poorly bound or no binding of MT-3, probably because dithionite reduced and broke the disulfide bond between MT-3 and β-Cys93. The COHbA complex structure also showed all four hemes ligated with CO. Inclusion of MT-3 and/or CO molecules, as well as water molecules to the protein models, and further refinements resulted in final Rfactor/Rfree of 19.8%/26.5% at 2.0 Å for the COHbA complex structure and 15.8%/19.7% at 1.7 Å for the deoxyHbA complex structure. Model buildings and corrections were carried out using COOT.30 In the crystal structure of COHbA complexed with TD-3, the locations of β-Lys144, β-Tyr145, and β-His146 could not be identified because of disorder. The atomic coordinates and structure factors from the COHbA and deoxyHbA complexed with TD-3 have been deposited in the RCSB Protein Data Bank with the accession codes 6BWU and 6BWP, respectively. Detailed crystallographic parameters are reported in Table S1.
Evaluation of the Ability of TD-3 To Reduce Sickling of SS RBCs in Vitro
SS blood was collected from many different patients, all of whom had homozygous SS disease. None of the blood donors were treated with hydroxyurea or chronic blood transfusions. The SS blood was suspended in HEMOX solution (100 μL) supplemented with glucose (10 mM) and bovine serum albumin (0.2 wt %) to a final hematocrit of 20%. The suspensions were incubated in a 96-well plate under air in the presence of vehicle (PBS with 30 vol % PEG400) or four different concentrations of TD-3 (0.5, 1.0, 1.5, and 2 mM) at 37 °C for 1 h. The concentration of PEG400 in the mixture of RBCs with vehicle or TD-3 was 2 vol %. The suspensions were exposed to a gas mixture of O2 (4%) and N2 (96%) at 37 °C for 2 h. After the incubation, aliquots (5 μL) of each sample were used to evaluate the sickling of RBCs, and the residual samples were used for HPLC and ODC studies to measure the modified Hb% and the affinity of HbS for oxygen.
After incubation in 4% O2, aliquots (5 μL) of each sample were collected without exposure to air and fixed in 2% glutaraldehyde. Cells were placed on glass microslides (Fiber Optic Center)21,31 so as to produce a single layer of cells and subjected to microscopic morphological analysis of bright field images (at 40× magnification) using an Olympus BX40 microscope fitted with an Infinity Lite B camera (Olympus, Center Valley, PA, USA) and Infinity Capture software. The percentage of sickled cells for each condition was determined using the computer-based image analysis system.32 The percentage of sickled cells (sickled%) induced by hypoxia was determined by subtracting the percentage of cells that were sickle shaped prior to hypoxia from the percentage that appeared sickled after hypoxia.
SS RBCs treated with TD-3 were lysed, and aliquots of the hemolysates were utilized for HPLC and ODC studies. HPLC studies were conducted as described for murine Hb. The ODC of the hemolysates was measured in potassium phosphate buffer (0.1 M phosphate, pH 7.0) supplemented with antifoaming agent at 25 °C using the HEMOX Analyzer.
Statistics
Statistical analysis was performed using GraphPad Prism 7 software (GraphPad Software, La Jolla, CA). A two-tailed, unpaired t test was used to compare the values of P50 or the percentage of sickled cells at two different concentrations of TD-3. The value of r (= |ΔlogP50/ΔlogpH|) of HbA with and without TD-3 was compared by analysis of covariance (ANCOVA). A two-tailed, paired t test was used to compare the values of P50 before and after the injection of TD-3 to mice. Statistical significance was defined as a p-value of less than 0.05.
Results
Solubility of TD-3 in Aqueous Solution Is Greater than that of TD-1
Although TD-1 was able to increase the affinity of Hb for oxygen, the further use of this compound is limited by its lack of aqueous solubility. In contrast to TD-1, when a solution of TD-3 was warmed to 37 °C, it was readily soluble in Dulbecco’s phosphate buffered saline (PBS) containing polyethylene glycol (PEG) 400 (30 vol %) at a concentration of 80 mM (compared to 4 mM for TD-1). The increased solubility of TD-3 permits intravenous administration of TD-3.
TD-3 Increases the Affinity of Human and Murine Hemoglobin and Red Blood Cells for Oxygen
We tested the ability of TD-3 to alter the affinity of purified Hb for oxygen, under the same conditions that were previously used to evaluate TD-1.21 In these in vitro studies, TD-3 was dissolved in DMSO and added to HbA (TD-3/Hb tetramer = 0, 1, 2, 4, and 6 mol/mol). In the mixture of HbA and TD-3, the concentration of Hb was 20 μM (as Hb tetramer) and the fraction of DMSO was 5 vol %. After a 10 min incubation at 37 °C, the oxygen dissociation curve (ODC) of the mixture was measured and was used to determine the partial pressure of oxygen at which 50% of Hb is oxygenated (P50). Incubation of TD-3 with HbA shifted the ODC to the left with increasing concentrations of TD-3 (Figure 1B), resulting in a marked reduction in P50. TD-3 decreased the P50 of Hb from 16 ± 0.2 mmHg (without TD-3, mean ± standard deviation) to 4.5 ± 0.07 mmHg (TD-3/Hb tetramer = 6 mol/mol, Figure 1C). The effect of TD-3 on the P50 of HbA was dose-dependent and was similar to that observed with TD-1 (Figure 1C). When the molar ratio of TD-3:HbA tetramer was decreased to 1:1, the P50 of TD-3-treated HbA was still lower than the P50 of HbA alone (TD-3-treated vs DMSO-treated HbA; 16 ± 0.2 mmHg vs 11 ± 0.2 mmHg, p-value <0.001).
