

Abstract
Historically, it has been widely presumed that differentiated cells are determined during development and become irreversibly committed to their designated fates. In certain circumstances, however, differentiated cells can display plasticity by changing their identity, either by dedifferentiation to a progenitor-like state or by transdifferentiation to an alternative differentiated cell type. Such cellular plasticity can be triggered by physiological or oncogenic stress, or it can be experimentally induced through cellular reprogramming. Notably, physiological stresses that promote plasticity, such as severe tissue damage, inflammation, or senescence, also represent hallmarks of cancer. Furthermore, key drivers of cellular plasticity include major oncogenic and tumor suppressor pathways and can be exacerbated by drug treatment. Thus, plasticity may help cancer cells evade detection and treatment. We propose that cancer can be considered as a disease of excess plasticity, a notion that has important implications for intervention and treatment.
Keywords: lineage plasticity, differentiation, transdifferentiation, reprogramming, cancer progression, drug resistance
INTRODUCTION: ROLLING BACK UP WADDINGTON’S HILL
Since the nineteenth century, studies of classical embryology supported a central dogma that the process of cellular differentiation is progressive, unidirectional, and essentially irreversible. Thus, starting with a pluripotent progenitor cell that can generate all somatic cell types within the embryo, specification events progressively restrict cell fates and ultimately lead to the generation of fully differentiated cell types. Through this process, cell fates become determined and can no longer be reversed or altered. Thus, the process of cell fate determination can be metaphorically likened to a ball rolling down a hill: A common starting point leads to multiple distinct paths, but once a certain path has been selected, there is no turning back (Waddington 1957) (Figure 1).
Figure 1.
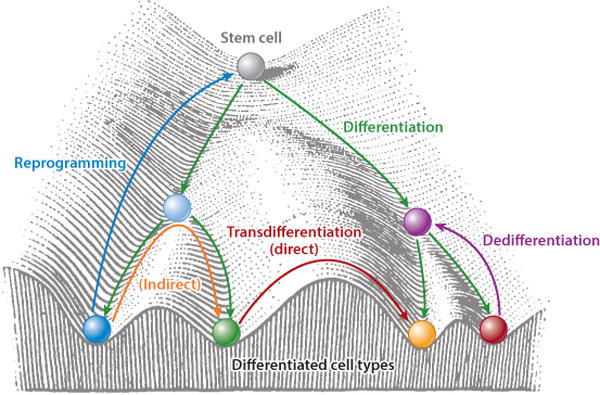
Waddington’s (1957) landscape of differentiation, depicting the process of differentiation of a stem cell into distinct cell types, visualized as a ball rolling down a hill. Pathways of dedifferentiation, transdifferentiation (both direct and indirect), and reprogramming are indicated. Adapted from Waddington (1957, p. 29, figure 4) with permission from Taylor & Francis.
However, studies over the past several decades have revealed that many differentiated cell types have a greater potential for altering their identity than previously appreciated. It is now realized that cellular plasticity, or the ability of differentiated cells to change their identity, is relatively common in normal physiological contexts, as well as in cancer. Since the term “plasticity” is not always used appropriately in the literature, we restrict our discussion of plasticity to scenarios in which meaningful experimental evidence supports a phenotypic alteration in differentiation status. Cellular plasticity may occur in response to physiological stresses, such as injury, inflammation, or senescence, or may be a consequence of oncogenic stimuli. Such plasticity may have profound implications for tumor progression, since it can provide a mechanism for cancer cells to evade detection and treatment or to escape from the confines of the primary tumor.
The conceptual framework underlying cellular determination and plasticity was established using experimental models that are amenable for direct analysis of lineage relationships in vivo. In contrast, it is more challenging to unequivocally demonstrate lineage plasticity in human tumors. Therefore, we first introduce conceptual aspects of lineage plasticity as understood in developmental contexts, and then discuss their relationship to cancer. We describe the advantages and disadvantages of experimental approaches that have been used to study plasticity and their application to cancer biology. Finally, we discuss examples of lineage plasticity in cancer initiation and progression, as well as treatment response, and consider how a deeper understanding of plasticity can improve cancer diagnosis and treatment. For additional discussion, we refer the reader to excellent reviews by Jessen et al. (2015), Jopling et al. (2011), Merrell & Stanger (2016), Pisco & Huang (2015), Roy & Hebrok (2015), Slack (2007), and Tata & Rajagopal (2016).
BASIC CONCEPTS IN LINEAGE PLASTICITY
Lineage plasticity is a broad concept that encompasses many cellular processes including trans-differentiation, dedifferentiation, and cellular reprogramming.
Transdetermination
One of the earliest demonstrations that determined cells can change their fates emerged from studies of Drosophila development in the 1960s. Imaginal discs are larval tissues that are primordia for specific structures of the adult fly and that can stably maintain their identity during long-term culture. However, at low frequencies, cultured imaginal discs can alter their fate and form alternative tissue structures, termed transdetermination (Worley et al. 2012). These early studies established the basic principle that determined cells could change their fate and offered hints about the importance of the native tissue microenvironment in maintaining stable cell fate.
Dedifferentiation, Transdifferentiation, and Metaplasia
We can distinguish two basic categories of plasticity that can occur at the cellular level in normal tissues. Dedifferentiation refers to the transition from a fully determined cell type to a less differentiated state, perhaps corresponding to an endogenous stem/progenitor. Although dedifferentiation is considered a distinguishing feature of tumor cells, it is not common in normal physiological contexts but appears to play a major role in tissue repair in response to injury. For example, in the Drosophila germline, exogenous stimuli or cell depletion can induce adult cells to dedifferentiate to functional stem cells during tissue regeneration (Brawley & Matunis 2004, Kai & Spradling 2004). In a mammalian context, a loss-of-function mutation of the Foxo1 transcription factor promotes diabetes by inducing the dedifferentiation of β cells (Talchai et al. 2012b).
In contrast, transdifferentiation represents a change in cellular identity from one differentiated cell type to an alternative differentiated state. Transdifferentiation may result from dedifferentiation to a progenitor state followed by differentiation to a distinct cell type, or it may instead correspond to direct conversion from one cell fate to another through a pathway that does not occur in normal development. For example, in the pancreas, following the near-total ablation of β cells, α cells can be converted to insulin-producing β cells (Thorel et al. 2010). Additionally, deletion of Foxo1 in the gut epithelium results in conversion to insulin-producing β-like cells (Talchai et al. 2012a). Although rare in normal physiological contexts, it is becoming increasingly apparent that transdifferentiation plays an important role in cancer and treatment response, as discussed below.
The phenomenon of metaplasia refers to tissue plasticity that may not necessarily occur at the cellular level. This term is generally used in circumstances in which multiple cell types within a tissue are replaced with other cell types, although the experimental evidence is not sufficient to ascertain whether such phenotypes reflect cellular plasticity or alternative mechanisms (Slack 2007). Metaplasia is rare in nontumorigenic contexts but can occur in cancer, as exemplified by Barrett’s esophageal cancer, in which the normal squamous epithelium is replaced with an intestinal-like columnar epithelium (Jankowski et al. 2000).
Cellular Reprogramming
Among the first experimental demonstrations of cellular plasticity at the molecular level was the classic work of Weintraub and colleagues, who showed that expression of a single gene, MyoD, could convert fibroblasts into muscle cells (Davis et al. 1987). Such conversions of cell types that arise through experimental manipulations represent reprogramming of cell fate, resulting in either dedifferentiation or transdifferentiation. Further interest in reprogramming was greatly stimulated by the demonstration that a combination of four transcriptional regulators, encoded by Oct4, Sox2, Klf4, and c-Myc (termed OSKM factors) could convert differentiated fibroblasts into induced pluripotent (iPS) cells (Takahashi & Yamanaka 2006, Takahashi et al. 2007). Notably, aberrant expression of each of the OSKM factors has been linked to cancer, which underscores the intimate relationship of plasticity mechanisms and oncogenesis, as discussed below.
