

Abstract
Chronological age represents the single greatest risk factor for human disease. One plausible explanation for this correlation is that mechanisms that drive ageing might also promote age-related diseases. Cellular senescence, which is a permanent state of cell cycle arrest induced by cellular stress, has recently emerged as a fundamental ageing mechanism that also contributes to diseases of late life, including cancer, atherosclerosis and osteoarthritis. Therapeutic strategies that safely interfere with the detrimental effects of cellular senescence, such as the selective elimination of senescent cells (SNCs) or the disruption of the SNC secretome, are gaining significant attention, with several programmes now nearing human clinical studies.
In his 1881 essay ‘The Duration of Life’, evolutionary biologist A. Weismann made an assertion considered radical in his day. “Death takes place because a worn-out tissue cannot forever renew itself, and because a capacity for increase by means of cell division is not ever-lasting but finite” (REF. 1). Weismann’s far-reaching idea — that an inherent limit to cell division contributed to ageing — lay dormant for more than 80 years until L. Hayflick’s work in 1961 demonstrated that mammalian cells do indeed have a finite capacity for cell division, which is a concept now referred to as the ‘Hayflick Limit’ (REF. 2) (FIG. 1).
Figure 1. Timeline of milestones relevant to senotherapy.
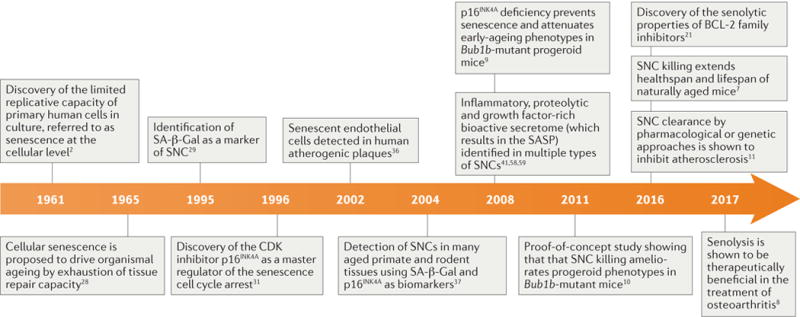
Selected events related to the developing field of senotherapy are highlighted. CDK, cyclin-dependent kinase; SA-β-Gal, senescence-associated β-galactosidase; SASP, senescence-associated secretory phenotype; SNCs, senescent cells.
Weismann and Hayflick both argued that tissues age because non-dividing cells within that tissue lose the ability to participate in repair. What could not be appreciated at the time was that a class of non- dividing cells induced by cellular stress, now referred to as ‘senescent cells’ (SNCs), are also harmful for an entirely different reason. In addition to not being able to contribute to tissue repair through proliferation, SNCs disrupt normal tissue function by secreting factors that recruit inflammatory cells, remodel the extracellular matrix, trigger unwanted cell death, induce fibrosis, and inhibit stem cell function3–6. SNCs that accumulate over time actively damage the tissues in which they reside and can be directly linked to features of natural ageing7.
Work over the past decade in animals has demonstrated that the selective elimination of SNCs (also known as ‘ senolysis’) lengthens ‘healthspan’, the period of time an organism lives free of chronic diseases of ageing. Senolysis in mouse models of disease has been shown to ameliorate atherosclerosis, osteoarthritis, cataracts, tumorigenesis, cardiac hypertrophy, renal dysfunction, lipodystrophy, and sarcopenia7–11. In addition to extending healthspan, senolysis also increases median mouse lifespan7 (FIG. 1).
