

SUMMARY
Immunity following an acutely resolved infection or the long-term equipoise of chronic viral infections often depends on the maintenance of antigen-specific CD8+ T cells, yet the ongoing transcriptional requirements of these cells remain unclear. We show that active and continuous programming by FOXO1 is required for the functional maintenance of a memory population. Upon Foxo1 deletion following resolution of an infection, memory cells rapidly lost their characteristic gene expression, gradually declined in number, and were impaired in self-renewal. This was extended to chronic infections, as a loss of FOXO1 during a persistent viral infection led to a rapid decline of the TCF7 (a.k.a. TCF1)-expressing memory-like subset of CD8+ T cells. We further establish FOXO1 regulation as a characteristic of human memory CD8+ T cells. Overall, we show that the molecular and functional longevity of a memory T cell population is actively maintained by the transcription factor FOXO1.
In Brief
Utzschneider et al. find that hallmarks of CD8+ T cell memory such as longevity, self-renewal, and the ability to cycle between quiescence and cell division depend on continued expression of FOXO1. Loss of FOXO1 during any of these stages leads to the interruption of T cell memory.
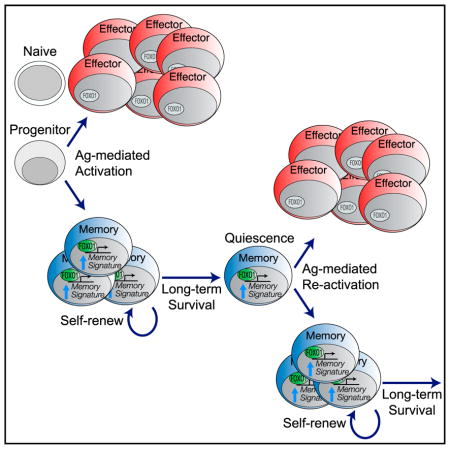
INTRODUCTION
Functional immune memory governed by CD8+ T cells is indispensable for resistance to bacterial and viral re-infection. The ability to provide such protection relies on the longevity of a memory population and its ability to mount a robust recall response when re-exposed to antigen derived from the same pathogen. In order to survive over long periods, memory CD8+ T cells persist at a population level by slow but constant self-renewal balanced against programmed cell death. Along with the unusual property of self-renewal, memory CD8+ T cells display the unique ability to serially transit through phases of activation, growth, and proliferation followed by quiescence. In essence, they exhibit characteristics of multipotent stem cells that simultaneously self-renew and produce progenitors of terminally differentiated cells (Gattinoni et al., 2017; Fearon et al., 2001). The ongoing transcriptional requirements for the homeostasis of memory cells through these phases are still under investigation.
The transcriptional network responsible for the generation of memory CD8+ T cells has been widely studied and found to include the evolutionarily conserved family of Forkhead box O (FOXO) transcription factors. The known cell-type-specific FOXO target genes profoundly affect survival, homing, proliferation, and differentiation of CD8+ T cells and constitute a large proportion of the memory gene expression signature. In particular, the transcription factor FOXO1 has been shown to positively regulate several genes associated with T cell survival and trafficking including Il7ra, Ccr7, Klf2, Sell (CD62L), Tcf7, Eomes, and Bcl2 (Hedrick et al., 2012). Moreover, FOXO1 has been shown to play an essential role in the generation of functional memory T cells by the direct or indirect repression of Tbx21 (T-BET), Ifng, and Gzmb (GRANZYME B), hallmarks of effector T cells (Hess Michelini et al., 2013; Rao et al., 2012; Ouyang et al., 2009). This is in part extrinsically governed by a variety of FOXO1 post-translational modifications (Klotz et al., 2015), which in turn impact its cellular localization such that nuclear FOXO1 has been shown to strongly correlate with a memory fate (Lin et al., 2015; Verbist et al., 2016; Zhang et al., 2016). Furthermore, a recent study has proposed that FOXO1 potentially shields memory precursors from deposition of repression-associated histone 3 lysine 27 trimethyl (H3K27me3) chromatin modifications (Gray et al., 2017). Importantly, many experimental efforts to study the role of a specific transcription factor on T cell differentiation have been based on gene deletion, and such studies have provided insights into the transcriptional and molecular mechanisms leading to an effector or memory T cell. However, whether a transcription factor, such as FOXO1, dynamically regulates the course of T cell activation, survival, and differentiation is not well understood.
Here, we show by using an inducible gene deletion system that FOXO1 must be continuously present for the homeostatic proliferation required to maintain a functional memory population. Upon Foxo1 deletion after the establishment of memory, there occurred a rapid loss of gene expression characteristic of memory cells combined with a deficiency in homeostatic (lymphopenia-induced) proliferation leading to a continuous decline of the memory T cell population. Still, early on, FOXO1-deficient memory T cells were capable of proliferation in response to a secondary infection, but these remaining functional memory cells gradually declined, and eventually, the progeny of these cells were impaired in their ability to mount a robust secondary response. Thus, we conclude that FOXO1 has to be present in at least two phases of the perduring cycle of T cell memory: long-term survival and stem cell-like self-renewal.
Moreover, the characteristics of memory CD8+ T cells are equally manifest in the ongoing immune response associated with a chronic viral infection. Loss of FOXO1 in the chronic phase led to a rapid decline of the proliferative TCF7+ CD8+ T cell subset responsible for sustaining the effector T cell response and thus mediating viral control. Finally, we underscore the broad importance of FOXO1 in memory by showing that it serves as a functional marker to delineate human memory CD8+ T cells specific for influenza A (FLU) and cytomegalovirus (CMV).
RESULTS
Resting Memory T Cells Display Transcriptional Plasticity after Foxo1 Deletion
FOXO1 has been shown to target a wide variety of molecules associated with memory T cell differentiation (Hedrick et al., 2012; Hess Michelini et al., 2013; Kim et al., 2013; Rao et al., 2012). We were interested in determining the role of continuous FOXO1 expression in memory CD8+ T cells and how this impacts cardinal aspects of a memory T cell population including longevity and the ability to mount a recall response. To study this, we crossed Foxo1f/f mice (Paik et al., 2007) to Rosa26Cre-ERT2 mice (termed herein iFx1f/f) in which the chimeric estrogen receptor-cre recombinase as defined for the Gt(ROSA)26Sortm2(cre/ERT2)Brn mice (ER-Cre) protein is activated upon treatment with tamoxifen (TAM). By further crossing these mice to include the P14 T cell receptor (TCR) transgene specific for the lymphocytic choriomeningitis virus (LCMV)-derived gp33-41 (gp33) epitope, we were able to create the experimental means to delete Foxo1 in LCMV-specific P14-transgenic cells at any time-point prior to, or after infection.
To determine the impact of Foxo1 deletion in memory T cells, we transferred either iFx1f/f-P14 or Rosa26Cre-ERT2− P14 T cells (either Foxo1+/+, Foxo1f/+, or Foxo1f/f; herein referred to as wild-type [WT]-P14) into congenic C57BL/6 hosts prior to challenge with LCMV Armstrong (LCMV Arm) (Figure 1A). Four weeks post-infection, we treated mice with TAM and validated efficient Foxo1 deletion in TAM-treated iFx1f/f-P14 (referred as iFx1−/−) compared to WT-P14 or endogenous host CD8+ T cells in the spleen (Figure 1B).
Figure 1. Memory T Cells Display Transcriptional Plasticity after Acute Foxo1 Deletion.