As with transdifferentiation, experimental approaches for reprogramming can be broadly separated into two distinct categories. Direct conversion approaches generate a differentiated cell type from a distinct cell type by transient expression of a cocktail of specification genes. Notably, a cocktail of three genes (Ascl1, Brn2, and Myt1l) can direct conversion of fibroblasts into neurons, whereas a different set of three genes (Gata4, Mef2c, and Tbx5) can mediate direct conversion into cardiomyocytes (Ieda et al. 2010, Vierbuchen et al. 2010). Such direct conversion approaches can also be used for reprogramming in vivo, as forced expression of a combination of three genes (Pdx1, Ngn3, and Mafa) can reprogram exocrine cells in the adult pancreas to insulin-producing endocrine β cells (Zhou et al. 2008).
In contrast, primed or indirect lineage conversion approaches use transient expression of OSKM pluripotency factors to induce a plastic developmental state that allows the respecification of desired cell fates after exposure to appropriate external cues, such as specific cell culture conditions (Morris & Daley 2013, Sancho-Martinez et al. 2012). Notably, OSKM-reprogrammed fibroblasts can be induced to generate cardiomyocytes, neurons, or prostate cells, depending on the cocktails of genes introduced (Ieda et al. 2010, Talos et al. 2017, Vierbuchen et al. 2010). Interestingly, studies examining whether such indirect lineage conversions require dedifferentiation into a transient iPS-like state followed by differentiation into the desired cell type have shown that in neural and cardiac indirect conversions, reprogramming occurs by traversing an intermediate pluripotent state (Bar-Nur et al. 2015, Maza et al. 2015), whereas reprogramming fibroblasts to prostate epithelial cells does not appear to involve a pluripotent intermediate (Talos et al. 2017).
EXPERIMENTAL APPROACHES FOR STUDYING LINEAGE PLASTICITY
It is often difficult to conclusively demonstrate that a given cell arises from a distinct cell type, while excluding a myriad of alternative possibilities. In particular, it is often difficult to establish that distinct cell types arise from changes in cellular identity rather than by the generation of new cells from stem/progenitors. Therefore, our understanding of lineage plasticity is profoundly influenced by the experimental approaches employed, which, in turn, depend on the model system studied. As described below, lineage tracing studies can precisely define lineage relationships in vivo, transplantation methods can provide insights into cell autonomous and nonautonomous mechanisms, and cell culture approaches allow functional analyses of putative specification/reprogramming genes. However, achieving a complementarity of in vivo and ex vivo approaches can be difficult for human tissues, and, ultimately, lineage relationships in human tissues and cancer are often inferred rather than directly demonstrated.
Lineage Tracing
The gold standard for defining lineage relationships in vivo is lineage tracing, which refers to the indelible genetic marking of cells and their progeny (clones), elucidating their fate (Alcolea & Jones 2013, Kretzschmar & Watt 2012). Traditional methods of lineage tracing in experimental embryology often used vital dye injection to follow the fates of marked cells in embryos or tissues in culture (Thomas & Beddington 1996). More recently, powerful methods of lineage tracing in vivo have used genetically engineered mouse models that express Cre recombinase together with Cre-activatable fluorescent reporter alleles (Soriano 1999, Srinivas et al. 2001), resulting in the irreversible marking of cells in vivo. Furthermore, the use of inducible Cre drivers facilitates lineage tracing in precise spatial and temporal contexts (Metzger et al. 1995). Notably, lineage tracing can be performed in combination with conditional alleles that result in gain- or or loss-of-function mutations of oncogenic drivers to evaluate lineage relationships in the context of cancer (Alcolea & Jones 2013, Aytes et al. 2013, Blanpain 2013, Rhim et al. 2012).
More recently, enhanced lineage tracing methods in vivo have developed Cre-activatable reporters that can express multiple fluorescent proteins, such as Brainbow (Livet et al. 2007), Confetti (Snippert et al. 2010), and Hue (Yu et al. 2017), which allow for a higher-resolution assessment of clonal lineage relationships. Two-color fluorescent protein systems also facilitate methods for mosaic analysis, such as mosaic analysis with double markers, which combine lineage tracing with the ability to distinguish daughter clones arising from the same cell division (Zong et al. 2005). Such clonal marking approaches can be advantageous for studying gene function and clonal competition in cancer (Liu et al. 2011). Finally, genome editing approaches for bar coding of individual clones in vivo combined with deep sequencing can provide precise details of lineage relationships (McKenna et al. 2016, Woodworth et al. 2017), although these approaches have not yet been integrated for lineage analysis.
Transplantation Studies
Another approach to study lineage relationships uses grafting of dissociated cells or tissue fragments into heterologous sites of host organisms. Such transplantation assays can be advantageous since they can be used to study human cells, although this requires an immunodeficient host, and because the grafted cells or tissues can be genetically manipulated ex vivo to study the functional roles of putative regulatory genes. However, unlike lineage tracing, which assesses lineage relationships within the native tissue microenvironment, transplantation assays implant cells in a heterologous tissue microenvironment. This type of experimental manipulation can induce cellular plasticity and represents the basis for an important distinction between cellular specification versus determination. Thus, a cell fate may be specified within a normal tissue context but may not be determined if its fate is altered by placement within a heterologous environment. Conversely, a cell fate may be specified as well as determined if transplantation does not change its fate.
For example, transplantation assays have been used to assess the stem cell potential of mammary epithelial cells by determining whether they can form an epithelial tree after grafting into a fat pad (Deome et al. 1959, Visvader & Stingl 2014). For studies of prostate growth, grafting under the renal capsule has been used for tissue recombination assays that combine epithelial cells and embryonic mesenchyme (Cunha et al. 1987, Goldstein et al. 2010, Wang et al. 2014). Notably, such transplantation studies can sometimes reveal differences in cellular plasticity between in vivo and ex vivo assays. For instance, adult prostate basal epithelial cells rarely generate luminal cells in their native tissue microenvironment in vivo, but they can readily form luminal cells following tissue recombination and renal grafting (Wang et al. 2013).
Cell Culture Approaches
Among the advantages of studying lineage plasticity in cell culture are the relative ease of investigating functional roles of putative plasticity regulators and the ability to assess cell autonomous effects. However, established cell lines may not be representative of the plasticity of the primary epithelium. Recent advances in the generation of three-dimensional (3D) culture models, such as organoid and organotypic models, have significantly facilitated in vitro studies of lineage relationships in a wide range of lineages (Fatehullah et al. 2016). However, these ex vivo organoid models may also evoke lineage plasticity that is not observed in vivo. For example, luminal progenitors can propagate mouse prostate organoids and can give rise to basal cells in culture, which is not observed for luminal cells in vivo (Chua et al. 2014, Karthaus et al. 2014). Further advances in these 3D models, including the incorporation of stromal components to study the contribution of the tissue microenvironment, may provide insights into stromal effects on lineage plasticity.