Interfering with the pro-ageing effects of SNCs, either by eliminating SNCs entirely or by shutting down the SNC secretory machinery, is now being considered as a potential strategy to treat diseases of ageing. However, to make such therapies a reality, a variety of technical challenges must be overcome. First, cellular senescence is not uniformly negative and supports a variety of beneficial functions, including tumour suppression, wound healing, embryonic development12–16, tissue regeneration17 and the promotion of insulin secretion by pancreatic β-cells during ageing18 (BOX 1). Care must therefore be taken as to when, where and how SNCs are targeted. Second, although a variety of molecules have been identified that eliminate SNCs in vivo, some of these compounds have undesirable off-target effects, such as thrombocytopenia19. Third, SNCs of different tissue origins have different vulnerabilities, which would require the design of unique molecules that not only target the appropriate cell type in vivo but also accumulate in the tissue in which these SNCs reside. Last, molecules that blunt the SNC secretome would need to be taken continuously, as SNCs would continue to persist after treatment. The long-term use of such molecules could present safety challenges because these molecules have been shown to be immunosuppressive20.
Box 1. Beneficial functions of SNCs.
The senescent cell (SNC) fate has been linked to several beneficial processes, including wound resolution15,163, embryogenesis12,14 and cancer prevention87. Permanent withdrawal from the cell cycle in response to potentially transforming insults is tumour- suppressive, as shown by high cancer rates in mice lacking the cyclin-dependent kinase inhibitors p16INK4A or p19ARF (REFS 85,161,164). Indeed, p16INK4A depletion or epigenetic silencing is a common event in human malignancy165–167. It seems that the senescence cell cycle arrest also limits other proliferative responses to injury, such as fibrosis108,163, and perhaps macrophage proliferation in atherosclerosis168,169.
The current working model is that these beneficial SNCs — termed acute SNCs5 — serve a temporospatially restricted signalling role through the senescence-associated secretory phenotype (SASP) and are subject to prompt immune clearance. These signals may act on parenchymal cells, progenitor cells or the immune system, and can play a modulatory role in tissue repair and regeneration. SNCs arising in wound healing, for example, release platelet-derived growth factor AA (PDGF-AA) as part of the SASP to support wound closure15. Similarly, SNC-derived interleukin-6 (IL-6) promotes in vivo skeletal muscle repair following injury170. Keratinocyte stemness can also be induced by SNCs, leading to enhanced skin engraftment71. SASP factors arising from SNCs in neoplasms may initially enhance antitumour immunity in addition to enforcing a paracrine senescence on neighbouring transformed cells64.
However, the biological significance of SNC-induced cancer surveillance is currently unclear, as long-term removal of SNCs — using the SNC elimination system in the INK-ATTAC (INK-linked apoptosis through targeted activation of caspase) mouse model7 — leads to a delay, rather than an acceleration, of the onset of cancer in mice. Perhaps as the immune system ages, the balance between SNC-mediated tumour-suppression and SNC-mediated tumour-promotion gradually shifts towards cancer.
Although not known for certain, it is possible that acute SNCs may convert into persistent chronic SNCs5 following immune system deterioration in ageing and that this type of chronic SNC could be deleterious through inappropriate expression of a signalling programme intended to be short-term. Presumably, these chronic SNCs are being removed to beneficially affect healthspan and lifespan in mice7. By contrast, some types of chronic SNCs may be beneficial. SNCs, in general, resist apoptosis75. In cell types whose depletion over time is dangerous, such as pancreatic β-cells, senescence may salvage organ functionality through cell death resistance. Indeed, pancreatic β-cells that are driven into senescence through p16INK4A overexpression or ageing secrete more insulin than their non-senescent counterparts18. Senescence as a cell salvage pathway in other secretory organs remains an intriguing, underexplored possibility.
It is a sufficiently attractive concept to blunt the deleterious effects of SNCs with a drug, and numerous groups are now attempting to develop senotherapies, that is, drugs that safely interfere with the pro-ageing effects of SNCs. The first molecular entities with senolytic potential have been reported8,21–23, and the properties of one class of these agents, B cell lymphoma 2 (BCL-2) family inhibitors, have been confirmed by multiple independent laboratories11,21,24. One of these molecules, navitoclax (also known as ABT-263), has been shown to exert a therapeutic effect in models of atherosclerosis through selective elimination of SNCs11 (FIG. 1). However, senolytic molecules in this class have side effects, which makes their clinical development for diseases of ageing challenging (FIG. 1). A second senolytic molecule, the proprietary agent UBX0101, has been shown to clear SNCs in a mouse model of osteoarthritis, which significantly attenuates both disease progression and pain8 (FIG. 1).
