

Abstract
TERT promoter mutations (TPMs) are the most common non-coding mutations in cancer. The timing and consequences of TPMs have not been fully established. Here we show that TPMs acquired at the transition from benign nevus to malignant melanoma do not support telomere maintenance. In vitro experiments revealed that TPMs do not prevent telomere attrition, resulting in cells with critically short and unprotected telomeres. Immortalization by TPMs requires a gradual upregulation of telomerase, coinciding with telomere fusions. These data suggest that TPMs contribute to tumorigenesis by promoting immortalization and genomic instability in two phases. In an initial phase, TPMs do not prevent bulk telomere shortening but extend cellular life-span by healing the shortest telomeres. In the second phase, the critically short telomeres lead to genome instability and telomerase is further upregulated to sustain cell proliferation.
Mutations in the promoter region of the gene coding for the reverse transcriptase component of telomerase (TERT) (1, 2) are found at high frequency in various human tumor types (3–6). These TERT promoter mutations (TPMs) have been detected both in early- and late-stage tumors, consistent with the possibility that they play a role early in tumorigenesis (3–7). However, tumor cells with TPMs have short telomeres despite exhibiting increased telomerase expression (8–10), an observation arguing that the critical functional effects of TPMs may occur at a later time point after telomeres have become critically short. Critically short telomeres can trigger a DNA damage response and replicative senescence, which can be bypassed by the loss of DNA damage checkpoints. This allows cells to continue proliferating until they enter a state called crisis, where telomeres become dysfunctional leading to chromosome end-to-end fusions and cell death. Cancer cells can emerge from crisis by reactivating telomerase (11). Through the genomic instability that occurs during crisis, short telomeres can drive cancer (11–14). Nevertheless, individuals with constitutionally shorter telomeres have a decreased cancer risk (15–17). The current model of telomerase reactivation during crisis fails to explain these contradictory observations. Moreover, it is not understood why tumors with TPMs are associated with a poor prognosis (8, 18).
To determine the role of TPMs in telomere maintenance during tumor formation, we followed the intratumoral changes in telomere length in tumors with TPMs. TPMs occur with high frequency (60–85%) in cutaneous melanoma (3, 4, 19). We took advantage of a subset of human melanomas, which arose from adjacent pre-neoplastic lesions such as nevi (7). This allowed us to compare genetic alterations and telomere length in four melanomas that acquired TPMs as they arose from nevi (Fig. 1 and Fig. S1). Using structured illumination microscopy, we acquired large, high-resolution telomere and centromere fluorescence in situ hybridization images of melanoma and nevus in the same tissue section and quantitatively analyzed telomere length (Fig. 1A, 1B and Fig. S1). Multiple iterations of random sampling of normalized telomeric signals showed that in all four cases telomeres were significantly shorter in the melanoma than in the nevus (reduced normalized signal intensity in melanoma compared to the nevus, Fig. 1C and 1D, Fig. S1D). These data highlight that in vivo, TPMs do not induce TERT expression levels sufficiently to counteract or reverse telomere shortening.
Fig. 1. Telomeres shorten during progression from nevus to melanoma despite acquisition of a TPM.