Moving from Model Organisms to Humans: Genomic and Clonal Analyses
In contrast to model organisms, it is more challenging to assess lineage relationships in human tissues since direct lineage tracing in the context of the whole organism in vivo is not feasible. However, in a recent study (Cortina et al. 2017), genome editing was used to lineage-trace Lgr5-positive colorectal cancer cells in patient-derived organoids to study their tumor-initiating ability and stem cell properties in vivo. Furthermore, lineage relationships in human tissues can be retrospectively inferred by analysis of naturally occurring lineage marks, such as somatic mutations or translocations (Woodworth et al. 2017). Similar types of retrospective lineage analyses can be performed using genomic mutations in mitochondrial DNA (Blackwood et al. 2011, Fellous et al. 2009). Combined with technologies such as single-cell sequencing (Navin et al. 2011), such analyses can be used to reconstruct the genealogy of a tissue, as shown in studies of lineage relationships of postmitotic cells in the human brain (Evrony et al. 2015, Lodato et al. 2015).
CELLULAR SPECIFICATION AND CANCER
Most specification and reprogramming factors responsible for defining cell fate correspond to transcriptional regulatory proteins, implicating the control of gene expression as an essential mechanism by which cell identity is determined (see Supplemental Table 1). Notably, many specification factors are homeobox genes, a multigene family whose members have been highly conserved throughout evolution and are known to play essential roles in developmental patterning and cell fate determination (Burglin & Affolter 2016, Gehring et al. 1994). A plethora of experimental evidence based on gain- or loss-of-function studies in a range of model organisms has established that homeobox genes are essential master regulators of cellular differentiation in embryonic and adult contexts. For example, Pax4 is essential for specification of insulin-producing β cells in the pancreas during development (Sosa-Pineda et al. 1997), while its forced expression in pancreatic progenitor cells promotes their conversion to β cells (Collombat et al. 2009). Similarly, Pdx1 is necessary for specification of insulin-producing β cells, while its forced expression in adult pancreas converts exocrine cells to the insulin-producing endocrine β cells (Zhou et al. 2008) and is sufficient to convert liver to pancreas (Horb et al. 2003).
Notably, homeobox genes are often deregulated in cancer, providing a link between the molecular programs responsible for cellular specification and cancer (Abate-Shen 2002). For example, members of the Caudal-type homeobox (Cdx) family have essential roles in specification of the trophectoderm during preimplantation development and later in specification of the definitive endoderm that gives rise to gut epithelium (Strumpf et al. 2005), while their aberrant expression is cancer promoting, as exemplified by CDX2, which is a key driver of Barrett’s metaplasia (Colleypriest et al. 2010) and a prognostic biomarker for colon cancer (Dalerba et al. 2016).
Nkx homeobox genes have been shown to be essential for cell type specification in multiple tissues, while their dysregulated expression leads to aberrant specification and cancer (Abate-Shen 2002). This is exemplified by Nkx2.1, which is required for specification of lung epithelium, while its aberrant expression unmasks a latent gastric phenotype and promotes cancer in the lung (Snyder et al. 2013, Watanabe et al. 2013, Winslow et al. 2011). Similarly, Nkx3.1 is required for the proper differentiation of luminal epithelial cells during prostate organogenesis as well as for the proper function of luminal stem/progenitors in prostate regeneration, while its loss-of-function mutation promotes cancer initiation (Abate-Shen et al. 2008, Wang et al. 2009). Notably, Nkx3.1 is necessary and sufficient for prostate epithelial specification, since its forced expression in nonprostatic epithelium of the seminal vesicle results in prostate differentiation in vivo (Dutta et al. 2016).
Another important family of transcriptional regulators are Sox genes, which encode HMG-binding domain proteins that function in many tissues to regulate progenitors and cell fate decisions (Kamachi & Kondoh 2013, Sarkar & Hochedlinger 2013). Sox2, a member of the SoxB subfamily, is necessary for maintaining self-renewal and pluripotency in embryonic stem cells and is expressed in progenitors in multiple tissue during development (Arnold et al. 2011, Fong et al. 2008). Furthermore, Sox2 has roles in cancer that have been linked to cellular plasticity, particularly in collaboration with other Sox genes such as Sox9 (see below).
EPITHELIAL-TO-MESENCHYMAL TRANSITION IN DEVELOPMENT AND CANCER
One of the most well-studied examples of lineage plasticity is epithelial-to-mesenchymal transition (EMT), which occurs in several contexts throughout normal development (Kalluri & Weinberg 2009, Nieto et al. 2016, Thiery et al. 2009). During EMT, polarized epithelial cells that are attached to a basement membrane convert to a mesenchymal phenotype, are thereby no longer tightly adherent, and become capable of migrating to distant sites (Hay 1995). For example, during gastrulation, epiblast cells undergo EMT to emerge from the primitive streak to form the mesoderm and definitive endoderm. During development of the nervous system, cells in the dorsal neural tube undergo EMT to generate migratory neural crest cells that will form the peripheral nervous system. In adults, EMT has been shown to play an important role in tissue repair following fibrosis of the liver and heart. Notably, despite these fundamentally different biological contexts, the underlying molecular mechanisms of EMT are mediated by a network of transcription factors (EMT-TFs), including Twist, Snail, and Zeb1, which regulate expression of cell adhesion molecules and promote cytoskeletal reorganization (Thiery et al. 2009).
Establishing a definitive role for EMT in human cancer has been more elusive, in part due to the difficulty of unequivocally demonstrating lineage relationships in human tumors. Nonetheless, several studies have used lineage tracing to demonstrate the occurrence of EMT in mouse models of cancer (Rhim et al. 2012). EMT is believed to be particularly significant for metastasis by promoting dissociation of cells from the confines of the tumor microenvironment to allow circulation to distant sites (Chaffer et al. 2016). Subsequent reversion to an epithelial phenotype by the reverse process of mesenchymal-to-epithelial transition (MET) may then promote metastatic colonization in secondary sites (Ocana et al. 2012, Tsai et al. 2012). However, recent studies have also suggested that EMT is not required for metastasis, at least in some circumstances (Fischer et al. 2015, Zheng et al. 2015).
Thus, EMT can be considered a specialized form of transdifferentiation that occurs in distinct contexts during normal development and can be reactivated in cancer. Notably, many aspects of EMT in cancer are highly conserved with those for EMT during development (Yang et al. 2004). In addition, the process of EMT in cancer may have some similarities to reprogramming, since EMT has been reported to confer stem cell–like phenotypes (Mani et al. 2008, Shibue & Weinberg 2017). Despite these similarities, the molecular mechanisms underlying EMT and reprogramming appear to differ, since EMT-TFs are defined by their ability to regulate cell adhesion, whereas OSKM factors are defined by their ability to promote pluripotency.
LINEAGE PLASTICITY IN HOMEOSTASIS AND CANCER INITIATION
Under normal physiological conditions, differentiated cell types in adult tissues are situated in stable microenvironments that help maintain their identities, and their replenishment is maintained by stem/progenitor cells. However, under conditions of profound physiological stress, such as injury, inflammation, oxidative stress, or senescence, tissue repair may exceed the capacity of the stem/progenitors, leading to plasticity and respecification of differentiated cells (Blanpain & Fuchs 2014) (Figure 2). Notably, the stresses that promote cellular plasticity are also hallmarks of cancer (Hanahan & Weinberg 2011).
Figure 2.
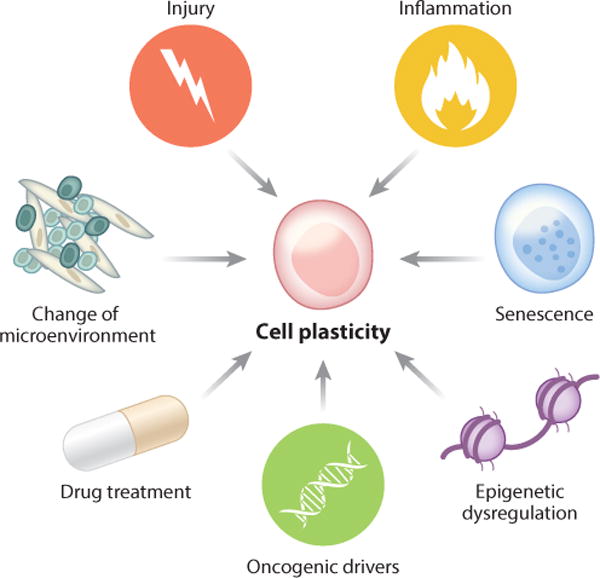
The intrinsic and extrinsic factors that can promote cell plasticity in homeostasis and cancer.