(A) C57BL/6 mice were engrafted with similar numbers (4 × 103) of WT- and iFx1f/f-P14 or co-injected with both populations (D, middle) and infected with LCMV Armstrong (LCMV Arm). 4 weeks post-infection, mice were treated with tamoxifen and splenocytes analyzed.
(B) FOXO1 expression levels of WT- and iFx1−/−-P14 compared to total host CD8+ T cell population.
(C) Expression of KLRG1 and CD127 of WT- and iFx1−/−-P14 and the percentage of CD127+KLRG1− P14 for all mice (right).
(D) Frequency of CD62L+, CD43+CD27+, and CXCR3+ P14.
(E and F) Splenocytes were ex vivo re-stimulated with gp33 peptide; intracellularly stained for IFN-γ, TNF, IL-2, and CD107α; and analyzed by flow cytometry. (E) IFN-γ and IL-2 production of WT- (black, left) and iFx1−/−-P14 cells (red, middle) and calculated fraction of IFN-γ+ IL-2-producing P14 cells (right). (F) Fraction of IFN-γ+ P14 cells that co-produce TNF, and the frequency of P14 that produce IFN-γ or CD107α.
(G) Expression levels of TCF7, EOMES, and T-BET of WT- (black) and iFx1−/−-P14 (red) compared to total host CD8+ T cell population (gray shadow) and mean fluorescence intensity (MFI) of P14 of all analyzed mice. CD4−CD8− cells (dashed line) served as internal negative staining control for TCF7.
Symbols in (C)–(F) represent individual mice; horizontal lines indicate mean. Data are representative of at least one experiment with deletion 4 weeks post-infection and two experiments with P14 co-transfer and deletion 4 or 5 weeks post-infection, with each at least four mice per group. Error bars in (G) indicate SD. Statistical analysis was by unpaired Student’s t test. ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant.
iFx1−/− -P14 T cells displayed a smaller fraction of memory cells based on the expression of killer cell lectin-like receptor G1 (KLRG1) and the interleukin-7 receptor α (IL-7Rα) chain (CD127), L-selectin (CD62L), CD43 and CD27, and CXCR3 (Figures 1C and 1D)—characteristics used to define CD8+ T cell memory (Hu et al., 2011; Kaech and Cui, 2012). They also exhibited a reduced ability to produce IL-2 following ex vivo re-stimulation with the gp33 peptide (Figure 1E), which is preferentially produced by central memory T cells (TCM) (Mueller et al., 2013). In contrast, effector function in the form of tumor necrosis factor (TNF), interferon-γ (IFN-γ), and CD107α production was not impacted by acute FOXO1 deficiency (Figure 1F). We also noted a decline in the expression of transcription factors associated with memory, such as T cell factor 7 (TCF7; also known as TCF1) and the T-box transcription factor Eomesodermin (EOMES) (Figure 1G) (Kaech and Cui, 2012; Zhou et al., 2010). This is in line with previous observations showing that FOXO1 binds directly to Tcf7 and Eomes and, in particular, is required for their expression in memory cells (Hess Michelini et al., 2013).
Expression of the T-box transcription factor T-BET, which promotes effector cell development (Intlekofer et al., 2005), was not substantially altered (Figure 1G), contrasting with previous studies of in-vitro-activated FOXO1-deficient T cells (Rao et al., 2012).
To confirm that we were accurately looking at a quiescent memory T cell population, we induced Foxo1 deletion late (12 weeks) following a resolved LCMV Arm infection (Figure S1A). Similar to what we observed upon deletion 4 weeks post-infection (Figure 1), acute deletion of Foxo1 in a late memory T cell population showed a similar decline in expression of memory-associated molecules (Figure S1). Collectively, acute deletion of Foxo1 showed that continuous FOXO1 expression is essential to sustain several phenotypic manifestations of T cell memory.
FOXO1-Induced Transcriptional Alterations Are Specific to Antigen-Activated T Cells
Naive T cells are sustained in a default state of quiescence (Hamilton and Jameson, 2012). Similar to a memory population, naive T cells express genes encoding CD127 and CD62L, which have been shown to be directly regulated by FOXO1 (Kerdiles et al., 2009; Ouyang et al., 2009). To study how acute FOXO1 deletion impacts the expression of these molecules in antigen-inexperienced T cells, we directly treated unimmunized iFx1f/f mice with TAM and phenotypically characterized FOXO1-deficient CD8+ T cells 5 days post-treatment. Specific-pathogen-free mice contain naive CD44− and CD44+ T cells, which are mainly antigen-inexperienced virtual memory T cells (TVM) arising as a result of cytokine stimulation (White et al., 2017). In line with previous studies (Kerdiles et al., 2009), acute loss of FOXO1 had no substantial effect on the proportion of naive and TVM cells, as Foxo1-deleted mice showed a similar proportion of CD44+CD8+ T cells compared to FOXO1-expressing control mice (Figure 2A). The activation of ER-Cre also did not diminish the proportion of CD44+ splenic T cells.
Figure 2. Acute Foxo1 Deletion in Antigen-Inexperienced T Cells.

Unimmunized iFx1f/f mice were treated with tamoxifen and splenocytes analyzed 5 days after the last treatment. Controls were untreated iFx1f/f as well as TAM-treated Rosa26Cre-ERT2neg and iFx1+/+ mice.
(A and B) Frequency of CD44+ (A) and KLRG1+ (B) CD8+ T cells.
(C–H) Naive (CD44−), virtual memory (CD44+ KLRG1−), and CD44+KLRG1+ T cells were phenotyped for expression of CD127 (C) CD62L (D), CXCR3 (E), TCF7 (F), EOMES (G), and T-BET (H). Shown are the mean fluorescent intensity (C and F–H) or the frequency of positive cells among the indicated T cell subsets (D and E). Symbols in (A), (B), (D), and (E) represent individual mice; horizontal lines indicate the mean. Data are cumulative from two independent experiments with at least two mice per group. Error bars in (C) and (F)–(H) indicate SD.
Statistical analysis was by Dunnett’s multiple comparisons test. ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant.
In contrast to the alterations in an LCMV-memory population induced by FOXO1 deletion, we did not observe an increase in the fraction of KLRG1-expressing T cells in unimmunized mice (Figure 2B). We further compared the impact of acute FOXO1 deficiency upon the expression of a variety of memory-signature molecules among naive and CD44+KLRG1+/− T cells. Similar to the alterations in memory T cells (Figure 1), we detected a significant loss of CD127 expression among TVM and naive iFx1−/− T cells (Figure 2C), consistent with Il7r being a direct FOXO1 target in naive, TVM, and memory T cells (Kerdiles et al., 2009). However, we did not detect a decline in expression of CD62L or CXCR3 or of the transcription factors TCF7 and EOMES in acutely Foxo1-deleted CD8+ T cells (Figures 2D–2G), yet Foxo1-deleted CD44+KLRG1−CD8+ T cells showed enhanced expression of T-BET (Figure 2H). Overall, these observations suggest that FOXO1 is not required to actively maintain the expression of characteristic memory-associated molecules at every phase of T cell activation and differentiation. A possibility is that FOXO1 directly controls expression of some of these genes, such as Il7r, but indirectly affects others, such that an acute loss of FOXO1 in the memory population causes an overall shift to an effector phenotype. We note that addition of TAM did not affect the T cell phenotype regardless of the presence of ER-Cre.