In this Review, we assess both the progress made to date towards human senotherapy and the challenges faced by investigators working in this field. It is our hope that this conceptual framework will help guide this nascent area of drug discovery towards clinical success.
Discovery and detection of SNCs
In 1961, Hayflick and Moorehead demonstrated that normal human cells possess limited replicative potential when grown in cell culture2 (FIG. 1). After approximately 50 divisions, most cells in culture have, at some point, lost the ability to divide but remain alive and metabolically active25–27. This finite proliferative capacity, now called replicative senescence, was thought to underlie the decline in cell replacement and tissue repair that accompanies ageing28. This idea was difficult to prove at the time, as there were no methods available to detect or manipulate SNCs in vivo.
The first tool to identify SNCs in vivo came from the discovery of a SNC-enriched enzymatic activity: SNCs display β-galactosidase enzymatic activity at pH 6, whereas more common β-galactosidase isoforms show peak enzymatic activity at pH 4–4.5 (REF. 29). This β-galactosidase activity is now referred to as senescence-associated β- galactosidase (SA-β-Gal), and this insight enabled the development of a simple colorimetric assay now in widespread use for the labelling and detection of SNCs (FIG. 1). A second key tool was the identification of p16INK4A, which is a cyclin-dependent kinase inhibitor (CDKi) that serves as a master regulator of cell cycle arrest in SNCs30,31. Mice lacking p16INK4A are tumourprone32, demonstrating that this critical senescence regulator has an important role in tumour suppression. Shortly thereafter, p19ARF-mediated stabilization of the transcription factor p53 was identified as a feature of SNCs, providing another valuable in vivo marker of SNCs9,33,34. These SNC detection tools as well as additional markers, such as intracellular lipofuscin accumulation35, were instrumental in establishing that senescence is a consequential in vivo programme and not merely a cell culture phenomenon. For example, SA-β-Gal activity and CDKi levels were found to be elevated in both human atherosclerotic plaques36 and in numerous aged rodent and aged primate tissues37 (FIG. 1). In addition, several ‘pro-senescence’ stresses were discovered, including robust oncogenic RAS signalling38, DNA double-strand breaks and critically shortened telomeres39,40. The utilization of these stresses as senescence- inducing tools has been invaluable in establishing that cellular senescence is in fact a bona fide cell state that occurs in vivo.
Characteristics of SNCs
Rather than existing as a single, discrete cellular state, SNCs are diverse: SNCs of different origins secrete different senescence-associated secretory phenotype (SASP) factors41–44, drive disease pathogenesis through varying mechanisms and can be triggered to enter apoptosis through distinct senolytic mechanisms45. Some of these differences may arise as a consequence of how these cells became senescent in the first place (that is, what stressor induced senescence, such as oncogene overexpression versus irradiation) or the tissue origin of the cell before senescence induction. Below, we focus on features that are sufficiently common to all SNCs that they can potentially be exploited for senotherapy. Our current knowledge of SNCs has been acquired largely using cell culture systems, as efforts to characterize SNCs in vivo have been limited by difficulties in detecting, tracking and collecting SNCs from aged or diseased tissues.