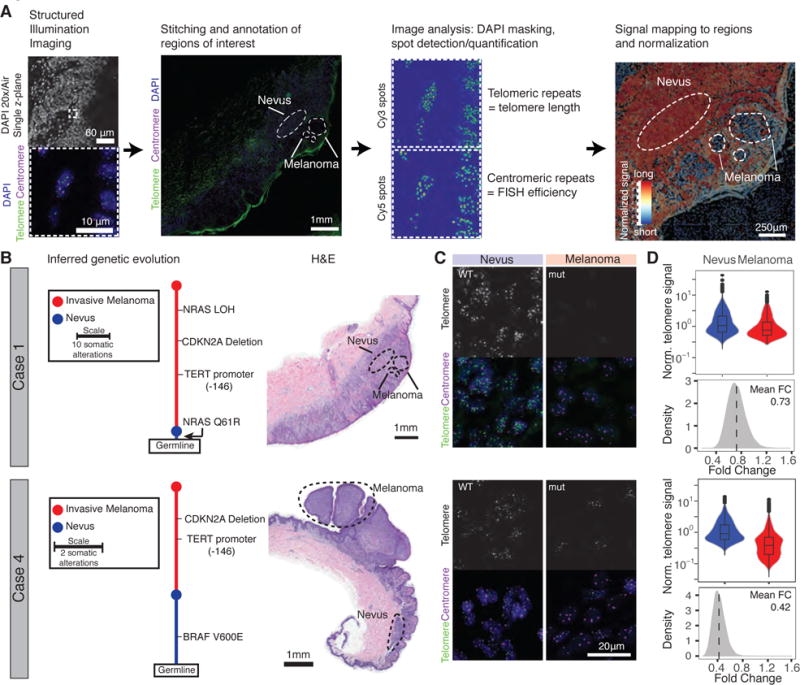
(A) Schematic overview of workflow. Melanoma progression samples from four patients were imaged using structured illumination microscopy. Acquired images were stitched and regions of interest were annotated. Telomeric and centromeric repeat FISH signals were detected and mapped back on pre-defined regions of interest. The relative telomere length was calculated by normalization to the closest centromeric signals. (B) The genetic evolution of each progression case, rooted at the germline shows how each case evolves from a wild-type nevus to a melanoma with TPM (nevus in blue, melanoma in red). Adjacent is the H&E staining of the analyzed sections of each case (Scale bar 1mm). (C) Magnification of the FISH image resolving individual telomere and centromere spots in nevus/melanoma area (DAPI = blue, telomere = green, centromere = magenta). TPM status is noted as wild-type (WT) or mutant (mut) (Scale bar 20 μm). (D) Violin plots of normalized telomere signal (telomere/centromere) with an inset boxplot indicating the median and quartiles of the signal. Histograms show the fold change of telomere spot intensity after random sampling (fold change melanoma/nevus).
To explore the long-term effects of TPMs on telomere length and genomic stability in a genetically defined setting, we engineered the three most frequent TPMs de novo into human embryonic stem cells (hESCs) using CAS9 mediated genome editing (20) (Fig. 2A and Fig. S2A–D). We started the “experimental clock” by differentiating the parental cell lines into fibroblasts, which are normally telomerase-negative. We assayed the proliferative capacity of hESC-derived fibroblasts with and without a single TPM using three distinct conditions: (i) fibroblasts transduced with Simian virus 40 large T antigen (SV40 TAg, inactivating pRb and p53 signaling) (Fig. 2B, S2B), (ii) fibroblasts with intact cell cycle and DNA damage checkpoints (Fig. 2C), and (iii) fibroblasts with inactivated cell cycle and DNA damage checkpoints, achieved by deletion of p14/p16 function (CDKN2AΔ/Δ) (Fig. 2D, Fig. S2C). In all cell lines the TPMs extended the proliferative capacity significantly past the proliferative barrier of wild-type cells. Cells with TPMs proliferated without signs of crisis or a strong decrease in doubling rate at the time when wild-type cells arrested in a telomere length dependent manner (21) (Fig. 2B–D, Fig. S2E and S2F). Overall, these data demonstrate that TPMs promote the immortalization of bulk cell populations.
Fig. 2. TPMs support cellular immortalization in vitro but do not prevent telomere shortening.
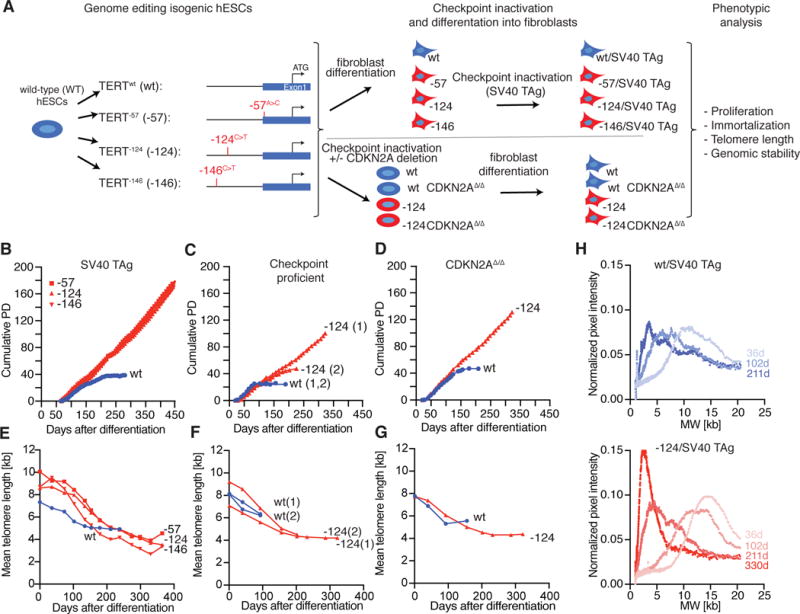
(A) Experimental overview: Isogenic hESCs with the TPMs were differentiated into fibroblasts. To inactivate cell cycle and DNA damage checkpoints, either CDKN2A function was deleted in hESCs prior differentiation or fibroblasts were infected with SV40 TAg. (B-D) Growth curves of cumulative PDs over days after differentiation. (B) SV40 TAg fibroblasts with (red, -57, -124, -146) or without (blue, wt) TPMs (C) DNA damage checkpoint proficient cells with (red, -124) or without (blue, wt) TPM. (1), (2) indicate two independent experiments. (D) CDKN2AΔ/Δ cells with (red, -124) or without (blue, wt) TPM (E-G) Quantification of mean telomere length over time after differentiation. (H) Accumulation of shorter telomeres over time shown by visualization of telomere length distribution of images shown in Fig. S3A. Quantification of the normalized pixel intensity over molecular weight per lane for the indicated time points after differentiation.