Following severe tissue damage, such as treatment with chemical agents or massive cell ablation, cells can transdifferentiate to assume new identities. For example, following genetic ablation or irradiation to induce death of Lgr5-positive adult stem cells that reside at the bottom of intestinal crypts, this Lgr5-positive population is rapidly replenished by cells at higher positions in the crypt expressing Bmi1 (Tian et al. 2011) or high levels of the Notch ligand Dll1 (van Es et al. 2012). Similarly, following chemical injury in the liver, differentiated hepatocytes can exhibit significant phenotypic plasticity by converting to bipotential progenitors and then reverting to hepatocytes after recovery (Tarlow et al. 2014); this process of dedifferentiation is dependent on Notch signaling (Yanger et al. 2013). Notably, lineage plasticity of hepatocytes in a form of liver cancer known as intrahepatic cholangiocarcinoma is also dependent on Notch signaling (Pribluda et al. 2013, Sekiya & Suzuki 2012).
Lineage plasticity in response to injury is not restricted to tissues that have a high capacity for self-renewal, such as the gastric epithelium and liver. In the adult lung, ablation of tracheal basal stem cells results in dedifferentiation of luminal secretory cells to basal stem cells that can repair the tissue (Tata et al. 2013). Conversely, depletion of luminal cells in the prostate induces basal epithelial cells to form luminal cells (Toivanen et al. 2016), which rarely occurs under normal physiological conditions. The degree of tissue damage may also affect the extent of cellular plasticity; as noted above, in the pancreas, nearly complete ablation of β cells is required for their replacement by α cells through transdifferentiation (Thorel et al. 2010).
A frequent consequence of tissue damage following injury is senescence, which can have context-dependent effects for both cellular plasticity and cancer. In particular, senescence can promote injury-induced plasticity in vivo, including in lung, skin, and muscle tissue (Chiche et al. 2017, Mosteiro et al. 2016, Ritschka et al. 2017). Senescence also promotes reprogramming in a mouse model that inducibly expresses OSKM factors in vivo (Mosteiro et al. 2016). Interestingly, these in vivo findings differ from conclusions of in vitro studies in which senescence impaired formation of iPS cells by OSKM (Banito et al. 2009), which may reflect the contribution of the microenvironment or the duration of senescence (Ritschka et al. 2017).
Inflammation provides an important link between senescence, cellular plasticity, and cancer. In particular, chronic pancreatitis is a significant risk for pancreatic cancer (Lowenfels et al. 1993) and has been shown to promote the progression from preinvasion to pancreatic adenocarcinoma by abrogating the senescence barrier (Guerra et al. 2007, Guerra et al. 2011) and by promoting the transdifferentiation of endocrine cells to adenocarcinoma cells (Gidekel Friedlander et al. 2009). In the prostate, where chronic inflammation is a risk factor for aggressive prostate cancer (Sfanos & De Marzo 2012), inflammation promotes the generation of luminal epithelial cells from basal cells and is associated with prostate cancer initiation (Kwon et al. 2014, 2016; Liu et al. 2016).
LINEAGE PLASTICITY IN TUMOR PROGRESSION AND DRUG RESISTANCE
As exemplified by the relationship between EMT and metastasis, lineage plasticity is often associated with the progression from less aggressive to more aggressive cancer phenotypes. A well-studied example of cellular plasticity is phenotype switching in the progression from nonmetastatic to metastatic melanoma (Hoek & Goding 2010, Hoek et al. 2008, Quintana et al. 2010). During this reversible phenotypic change, melanoma cells acquire invasive and migratory properties that promote metastasis, suggesting a partial dedifferentiation toward the neural crest origin of melanocytes. Notably, phenotypic switching requires the MITF transcriptional regulator, a master regulator of melanocyte fate during development (Cheli et al. 2011, Garraway & Sellers 2006, Hoek & Goding 2010).
Consistent with its association with more aggressive cancer phenotypes, lineage plasticity has also emerged as an important mechanism of drug resistance (Konieczkowski et al. 2014, Mu et al. 2017, Oser et al. 2015, Sequist et al. 2011, Zou et al. 2017). Although it is challenging to provide direct evidence of plasticity in treatment resistance in human cancer, indirect evidence can be augmented by lineage tracing and functional analyses in model systems. For example, in advanced prostate cancer, this combination of analyses of human tissues and experimental models has generated considerable evidence that treatment resistance can occur by transdifferentiation of adenocarcinoma into neuroendocrine-like cells.
In particular, advanced prostate cancer is commonly treated with androgen deprivation therapy, which leads to resistance and progression to aggressive phenotypes (Watson et al. 2015). While the recent development of second-generation antiandrogens such as abiraterone and enzalutamide has enhanced the efficacy of treatment, a subset of patients develop resistance to these treatments (Watson et al. 2015). Treatment failure is often associated with highly aggressive variants that display characteristics of small cell carcinoma and exhibit features of neuroendocrine differentiation (Beltran et al. 2014). Analyses of treatment-resistant human prostate cancer by histological and whole-exome sequencing approaches suggest that treatment-resistant aggressive variants are derived from prostate adenocarcinoma (Beltran et al. 2011, 2016; Lotan et al. 2011). In particular, genomic rearrangements of the ERG gene, a frequent early event in prostate adenocarcinoma, are also found in the neuroendocrine-like cells of advanced tumors (Beltran et al. 2011, Lotan et al. 2011), supporting their origin from adenocarcinoma cells. Furthermore, lineage tracing studies in mouse models have directly demonstrated that focal neuroendocrine differentiation, as well as overt neuroendocrine disease, arises via transdifferentiation from luminal adenocarcinoma cells (Zou et al. 2017).
Functional analyses have shown that cellular plasticity in prostate cancer is promoted by the combined loss-of-function mutation of TP53, RB1, or PTEN, which are significantly more prevalent in advanced than primary disease (Cancer Genome Atlas Res. Netw. 2015, Robinson et al. 2015). Analyses of the cooperativity of RB1 and TP53 loss in promoting resistance to the antiandrogen enzalutamide in cell lines and mouse models have shown that plasticity is mediated at least in part by SOX2 (Ku et al. 2017, Mu et al. 2017). Interestingly, cooperativity of Trp53 and Pten loss in mouse models is not mediated by Sox2 but instead by Sox11, a member of the SoxC subgroup that promotes neural differentiation (Zou et al. 2017), suggesting that distinct Sox genes can promote cellular plasticity in different tumor contexts.
Another well-studied example of therapy-induced plasticity occurs in lung cancer, which is the most common cause of cancer deaths worldwide. Notably, lung cancer has two broad subtypes, the predominant of which is the non-small-cell subtype, including lung adenocarcinoma, while small-cell lung cancer represents about 15% of cases. These broad subtypes differ significantly in terms of aggressiveness, overall survival, and treatment options; while both subtypes can be treated with chemotherapy, albeit different regimens, non-small-cell lung cancer can also be treated with targeted agents, such as epidermal growth factor receptor (EGFR) inhibitors. Non-small-cell lung tumors with EGFR mutations usually relapse, and a subset of resistant tumors display a phenotypic transition to small-cell neuroendocrine lung cancer (Oser et al. 2015, Sequist et al. 2011). These resistant tumors harbor the original EGFR mutation, suggesting transdifferentiation from the initial adenocarcinoma, and display loss of the RB1 suppressor gene, which also occurs in classical (nontreatment-induced) small-cell lung cancer (Niederst et al. 2015).