Acute Foxo1 Deletion Primarily Impacts the TCM Subset
To determine whether acute FOXO1 deletion following infection impacts the abundance of a memory T cell population, we measured the proportion of congenic iFx1−/−-P14 to WT-P14 within the same LCMV-Arm-infected host comparing TAM-treated and untreated controls. In contrast to the rapid molecular changes, we did not detect alterations in the ratio of iFx1−/−-P14 compared to WT-P14 caused by TAM deletion in the blood, spleen, or intestinal epithelium (IEL), despite a decline in CD127 and TCF7 expression (Figures 3A–3D). The only change in maintenance induced by TAM was in the peripheral lymph node (pLN) population (Figures 3A and 3B), suggesting an increased impact of FOXO1 deletion on TCM that localize via L-selectin and CCR7 (Tomura et al., 2008; Masopust et al., 2001).
Figure 3. Acute Foxo1 Deletion in a Quiescent Memory T Cell Population Primarily Impacts TCM Subset.

C57BL/6 mice were co-injected with similar numbers (1.5 × 104) of WT- and iFx1f/f-P14 and infected with LCMV Arm.
(A–H) 4 weeks post-infection, mice were treated with tamoxifen and 5 days after the last treatment (day 34 post infection) sacrificed and analyzed. (A) Representative flow cytometry gated on total P14 in TAM-treated mice showing WT- (CD45.1+CD45.2+) and iFx1−/−-P14 (CD45.2+) percentage in indicated organs.
(B) WT/iFx1−/−-P14 ratio compared to the ratio of untreated controls.
(C and D) CD127 (C) and TCF7 (D) expression among P14 cells from indicated organs.
(E) Representative flow cytometry of intravascular staining pattern distinguishing between P14 in red pulp (RP; CD8α+) and white pulp (WP; CD8α−), and the WT (CD45.1+CD45.2+) and iFx1−/−-P14 (CD45.2+) percentage within the RP (top) and WP (bottom). Calculated WT/iFx1−/−-P14 ratio in the RP and WP compared to untreated controls (right).
(F and G) Expression of KLRG1 and CD127 of WT- and iFx1−/−-P14 in the red pulp (RP; CD8α+) and white pulp (WP; CD8α−), and the percentage of CD127+KLRG1− P14 for all mice (G).
(H) TCF7 expression levels of WT- and iFx1−/−-P14 in the red pulp (RP; CD8α+) and white pulp (WP; CD8α−).
(I) Experimental design used in (J)–(P). C57BL/6 mice grafted with similar numbers (5 × 104) of WT- and iFx1f/f-P14 cells were infected with LCMV Arm. 4 weeks post-infection, KLRG1− WT- and KLRG1− iFx1f/f-P14 were FACS purified from spleen and lymph nodes, labeled with CellTrace violet (CTV), and co-transferred into naive C57BL/6 hosts, which were 1 day later treated with TAM.
(J) Purification control of transferred KLRG1− WT- and KLRG1− iFx1f/f-P14.
(K–P) Splenocytes were stained for expression of KLRG1 and CD62L. (K–M) Representative FACS plots illustrating the fraction of undivided KLRG1-expressing KLRG1− WT- and KLRG1− iFx1−/−-P14 (K) as well as the calculated fraction (L) and absolute numbers (M) for all mice. (N–P) Fraction (N and O) and absolute numbers (P) of undivided CD62L− WT- and CD62L− iFx1−/−-P14.
Data are representative of at least two experiments with at least four mice per treated group. Error bars in (B) and (D) indicate SD. Statistical analysis was by paired Student’s t test. ***p < 0.001; **p < 0.01; ns, not significant.
Effector memory T cells (TEM) preferentially locate to the red pulp of the spleen, whereas TCM preferentially reside in the white pulp (Jung et al., 2010; Mueller et al., 2013). To see whether FOXO1 deletion differentially alters the maintenance within these splenic niches irrespective of the expression of canonical memory markers, we used an intravascular staining protocol that distinguishes T cells by location within the red and white pulp (Galkina et al., 2005) (Figure 3E, left). We then determined within each T cell subset the WT/iFx1−/−-P14 ratio and compared it to the WT/iFx1f/f-P14 ratio of an untreated control group (Figure 3E). Using this separation, we noted that iFx1−/− memory P14 within the white pulp displayed a numerical discrepancy similar to what was seen in the pLNs. In contrast, the TEM subset within the red pulp displayed only minor alterations in favor of the iFx1−/−-P14 population (Figure 3E). Notably, both subsets showed significant phenotypic alterations in regards of expression of KLRG1, CD127, and TCF7 (Figures 3F–3H). These observations indicate that besides a loss of a memory signature, acute FOXO1-deletion impacts the maintenance of TCM subset as evidenced by their exclusion from the white pulp of the spleen and pLNs.
To directly determine whether this proportional shift following Foxo1 deletion arises from a loss of TCM or a de-differentiation of TCM into TEM, we sorted KLRG1− WT and KLRG1− iFx1f/f-P14 cells 4 weeks post-LCMV Arm infection and transferred them into naive hosts, which were then treated with TAM (Figures 3I and 3J). Following the deletion of Foxo1, undivided iFx1−/−-P14 T cells significantly increased expression of KLRG1 and decreased expression of CD62L compared to co-transferred WT-P14 cells (Figures 3K–3P). Thus, sustained expression of FOXO1 is essential to actively maintain the continuous programming and thus the phenotypic characteristics of T cell memory.
Deletion of Foxo1 in Memory T Cells Impairs Maintenance
Longevity is a signature attribute of T cell memory allowing re-expansion upon reinfection, yet it is unknown how the loss of FOXO1 expression impacts the maintenance and thus the longevity of a memory population. Following Foxo1 deletion in a P14 memory population, we noted a rapid loss of the anti-apoptotic proteins BCL2 among both the KLRG1− and KLRG1+ iFx1−/− subsets (Figure 4A). Furthermore, we observed a decreased proportion of KLRG1− and KLRG1+ iFx1−/−-P14 in cycle based on the expression of KI67 or the incorporation of bromodeoxyuridine (BrdU) (Figures 4B and 4C). We also noted reduced expression of CD127 in both KLRG1−/+ subsets (Figure 4D), which lead to an increased fraction of iFx1−/− P14 unable to induce STAT5 phosphorylation following ex vivo stimulation with IL-7 (Figures 4E and 4F). Notably, STAT5 phosphorylation was still inducible by IL-15 stimulation (data not shown).
Figure 4. Foxo1 Deletion Impairs Memory Homeostasis.

C57BL/6 mice were grafted with similar numbers (4 × 103–104) of WT- and iFx1f/f-P14 prior to infection with LCMV Arm. Mice were treated from days 27 to 31 with tamoxifen (TAM).
(A–F) Mice were administered BrdU for 6 days (days 36–42) (A–C) and spleens analyzed (day 42 post-LCMV Arm). BCL2 expression levels among KLRG1+/− WT-and iFx1−/−-P14 (A). Fraction of KI67+ (B) and BrdU+ (C) KLRG1+/− WT- and iFx1−/−-P14. CD127 expression among KLRG1+/− WT- and iFx1−/−-P14 (D). Representative histogram of STAT5 phosphorylation (E) following IL-7 stimulation of KLRG1+/− WT- and iFx1−/−-P14 compared to unstimulated KLRG1+/− WT-P14, which were representative of unstimulated KLRG1+/− iFx1−/−-P14 (data not shown). Percentage of pSTAT5+ among KLRG1+/− WT- and iFx1−/−-P14 (F).
(G) P14 kinetics following TAM treatment in the blood of the primary host.