To test the effect of the disulfide bond of TD-3 on the ability of TD-3 to increase the affinity of Hb for oxygen, 5-mercapto-1H-1,2,3-triazole (MT-3) was added to HbA at the same molar ratios as we used to test TD-3. MT-3 is a monomeric thiol form of TD-3 and does not have a disulfide bond (Figure 1A). Incubation of HbA with MT-3 for 10 min at 37 °C did not alter the P50 of HbA (Figure 1C). These results indicate that, as with TD-1,21 the disulfide bond in TD-3 is required to increase the affinity of Hb for oxygen.
To test the ability of TD-3 to alter the affinity of intact RBCs for oxygen, TD-3 was dissolved in PBS containing 30 vol % PEG400 and was added to normal human whole blood at increasing molar ratios (TD-3/Hb tetramer = 0, 1, 2, and 3 mol/mol). In the mixture of TD-3 and whole blood, the fraction of PEG400 was 2 vol %. After a 10 min incubation at 37 °C, the mixture was diluted with HEMOX solution, and the ODC of the diluted blood sample was measured to determine the P50 of the RBCs. Incubation of TD-3 with whole blood, at a TD-3:Hb tetramer molar ratio of 3:1, shifted the ODC to the left (Figure 1D), resulting in a reduction in P50 from 27 ± 0.4 to 7.4 ± 0.1 mmHg (Figure 1E). The effect of TD-3 on the P50 of RBCs was dose-dependent (Figure 1E). When the molar ratio of TD-3:Hb tetramer was decreased to 1:1, the P50 of TD-3-treated RBCs was still lower than the P50 of RBCs in the absence of TD-3 (TD-3-treated vs vehicle-treated normal RBCs; 27 ± 0.4 mmHg vs 23 ± 0.2 mmHg, p-value <0.001).
To investigate whether the effect of TD-3 on SS RBC was similar to its effect on normal RBCs, we tested the ability of TD-3 to decrease the P50 of SS RBCs. Incubation of TD-3 with SS blood, at a TD-3:Hb tetramer molar ratio of 3:1, shifted the ODC to the left (Figure 1F), resulting in a reduction in P50 from 29 ± 0.3 to 7.8 ± 0.4 mmHg (Figure 1G). The effect of TD-3 on the P50 of SS RBCs was dose-dependent (Figure 1G) and was similar to the effect of TD-3 on the P50 of normal RBCs (Figure 1E). When the molar ratio of TD-3:Hb tetramer was decreased to 1:1, the P50 of TD-3-treated SS RBCs was still lower than the P50 of SS RBCs in the absence of TD-3 (TD-3-treated vs vehicle-treated SS RBCs; 29 ± 0.3 mmHg vs 24 ± 0.4 mmHg, p-value <0.001, Figure 1G).
To investigate whether TD-3 increases the affinity of RBCs for oxygen in vivo, TD-3 (50 or 100 mg/kg) or vehicle (DPBS with 30 vol % PEG400) alone was administered intravenously to C57BL/6 mice. Blood was collected before (as baseline) and 1 h after the administration of TD-3. The collected blood was diluted with HEMOX solution and the ODC of the diluted solution was measured to determine the P50 of the RBCs. One hour after the administration of vehicle, the P50 of the RBCs was not changed from baseline. In mice treated with a low dose of TD-3 (50 mg/kg), there was a trend toward a decrease in the P50 of the RBCs 1 h after administration (baseline vs 1 h; 41 ± 1.5 mmHg vs 39 ± 0.5 mmHg, p-value = 0.064). Administration of a higher dose of TD-3 (100 mg/kg) produced a more substantial shift of the ODC to the left (Figure 2A), reflecting a 19% decrease in the P50 of murine RBCs (baseline vs 1 h; 42 ± 1.3 mmHg vs 34 ± 2.5 mmHg, p-value = 0.011). All of the mice treated intravenously with vehicle or either dose of TD-3 appeared normal 24 h after administration of TD-3.
Figure 2.

Increase of the affinity of murine Hb for oxygen and covalently modified murine Hb by administration of TD-3 (100 mg/kg) to C57BL/6 mice. The oxygen dissociation curve of C57BL/6 murine RBCs (A) and murine hemolysate (B) measured at 37 °C before and 1 h after intravenous administration of TD-3 (100 mg/kg). The effect of time on the P50 of murine Hb (C) and the percentage of modified Hb (D) before and after administration of TD-3. (E) Correlation between the percentage of modified Hb and the change in P50 of the hemolysate. ΔP50% = |(P50 at a time point) – (baseline P50)|/(baseline P50). Each data point in C, D, and E represents the mean value, and error bars represent the standard deviation of the P50 or modified Hb%. Four mice were used in each of the vehicle- and TD-3-treated group.
To investigate the ability of TD-3 to covalently modify Hb in murine blood, blood was collected before and 1 h after the administration of TD-3, and the fraction of Hb that was modified by TD-3 was assessed using cation-exchange HPLC. Before the administration of TD-3, HPLC analysis of collected blood showed a major peak produced by unmodified murine Hb, with a retention time of 4.2 min (Figure S1). HPLC analyses of murine blood collected 1 h after the administration of TD-3 revealed a new peak (Hb modified by TD-3), which had a retention time of 3.7 min (Figure S1C). The percentage of modified Hb was determined by dividing the area of the peak of modified Hb by the area of the peaks produced by both unmodified and modified Hb. In mice administered with the lower concentration of TD-3, the percentage of modified Hb was 9.0% ± 2.5 (mean value ± sd). After treatment with the higher concentration of TD-3 (100 mg/kg), the percentage of modified Hb increased to 19% ± 3.9. These results indicate that intravenous administration of TD-3 produces a dose-dependent increase in modified Hb.