Although cellular plasticity in cancer progression and drug resistance is frequently mediated by transcriptional regulators, including TP53 and RB1, there is also increasing evidence for a key role of epigenetic regulators in promoting plasticity (Easwaran et al. 2014). For example, studies of EGFR-mutant lung cancer cell lines have shown that a reversible drug-tolerant state is mediated by the histone demethylase KDM5A together with IGF-1 receptor activity (Sharma et al. 2010). Similarly, treatment of glioblastoma with receptor tyrosine kinase inhibitors promotes phenotypic plasticity that is mediated by the histone demethylases KMD6A and KDM6B as well as by Notch signaling (Liau et al. 2017).
ONCOGENIC DRIVERS OF CELLULAR REPROGRAMMING
The intimate relationship between cellular plasticity and cancer is perhaps best underscored by analyses of the OSKM reprogramming factors. Of these four factors, Myc represents one of the most prevalent oncogenes in human cancer, and concerns about its oncogenic potential prompted the development of reprogramming cocktails for induced pluripotency lacking Myc (Nakagawa et al. 2010, Wernig et al. 2008). Moreover, several studies have suggested that Myc can promote cellular plasticity in cancer. For example, Myc can collaborate with Kras to induce expression of EMT factors and metastatic phenotypes in pancreatic cancer cells (Ischenko et al. 2013). Furthermore, a recent analysis of a Kras mouse model of pancreatic cancer has shown that loss of the SWI/SNF chromatin factor SMARCB1 leads to a Myc-driven transition to a mesenchymal phenotype (Genovese et al. 2017).
In addition to Myc, the other three OSKM factors also display oncogenic activities when expressed in inappropriate contexts. Thus, ectopic expression of the POU-domain homeodomain transcription factor Oct4 in differentiated adult cell types leads to widespread dysplasia (Hochedlinger et al. 2005). Similarly, overexpression of Klf4, a member of the Krüppel-like family of transcription factors, collaborates with Kras in a mouse model of pancreatic cancer and promotes reprogramming from an acinar to a ductal epithelial phenotype (Wei et al. 2016). Finally, Sox2 expression has been linked to cellular plasticity in several cancers, as noted above for prostate cancer (Ku et al. 2017, Mu et al. 2017). In the lung, for instance, expression of Sox2 promotes conversion from adenocarcinoma to squamous cell carcinoma (Ferone et al. 2016), while it acts coordinately with Sox9 to promote EMT (Lin et al. 2016).
Similar to OSKM factors, tumor suppressors such as TP53, RB1, and PTEN have essential roles in regulating differentiation programs in normal development (e.g., Cam et al. 2006, Hamada et al. 2005, Wang et al. 2017, Wu et al. 2003), while their dysregulation is associated with lineage plasticity in cancer. For example, p53 has been shown to protect glioblastoma cells from acquiring a mesenchymal phenotype in response to radiation therapy (Halliday et al. 2014). In the liver, loss of p53 promotes dedifferentiation of hepatocytes (Tschaharganeh et al. 2014), while p53 loss in colorectal cancer promotes an inflammatory microenvironment that leads to increased EMT (Schwitalla et al. 2013). Although Pten has been less studied in the context of plasticity, deletion of Pten in prostate basal cells promotes basal-to-luminal differentiation during tumor initiation (Choi et al. 2012; Wang et al. 2013, 2014), which can be considered as transdifferentiation in this context. Furthermore, collaboration of tumor suppressors plays a key role in mediating cellular plasticity in several tumor types. In particular, combined loss-of-function mutations of TP53, RB1, or PTEN promote transdifferentiation from adenocarcinoma to small-cell neuroendocrine carcinoma in both the lung and prostate (Ku et al. 2017, Meuwissen et al. 2003, Zhou et al. 2006, Zou et al. 2017).
Notably, there is a strong mechanistic link between the p53 and Rb tumor suppressor pathways and induced pluripotency. Several studies have shown that inhibition of p53 pathway activity results in increased efficiency of iPS cell formation from fibroblasts, and conversely p53 expression restricts the reprogramming of differentiated cells (Kawamura et al. 2009, Marion et al. 2009, Rasmussen et al. 2014, Utikal et al. 2009, Yi et al. 2012). One possible explanation of this linkage is that reprogramming induces DNA damage, and p53-dependent mechanisms eliminate damaged cells; thus, p53 pathway alterations would increase reprogramming efficiency (Gonzalez et al. 2013). Consequently, tumors with p53 pathway alterations may indirectly provide a permissive environment for reprogramming. Alternatively, there may be a direct regulatory relationship between tumor suppressor pathways and pluripotency regulators. In this regard, Rb loss also increases iPS cell reprogramming efficiency, and Rb protein directly binds to the promoters of Sox2, Oct4, and the pluripotency regulator Nanog to repress their expression (Kareta et al. 2015). Furthermore, pituitary tumor formation upon loss of Rb in mice requires Sox2 activity, indicating the functional significance of this regulatory relationship (Kareta et al. 2015).
PLASTICITY AND CANCER STEM CELLS
The prevalence of cellular plasticity in cancer has profound implications for the notion of cancer stem cells (CSCs), which are described as a subset of cells within a tumor that are endowed with stem-like properties such as self-renewal and the exclusive ability to initiate and propagate tumor growth (Clevers 2011, Reya et al. 2001). Furthermore, due to their potential quiescence, CSCs may be more resistant to therapy and are consequently able to regrow the tumor after treatment (Meacham & Morrison 2013, Nassar & Blanpain 2016, Reya et al. 2001). Thus, the origin and properties of CSCs are a major topic of investigation, yet definitive evidence for their existence in many solid tumors remains controversial.
Notably, the term “cancer stem cell” has generated much confusion since it implies that such tumor cells may directly derive from normal adult stem cells. While this hypothesis remains plausible, it is also likely that putative CSCs may correspond to non-stem-like tumor cells that have acquired plasticity due to environmental stresses or treatment. The ability of tumor cells undergoing EMT to acquire stem-like properties (Mani et al. 2008) is consistent with this alternative view. Furthermore, the permissive environment for plasticity generated by loss of p53, Rb, or Pten also correlates with experimental observations that CSC activity is more readily detected in advanced tumors (Quintana et al. 2008). Thus, cellular plasticity may represent a cardinal feature of putative CSCs in many tumors. The validity of this hypothesis has been strengthened by recent studies demonstrating that differentiated cancer cells may be able to reverse to a less mature stage and compensate for the loss of CSCs if needed (de Sousa e Melo et al. 2017, Shimokawa et al. 2017). These studies in both mouse and human models, respectively, show that differentiated colon cells have the intrinsic capacity to dedifferentiate into CSCs upon ablation of pre-existing LGR5-positive CSCs. From a therapeutic perspective, this suggests that targeting CSCs may be even more challenging than previously thought, as its efficacy may be transient as a result of acquired plasticity.
PERSPECTIVES: CANCER AS A DISEASE OF EXCESS PLASTICITY
In contrast with physiological contexts, in which cellular plasticity allows cells to respond to external stresses and adapt to their environment, cancer cells use plasticity to escape the normal parameters that limit growth and survival. In this regard, we can envision tumor cells as inappropriately plastic, and cancer as a disease of excess plasticity. Historically, this notion has been described by pathologists in terms of the dedifferentiated histological appearance of advanced cancers, which we can now reframe as a caricature of excess plasticity.