(H) Ratio of WT to iFx1−/−-P14 over time in the blood compared to untreated controls.
(I and J) 100 days post-TAM treatment (day 130 post-LCMV Arm), mice were sacrificed and the ratio of WT- to iFx1−/−-P14 in different organs determined. Representative flow cytometry gated on total P14 showing WT- (CD45.1+CD45.2+) and iFx1−/−-P14 (CD45.2+) percentage (I) and WT/iFx1−/−-P14 ratio compared to untreated controls (J) in indicated organs 130 days post-infection.
(K) Experimental design used in (L) and (M): C57BL/6 mice grafted with similar numbers (5 × 104) of WT- and iFx1f/f-P14 were infected with LCMV Arm and treated with TAM 4 weeks post-infection. 5 days later, KLRG1− WT- and KLRG1− iFx1−/−-P14 were FACS purified from spleen, labeled with CTV, and co-transferred into sublethally irradiated naive C57BL/6 hosts.
(L) Lymphopenia-induced proliferation of transferred KLRG1− WT- (black) and KLRG1− iFx1−/−-P14 (red) 15 days post-transfer in the spleen. Host cells (gray shadow) served as CTV− controls.
(M) Calculated proliferation index of transferred P14.
Error bars in (A), (G), (H), and (J) indicate SD. Symbols in (B), (C), (F), and (M) represent individual mice and in (G) and (H) the mean of four mice per treated group and at least two untreated mice; horizontal lines indicate the mean. Data are representative of at least two experiments, with at least four mice per treated group. Statistical analysis was by paired Student’s t test. ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant.
This reduced fraction of cycling cells progressively yet slowly impacted the overall abundance of the memory iFx1−/−-P14 population (Figure 4G). Around 60 days post-infection (30 days post-TAM treatment), the WT-P14 population leveled out, whereas the FOXO1-deleted P14 population continuously declined (Figures 4G and 4H).
Similar observations were made regarding the WT/iFx1−/−-P14 ratio in the spleen 100 days post TAM-treatment (day 130 post infection) (Figures 4I and 4J) whereas the circulating T cells in the red pulp were less affected than the residing memory T cells in the white pulp despite a clear decrease in CD127 and TCF7 expression within both subsets (Figures S2A–S2E). Similarly, the fraction of FOXO1-deficient P14 able to traffic to the pLNs revealed the most drastic impairment compared to WT-P14 (Figures 4I and 4J), but surprisingly, FOXO1 deletion had no impact on T cell maintenance within the IEL, as the WT/iFx1−/−-P14 ratio in TAM-treated mice was similar to the WT/iFx1f/f-P14 ratio in untreated mice (Figures 4I and 4J). This was specific to tissue-resident memory T cells (TRM) in the IEL, as the iFx1−/− in the lung, liver, and kidney displayed a similar impaired maintenance as seen in the blood or the spleen (Figure S2D). Interestingly, long-living TRM in the IEL display a lower turnover rate than circulating memory T cells (Figure S3), and thus, the impact of Foxo1 deletion on the maintenance of TRM in the IEL might be of lower significance.
To directly determine the role of FOXO1 in homeostatic proliferation of circulating memory T cells, we fluorescence-activated cell sorting (FACS)-purified KLRG1− WT- and iFx1−/−-P14 memory cells, labeled them with CellTrace violet (CTV), and co-transferred into naive sublethally irradiated hosts to promote lymphopenia-induced proliferation, a manifestation of homeostatic turnover (Figure 4K). 2 weeks post-transfer, we harvested mice and compared turnover of WT- and iFx1−/− T cells. Based on CTV dilution, FOXO1-deficient memory cells were still able to proliferate yet underwent fewer rounds of lymphopenia-induced proliferation than WT-P14 (Figures 4L and 4M).
Aside of actively sustaining a memory program, these results indicate that continuous FOXO1 expression is also essential for the longevity of a memory population by sustaining expression of mechanistically important pro-survival molecules and enabling homeostatic turnover. Thus, the impaired abundance of memory T cells in the lymph nodes and the white pulp of the spleen might impact the overall longevity of the anamnestic T cell response despite apparently unimpaired persistence of Foxo1 null IELs.
Foxo1-Deleted Memory Cells Display Impaired Self-Renewal
Another hallmark of T cell memory is the ability to rapidly re-expand following TCR re-stimulation. To determine the impact of Foxo1-deletion on the re-expansion potential, we FACS-purified the KLRG1− subsets of TAM-treated WT- and iFx1−/−-P14 memory cells and co-transferred equal numbers into naive congenic C57BL/6 hosts followed by infection with LCMV Arm (Figure 5A). Interestingly, the presence or absence of FOXO1 had no impact on the proliferation of the transferred memory populations, as both P14 subsets underwent similar re-expansion (Figures 5B–5D). However, by monitoring the T cell response over time in the blood, we slowly observed a difference between the two cell populations in favor of the WT population (Figures 5B and 5C). An impaired maintenance of iFx1−/− T cells was also visible in the spleen, yet to a lesser magnitude (Figure 5D). Despite the unimpaired initial expansion, the ability of T cells to re-differentiate into secondary memory T cells was completely abolished in the absence of FOXO1 as seen by the impaired re-expression of TCF7 and CD62L (Figure 5E). These observations are consistent with the differentiation and expansion kinetics of FOXO1 knockout CD8+ T cells (Hess Michelini et al., 2013; Kim et al., 2013), demonstrating that FOXO1 is essential for the generation or regeneration of (secondary) memory T cells.
Figure 5. T Cells Originating from FOXO1 Null Memory Cells Display Impaired Re-expansion Capacity.

(A and F) C57BL/6 mice grafted with similar numbers (2 × 104) of WT- and iFx1f/f-P14 were infected with LCMV Arm. 4 weeks post-infection, mice were treated with TAM. 5 days (A–E) or 100 days (F–I) post-TAM treatment, KLRG1− WT- and KLRG1− iFx1−/−-P14 splenocytes were individually FACS purified and co-transferred (A, 1.6–3.3 × 103 P14; F, 1.5–5.5 × 103 P14) into naive secondary C57BL/6 hosts followed by an LCMV Arm infection.
(B and G) Representative flow cytometry gated on P14 showing frequency of WT- (CD45.1+CD45.2+) and iFx1−/−-P14 (CD45.2+) transferred (injection control) and on day 7 (middle) and 70–77 (right) post-infection in the blood and calculated ratio for all mice (C and H).
(D and I) WT/iFx1−/− ratio in the spleen on days 6 and 80–84 compared to ratio of injection control (day 0).
(E) TCF7 and CD62L expression on WT- and iFx1−/−-P14 obtained from the spleen 84 days post-infection.
Symbols in (C) and (G) represent the mean of four mice. Symbols in (D) and (I) represent individual mice. Error bars in (C), (D), (H), and (I) indicate SD. Data are representative of at least two experiments with four mice per group. Statistical analysis was by paired Student’s t test. ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant.