SNCs are permanently growth-arrested
SNCs are characterized by a state of permanent growth arrest that promotes organism survival by suppressing cancer4,46. Although DNA damage, telomere shortening and oncogene activation are the most studied pro-senescence stimuli, several other stressors can induce this cell fate, including phosphatase and tensin homologue (PTEN) insufficiency47, mitotic stress9, stalled DNA replication48 and the unfolded protein response49. Two main signalling pathways initiate and maintain this growth arrest: p53–p21–retinoblastoma protein (RB) and p16INK4A–RB (REF. 5). In response to DNA double-strand breaks or uncapped telomeres, the DNA damage response (DDR) is activated, which results in stabilization of p53 through phosphorylation by the kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and RAD3-related protein (ATR) as well as p14ARF- or p19ARF-mediated inhibition (human or mouse, respectively) of the ubiquitin ligase mouse double minute 2 (MDM2)50,51. Enhanced p53 transcription factor activity increases expression of the CDKi p21, which initially arrests the cell cycle52. Permanent arrest is mediated by p16INK4A, which is transcriptionally upregulated by BMI1, and ETS1 and ETS2 under the control of p38 mitogen-activated protein kinase (p38 MAPK)53 and extracellular signal-regulated kinase (ERK) signalling54, respectively. p16INK4A inhibits CDK4 and CDK6 activity, which leads to RB hypophosphorylation, blockade of S-phase entry, and cell cycle arrest. Furthermore, p16INK4A cooperates with mitogenic signalling to trigger a ROS–protein kinase C δ-type (PKCδ)-positive feedback loop that stably inhibits cytokinesis55. Accordingly, SNCs in vitro can exhibit polyploidy, being left with a 4n genome following failed cytokinesis56.
SNCs possess a bioactive secretome
SNCs secrete a bioactive SASP consisting of inflammatory cytokines, chemokines, growth factors and proteases41–43,57–59 (FIG. 1). Although no exhaustive comparison of SASPs from distinct cell origins exists, the precise molecular composition of one SASP in particular (that of the primary fetal lung fibroblastic cell line, IMR90, which had been made senescent via oncogene overexpression) has been characterized in detail using stable isotope labelling with amino acids in cell culture (SILAC) and mass spectrometric analysis60. Its SASP consists of 103 distinct secreted proteins that are specifically upregulated after oncogene-induced senescence and includes several factors implicated as causal in specific chronic diseases of ageing: for example, vascular endothelial growth factor (VEGF) contributes directly to the pathophysiology of macular degeneration, and matrix metalloproteinase 3 (MMP3) and interleukin-6 (IL-6) have both been associated with osteoarthritis. As a consequence, the SASP has been proposed as a plausible link between cellular senescence and ‘inflammaging’ (REFS 61,62), the idea that chronic inflammation and proteolysis contribute to tissue dysfunction, age-related diseases and perhaps even ageing itself.
In vitro, the SASP is produced in a p16INK4A- independent fashion as a result of DDR-dependent and DDR-independent signalling through p38 MAPK and nuclear factor-κB (NF-κB)44,63. The early SASP is dominated by transforming growth factor-β (TGFβ) family members, which can induce senescence in nearby cells64. Rising levels of Notch activity then promote a switch to a pro-inflammatory SASP in which secreted and membrane-bound IL-1α acts in an autocrine fashion to reinforce the production of IL-6 and IL-8 (REFS 64,65) (FIG. 2). However, the chronology of these events in vivo is unclear. In cancer, one hypothesis is that the SASP of SNCs may initially drive cell cycle arrest of their transformed neighbours through juxtacrine signalling (that is, signalling to neighbours), and this ‘secondary senescence’ could have a role in cancer suppression. Following this, late SASP cytokines may encourage bulk clearance of neoplastic cells by recruiting the immune system64. However, a pro- inflammatory microenvironment can also be conducive to tumour proliferation20, angio genesis66, metastatic seeding67,68 and suppression of antitumour immunity69,70. Thus, the SASP can be both tumour-suppressive and tumour-promoting, possibly because an acute, time- limited SASP that effectively engages immune surveillance of SNCs may be beneficial, whereas a chronic, protracted SASP may not (BOX 1). Similarly, brief exposure to SNCs promotes keratinocyte- regenerative capacity in a skin transplantation model in vivo, but prolonged contact inhibits repopulation capacity71. This could indicate that, under conditions of injury requiring repair, SNCs produce a SASP that initially activates resident stem cells but is inherently self-limiting.
Figure 2. Hallmarks of SNCs.