Notably, many pathways and factors controlling cancer-related cell plasticity are similar across cancers, suggesting the conservation of key mechanisms in various tumor types. Moreover, mechanisms of cellular plasticity may largely account for the remarkable phenotypic heterogeneity of human tumors. Therefore, regulators of cellular plasticity may represent promising targets for future therapeutic approaches. However, since drug treatment may itself provoke plasticity and subsequent resistance, targeting approaches will require great specificity. Furthermore, the tumor microenvironment may also display plasticity or influence tumor cell plasticity, indicating that therapeutic approaches should target plasticity in both epithelial and stromal compartments.
Finally, the mechanistic relationship between therapy and tumor cell plasticity is still poorly understood. In most cases, it remains unclear whether cellular plasticity is a cause of therapeutic resistance or a consequence of selection for resistance. Further studies should also resolve whether cellular plasticity is required for both the establishment and maintenance of drug resistance, which will be essential to understand if therapeutic targeting of plasticity mechanisms is to be successful.
Supplementary Material
Acknowledgments
The authors apologize to the many colleagues whose work we were unable to cite because of space limitations. The research in C.A.-S.’s laboratory is supported in part by funding by NIH grants CA196662, CA193442, CA183929, and CA173481. The research in M.M.S.’s laboratory is supported in part by NIH grants DK076602, CA196662, and CA193313. C.L. is supported by Swiss National Science Foundation grants PBBSP3_146959 and P300P3_151158. C.A.-S. is an American Cancer Society Research Professor, supported in part by a generous gift from the F.M. Kirby Foundation.
Glossary
- Cell fate determination
process by which a cell assumes a specific differentiated state
- Differentiation
process by which a less specialized cell acquires properties of a more specialized cell, usually a mature functioning cell
- Progenitor cell
cell that can differentiate into more specialized cell types within a tissue
- Cellular identity
the features of a cell that are associated with its differentiation state
- Cellular plasticity
ability of a cell to change from one identity to another
- Senescence
process by which cells stop dividing and enter a state of permanent growth arrest without undergoing cell death
- Dedifferentiation
process by which a specialized cell converts to a less specialized phenotype, sometimes to a stem/progenitor cell
- Stem cell
cell that can maintain itself through self-renewal and generate multiple distinct cell types through differentiation of its progeny
- Transdifferentiation
conversion of a cell from one differentiated state to an alternative differentiated fate
- Metaplasia
conversion of a tissue from one differentiated state to an alternative state
- Reprogramming
experimental manipulation that results in dedifferentiation or transdifferentiation often refers to process of generating induced pluripotent stem cells
- Epithelial-to-mesenchymal transition (EMT)
process in which epithelial cells convert to mesenchymal cells and acquire their motility and invasive properties
- Mesenchymal-to-epithelial transition (MET)
process in which mesenchymal cells convert to epithelial cells
- Metastasis
the spread of cancer cells from the place where they initially formed to another part of the body
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Abate-Shen C. Deregulated homeobox gene expression in cancer: cause or consequence? Nat Rev Cancer. 2002;2:777–85. doi: 10.1038/nrc907. [DOI] [PubMed] [Google Scholar]
- Abate-Shen C, Shen MM, Gelmann E. Integrating differentiation and cancer: the Nkx3.1 homeobox gene in prostate organogenesis and carcinogenesis. Differentiation. 2008;76:717–27. doi: 10.1111/j.1432-0436.2008.00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcolea MP, Jones PH. Tracking cells in their native habitat: lineage tracing in epithelial neoplasia. Nat Rev Cancer. 2013;13:161–71. doi: 10.1038/nrc3460. [DOI] [PubMed] [Google Scholar]
- Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, et al. Sox2+ adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell. 2011;9:317–29. doi: 10.1016/j.stem.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytes A, Mitrofanova A, Kinkade CW, Lefebvre C, Lei M, et al. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. PNAS. 2013;110:E3506–15. doi: 10.1073/pnas.1303558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134–39. doi: 10.1101/gad.1811609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Nur O, Verheul C, Sommer AG, Brumbaugh J, Schwarz BA, et al. Lineage conversion induced by pluripotency factors involves transient passage through an iPSC stage. Nat Biotechnol. 2015;33:761–68. doi: 10.1038/nbt.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Rickman DS, Park K, Chae SS, Sboner A, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1:487–95. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res. 2014;20:2846–50. doi: 10.1158/1078-0432.CCR-13-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood JK, Williamson SC, Greaves LC, Wilson L, Rigas AC, et al. In situ lineage tracking of human prostatic epithelial stem cell fate reveals a common clonal origin for basal and luminal cells. J Pathol. 2011;225:181–88. doi: 10.1002/path.2965. [DOI] [PubMed] [Google Scholar]
- Blanpain C. Tracing the cellular origin of cancer. Nat Cell Biol. 2013;15:126–34. doi: 10.1038/ncb2657. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science. 2014;344:1242281. doi: 10.1126/science.1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawley C, Matunis E. Regeneration of male germline stem cells by spermatogonial dedifferentiation in vivo. Science. 2004;304:1331–34. doi: 10.1126/science.1097676. [DOI] [PubMed] [Google Scholar]
- Burglin TR, Affolter M. Homeodomain proteins: an update. Chromosoma. 2016;125:497–521. doi: 10.1007/s00412-015-0543-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cam H, Griesmann H, Beitzinger M, Hofmann L, Beinoraviciute-Kellner R, et al. p53 family members in myogenic differentiation and rhabdomyosarcoma development. Cancer Cell. 2006;10:281–93. doi: 10.1016/j.ccr.2006.08.024. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Res Netw The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35:645–54. doi: 10.1007/s10555-016-9648-7. [DOI] [PubMed] [Google Scholar]
- Cheli Y, Giuliano S, Botton T, Rocchi S, Hofman V, et al. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene. 2011;30:2307–18. doi: 10.1038/onc.2010.598. [DOI] [PubMed] [Google Scholar]
- Chiche A, Le Roux I, von Joest M, Sakai H, Aguin SB, et al. Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell. 2017;20:407–14.e4. doi: 10.1016/j.stem.2016.11.020. [DOI] [PubMed] [Google Scholar]
- Choi N, Zhang B, Zhang L, Ittmann M, Xin L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell. 2012;21:253–65. doi: 10.1016/j.ccr.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua CW, Shibata M, Lei M, Toivanen R, Barlow LJ, et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat Cell Biol. 2014;16:951–61. doi: 10.1038/ncb3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–19. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- Colleypriest BJ, Farrant JM, Slack JM, Tosh D. The role of Cdx2 in Barrett’s metaplasia. Biochem Soc Trans. 2010;38:364–69. doi: 10.1042/BST0380364. [DOI] [PubMed] [Google Scholar]
- Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, et al. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into α and subsequently β cells. Cell. 2009;138:449–62. doi: 10.1016/j.cell.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortina C, Turon G, Stork D, Hernando-Momblona X, Sevillano M, et al. A genome editing approach to study cancer stem cells in human tumors. EMBO Mol Med. 2017;9:869–79. doi: 10.15252/emmm.201707550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha GR, Donjacour AA, Cooke PS, Mee S, Bigsby RM, et al. The endocrinology and developmental biology of the prostate. Endocr Rev. 1987;8:338–62. doi: 10.1210/edrv-8-3-338. [DOI] [PubMed] [Google Scholar]
- Dalerba P, Sahoo D, Paik S, Guo X, Yothers G, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med. 2016;374:211–22. doi: 10.1056/NEJMoa1506597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature. 2017;543:676–80. doi: 10.1038/nature21713. [DOI] [PubMed] [Google Scholar]
- Deome KB, Faulkin LJ, Jr, Bern HA, Blair PB. Development of mammary tumors from hyperplastic alveolar nodules transplanted into gland-free mammary fat pads of female C3H mice. Cancer Res. 1959;19:515–20. [PubMed] [Google Scholar]