Next, we were interested to determine whether, despite the impaired maintenance (Figure 4), functional memory would be sustained in the absence of FOXO1. To test this, we FACS-purified the KLRG1− subset of WT- or iFx1−/−-P14 100 days after TAM-treatment (day 130 post LCMV Arm) and co-transferred equal numbers of KLRG1− P14 into congenic C57BL/6 hosts before challenging with LCMV Arm (Figure 5F). In contrast to the transfer immediately after Foxo1 deletion (Figures 5B–5D), the initial re-expansion magnitude was strongly diminished in long-term surviving memory T cells (Figures 5G and 5H). 7 days post-infection, we detected 4-fold fewer iFx1−/−-P14 cells than WT-P14, and this difference increased over time (Figures 5G and 5H). The iFx1−/−-P14 population was also immediately diminished in the spleen (Figure 5I). These data suggest that in addition to the vital role of FOXO1 for the longevity of a memory T cell population, continuous FOXO1 expression is indispensable to sustain the functionality of a memory population. Memory T cells remaining after a prolonged loss of FOXO1 displayed a greatly reduced potential for re-expansion compared to acutely FOXO1-deleted memory T cells. This impaired re-expansion capacity can either originate from a general defect in turnover or from an impaired ability to traffic to secondary lymphoid organs wherein re-expansion can efficiently occur.
Continuous FOXO1 Expression Is Essential to Sustain the Proliferative T Cell Subset during Chronic Infection
The long-term immune response to chronic viral infections is sustained by a memory-like T cell subpopulation distinguished by the expression of TCF7 and CXCR5 that exhibits constant self-renewal and generates effector progeny that control viral replication (Utzschneider et al., 2016b; Leong et al., 2016; Im et al., 2016; He et al., 2016). Thus, we hypothesized that an impaired ability to renew would lead to an exaggerated viral expansion and a poor outcome following infection with a persistent virus. For this, we co-transferred iFx1f/f- and WT-P14 into congenic C57BL/6 hosts that were subsequently infected with the LCMV strain clone-13 that causes a persistent viral infection. Again, we treated mice 4 weeks post-infection with TAM and monitored the WT/iFx1−/−-P14 ratio. In contrast to a conventional memory T cell population in time-matched LCMV-Arm-infected mice, the chronically stimulated T cell population immediately declined in the absence of FOXO1 (Figures 6A and 6B). In line with this population decline, we observed a strong deterioration of the CXCR5+TCF7+ memory-like T cell population (Figure 6C) responsible for sustaining a long-term CD8+ response to chronic viral infection (Utzschneider et al., 2016b). This was associated with a reduced proportion of CD127-expressing iFx1−/−-P14 as well as KI67+iFx1−/−-P14 (Figures 6D and 6E). Paradoxically, the remaining iFx1−/−-P14 expressed lower amounts of the inhibitory receptor PD-1 (Figure 6F), a hallmark of the chronic infection phenotype, but were more dysfunctional based on a reduced ability to produce IFN-γ and TNF (Figures 6G and 6H). These data reveal that continuous FOXO1 expression is essential for sustaining CD8+ immunity to a chronic infection by maintaining the memory-like T cell subset, which in turn is responsible for upholding an effector response.
Figure 6. Sustained FOXO1 Expression Is Essential to Uphold Long-Term Effector Response.

C57BL/6 mice grafted with similar numbers (1.5 × 103) of WT- and iFx1f/f-P14 were infected with LCMV clone 13 (c13) or Armstrong (Arm). 4 weeks post-infection, mice were treated with TAM.
(A) Representative flow plots showing frequency of WT- (CD45.1+CD45.2+) and iFx1−/−-P14 populations (CD45.2+) among total CD8+ T cells before (day 26) and after (day 63) TAM treatment in the blood of c13-infected mice.
(B) WT/iFx1−/− ratio over time in the blood of c13-infected mice (left) compared to time-matched TAM-treated Arm-infected mice (right).
(C–H) 60–65 days post-c13 infection, mice were sacrificed and P14 splenocytes analyzed.
(C) Representative flow plots showing TCF7 and CXCR5 expression among WT- and iFx1−/−-P14 and frequency of CXCR5+TCF7+ P14 for all mice (right).
(D and E) Frequency of CD127+ (D) and KI67+ (E) P14.
(F) Expression levels of PD-1 of WT- (black) and iFx1−/−-P14 (red) compared to total host CD8+ T cell population (gray shadow) and mean fluorescence intensity (MFI) of P14 of all analyzed mice (right).
(G–I) Splenocytes were ex vivo re-stimulated with gp33 peptide, intracellularly stained for IFN-γ and TNF, and analyzed by flow cytometry. (G) Shown are representative flow cytometry plots of IFN-γ and TNF production of WT- (black, left) and iFx1−/−-P14 (red, right), the frequency of P14 that produce IFN-γ (H), and the fraction of IFN-γ+ P14 that co-produce TNF (I). Data are combined from three independent experiments with at least three mice per group.
Symbols in (B) represent the mean of 11 (c13) or 14 (Arm) mice. Symbols in (C)–(E), (H), and (I) represent individual mice; connection lines indicate populations from same host. Error bars in (B) and (F) indicate SD. Statistical analysis was by paired Student’s t test. ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant.
FOXO1 Expression Demarcates Human FLU- and CMV-Specific Memory T Cells
Similar to what we observed in mice, we were interested to determine whether the amount of FOXO1 expression is indicative of a memory phenotype in human T cells. Indeed, we noted that FOXO1 expression positively correlated with expression of CCR7, TCF7, and CD127, whereas it negatively correlated with expression of KLRG1 in CD8+ T cells of healthy donors (Figure 7A). Moreover, FOXO1 expression was increased in TCM (CCR7+CD45RA−) compared to TEM (CCR7−CD45RA−) or TEMRA (CCR7−CD45RA+) CD8+ T cells (Figure 7B). As such, we noted that FOXO1 expression was increased among FLU-specific CD8+ T cells compared to CMV-specific CD8+ T cells (Figures 7C and 7D). Influenza A infection is an acute infection that typically resolves, leaving classical TEM and TCM, whereas CMV establishes a latent infection that results in an anatomically specific low-level constant stimulation of “inflationary” CMV-specific CD8+ T cells. This has been found to drive their differentiation to a predominantly TEM and TEMRA phenotype (Figures 7E and 7F)(Appay et al., 2002). Finally, FOXO1 expression was, in combination with CCR7, TCF7, or CD127, effective as a marker in demarcating FLU-specific memory CD8+ T cells (Figure 7G).
Figure 7. FOXO1 Expression Demarcates Human Memory T Cells.

(A) Total CD8+ T cells from healthy patients were stained for expression of FOXO1 versus CCR7, TCF7, CD127, or KLRG1.
(B) FOXO1 expression among naive (CCR7+CD45RA+), central memory (CM; CCR7+CD45RA−), effector memory (EM; CCR7−CD45RA−) and EMRA (CCR7−CD45RA+) CD8+ T cells. Shown are representative histograms (left) of the individual cell subsets and mean fluorescent intensity (MFI) of all analyzed patient samples (right).
(C–F) Cytomegalovirus (CMV) pp65495- and influenza A (FLU) M158-specific CD8+ T cells were enriched and stained for FOXO1 expression (C and D). Fraction of naive, CM, EM, and EMRA among enriched CMV- (E) or FLU-tetramer-specific CD8+ T cells (F). Subsets are categorized based on expression of CCR7 and CD45RA.
(G) Expression of FOXO1 versus CCR7, TCF7, or CD127 among total FLU M158-specific CD8+ T cells. Symbols in (B) and (D)–(F) represent individual patients. Error bars in (B) and (D)–(F) indicate SD. Statistical analysis was performed by Friedman and Dunn’s multiple comparison test (B) and Mann-Whitney test (D). **p < 0.01; *p < 0.05.