We investigated the effect of time on both the TD-3-induced change in affinity of murine Hb for oxygen and the percent of murine Hb modified by TD-3. TD-3 (100 mg/kg) or vehicle was administered intravenously to C57BL/6 mice, and blood was collected before and 1, 5, 10, and 24 h after administration. The ODC of the hemolysates was measured to determine the P50 values of murine Hb and the hemolysates were also subjected to HPLC to determine the percentage of modified Hb. The P50 of murine Hb before the administration of TD-3 was 21 ± 1.0 mmHg. The reduction in the P50 of murine Hb was greatest 1 h after treatment (baseline vs 1 h treatment; 21 ± 1.0 vs 18 ± 0.5 mmHg, p-value <0.01; Figure 2B,C), and a decrease in P50 was still detected 5 h after administration of TD-3 (baseline vs 5 h treatment, 21 ± 1.0 vs 19 ± 0.6 mmHg, p-value <0.05; Figure 2C). The P50 of Hb returned to baseline 10 h after administration of TD-3 (baseline vs 10 h treatment; 21 ± 1.0 vs 21 ± 0.7 mmHg, Figure 2C). The percentage of modified Hb was greatest 1 h after administration of TD-3 (Figure S2B); modified Hb was no longer detected 24 h after administration of TD-3 (Figures S2E and 2D). The percentage of modified Hb was positively correlated with the change in the P50 of the hemolysate (Figure 2E).
Thiol of β-Cys93 Is Required for TD-3 To Increase the Affinity of Hemoglobin for Oxygen
In previous studies, we showed that TD-1 interacted with the thiol of Hb β-Cys93 to increase the affinity of Hb for oxygen. In this study, we used NEM, a small molecule that covalently binds to Hb β-Cys9322,33−35 to investigate whether TD-3, like TD-1, also interacts with this residue. NEM was added to HbA (NEM/Hb tetramer = 20/1 mol/mol) in PBS and the mixture was incubated at room temperature for 1 h. After incubation, the remaining free NEM was removed from NEM-treated HbA using size exclusion column chromatography. Increasing concentrations of TD-3 were added to NEM-treated HbA (20 μM), and the mixtures were incubated at 37 °C for 10 min. After incubation, the ODC of NEM-treated HbA was measured to determine the P50. In the absence of TD-3, the P50 of NEM-treated HbA was 7.8 ± 0.6 mmHg. Increasing concentrations of TD-3 did not further reduce the P50 of NEM-treated HbA (Figure 3A). When NEM-treated HbA was incubated with TD-3, at a TD-3 to Hb tetramer molar ratio of 6:1, the P50 was 8.3 ± 0.2 mmHg. The results suggest that prior modification of the thiol of β-Cys93 by NEM blocks the ability of TD-3 to increase the affinity of Hb for oxygen.
Figure 3.

Effect of TD-3 on the oxygen affinity of NEM-modified Hb (A) and Hb C93A (B). NEM treatment blocked the ability of TD-3 to further decrease the P50 of HbA. TD-3 did not decrease the P50 of Hb C93A. Each data point represents the mean P50, and error bars represent standard deviation (sd) of the P50 measured at 37 °C in triplicate. Most error bars are too short to visualize because the sd was less than 0.7 mmHg.
A HbA mutant with an alanine replacing β-Cys93 (HbC93A) was used to further investigate the mechanism by which TD-3 alters the affinity of Hb for oxygen. In these studies, HbC93A was purified from blood that was collected from knock-in mice that exclusively express HbA with β-chain C93A (“HbC93A mice”).23 As a control, Hb was purified from blood that was collected from knock-in mice (“HbC93 mice”)23 that express human wild-type HbA (“wt Hb” or HbC93). The P50 values of wt Hb and HbC93A, in the absence of TD-3, were 16 ± 0.1 and 16 ± 0.2 mmHg, respectively. These P50 values were similar to the P50 of HbA purified from human blood (17 ± 0.3 mmHg). When wt Hb was incubated with TD-3 at a TD-3:Hb tetramer molar ratio of 6:1, the P50 of wt Hb was reduced from 16 ± 0.1 to 6.2 ± 0.6 mmHg (p-value <0.001, Figure 3B). This change was similar to that observed using HbA purified from normal human blood (Figure 1c). In contrast, the P50 of HbC93A was unchanged by increasing the concentration of TD-3 (Figure 3B). These results, taken together with the results using NEM-treated Hb, support the hypothesis that the thiol of Hb β-Cys93 is required for the ability of TD-3 to increase the affinity of Hb for oxygen.
TD-3 Decreases the Magnitude of the Bohr Effect
The Bohr effect is the pH dependency of the affinity of Hb for oxygen, a property that results from effects of protons on the equilibrium between the T-state and R-state structures.17,18,36−38 The equation r = |ΔlogP50/ΔlogpH| is a measure of the magnitude of the Bohr effect.38 Disrupting the salt-bridge between β-Asp94 and β-His146 destabilizes the T-state of Hb and significantly contributes to decreasing r (making the P50 of Hb less dependent on pH).17,18,37,39−42 In previous studies, we showed that treatment of HbA with TD-1 decreases r when compared to untreated HbA (Figure S3), suggesting that TD-1 inhibited salt-bridge formation between β-Asp94 and β-His146.21
We investigated whether TD-3, like TD-1, changes the magnitude of the Bohr effect. HbA in phosphate buffer (phosphate 0.1 M) was treated with TD-3 (TD-3/Hb tetramer = 6/1 mol/mol) at pH 6.0, 6.5, 7.0, 7.5, and 8.0, and the ODC of each sample was measured at 37 °C. Compared to untreated HbA, treatment of HbA with TD-3 decreased r (HbA with TD-3 vs HbA alone; 0.23 ± 0.013 vs 0.37 ± 0.012, p-value <0.001, Figure S3). This result indicates that TD-3 makes the P50 of HbA less dependent on pH. As with TD-1, the ability of TD-3 to reduce the pH dependency of the P50 of HbA is likely caused by inhibiting salt-bridge formation between β-Asp94 and β-His146.