Telomere maintenance is essential for cancer cell immortalization (22). To investigate whether TPMs prevented telomere shortening, we measured telomere length over time in the fibroblast model system (Fig. 2E–G and Fig. S3A and S3B). In wild-type cells, telomeres shortened progressively. Surprisingly, telomeres in cells with TPMs initially also shortened and stabilized only after approximately 70 population doublings (PD) at a length of 2.5–4.5 kb. The stabilized mean telomere length in the TPM cells was shorter than telomere length in wild-type control cells at the time of crisis (Fig. 2E–G). The analysis of the telomere length distribution of cells with TPMs revealed that the mean length shifted from long to short telomeres and then stabilized (Fig. 2H and Fig. S3C), suggesting that short telomeres were preferentially maintained, while long telomeres continued to shorten.
Unprotected short telomeres can lead to genomic instability through telomere fusion events, which have been detected in a variety of tumor samples and cell lines (11–13, 23–25). To assess whether the chromosome ends are protected in cells with TPMs, we used a PCR-based method to quantify telomere-telomere fusion events over the course of a year after cell differentiation (23). We detected fusions in SV40 TAg cells with the -124 and -146 TPMs when telomere length stabilized (Fig. 3A and 3B and Fig. S4A and S4B). These fusion events were suppressed by overexpression of TERT (Fig. S4C), showing that they were driven by telomere attrition. Cells with the -57 TPM showed fewer fusions, presumably due to their slightly longer telomeres (Fig. 2E). Thus, TPMs not only promote cancer cell immortalization but might also drive cancer cell evolution via dysregulation of telomere protection.
Fig. 3. TPMs do not fully protect against genomic instability and mutant cells gradually increase telomerase expression.
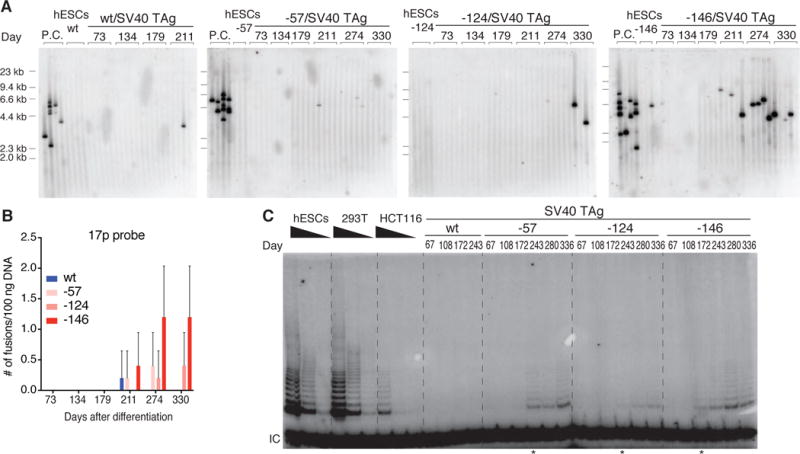
(A) Detection of interchromosomal telomere fusions over time in SV40 TAg fibroblasts by PCR of specific subtelomeric regions. Fusions were detected by probes against the 17p subtelomeric regions (23). DNA from cells in crisis served as a positive control (P.C.: TERT, p53 and p16 triple knockout fibroblasts cultured into crisis). (B) Quantification of fusion events shown in Fig. 3A. (C) Telomerase activity assay of the indicated cell lines over time. IC: internal control. * indicates time points when telomerase activity became detectable.