DISCUSSION
The formation of memory T cells is fundamental to cellular-based immunity arising from a previous infection or vaccination. In addition, the very same cell subsets appear to contribute an essential aspect of the host-pathogen equilibrium that describes non-pathogenic persistence of chronic viral or bacterial infections. The origin, development, and phenotypic characteristics of such memory cells thus define an essential aspect of the physiological responses to many forms of infectious agents.
The hallmarks of memory T cells include (1) longevity, (2) self-renewal, and (3) an ability to cycle between quiescence and cell division (Gattinoni et al., 2017; Fearon et al., 2001), but how these attributes are conveyed and maintained in memory T cells is not entirely understood. One key component is the nuclear retention of FOXO1 and its direct and indirect effects on gene expression (Klebanoff et al., 2017; Hedrick et al., 2012; Lin et al., 2015, Verbist et al., 2016; Zhang et al., 2016). We and others previously presented experiments consistent with a need for FOXO1 in the initiation of memory T cells (Delpoux et al., 2018; Hess Michelini et al., 2013; Zhou et al., 2010), and here, we expand on these results in two ways. We sought to determine whether FOXO1 is needed for both initiation and maintenance of memory T cells and whether it is selectively involved in one or more memory phenotypic characteristics.
FOXO1 positively regulates several genes in naive and memory T cell survival and trafficking, including Il7ra, Sell (CD62L), Tcf7, Eomes, and Bcl2 (Hedrick et al., 2012). By acutely deleting Foxo1 specifically in antigen-inexperienced or in memory T cells, we were able to show that FOXO1 is not absolutely required for sustained expression of most of these molecules. For example, Foxo1 inactivation in T cells from unimmunized mice, either following positive selection driven by the distal promoter of the lymphocyte protein tyrosine kinase (Lck) (Delpoux et al., 2017) or acutely by TAM treatment, as we show here, had no impact on the expression of TCF7, whereas FOXO1 is crucial to sustain TCF7 expression in a memory population (Delpoux et al., 2018). We also noted an increased fraction of KLRG1-expressing virus-specific T cells following Foxo1 deletion, similar to previous studies (Hess Michelini et al., 2013; Delpoux et al., 2018); however, this shift was not observed for acutely Foxo1-deleted naive T cells. Of note, the role of KLRG1 has been shown to be dispensable for T cell differentiation (Gründemann et al., 2010) and rather serves as a subset defining marker. One possibility is that FOXO1 is differentially required for the expression of a number of different genes in antigen-inexperienced versus memory T cells; however, another more likely possibility is that most of the gene expression changes seen following the deletion of FOXO1 in memory cells occur indirectly as a result of its role in determining and maintaining the memory T cell differentiation state. As such, FOXO1 is absolutely essential for post-activation TCF7 expression and consequently influences EOMES expression (Delpoux et al., 2017, 2018; Hess Michelini et al., 2013). We now extend upon these previous observations by showing that these transcription factors are actively mediated by continuous FOXO1 expression. In the absence of FOXO1, KLRG1− memory T cells rapidly de-differentiated into KLRG1+CD62L− T cells, without the presence of cell division. In addition, FOXO1 is continuously required to maintain a constant number of memory cells by facilitating survival and turnover (longevity), and to maintain memory function throughout this process (cell renewal). Finally, FOXO1 is dispensable during antigen-mediated expansion (or re-expansion) and acquisition of effector function (Hess Michelini et al., 2013), but it must be present in order to generate quiescent cells capable of re-expansion (serial activation and quiescence). This might occur through de-differentiation from effector T cells after the resolution of an infection (Youngblood et al., 2017). This would be in line with the hypothesis of FOXO1 preventing the deposition of the H3K27me3 at that stage on certain pro-memory genes in CD8+ T cells and thus facilitating the expression and perhaps the maintenance of an extended memory signature (Gray et al., 2017). Further studies are necessary to precisely define the role(s) of FOXO1 in these early events. Nevertheless, we conclude that FOXO1 expression constitutes an ongoing contingency that affects a program of gene expression touching on all phenotypic aspects of memory T cells. Whether FOXO1 activity in uncommitted activated T cells is sufficient for a memory phenotype is unknown.
Interestingly, this continuous requirement for FOXO1 was dispensable for the maintenance of TRM in the intraepithelial layer. TRM are transcriptionally distinct from circulating memory T cells (Milner et al., 2017; Mackay et al., 2013), suggesting an alternate or less prominent role of FOXO1 for TRM maintenance. As such, TRM in the IEL do not get replenished by circulating memory T cells (Masopust et al., 2010), and they display a lower turnover rate than circulating memory T cells (Masopust et al., 2006), indicating that the per cell longevity (of TRM) is independent of FOXO1 expression.
Finally, FOXO1 appears to be essential for the maintenance of effective antigen-specific T cells in the presence of a persistent LCMV infection. The deletion of FOXO1 interrupted the persisting CD8+ T cell response, leading to a rapid decline in cell numbers. Notably, the remaining T cells expressed less PD-1 than WT T cells, possibly due to a putative FOXO1 binding site in the “C-region” upstream of Pdcd1 (PD-1) (Staron et al., 2014). These FOXO1 null T cells, which expressed less PD-1 than WT T cells, displayed a more dysfunctional cytokine profile, which is another hallmark of the chronic infection “exhausted” phenotype. This might seem controversial at first, yet PD-1 has been shown to preserve T cells from undergoing severe exhaustion (Odorizzi et al., 2015). Thus, the decline in PD-1 expression following acute Foxo1 deletion might remove the inhibitory signal responsible for sustaining T cell functionality during chronic infection. However, these phenotypic alterations are modest compared to those resulting from the ablation of proliferative CXCR5+TCF7+ T cells, the cells responsible for sustaining the overall effector T cell response (Speiser et al., 2014; Utzschneider et al., 2016b). This minor subpopulation contains the ability to differentiate into TCF7− effector T cells, which mediate viral control, as well as TCF7+ memory-like T cells that are capable of self-renewal. The maintenance of the proliferative capacity in chronic infections has been shown to be uncoupled from the phenotype of a T cell population (Utzschneider et al., 2016a). This presents the possibility that the same T cell program of gene expression important for immunity to an acute reinfection is continuously maintained and active during a chronic infection (Speiser et al., 2014). Furthermore, based solely on FOXO1 expression amounts, the suggestion is that this is true for human chronic virus infections as well. In line with a recent study (Klebanoff et al., 2017), these observations underpin FOXO1 as a clinically relevant target for future immunotherapeutic interventions.
EXPERIMENTAL PROCEDURES
Mice and Tamoxifen Treatment
C57BL/6 CD45.1+ mice were obtained from The Jackson Laboratory. CD45.1+CD45.2+ mice were bred in house. Foxo1f/f (Foxo1tm1Rdp) × Rosa26Cre-ERT2 (Gt(ROSA)26Sortm2(cre/ERT2)Brn) mice have been previously described (Kerdiles et al., 2009). Rosa26Cre-ERT2-negative P14 transgenic mice (Tg(TcrLCMV)327Sdz), which were either Foxo1+/+, Foxo1+/f, or Foxo1f/f, served as WT controls. Foxo1 deletion was induced by intraperitoneal injection of 2 mg TAM emulsified in 200 μl sunflower seed oil (both from Sigma-Aldrich) daily for 5 days, followed by 5 days of rest. Mice were maintained in a specific-pathogen-free vivarium. All experiments were carried out in 6-week-old or older mice in accordance to the Institutional Animal Care and Use Committee of the University of California, San Diego.