- Dutta A, Le Magnen C, Mitrofanova A, Ouyang X, Califano A, Abate-Shen C. Identification of an NKX3.1-G9a-UTY transcriptional regulatory network that controls prostate differentiation. Science. 2016;352:1576–80. doi: 10.1126/science.aad9512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–27. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrony GD, Lee E, Mehta BK, Benjamini Y, Johnson RM, et al. Cell lineage analysis in human brain using endogenous retroelements. Neuron. 2015;85:49–59. doi: 10.1016/j.neuron.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatehullah A, Tan SH, Barker N. Organoids as an in vitro model of human development and disease. Nat Cell Biol. 2016;18:246–54. doi: 10.1038/ncb3312. [DOI] [PubMed] [Google Scholar]
- Fellous TG, McDonald SA, Burkert J, Humphries A, Islam S, et al. A methodological approach to tracing cell lineage in human epithelial tissues. Stem Cells. 2009;27:1410–20. doi: 10.1002/stem.67. [DOI] [PubMed] [Google Scholar]
- Ferone G, Song JY, Sutherland KD, Bhaskaran R, Monkhorst K, et al. SOX2 is the determining oncogenic switch in promoting lung squamous cell carcinoma from different cells of origin. Cancer Cell. 2016;30:519–32. doi: 10.1016/j.ccell.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–76. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong H, Hohenstein KA, Donovan PJ. Regulation of self-renewal and pluripotency by Sox2 in human embryonic stem cells. Stem Cells. 2008;26:1931–38. doi: 10.1634/stemcells.2007-1002. [DOI] [PubMed] [Google Scholar]
- Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer. 2006;6:593–602. doi: 10.1038/nrc1947. [DOI] [PubMed] [Google Scholar]
- Gehring WJ, Affolter M, Burglin T. Homeodomain proteins. Annu Rev Biochem. 1994;63:487–526. doi: 10.1146/annurev.bi.63.070194.002415. [DOI] [PubMed] [Google Scholar]
- Genovese G, Carugo A, Tepper J, Robinson FS, Li L, et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature. 2017;542:362–66. doi: 10.1038/nature21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379–89. doi: 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329:568–71. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez F, Georgieva D, Vanoli F, Shi ZD, Stadtfeld M, et al. Homologous recombination DNA repair genes play a critical role in reprogramming to a pluripotent state. Cell Rep. 2013;3:651–60. doi: 10.1016/j.celrep.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–39. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Halliday J, Helmy K, Pattwell SS, Pitter KL, LaPlant Q, et al. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. PNAS. 2014;111:5248–53. doi: 10.1073/pnas.1321014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada K, Sasaki T, Koni PA, Natsui M, Kishimoto H, et al. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 2005;19:2054–65. doi: 10.1101/gad.1308805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–77. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Hoek KS, Eichhoff OM, Schlegel NC, Dobbeling U, Kobert N, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68:650–56. doi: 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- Hoek KS, Goding CR. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010;23:746–59. doi: 10.1111/j.1755-148X.2010.00757.x. [DOI] [PubMed] [Google Scholar]
- Horb ME, Shen CN, Tosh D, Slack JM. Experimental conversion of liver to pancreas. Curr Biol. 2003;13:105–15. doi: 10.1016/s0960-9822(02)01434-3. [DOI] [PubMed] [Google Scholar]
- Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–86. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischenko I, Zhi J, Moll UM, Nemajerova A, Petrenko O. Direct reprogramming by oncogenic Ras and Myc. PNAS. 2013;110:3937–42. doi: 10.1073/pnas.1219592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski JA, Harrison RF, Perry I, Balkwill F, Tselepis C. Barrett’s metaplasia. Lancet. 2000;356:2079–85. doi: 10.1016/S0140-6736(00)03411-5. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R, Arthur-Farraj P. The role of cell plasticity in tissue repair: adaptive cellular reprogramming. Dev Cell. 2015;34:613–20. doi: 10.1016/j.devcel.2015.09.005. [DOI] [PubMed] [Google Scholar]
- Jopling C, Boue S, Izpisua Belmonte JC. Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. Nat Rev Mol Cell Biol. 2011;12:79–89. doi: 10.1038/nrm3043. [DOI] [PubMed] [Google Scholar]
- Kai T, Spradling A. Differentiating germ cells can revert into functional stem cells in Drosophila melanogaster ovaries. Nature. 2004;428:564–69. doi: 10.1038/nature02436. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Investig. 2009;119:1420–28. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamachi Y, Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development. 2013;140:4129–44. doi: 10.1242/dev.091793. [DOI] [PubMed] [Google Scholar]
- Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell. 2015;16:39–50. doi: 10.1016/j.stem.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 2014;159:163–75. doi: 10.1016/j.cell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–44. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4:816–27. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar K, Watt FM. Lineage tracing. Cell. 2012;148:33–45. doi: 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon OJ, Zhang B, Zhang L, Xin L. High fat diet promotes prostatic basal-to-luminal differentiation and accelerates initiation of prostate epithelial hyperplasia originated from basal cells. Stem Cell Res. 2016;16:682–91. doi: 10.1016/j.scr.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Kwon OJ, Zhang L, Ittmann MM, Xin L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. PNAS. 2014;111:E592–600. doi: 10.1073/pnas.1318157111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell. 2017;20:233–46.e7. doi: 10.1016/j.stem.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Chou YT, Jiang SS, Chang JL, Chung CH, et al. Epigenetic switch between SOX2 and SOX9 regulates cancer cell plasticity. Cancer Res. 2016;76:7036–48. doi: 10.1158/0008-5472.CAN-15-3178. [DOI] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209–21. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Grogan TR, Hieronymus H, Hashimoto T, Mottahedeh J, et al. Low CD38 identifies progenitor-like inflammation-associated luminal cells that can initiate human prostate cancer and predict poor outcome. Cell Rep. 2016;17:2596–606. doi: 10.1016/j.celrep.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livet J, Weissman TA, Kang H, Draft RW, Lu J, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350:94–98. doi: 10.1126/science.aab1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotan TL, Gupta NS, Wang W, Toubaji A, Haffner MC, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol. 2011;24:820–28. doi: 10.1038/modpathol.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, et al. Pancreatitis and the risk of pancreatic cancer. N Engl J Med. 1993;328:1433–37. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion RM, Strati K, Li H, Murga M, Blanco R, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–53. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maza I, Caspi I, Zviran A, Chomsky E, Rais Y, et al. Transient acquisition of pluripotency during somatic cell transdifferentiation with iPSC reprogramming factors. Nat Biotechnol. 2015;33:769–74. doi: 10.1038/nbt.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Findlay GM, Gagnon JA, Horwitz MS, Schier AF, Shendure J. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science. 2016;353:aaf7907. doi: 10.1126/science.aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–37. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrell AJ, Stanger BZ. Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol. 2016;17:413–25. doi: 10.1038/nrm.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger D, Clifford J, Chiba H, Chambon P. Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre recombinase. PNAS. 1995;92:6991–95. doi: 10.1073/pnas.92.15.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–89. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- Morris SA, Daley GQ. A blueprint for engineering cell fate: current technologies to reprogram cell identity. Cell Res. 2013;23:33–48. doi: 10.1038/cr.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosteiro L, Pantoja C, Alcazar N, Marion RM, Chondronasiou D, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016;354:aaf4445. doi: 10.1126/science.aaf4445. [DOI] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M, Takizawa N, Narita M, Ichisaka T, Yamanaka S. Promotion of direct reprogramming by transformation-deficient Myc. PNAS. 2010;107:14152–57. doi: 10.1073/pnas.1009374107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol. 2016;11:47–76. doi: 10.1146/annurev-pathol-012615-044438. [DOI] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. 