To determine the effect of TPMs on telomerase levels, we assayed enzymatic activity and TERT mRNA expression over time (Fig. 3C, Fig. S4D and S4E). Shortly after differentiation, telomerase activity and TERT mRNA expression levels were higher in cells with TPMs than in wild-type cells, consistent with previous reports (20). However, telomerase levels rapidly fell below the detection limit of the assay in both cell types (Fig. 3C and Fig. S4D). Nevertheless, cells with the TPM proliferated at a similar rate as cells overexpressing TERT (Fig. S5A–C), demonstrating that TERT expression levels were not limiting for proliferation when telomeres were long. To examine whether TERT was fully transcriptionally silenced, we overexpressed POT1ΔOB, an allele sensitizing cells to low telomerase levels (26). The expression of the allele induced significant telomere elongation specifically in cells with TPMs (Fig. S5D–G). Therefore telomerase was not fully transcriptionally silenced but TPMs initially lead to telomerase activity below the detection threshold. At the time when telomere length in TPM cells had stabilized, telomerase expression became detectable again and activity rose as high as 5–10% of that found in hESCs and established cancer cell lines (Fig. 3C and Fig. S6). To exclude the possibility that this upregulation of telomerase was the result of an outgrowth of a small subpopulation of cells with preexisting high telomerase activity, we performed sub-cloning experiments. Individual cell clones recapitulated the behavior of the bulk population (Fig. S7). Like the bulk cell population, individual clones also first had low telomerase levels, which became elevated with increasing PDs. This confirmed that TERT upregulation was the consequence of an additional event occurring when telomeres became critically short.
At the time point of the acquisition of a TPM telomeres did not immediately stop shortening. The time lag until telomerase activity increased sufficiently to maintain all telomeres could provide a rationale for the cancer-protective function of constitutionally short telomeres, and explain how the cellular context influences the immortalization process. We hypothesize that: (i) telomere length at the time when a cell acquires a TPM determines the number of cell divisions a cell can undergo before telomeres become critically short and (ii) once telomeres have become critically short the ability to upregulate telomerase determines whether a cell with a TPM becomes immortalized or ceases to proliferate. To test these hypotheses, we generated hESCs with shorter telomeres by modulating TERT expression. We attenuated the effect of TPMs by mutating the three endogenous ETS-factor binding sites in the TERT promoter (E3), which facilitate binding of the GABPα - as a heterodimer with GABPβ w(27, 28) (Fig. 4A), and which we show to be essential in hESCs (Fig. S8A and S8B). hESCs with a TPM and the E3 mutation (E3-124 and E3-146) had lower TERT expression and stably maintained shorter telomeres in comparison to hESCs with only a TPM (-124 and -146) (Fig. 4B, 4C and S8C–F). From this allelic series we generated SV40 Tag-transduced fibroblasts and monitored proliferation, telomere length, and frequency of telomere fusions (Fig. 4B, and 4D–E). Fibroblasts without the TPM, but with the three ETS mutations (wt-E3), had the shortest telomeres and died shortly after differentiation. In accordance with our first hypothesis, -124-E3 and -146-E3 cells with constitutionally shorter telomeres showed a high frequency of fusion events, much earlier than -124/-146 cells (Fig. 4D and S8G). Both -124-E3 and -146-E3 cells initially proliferated despite their short telomeres (Fig. 4E), arguing that they retained low levels of telomerase activity. Although they harbored TPMs, both -124-E3 and -146-E3 cells upregulated telomerase to only low levels; these cells entered crisis and did not become immortalized, confirming our second hypothesis.
Fig. 4. Short telomeres and low telomerase levels protect cells from immortalization by TPMs.