Crystal Structures of R-State and T-State HbA, Each in a Complex with TD-3 Show a Disulfide Bond between MT-3 and the Thiol of β-Cys93
To further investigate the mechanism by which TD-3 increases the affinity of Hb for oxygen, we determined the crystal structures of COHbA and deoxyHbA, each in a complex with TD-3. DeoxyHbA was prepared from a solution containing oxyHbA by using a vacuum to remove oxygen. COHbA was prepared by adding carbon monoxide to a solution containing deoxyHbA. COHbA and deoxyHbA were each cocrystallized with a 10-fold molar excess of TD-3 in buffer containing a high concentration of phosphate and/or sulfate salt (3.2–3.4 M) using a modification of the batch method.25
The crystal of COHbA complexed with TD-3 was isomorphous with the crystal of native COHbA (PDB ID: 1LJW(27)) with the space group of P41212, while the crystal of deoxyHbA complexed with TD-3 was isomorphous with the crystal of native deoxyHbA (PDB ID: 2DN2(28)) with the space group P21. The crystal structures of COHbA and deoxyHbA (1LJW and 2DN2, respectively) were used to phase and refine the respective complexes, and detailed crystallographic parameters are reported in Table S1. The COHbA complexed with TD-3 showed CO molecules binding at all four heme irons. Both structures revealed one molecule of MT-3 (a monomeric thiol form of TD-3) covalently binding at the thiol of each β-Cys93.
MT-3 Binds to COHbA and Stabilizes the R-State of HbA
The crystal structure of COHbA complexed with TD-3 revealed that MT-3 forms a disulfide bond with the thiol of β-Cys93 (Figures 4 and S4). The N1 atom of the triazole ring formed a hydrogen-bond interaction with the peptide oxygen atom of β-Ala142 (Figure 4B), while the N2 and N3 atoms of the triazole ring formed water-mediated hydrogen-bond interactions with α-Ser35, α-Thr38, β-Asp94, and β-His143. MT-3 occupied the position of β-Tyr145 in the native COHbA (1LJW) (Figure S5), suggesting that MT-3 causes a significant change in the location of the C-terminus of the β-chain, including β-Lys144, β-Tyr145, and β-His146. The new locations of β-Tyr145 and β-His146 could not be identified because of disorder. The structure of COHbA complexed with TD-3 suggests that the binding of MT-3 at β-Cys93 prevents β-His146 from forming a salt-bridge interaction with β-Asp94 when Hb is converted from the R- to the T-state. Disruption of the salt-bridge interaction explains, in part, the reduced magnitude of the Bohr effect and the increased affinity of Hb for oxygen in the presence of TD-3.
Figure 4.
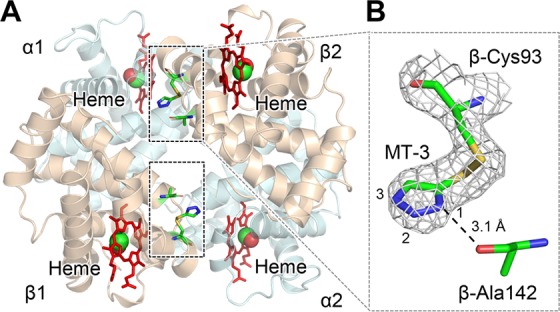
Crystal structure of COHbA in a complex with TD-3. MT-3 formed a disulfide bond with the thiol of COHbA β-Cys93. (A) The binding sites of MT-3 in COHbA. Hb α and β subunits are shown as pale blue and brown, respectively. Carbon monoxide bound to heme is shown as a red and green sphere. The locations of MT-3, β-Cys93, and β-Ala142 are shown as sticks within the dashed rectangles. (B) The electron density of MT-3 and β-Cys93 indicated by the 2Fo-Fc map (gray mesh, contoured at 1.0σ). The N1 atom of MT-3 at β-Cys93 formed a hydrogen bond with the oxygen atom of β-Ala142, which is shown as a dotted line.
MT-3 Binds to DeoxyHbA and Destabilizes the T-State of HbA
The crystal structure of deoxyHbA complexed with TD-3 confirms that MT-3 forms a disulfide bond with the thiol of β-Cys93 (Figures 5A,B and S6). The N3 atom of the triazole ring formed a hydrogen bond with the side chain of β-His146, while the N2 atom of the triazole ring formed a hydrogen bond with the oxygen atom of β-Lys144 (Figure 5B). As noted previously, in native deoxyHbA (e.g., 2DN2, Figure 5C), there is a salt-bridge interaction between β-Asp94 and β-His146, which stabilizes the T-state.43 In deoxyHbA complexed with TD-3, the covalently bound MT-3 at β-Cys93 localized between β-Asp94 and β-His146 and moved the two residues away from each other, preventing the salt-bridge interaction (Figures 5D and S7). The distance between β-Asp94 and β-His146 increased from 3 to 12 Å (native deoxyHbA vs deoxyHbA complexed with TD-3, Figure 5C,D). The disruption of the salt-bridge formation is consistent with the TD-3-induced increase in the affinity of Hb for oxygen and the decrease in the magnitude of the Bohr effect.
Figure 5.