LCMV Infections
LCMV Arm and clone 13 were propagated and quantified as previously described (Utzschneider et al., 2013). Frozen stocks were diluted in PBS; 2 × 105 plaque-forming units (PFUs) LCMV Arm were injected intraperitoneally, and 2 × 106 PFUs LCMV clone 13 were injected intravenously.
Purification of Mouse T Cells and Adoptive T Cell Transfer
Single-cell splenocyte suspensions were obtained by mashing total spleens through a 70-μm nylon cell strainer (BD), and red blood cells were lysed with a hypotonic ammonium chloride-potassium bicarbonate (ACK) buffer. Lymph nodes were homogenized by teasing between frosted glass slides followed by filtration through nylon cell strainer. IEL cells were isolated as previously described (Lefrancois and Lycke, 2001). Liver was homogenized and cells recovered after Percoll separation. Lungs and kidneys were cut in small pieces and digested in 1 mg/mL collagenase D and 0.1 mg/mL DNase I (Roche) for 2 hr at 37°C. Tissues were then homogenized and passed through a nylon strainer followed by an ACK lysis.
Transgenic naive iFx1f/f- and WT-P14 were isolated using the mouse CD8+ T cell enrichment kit (Miltenyi Biotech), and 103–105 P14 were transferred into naive CD45.1+ C57BL/6 mice. P14 cells were re-isolated from infected mice by staining total splenocytes in 10% FBS RPMI media with anti-CD8α (53-6.7), CD45.1 (A20), CD45.2 (104), and KLRG1 (2F1; all from ThermoFisher).
Lymphopenia-Induced Proliferation and Proliferation Index
FACS-purified P14 were labeled with 5 μM CellTrace Violet (Invitrogen) for 10 min at 37°C and transferred into sublethally irradiated (6 Gy) mice. The proliferation index was calculated as defined by FlowJo (Tree Star).
BrdU Administration and Staining
Mice were fed with 0.8 mg/mL BrdU (Sigma-Aldrich) in drinking water starting 1 day after the final TAM treatment. Mice also received a 2-mg bolus intraperitoneal injection of BrdU (BD) on days 36 and 39 post-LCMV Arm. On day 42, splenocytes were stained with the BD BrdU staining kit.
Surface and Intracellular Antibody Staining of Mouse Cells
Surface staining was performed for 30 min at 4°C in PBS supplemented with 2% fetal calf serum (FCS) and 0.01% azide using (FACS buffer) the following antibodies: CD4 (GK1.5), CD8α (53-6.7), CD62L (MEL-14; all from BioLegend), CD8β (53-5.8), CD45.1 (A20), CD45.2 (104), CD44 (IM7), KLRG1 (2F1), CD127 (A7R34), CD43 (ebioR2/60), CD27 (LG.7F9), CXCR3 (CXCR3-173), PD-1 (J43), Lag-3 (ebioC9B7W; all ThermoFisher). CXCR5 staining was done as described (Shaw et al., 2016) using anti-CXCR5 (2G8; BD). Each cell-staining reaction was preceded by a 10-min incubation with a purified anti-mouse CD16/32 Ab (FcγRII/III block; 2.4G2). For intracellular cytokine staining, cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD) and stained with anti-IFN-γ (XMG1.2), TNF (MP6-XT22), IL-2 (JES6-5H4; all ThermoFisher), and CD107α (1D4B; BD). Intracellular transcription factor staining was performed with a transcription factor staining kit (eBioscience) and stained with TCF7 (C63D9), FOXO1 (C29H4; both Cell Signaling), T-BET (4B10; BioLegend), KI67 (B56; BD), BCL2 (10C4), and EOMES (Dan11mag; both from ThermoFisher).
For intravascular cell staining, 3 μg anti-CD8α antibody was intravenously injected 3 min before harvesting the mice.
For pSTAT5 staining, splenocytes were starved for 2–3 hr in 10% FBS RPMI media followed by extracellular surface staining. Cells were then washed and stimulated with IL-7 or IL-15 for 20 min at 37°C before being fixed with 10× vol pre-warmed Lyse/Fix Buffer (BD) for 15 min at 37°C. Cells were then permeabilized with pre-chilled Perm Buffer III (BD) for 30 min at 4°C, followed by two washing steps with FACS buffer, and stained for pSTAT5 (pY694; BD) for 60 min at room temperature.
Human Samples
Human blood was obtained from HLA-A*02-positive healthy donors visiting the outpatient clinic of the University Hospital Freiburg. In all cases, written informed consent was obtained. The study was conducted according to federal guidelines, local ethics committee regulations (Albert-Ludwigs-Univer-sität, Freiburg, Germany), and the Declaration of Helsinki (1975).
Peripheral blood mononuclear cells (PBMCs) were isolated using density gradient centrifugation (Pancoll; Pan-Biotech, Germany). Frozen PBMCs were thawed in complete medium (RPMI 1640 with 10% FBS, 1% penicillin-streptomycin, and 1.5% 1 M HEPES; all from Life Technologies) followed by peptide/HLA-A*02 tetramer-based enrichment (Alanio et al., 2010). Briefly, PBMCs were labeled with phycoerythrin (PE)-coupled peptide/HLA-A*02 tetramers and enriched with anti-PE microbeads applying MACS technology (Miltenyi Biotec, Germany). The following peptide/HLA-A*02 tetramers were used for enrichment: HLA-A*02/CMV pp65495 (NLVPMVATV) and HLA-A*02/FLU M158 (GILGFVFTL). Surface staining was performed on enriched samples for 15 min at room temperature in PBS supplemented with 1% FBS using the following antibodies: CD14 (61D3), CD19 (HIB19), KLRG1 (13F12F2, all ThermoFisher), CD127 (A019D5), CD45RA (HI100), CCR7 (G043H7, all from BioLegend) and CD8 (RPA-T8, BD). For live/dead discrimination, Fixable Viability Dye eFluor 780 (ThermoFisher) was used. Cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD), followed by fixation, nuclear permeabilization, and intracellular staining with Foxp3 transcription factor staining kit (ThermoFisher) according to the manufacturers’ protocols. The following intracellular antibodies were used: TCF7 (7F11A10), anti-rabbit immunoglobulin G (IgG) (Poly4064, both from BioLegend), and FOXO1 (C29H4, Cell Signaling). Cells were fixed with 2% paraformaldehyde (PFA) before sample acquisition.
Data Analyses
Flow cytometry measurements of cells were performed on a BD Fortessa or BD Fortessa X-20 and sort-purified on a BD FACSAria IIu. All data were analyzed using FlowJo 10 (Tree Star). Graphs were prepared with Prism 6 (GraphPad Software).
Supplementary Material
Highlights.
Continuous FOXO1 expression actively sustains T cell memory
Memory survival and turnover decline in the absence of FOXO1
FOXO1 sustains a TCF7+ memory-like subset found in chronic infections
FOXO1 functions as a marker for human memory CD8+ T cells
Acknowledgments
We thank members of the Goldrath lab for helpful discussions. D.T.U. was supported by Early Postdoc. Mobility fellowship P2LAP3_161942 from the Swiss National Science Foundation. M.H. was supported by grant TRR179-TP01 from the DFG. This work was supported by NIH grant R01AI103440 to S.M.H.
Footnotes
Supplemental Information includes three figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.020.