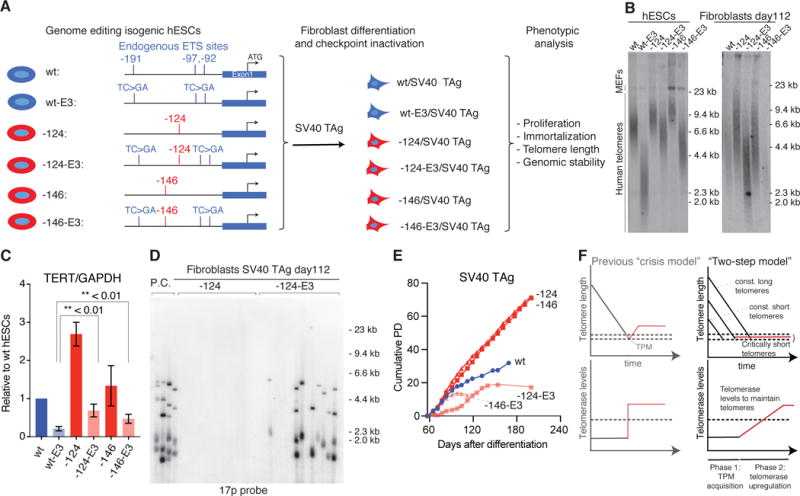
(A) Schematic overview of allelic series of cells carrying TPMs (red) and/or endogenous ETS site mutations (E3: mutated TC>GA, blue) in the TERT promoter. E3 refers to the simultaneous mutations of all three endogenous ETS sites (-191, -97 and -92 from the translational start site: ATG). (B) Telomere length analysis for hESCs and fibroblasts with TPMs and ETS mutation combinations. (C) TERT expression normalized to GAPDH relative to wt hESC. Error bars: SEM, two-sided student t-test, n = 3. (D) Telomere fusions detected in -124 and -124 E3 fibroblasts with 17p. (E) Growth curve of fibroblasts with or without TPMs and/or ETS mutations. (F) Telomere length (top) and TERT expression (bottom) in a revised model for immortalization by TPMs. In the classic crisis model, TERT expression is acquired during crisis as a single event (left panel). In the biphasic model (right panel), TPMs can be acquired in a telomere length independent manner at any time (Phase 1). In phase 2 cells are required to gradually upregulate telomerase. Due to unprotected telomeres cells can acquire genomic instability.
In summary, we propose that TPMs drive cell immortalization in two phases (Fig. 4F). TPMs that arise in early phases of tumorigenesis result in telomerase activity that is insufficient to prevent bulk telomere shortening, preferentially acting on critically short telomeres and thereby delaying replicative senescence (Phase 1). During subsequent divisions, the number of critically short telomeres increases, and telomerase activity becomes rate-limiting, causing telomere-driven genomic instability and selective pressure for telomerase upregulation by yet to be characterized factors (Phase 2). In contrast to crisis, where genomic instability arises as an acute and pronounced phenomenon, the genomic instability in cells with TPMs is reduced and persists over a longer period. Thus, TPMs play a dual role in tumorigenesis by enabling not only cancer cell immortalization via upregulation of TERT but also by promoting genome instability.
The finding that TPMs can become selected at the transition from pre-neoplastic to cancerous lesions indicates that telomere shortening acts as a critical barrier early during the neoplastic transformation of some cancers (7). Together with our findings that TPMs are necessary but not sufficient to maintain telomere length this might explain why individuals with constitutionally short telomeres have a decreased rate of melanoma incidence (15–17). Variation in constitutional telomere length likely limits the numeric expansion of early neoplastic cells at a precancerous stage. By characterizing the TPM-driven immortalization process over extended periods of time, we were able to reconcile the seemingly contradictory observations that short telomeres can initially protect against cancer formation (29, 30), yet have a cancer promoting effect and can be associated with a poor prognosis (8, 18) by later fueling genomic instability (11, 12, 24).
Supplementary Material
Acknowledgments
We would like to thank G. Steven Martin, Robert Tjian and the members of the Hockemeyer lab for critical comments on the manuscript. K.C. is supported by a Nakajima foundation fellowship. D.H. is a Pew-Stewart Scholar for Cancer Research supported by the Pew Charitable Trusts and the Alexander and Margaret Stewart Trust. D.H. supported by the Siebel Stem Cell Institute and NIH R01-CA196884. A.H.S is supported by NIH 5T32 CA177555. B.C.B is supported by R01 - CA142873. X.D. is supported by the California Institute for Regenerative Medicine grant LA1-08013. StellarVision is an imaging instrument developed by Optical Biosystems, and A.O. and J.R. are full time employees by this company. MATLAB code used for image analysis is available under https://gitlab.com/tjian-darzacq-lab. Patient sequencing data is available within the Short Read Archive with the accession number SRP113411.