Crystal structure of deoxyHbA in a complex with TD-3. MT-3 formed a disulfide bond with the thiol of deoxyHbA β-Cys93. (A) The binding sites of MT-3 in deoxyHbA. Hb α and β subunits are shown in pale blue and brown, respectively. The locations of MT-3, β-Cys93, β-Lys144, and β-His146 are shown as sticks within the dashed rectangular areas. (B) The electron density of MT-3 and β-Cys93 indicated by the 2Fo-Fc map (gray mesh, contoured at 1.0σ). (C) β-subunit of the crystal structure of native deoxyHbA (PDB ID: 2DN2). β-Asp94 and β-His146 formed the characteristic salt-bridge to stabilize the T-state Hb. (D) β-subunit of the crystal structure of deoxyHbA in a complex with TD-3. The binding of MT-3 at the thiol of β-Cys93 disrupted the salt-bridge interaction between β-Asp94 and β-His146.
TD-3 Reduces Hypoxia-Induced Sickling of Human SS Red Blood Cells in Vitro
TD-3 significantly increased the affinity of human SS RBCs for oxygen (Figure 1F,G). To investigate the ability of TD-3 to reduce sickling, SS RBCs were incubated with TD-3 and then exposed to a low concentration of oxygen (hypoxia) to induce sickling. Human SS RBCs (hematocrit∼ 20%) were mixed with vehicle or TD-3 (0.5, 1.0, 1.5, and 2 mM) in air and then incubated in a gas mixture of oxygen (4%) and nitrogen (96%) at 37 °C for 2 h. After incubation, SS RBCs were fixed using glutaraldehyde (2%), and the number of sickled cells and normal cells were counted. Treatment of SS RBCs with TD-3 in 4% oxygen changed the shape of the RBCs from discoid to sickled (Figure 6A) and increased the percentage of sickled cells from 7.4% ± 0.7 (air) to 93% ± 2.5 (mean value ± sd, p-value <0.001, Figure 6A). When SS RBCs were treated with increasing concentrations of TD-3 and then exposed to 4% O2, the percentage of sickled cells decreased in a dose-dependent manner (Figure 6B–E). At the highest dose of TD-3 tested, the percentage of sickled cells decreased from 93% ± 2.5 (without TD-3) to 17% ± 3.5 (p-value <0.001, Figure 6E and F). Hemolysis of RBCs was not detected at any concentration of TD-3 (0–2 mM).
Figure 6.

Inhibition of hypoxia-induced sickling of SS RBCs by TD-3 in vitro. The morphology of SS RBCs treated with vehicle (A) or with TD-3 (0.5–2 mM, B–E) and incubated with 4% oxygen at 37 °C for 2 h. (F) The effect of TD-3 on the percentage of sickled cells (sickled cells%) in cells exposed to hypoxia. (G) Representative oxygen dissociation curves (ODCs) of the hemolysate of SS RBCs with or without TD-3. The ODCs were measured in phosphate buffer (0.1 M phosphate, pH 7.0) at 25 °C. (H) HPLC chromatograms of hemolysates prepared from SS RBCs treated with TD-3 (0–2 mM). (I) The effect of treating SS RBCs with TD-3 on the percentage of modified Hb (modified Hb%). (J) The relationship between the modified Hb% and sickled cell% of SS RBCs treated with TD-3. Each data point represents the mean value of modified Hb% or sickled cells% measured in triplicate. Error bars represent standard deviation. Panel J was generated using the data from panels F and I.
To determine whether addition of TD-3 to SS RBCs also increased the affinity of HbS for oxygen, hemolysates were prepared from blood samples, prior to adding glutaraldehyde, and the ODC was measured to determine the P50 of HbS. Increasing the concentration of TD-3 shifted the ODC of the hemolysate to the left and decreased the P50 of the hemolysate (Figure 6G). The treatment of SS RBCs with TD-3 (2 mM) significantly reduced the P50 of the hemolysate from 7.8 ± 1.3 (vehicle treated) to 2.0 ± 0.5 mmHg (TD-3 treated; p-value <0.01). The results indicate that TD-3 increases the affinity of HbS for oxygen while also reducing the percentage of sickled SS RBCs.
To investigate the ability of TD-3 to modify HbS in SS RBCs, HPLC was performed on hemolysates prepared from SS RBCs. In cells treated with vehicle alone, HPLC analyses of SS RBC lysate showed a major peak with a retention time of 4.2 min (Figure 6H). In the presence of TD-3, HPLC analyses of the hemolysate showed a new peak with a retention time of 4.1 min (Figure 6H). The percentage of modified Hb increased with increasing concentrations of TD-3 (Figure 6I). When SS RBCs were treated with the highest concentration of TD-3 (2 mM), HPLC analyses of HbS did not show the peak of unmodified HbS (Figure 6H), suggesting that this concentration of TD-3 modified all HbS in SS RBCs. As the fraction of modified HbS in TD-3-treated SS RBCs increased, the percentage of sickled RBCs decreased (Figure 6J).
Discussion
In previous studies, we showed that a triazole disulfide compound, TD-1, increased the affinity of Hb for oxygen. The potential use of TD-1 as a drug to treat SCD was limited by the low aqueous solubility of TD-1. In this study, we describe a derivative of TD-1, designated TD-3, that retains the ability to increase the affinity of Hb for oxygen but has increased solubility. Covalent modification of the thiol of β-Cys93 was required for TD-3 to increase the affinity of Hb for oxygen. Intravenous administration of TD-3 (100 mg/kg) to C57BL/6 mice increased both the affinity of murine RBCs for oxygen and the percentage of modified Hb. TD-3 markedly increased the affinity of SS RBCs for oxygen and reduced hypoxia-induced sickling of RBCs.