Based predominately on in vitro experimentation, senescent cells (SNCs) possess several key features, namely engagement of a permanent cell cycle arrest, resistance to cell death signalling and production of a bioactive secretome, known as the senescence-associated secretory phenotype (SASP). a | In response to pro-senescence stresses, including reactive oxygen species (ROS), DNA damage and others, the p53–p21 and p16INK4A cell cycle arrest pathways are activated, which inhibits cyclin-dependent kinase 2 (CDK2), CDK4 and CDK6, respectively. Consequently, retinoblastoma protein (RB) is maintained in a hypophosphorylated state that suppresses expression of S-phase genes by binding to and sequestering the transcription factors E2F, DP1 and DP2 as well as recruiting histone deacetylases (HDACs) that act on heterochromatin. b | SNCs resist mitochondria-mediated apoptosis, in part by upregulating B cell lymphoma 2 (BCL-2) family members (BCL-2, BCL-XL and BCL-W), which bind to and sequester BAX and BCL-2-associated agonist of cell death (BAD). This sequestration blocks pore formation by BAX–BAD, which inhibits mitochondrial outer membrane permeabilization (MOMP) and release of pro-apoptotic cytochrome c, second mitochondria-derived activator of caspase (SMAC; also known as DIABLO) and OMI (also known as HTRA2). SNCs also resist extrinsic apoptosis by overexpressing decoy receptor 2 (DCR2), which intercepts FAS ligand expressed on cytotoxic immune cells. c | SNCs produce a dynamic, bioactive secretome. SASP production is initiated and sustained by a chronic DNA damage response (DDR) called ‘DNA-SCARS’ (DNA segments with chromatin alterations reinforcing senescence). Initially, Notch signalling drives transforming growth factor-β (TGFβ) secretion (‘early SASP’), which acts in a cell-autonomous manner to promote cell cycle arrest. A subsequent decrease in Notch signalling promotes a shift to a DDR-dependent ‘transitional SASP’ that is enhanced by mechanistic target of rapamycin (mTOR), in which cell surface-associated interleukin-1α (IL-1α) binds to interleukin-1 receptor (IL-1R). Either this cell-autonomous IL-1α signal or p38 mitogen-activated protein kinase (p38 MAPK) activity is transmitted through nuclear factor-κB (NF-κB), resulting in the secretion of a ‘late SASP’, which contains metalloproteinases (MMPs), IL-6, IL-8 and numerous other factors. At this stage, IL-6 and IL-8 can also reinforce the cell cycle arrest. Crucially, many of the specific details of SNC growth arrest, death resistance and SASP have not been confirmed in vivo and are likely to show substantial differences. ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and RAD3-related protein; CHK2, checkpoint kinase 2; NBS1, Nijmegen breakage syndrome protein 1.
The biological relevance of individual SASP factors has been explored in limited ways in vivo. For example, platelet-derived growth factor AA (PDGF-AA) released by senescent myofibroblasts in wounds promotes wound closure72. The serpin (serine protease inhibitor) plasminogen activator inhibitor 1 (PAI1; also known as SERPINE1) is necessary for several accelerated ageing phenotypes in klotho-deficient progeroid mice73. MMP activity and hepatocyte growth factor have indirect and direct effects on tumour proliferation, respectively74. Intriguingly, the SASP has been shown to induce stem cell features in a paracrine manner71, which may explain the observations of SNC-driven carcinogenesis and tissue repair. In other cases, SNCs — presumably through the SASP — have been shown to drive age-related diseases or disorders8,10,11, but a link between a specific SASP factor (or factors) and these phenotypes is more speculative.