AUTHOR CONTRIBUTIONS
D.T.U. conceived the study, performed the experiments, analyzed data, and wrote the manuscript. A.D., X.H., and C.-Y.L. assisted in mouse experiments. D.W. designed and performed experiments and analyzed data from the human study. M.H. and R.T. designed the human study. S.M.H. conceived and supervised the study and wrote the manuscript. All authors reviewed the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Alanio C, Lemaitre F, Law HK, Hasan M, Albert ML. Enumeration of human antigen-specific naive CD8+ T cells reveals conserved precursor frequencies. Blood. 2010;115:3718–3725. doi: 10.1182/blood-2009-10-251124. [DOI] [PubMed] [Google Scholar]
- Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8:379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- Delpoux A, Lai CY, Hedrick SM, Doedens AL. FOXO1 opposition of CD8+T cell effector programming confers early memory properties and phenotypic diversity. Proc Natl Acad Sci USA. 2017;114:E8865–E8874. doi: 10.1073/pnas.1618916114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpoux A, Michelini RH, Verma S, Lai CY, Omilusik KD, Utzschneider DT, Redwood AJ, Goldrath AW, Benedict CA, Hedrick SM. Continuous activity of Foxo1 is required to prevent anergy and maintain the memory state of CD8+T cells. J Exp Med. 2018;215:575–594. doi: 10.1084/jem.20170697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon DT, Manders P, Wagner SD. Arrested differentiation, the self-renewing memory lymphocyte, and vaccination. Science. 2001;293:248–250. doi: 10.1126/science.1062589. [DOI] [PubMed] [Google Scholar]
- Galkina E, Thatte J, Dabak V, Williams MB, Ley K, Braciale TJ. Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. J Clin Invest. 2005;115:3473–3483. doi: 10.1172/JCI24482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. 2017;23:18–27. doi: 10.1038/nm.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SM, Amezquita RA, Guan T, Kleinstein SH, Kaech SM. Polycomb repressive complex 2-mediated chromatin repression guides effector CD8+T cell terminal differentiation and loss of multipotency. Immunity. 2017;46:596–608. doi: 10.1016/j.immuni.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründemann C, Schwartzkopff S, Koschella M, Schweier O, Peters C, Voehringer D, Pircher H. The NK receptor KLRG1 is dispensable for virus-induced NK and CD8+ T-cell differentiation and function in vivo. Eur J Immunol. 2010;40:1303–1314. doi: 10.1002/eji.200939771. [DOI] [PubMed] [Google Scholar]
- Hamilton SE, Jameson SC. CD8 T cell quiescence revisited. Trends Immunol. 2012;33:224–230. doi: 10.1016/j.it.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature. 2016;537:412–428. doi: 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol. 2012;12:649–661. doi: 10.1038/nri3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med. 2013;210:1189–1200. doi: 10.1084/jem.20130392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JK, Kagari T, Clingan JM, Matloubian M. Expression of chemokine receptor CXCR3 on T cells affects the balance between effector and memory CD8 T-cell generation. Proc Natl Acad Sci USA. 2011;108:E118–E127. doi: 10.1073/pnas.1101881108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–1244. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- Jung YW, Rutishauser RL, Joshi NS, Haberman AM, Kaech SM. Differential localization of effector and memory CD8 T cell subsets in lymphoid organs during acute viral infection. J Immunol. 2010;185:5315–5325. doi: 10.4049/jimmunol.1001948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, De-Pinho RA, Hedrick SM. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity. 2013;39:286–297. doi: 10.1016/j.immuni.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, Crompton JG, Leonardi AJ, Yamamoto TN, Chandran SS, Eil RL, Sukumar M, Vodnala SK, Hu J, Ji Y, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight. 2017;2:23. doi: 10.1172/jci.insight.95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz LO, Sánchez-Ramos C, Prieto-Arroyo I, Urbánek P, Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51–72. doi: 10.1016/j.redox.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrancois L, Lycke N. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer’s patch, and lamina propria cells. Curr Protoc Immunol. 2001;Chapter 3(Unit 3.19) doi: 10.1002/0471142735.im0319s17. [DOI] [PubMed] [Google Scholar]
- Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, Minnich M, Meckiff BJ, Wei Y, Hou Z, et al. CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol. 2016;17:1187–1196. doi: 10.1038/ni.3543. [DOI] [PubMed] [Google Scholar]
- Lin WH, Adams WC, Nish SA, Chen YH, Yen B, Rothman NJ, Kratchmarov R, Okada T, Klein U, Reiner SL. Asymmetric PI3K signaling driving developmental and regenerative cell fate bifurcation. Cell Rep. 2015;13:2203–2218. doi: 10.1016/j.celrep.2015.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- Masopust D, Vezys V, Marzo AL, Lefrançois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol. 2006;176:2079–2083. doi: 10.4049/jimmunol.176.4.2079. [DOI] [PubMed] [Google Scholar]
- Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. 2010;207:553–564. doi: 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, et al. Runx3 programs CD8+T cell residency in non-lymphoid tissues and tumours. Nature. 2017;552:253–257. doi: 10.1038/nature24993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013;31:137–161. doi: 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. 2015;212:1125–1137. doi: 10.1084/jem.20142237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Beckett O, Flavell RA, Li MO. An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity. 2009;30:358–371. doi: 10.1016/j.immuni.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity. 2012;36:374–387. doi: 10.1016/j.immuni.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LA, Bélanger S, Omilusik KD, Cho S, Scott-Browne JP, Nance JP, Goulding J, Lasorella A, Lu LF, Crotty S, Goldrath AW. Id2 reinforces TH1 differentiation and inhibits E2A to repress TFH differentiation. Nat Immunol. 2016;17:834–843. doi: 10.1038/ni.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser DE, Utzschneider DT, Oberle SG, Münz C, Romero P, Zehn D. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat Rev Immunol. 2014;14:768–774. doi: 10.1038/nri3740. [DOI] [PubMed] [Google Scholar]
- Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, Cui G, Li MO, Kaech SM. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41:802–814. doi: 10.1016/j.immuni.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, Kanagawa O. Monitoring cellular movement in vivo with photo-convertible fluorescence protein “Kaede” transgenic mice. Proc Natl Acad Sci USA. 2008;105:10871–10876. doi: 10.1073/pnas.0802278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Legat A, Fuertes Marraco SA, Carrié L, Luescher I, Speiser DE, Zehn D. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol. 2013;14:603–610. doi: 10.1038/ni.2606. [DOI] [PubMed] [Google Scholar]
- Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, Delorenzi M, Pinschewer DD, Zehn D. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J Exp Med. 2016a;213:1819–1834. doi: 10.1084/jem.20150598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity. 2016b;45:415–427. doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- Verbist KC, Guy CS, Milasta S, Liedmann S, Kamiński MM, Wang R, Green DR. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature. 2016;532:389–393. doi: 10.1038/nature17442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JT, Cross EW, Kedl RM. Antigen-inexperienced memory CD8+T cells: where they come from and why we need them. Nat Rev Immunol. 2017;17:391–400. doi: 10.1038/nri.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, Araki K, West EE, Ghoneim HE, Fan Y, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature. 2017;552:404–409. doi: 10.1038/nature25144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Tschumi BO, Lopez-Mejia IC, Oberle SG, Meyer M, Samson G, Rüegg MA, Hall MN, Fajas L, Zehn D, et al. Mammalian target of rapamycin complex 2 controls CD8 T cell memory differentiation in a Foxo1-dependent manner. Cell Rep. 2016;14:1206–1217. doi: 10.1016/j.celrep.2015.12.095. [DOI] [PubMed] [Google Scholar]
- Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33:229–240. doi: 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.