To understand the structural basis of how TD-3 decreased the Bohr effect and increased the affinity of Hb for oxygen, we determined the crystal structures of COHbA and deoxyHbA, each complexed with TD-3. The structures revealed that modification of the thiol of β-Cys93 by MT-3 inhibits formation of the salt-bridge interaction between β-Asp94 and β-His146. The salt-bridge interaction is very important in stabilizing the T-state Hb, as well as contributing to the Bohr effect. Disruption of the salt-bridge interaction is consistent with the ability of TD-3 to increase the affinity of Hb for oxygen and decrease the magnitude of the Bohr effect.
The changes in the affinity of Hb for oxygen and the magnitude of the Bohr effect are consistent with previous reports that disulfide compounds (cystine dimethylester and formamidine disulfide)43,44 and NEM33−35 have a similar effect. Formamidine disulfide reduces the P50 of HbA to a greater extent than cystine dimethylester,45 and we previously demonstrated that TD-1 reduces the P50 of HbA greater than formamidine disulfide and NEM.21 The ability of TD-3 to reduce the P50 of Hb was similar to that of TD-1. We speculate that the covalent binding of MT-3 to the thiol of β-Cys93 disrupts the salt-bridge between β-Asp94 and β-His146 to a greater extent than either NEM or formamidine disulfide.
Intravenous administration of TD-3 to mice increased the affinity of murine Hb for oxygen. Because murine Hb, like human Hb, has β-Cys93,46 the thiol of murine Hb β-Cys93 could react with TD-3 to make a disulfide bond between MT-3 and β-Cys93 to increase the affinity of Hb for oxygen. One potential limitation to the use of TD-3 as a treatment for SCD is that only a relatively small fraction of the Hb in murine blood was modified by TD-3. One hour after administration of TD-3 (100 mg/kg) the percentage of modified murine Hb increased to only 15–20%. This finding may suggest that TD-3 is insufficiently delivered to Hb in RBCs. Alternatively, it is possible that TD-3 may covalently or noncovalently bind to other molecules (including thiols) in blood, and thus may be unable to reach Hb β-Cys93. Modification of TD-3, so as to increase the interaction between the compound’s side chains and amino acids that are adjacent to Hb β-Cys93, may improve the specificity of the interaction between the compound and Hb. We anticipate that these changes would increase delivery of the molecule to Hb and would decrease potential off-target effects.
In conclusion, we identified a triazole disulfide compound (TD-3) that covalently modifies the thiol of Hb β-Cys93 and increases the affinity of Hb for oxygen. Covalent binding to the thiol of Hb β-Cys93 is required for TD-3 to increase the affinity of Hb for oxygen. Structural studies of both COHbA and deoxyHbA suggest that binding of TD-3 to β-Cys93 leads to stabilization of the R-state and destabilization of the T-state, resulting in the increased affinity of Hb for oxygen. Intravenous administration of TD-3 (100 mg/kg) to C57BL/6 mice increased the oxygen affinity of Hb; all of the TD-3 treated mice appeared normal 24 h after the administration of TD-3. TD-3 reduced in vitro hypoxia-induced sickling of human SS RBCs. The percentage of sickled RBCs and the P50 of human SS RBCs by TD-3 were inversely correlated with the fraction of Hb modified by TD-3. Taken together, our findings suggest that TD-3, and related triazole disulfide compounds that bind to Hb β-Cys93, may prove to be new treatments for patients with sickle cell disease.
Acknowledgments
The authors wish to acknowledge T. Townes, PhD, and K. Pawlik, PhD (University of Alabama, Birmingham), for kindly providing the authors with mice that express Hb C93 and C93A. The authors also wish to acknowledge L. McMahon, MD, and A. Akinbami (Boston Medical Center) for helping the authors to obtain blood from sickle cell disease patients. Structural biology resources were provided by a NIH Shared Instrumentation Grant S10-OD021756 and Virginia General Assembly Higher Education Equipment Trust Fund (HEETF) to Virginia Commonwealth University (M.K.S.). This work was supported by Eleanor and Miles Shore 50th Anniversary Fellowship Program for Scholars in Medicine, Harvard Medical School and Massachusetts General Hospital (A.N.), Sanofi innovation award program (iAwards, A.N.), NIH/NIMHD grant MD009124 (M.K.S. and O.A.), NIH/NIDDK grant DK082971 (D.B.B.), and funds from the Department of Anesthesia, Critical Care, and Pain Medicine at Massachusetts General Hospital (W.M.Z.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.8b00108.
Crystallographic data and refinement statistics, representative HPLC chromatograms, the effect of TD-3 on the pH dependency, electron density maps, and a comparison of the crystal structure of native COHbA and COHbA complexed with TD-3 (PDF)
Author Contributions
# D.B.B. and W.M.Z. contributed equally to this work.
The authors declare the following competing financial interest(s): The General Hospital Cooperation has filed a patent related to TD-1 and TD-3. The atomic coordinates and structure factor files have been submitted to the Protein Data Bank under an accession code for COHbA in a complex with TD-3 (PDB ID: 6BWU) and deoxyHbA in a complex with TD-3 (PDB ID: 6BWP).
Supplementary Material
References
- Rees D. C.; Williams T. N.; Gladwin M. T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- Center for Disease Control and Prevention. https://www.cdc.gov/ncbddd/sicklecell/data.html (accessed on Jan 17, 2018).