References
- 1.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 2.Greider CW, Blackburn EH. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell. 1987;51:887–898. doi: 10.1016/0092-8674(87)90576-9. [DOI] [PubMed] [Google Scholar]
- 3.Horn S, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 4.Huang FW, et al. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jafri MA, Ansari SA, Alqahtani MH, Shay JW. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016;8:69. doi: 10.1186/s13073-016-0324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Killela PJ, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shain AH, et al. The Genetic Evolution of Melanoma from Precursor Lesions. N Engl J Med. 2015;373:1926–1936. doi: 10.1056/NEJMoa1502583. [DOI] [PubMed] [Google Scholar]
- 8.Nagore E, et al. TERT promoter mutations in melanoma survival. Int J Cancer. 2016;139:75–84. doi: 10.1002/ijc.30042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ceccarelli M, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016;164:550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayward NK, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545:175–180. doi: 10.1038/nature22071. [DOI] [PubMed] [Google Scholar]
- 11.Maciejowski J, de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol. 2017;18:175–186. doi: 10.1038/nrm.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Artandi SE, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 13.Lin TT, et al. Telomere dysfunction accurately predicts clinical outcome in chronic lymphocytic leukaemia, even in patients with early stage disease. Br J Haematol. 2014;167:214–223. doi: 10.1111/bjh.13023. [DOI] [PubMed] [Google Scholar]
- 14.Stanley SE, Armanios M. The short and long telomere syndromes: paired paradigms for molecular medicine. Curr Opin Genet Dev. 2015;33:1–9. doi: 10.1016/j.gde.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han J, et al. A prospective study of telomere length and the risk of skin cancer. J Invest Dermatol. 2009;129:415–421. doi: 10.1038/jid.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iles MM, et al. The effect on melanoma risk of genes previously associated with telomere length. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nan H, et al. Shorter telomeres associate with a reduced risk of melanoma development. Cancer Res. 2011;71:6758–6763. doi: 10.1158/0008-5472.CAN-11-1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borah S, et al. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science. 2015;347:1006–1010. doi: 10.1126/science.1260200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.N. Cancer Genome Atlas. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiba K, et al. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife. 2015;4 doi: 10.7554/eLife.07918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bodnar AG, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 22.Kim NW, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 23.Capper R, et al. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007;21:2495–2508. doi: 10.1101/gad.439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feldser DM, Hackett JA, Greider CW. Telomere dysfunction and the initiation of genome instability. Nat Rev Cancer. 2003;3:623–627. doi: 10.1038/nrc1142. [DOI] [PubMed] [Google Scholar]
- 25.Plentz RR, et al. Hepatocellular telomere shortening correlates with chromosomal instability and the development of human hepatoma. Hepatology. 2004;40:80–86. doi: 10.1002/hep.20271. [DOI] [PubMed] [Google Scholar]
- 26.Loayza D, De Lange T. POT1 as a terminal transducer of TRF1 telomere length control. Nature. 2003;423:1013–1018. doi: 10.1038/nature01688. [DOI] [PubMed] [Google Scholar]
- 27.Bell RJ, et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. 2015;348:1036–1039. doi: 10.1126/science.aab0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Min J, Shay JW. TERT Promoter Mutations Enhance Telomerase Activation by Long-Range Chromatin Interactions. Cancer Discov. 2016;6:1212–1214. doi: 10.1158/2159-8290.CD-16-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bojesen SE, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–384. 384e371–372. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rode L, Nordestgaard BG, Bojesen SE. Long telomeres and cancer risk among 95 568 individuals from the general population. Int J Epidemiol. 2016;45:1634–1643. doi: 10.1093/ije/dyw179. [DOI] [PubMed] [Google Scholar]
- 31.Lengner CJ, et al. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell. 2010;141:872–883. doi: 10.1016/j.cell.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 32.Soldner F, et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hockemeyer D, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hockemeyer D, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiba K, et al. Endogenous Telomerase Reverse Transcriptase N-Terminal Tagging Affects Human Telomerase Function at Telomeres In Vivo. Mol Cell Biol. 2017;37 doi: 10.1128/MCB.00541-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hockemeyer D, Sfeir AJ, Shay JW, Wright WE, de Lange T. POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 2005;24:2667–2678. doi: 10.1038/sj.emboj.7600733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kimura M, et al. Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nat Protoc. 2010;5:1596–1607. doi: 10.1038/nprot.2010.124. [DOI] [PubMed] [Google Scholar]
- 38.Takai H, et al. A POT1 mutation implicates defective telomere end fill-in and telomere truncations in Coats plus. Genes Dev. 2016;30:812–826. doi: 10.1101/gad.276873.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serge A, Bertaux N, Rigneault H, Marguet D. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat Methods. 2008;5:687–694. doi: 10.1038/nmeth.1233. [DOI] [PubMed] [Google Scholar]
- 40.Ryu J, Hong SS, Horn BKP, Freeman DM, Mermelstein MS. Multibeam interferometric illumination as the primary source of resolution in optical microscopy. Appl Phys Lett. 2006;88 [Google Scholar]
- 41.Baumann P, Cech TR. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–1175. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.