2015;6:6377. doi: 10.1038/ncomms7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22:709–24. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16:e165–72. doi: 10.1016/S1470-2045(14)71180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘What does not kill me strengthens me’. Br J Cancer. 2015;112:1725–32. doi: 10.1038/bjc.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell. 2013;24:242–56. doi: 10.1016/j.ccr.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;18:510–23. doi: 10.1016/j.ccr.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–98. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen MA, Holst B, Tumer Z, Johnsen MG, Zhou S, et al. Transient p53 suppression increases reprogramming of human fibroblasts without affecting apoptosis and DNA damage. Stem Cell Rep. 2014;3:404–13. doi: 10.1016/j.stemcr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–61. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31:172–83. doi: 10.1101/gad.290635.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy N, Hebrok M. Regulation of cellular identity in cancer. Dev Cell. 2015;35:674–84. doi: 10.1016/j.devcel.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Martinez I, Baek SH, Izpisua Belmonte JC. Lineage conversion methodologies meet the reprogramming toolbox. Nat Cell Biol. 2012;14:892–99. doi: 10.1038/ncb2567. [DOI] [PubMed] [Google Scholar]
- Sarkar A, Hochedlinger K. The Sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12:15–30. doi: 10.1016/j.stem.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell. 2013;23:93–106. doi: 10.1016/j.ccr.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J Clin Investig. 2012;122:3914–18. doi: 10.1172/JCI63065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfanos KS, De Marzo AM. Prostate cancer and inflammation: the evidence. Histopathology. 2012;60:199–215. doi: 10.1111/j.1365-2559.2011.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–29. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature. 2017;545:187–92. doi: 10.1038/nature22081. [DOI] [PubMed] [Google Scholar]
- Slack JM. Metaplasia and transdifferentiation: from pure biology to the clinic. Nat Rev Mol Cell Biol. 2007;8:369–78. doi: 10.1038/nrm2146. [DOI] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–44. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- Snyder EL, Watanabe H, Magendantz M, Hoersch S, Chen TA, et al. Nkx2-1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol Cell. 2013;50:185–99. doi: 10.1016/j.molcel.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing β cells in the mammalian pancreas. Nature. 1997;386:399–402. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumpf D, Mao CA, Yamanaka Y, Ralston A, Chawengsaksophak K, et al. Cdx2 is required for correct cell fate specification and differentiation of trophectoderm in the mouse blastocyst. Development. 2005;132:2093–102. doi: 10.1242/dev.01801. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Kitamura T, DePinho RA, Accili D. Generation of functional insulin-producing cells in the gut by Foxo1 ablation. Nat Genet. 2012a;44:406–S1. doi: 10.1038/ng.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012b;150:1223–34. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos F, Mitrofanova A, Bergren SK, Califano A, Shen MM. A computational systems approach identifies synergistic specification genes that facilitate lineage conversion to prostate tissue. Nat Commun. 2017;8:14662. doi: 10.1038/ncomms14662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlow BD, Pelz C, Naugler WE, Wakefield L, Wilson EM, et al. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell. 2014;15:605–18. doi: 10.1016/j.stem.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata PR, Mou H, Pardo-Saganta A, Zhao R, Prabhu M, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503:218–23. doi: 10.1038/nature12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata PR, Rajagopal J. Cellular plasticity: 1712 to the present day. Curr Opin Cell Biol. 2016;43:46–54. doi: 10.1016/j.ceb.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Thomas P, Beddington R. Anterior primitive endoderm may be responsible for patterning the anterior neural plate in the mouse embryo. Curr Biol. 1996;6:1487–96. doi: 10.1016/s0960-9822(96)00753-1. [DOI] [PubMed] [Google Scholar]
- Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, et al. Conversion of adult pancreatic α-cells to β-cells after extreme β-cell loss. Nature. 2010;464:1149–54. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, et al. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478:255–59. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivanen R, Mohan A, Shen MM. Basal progenitors contribute to repair of the prostate epithelium following induced luminal anoikis. Stem Cell Rep. 2016;6:660–67. doi: 10.1016/j.stemcr.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–36. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell. 2014;158:579–92. doi: 10.1016/j.cell.2014.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145–48. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es JH, Sato T, van de Wetering M, Lyubimova A, Nee AN, et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol. 2012;14:1099–104. doi: 10.1038/ncb2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–41. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 2014;28:1143–58. doi: 10.1101/gad.242511.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. The Strategy of the Genes: A Discussion of Some Aspects of Theoretical Biology. London: Allen & Unwin; 1957. [Google Scholar]
- Wang Q, Zou Y, Nowotschin S, Kim SY, Li QV, et al. The p53 family coordinates Wnt and nodal inputs in mesendodermal differentiation of embryonic stem cells. Cell Stem Cell. 2017;20:70–86. doi: 10.1016/j.stem.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, Mitrofanova A, Bergren SK, Abate-Shen C, Cardiff RD, et al. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol. 2013;15:274–83. doi: 10.1038/ncb2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, Toivanen R, Bergren SK, Chambon P, Shen MM. Luminal cells are favored as the cell of origin for prostate cancer. Cell Rep. 2014;8:1339–46. doi: 10.1016/j.celrep.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Francis JM, Woo MS, Etemad B, Lin W, et al. Integrated cistromic and expression analysis of amplified NKX2-1 in lung adenocarcinoma identifies LMO3 as a functional transcriptional target. Genes Dev. 2013;27:197–210. doi: 10.1101/gad.203208.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D, Wang L, Yan Y, Jia Z, Gagea M, et al. KLF4 is essential for induction of cellular identity change and acinar-to-ductal reprogramming during early pancreatic carcinogenesis. Cancer Cell. 2016;29:324–38. doi: 10.1016/j.ccell.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Cassady JP, Jaenisch R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell. 2008;2:10–12. doi: 10.1016/j.stem.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, et al. Suppression of lung ade-nocarcinoma progression by Nkx2-1. Nature. 2011;473:101–4. doi: 10.1038/nature09881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodworth MB, Girskis KM, Walsh CA. Building a lineage from single cells: genetic techniques for cell lineage tracking. Nat Rev Genet. 2017;18:230–44. doi: 10.1038/nrg.2016.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley MI, Setiawan L, Hariharan IK. Regeneration and transdetermination Drosophila imaginal discs. Annu Rev Genet. 2012;46:289–310. doi: 10.1146/annurev-genet-110711-155637. [DOI] [PubMed] [Google Scholar]
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003;421:942–47. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27:719–24. doi: 10.1101/gad.207803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L, Lu C, Hu W, Sun Y, Levine AJ. Multiple roles of p53-related pathways in somatic cell reprogramming and stem cell differentiation. Cancer Res. 2012;72:5635–45. doi: 10.1158/0008-5472.CAN-12-1451. [DOI] [PubMed] [Google Scholar]
- Yu VW, Yusuf RZ, Oki T, Wu J, Saez B, et al. Epigenetic memory underlies cell-autonomous heterogeneous behavior of hematopoietic stem cells. Cell. 2017;168:944–45. doi: 10.1016/j.cell.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–30. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature. 2008;455:627–32. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006;66:7889–98. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–92. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Zou M, Toivanen R, Mitrofanova A, Floc’h N, Hayati S, et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017;7:736–49. doi: 10.1158/2159-8290.CD-16-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.