- Bunn H. F. Pathogenesis and treatment of sickle cell disease. N. Engl. J. Med. 1997, 337 (11), 762–9. 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- Eaton W. A.; Bunn H. F. Treating sickle cell disease by targeting HbS polymerization. Blood 2017, 129, 2719–2726. 10.1182/blood-2017-02-765891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg M. H.; McCarthy W. F.; Castro O.; Ballas S. K.; Armstrong F. D.; Smith W.; Ataga K.; Swerdlow P.; Kutlar A.; DeCastro L.; Waclawiw M. A. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell, A.; Follow-Up, M. S. H. P., The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am. J. Hematol. 2010, 85, 403–408. 10.1002/ajh.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg M. H.; Barton F.; Castro O.; Pegelow C. H.; Ballas S. K.; Kutlar A.; Orringer E.; Bellevue R.; Olivieri N.; Eckman J.; Varma M.; Ramirez G.; Adler B.; Smith W.; Carlos T.; Ataga K.; DeCastro L.; Bigelow C.; Saunthararajah Y.; Telfer M.; Vichinsky E.; Claster S.; Shurin S.; Bridges K.; Waclawiw M.; Bonds D.; Terrin M. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA 2003, 289, 1645–1651. 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- Eaton W. A.; Hofrichter J. Sickle cell hemoglobin polymerization. Adv. Protein Chem. 1990, 40, 63–279. 10.1016/S0065-3233(08)60287-9. [DOI] [PubMed] [Google Scholar]
- Akinsheye I.; Alsultan A.; Solovieff N.; Ngo D.; Baldwin C. T.; Sebastiani P.; Chui D. H.; Steinberg M. H. Fetal hemoglobin in sickle cell anemia. Blood 2011, 118, 19–27. 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg M. H.; Lu Z. H.; Barton F. B.; Terrin M. L.; Charache S.; Dover G. J. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter Study of Hydroxyurea. Blood 1997, 89, 1078–1088. [PubMed] [Google Scholar]
- Charache S.; Dover G. J.; Moyer M. A.; Moore J. W. Hydroxyurea-induced augmentation of fetal hemoglobin production in patients with sickle cell anemia. Blood 1987, 69, 109–116. [PubMed] [Google Scholar]
- Platt O. S. Hydroxyurea for the treatment of sickle cell anemia. N. Engl. J. Med. 2008, 358, 1362–1369. 10.1056/NEJMct0708272. [DOI] [PubMed] [Google Scholar]
- Wong T. E.; Brandow A. M.; Lim W.; Lottenberg R. Update on the use of hydroxyurea therapy in sickle cell disease. Blood 2014, 124, 3850–3857. 10.1182/blood-2014-08-435768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA approved l-glutamine powder for the treatment of sickle cell disease. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm566097.htm (accessed on Jan 17, 2018).
- FDA D.I.S.C.O.: l-glutamine for sickle cell disease. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm572213.htm (accessed on Jan 17, 2018).
- Niihara Y.; Zerez C. R.; Akiyama D. S.; Tanaka K. R. Increased red cell glutamine availability in sickle cell anemia: demonstration of increased active transport, affinity, and increased glutamate level in intact red cells. J. Lab. Clin. Med. 1997, 130, 83–90. 10.1016/S0022-2143(97)90062-7. [DOI] [PubMed] [Google Scholar]
- Niihara Y.; Zerez C. R.; Akiyama D. S.; Tanaka K. R. Oral L-glutamine therapy for sickle cell anemia: I. Subjective clinical improvement and favorable change in red cell NAD redox potential. Am. J. Hematol. 1998, 58, 117–121. . [DOI] [PubMed] [Google Scholar]
- Perutz M. F. Structure and mechanism of haemoglobin. Br. Med. Bull. 1976, 32, 195–208. 10.1093/oxfordjournals.bmb.a071363. [DOI] [PubMed] [Google Scholar]
- Perutz M. F.; Wilkinson A. J.; Paoli M.; Dodson G. G. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 1–34. 10.1146/annurev.biophys.27.1.1. [DOI] [PubMed] [Google Scholar]
- Oder E.; Safo M. K.; Abdulmalik O.; Kato G. J. New developments in anti-sickling agents: can drugs directly prevent the polymerization of sickle haemoglobin in vivo?. Br. J. Haematol. 2016, 175, 24–30. 10.1111/bjh.14264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safo M. K.; Kato G. J. Therapeutic strategies to alter the oxygen affinity of sickle hemoglobin. Hematol. Oncol. Clin. North Am. 2014, 28, 217–231. 10.1016/j.hoc.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa A.; Lui F. E.; Wassaf D.; Yefidoff-Freedman R.; Casalena D.; Palmer M. A.; Meadows J.; Mozzarelli A.; Ronda L.; Abdulmalik O.; Bloch K. D.; Safo M. K.; Zapol W. M. Identification of a Small Molecule that Increases Hemoglobin Oxygen Affinity and Reduces SS Erythrocyte Sickling. ACS Chem. Biol. 2014, 9, 2318–2325. 10.1021/cb500230b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Shiva S.; Kim-Shapiro D. B.; Patel R. P.; Ringwood L. A.; Irby C. E.; Huang K. T.; Ho C.; Hogg N.; Schechter A. N.; Gladwin M. T. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J. Clin. Invest. 2005, 115, 2099–2107. 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isbell T. S.; Sun C. W.; Wu L. C.; Teng X.; Vitturi D. A.; Branch B. G.; Kevil C. G.; Peng N.; Wyss J. M.; Ambalavanan N.; Schwiebert L.; Ren J.; Pawlik K. M.; Renfrow M. B.; Patel R. P.; Townes T. M. SNO-hemoglobin is not essential for red blood cell-dependent hypoxic vasodilation. Nat. Med. 2008, 14, 773–777. 10.1038/nm1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaguti P.; Melloni E.; Cavalletti E. Acute intravenous toxicity of dimethyl sulfoxide, polyethylene glycol 400, dimethylformamide, absolute ethanol, and benzyl alcohol in inbred mouse strains. Arzneimittelforschung 1994, 44, 566–570. [PubMed] [Google Scholar]
- Safo M. K.; Abraham D. J. X-ray crystallography of hemoglobins. Methods Mol. Med. 2003, 82, 001–019. 10.1385/1-59259-373-9:001. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safo M. K.; Burnett J. C.; Musayev F. N.; Nokuri S.; Abraham D. J. Structure of human carbonmonoxyhemoglobin at 2.16 Å: a snapshot of the allosteric transition. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, 58, 2031–2037. 10.1107/S0907444902015809. [DOI] [PubMed] [Google Scholar]
- Park S. Y.; Yokoyama T.; Shibayama N.; Shiro Y.; Tame J. R. 1.25 Å resolution crystal structures of human haemoglobin in the oxy, deoxy and carbonmonoxy forms. J. Mol. Biol. 2006, 360, 690–701. 10.1016/j.jmb.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L. W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, 58, 1948–1954. 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulmalik O.; Safo M. K.; Chen Q.; Yang J.; Brugnara C.; Ohene-Frempong K.; Abraham D. J.; Asakura T. 5-hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin and inhibits sickling of red blood cells. Br. J. Haematol. 2005, 128 (4), 552–561. 10.1111/j.1365-2141.2004.05332.x. [DOI] [PubMed] [Google Scholar]
- Hijiya N.; Horiuchi K.; Asakura T. Morphology of sickle cells produced in solutions of varying osmolarities. J. Clin. Lab. Med. 1991, 117, 60–66. [PubMed] [Google Scholar]
- Benesch R.; Benesch R. Chemistry of Bohr Effect 1. Reaction of N-ethyl Maleimide with Oxygen-Linked Acid Groups of Hemoglobin. J. Biol. Chem. 1961, 236, 405–410. [Google Scholar]
- Goldstein J.; Guidotti G.; Konigsberg W.; Hill R. J. The amino acid sequence around the “reactive sulfhydryl” group of the beta chain from human hemoglobin. J. Biol. Chem. 1961, 236, PC77–PC78. [PubMed] [Google Scholar]
- Riggs A. The binding of N-ethylmaleimide by human hemoglobin and its effect upon the oxygen equilibrium. J. Biol. Chem. 1961, 236, 1948–1954. [PubMed] [Google Scholar]
- Henderson L. J. The equilibrium between oxygen and carbonic acid in blood. J. Biol. Chem. 1920, 41, 401–430. [Google Scholar]
- Perutz M. F.; TenEyck L. F. Stereochemistry of cooperative effects in hemoglobin. Cold Spring Harbor Symp. Quant. Biol. 1972, 36, 295–310. 10.1101/SQB.1972.036.01.040. [DOI] [PubMed] [Google Scholar]
- Riggs A. The Nature and Significance of the Bohr Effect in Mammalian Hemoglobins. J. Gen. Physiol. 1960, 43, 737–752. 10.1085/jgp.43.4.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Shen T. J.; Simplaceanu V.; Ho C. Ligand binding properties and structural studies of recombinant and chemically modified hemoglobins altered at β93 cysteine. Biochemistry 2002, 41, 11901–11913. 10.1021/bi0202880. [DOI] [PubMed] [Google Scholar]
- Kilmartin J. V.; Fogg J. H.; Perutz M. F. Role of C-terminal histidine in the alkaline Bohr effect of human hemoglobin. Biochemistry 1980, 19, 3189–3193. 10.1021/bi00555a013. [DOI] [PubMed] [Google Scholar]
- Perutz M. F.; Kilmartin J. V.; Nishikura K.; Fogg J. H.; Butler P. J.; Rollema H. S. Identification of residues contributing to the Bohr effect of human haemoglobin. J. Mol. Biol. 1980, 138, 649–668. 10.1016/S0022-2836(80)80022-2. [DOI] [PubMed] [Google Scholar]
- Russu I. M.; Ho N. T.; Ho C. Role of the β146 histidyl residue in the alkaline Bohr effect of hemoglobin. Biochemistry 1980, 19, 1043–1052. 10.1021/bi00546a033. [DOI] [PubMed] [Google Scholar]
- Garel M. C.; Caburi-Martin J.; Domenget C.; Kister J.; Craescu C. T.; Poyart C.; Beuzard Y. Changes of polymerization and conformation of hemoglobin S induced by thiol reagents. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1990, 1041, 133–140. 10.1016/0167-4838(90)90056-L. [DOI] [PubMed] [Google Scholar]
- Antonini E.; Condo S. G.; Giardina B.; Ioppolo C.; Bertollini A. The effect of pH and D-glycerate 2,3-bisphosphate on the O2 equilibrium of normal and SH(β93)-modified human hemoglobin. Eur. J. Biochem. 1982, 121, 325–328. 10.1111/j.1432-1033.1982.tb05789.x. [DOI] [PubMed] [Google Scholar]
- Garel M.; Beuzard Y.; Thillet J.; Domenget C.; Martin J.; Galacteros F.; Rosa J. Binding of 21 Thiol Reagents to Human-Hemoglobin in Solution and Intact-Cells. Eur. J. Biochem. 1982, 123, 513–519. 10.1111/j.1432-1033.1982.tb06561.x. [DOI] [PubMed] [Google Scholar]
- Popp R. A. Sequence of amino acids in the chain of single hemoglobins from C57BL, SWR, and NB mice. Biochim. Biophys. Acta, Protein Struct. 1973, 303, 52–60. 10.1016/0005-2795(73)90147-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.