SNCs resist apoptosis
In cell culture, SNCs show alterations in apoptotic signalling that, in general, cause resistance to programmed cell death75,76. One key determinant of the senescent versus apoptotic cell fate choice is signalling through the p53 stress response pathway. Total p53 levels and post-translational modifications fine-tune target gene expression profiles to determine cell fate. With pro-senescent stresses, p53 accumulates but to a lesser extent than in apoptosis, which leads to less significant levels of pro-apoptotic p53-upregulated modulator of apoptosis (PUMA; also known as BBC3) and NOXA (also known as PMAIP1) and higher levels of BCL-2 family members77. BCL-2, BCL-XL (also known as BCL-2L1), and BCL-W (also known as BCL-2L2), which are overexpressed in SNCs, sequester pro-apoptotic BH3 domain-containing proteins to inhibit mitochondrial outer membrane permeabilization (MOMP) and apoptosis24 (FIG. 2).
p53 is also post-translationally modified distinctively in senescence and apoptosis78. The importance of differential p53 modification is illustrated by separation- of-function mutations that redirect the cellular stress response. For example, cells lacking the acetyl receptor residue K117 on p53 are unable to undergo apoptosis but retain the ability to enter into senescence through p53- dependent upregulation of p21 (REF. 17). By contrast, p53-K117R/K161R/K162R triple-mutant cells are deficient in both the senescent and apoptotic responses17. The disruption of p53-binding domain cooperativity through the E177R mutation similarly ablates pro- apoptotic but not pro-senescent p53 target gene expression79. p21 itself can block apoptosis80,81, and — intriguingly — may inhibit caspase 3 (CASP3) activity directly82. Indeed, apoptotic cells actively silence p21 expression via p53-dependent DNA (cytosine-5)- methyltransferase 3A (DNMT3A) activity80.
Senescence and Ageing
Studies performed on a rapidly ageing animal (a mouse model with deficiencies in the mitotic checkpoint protein BUBR1 (encoded by Bub1b)83 have demonstrated a causal link between SNCs and ageing. Prematurely aged tissues in this animal, including skeletal muscle, eye and adipose tissue, accumulate high numbers of p16INK4A- positive SNCs. The accumulation of SNCs has been shown to trigger natural features of mouse ageing, including sarcopenia, cataracts and lipodystrophy, as evidenced by experiments showing that the genetic inactivation of p16INK4A blocked the formation of SNCs and attenuated the development of these ageing phenotypes9 (FIG. 1).
The idea that senescence is a driver of age-related tissue dysfunction has been further tested in two studies using a transgenic SNC killing system called INK-ATTAC (INK-linked apoptosis through targeted activation of caspase), which selectively expresses a FK506-binding protein (FKBP)–CASP8 fusion protein in p16INK4A-postive SNCs and triggers apoptosis following administration of AP20187, a molecule that dimerizes the FKBP–CASP8 fusion protein (FIG. 1). The first study demonstrated that intermittent removal of SNCs from Bub1b-mutant progeroid mice mimicked the effects of genetic elimination of p16INK4A, blunting sarcopenia, cataracts and lipodystrophy10. Although conducted in progeroid mice, this study spurred wide interest in exploring senolysis as a potential therapy to treat ailments of old age. However, the inherent limitations of such progeroid models, including the inability to assess the effect of senolysis on lifespan, natural ageing or tumorigenesis, motivated a second study in which SNCs were eliminated from naturally aged, non-progeroid mice from midlife onwards7. In this study, SNC clearance was achieved using biweekly AP20187 (or vehicle) administration to INK-ATTAC mice. In two genetically distinct backgrounds, SNC killing resulted in extended median lifespan. Healthspan analysis showed that SNC removal blunted multiple features of ageing, including glomerulosclerosis, cardiomyocyte hypertrophy, diminished cardiac stress tolerance, cataract formation and lipodystrophy as well as a key age-related disease, cancer. The observation that senolysis suppresses cancer was encouraging in regard to therapeutic development, because it suggested that the elimination of SNCs — once they arise — does not have the tumour-promoting side-effects observed upon genetic obstruction of the senescence programme84,85.
Senescence and age-related diseases
Numerous age-related diseases have now been associated with cellular senescence, including atherosclerosis, osteoarthritis, cancer, Alzheimer disease, chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis8,11,86–90 (TABLE 1). SNCs accumulate at sites of disease and — once present in sufficient numbers in a tissue — may actively drive diseases of ageing. However, currently, only a small number of studies definitively demonstrate that SNCs cause disease8,11.
Table 1.
Age-related diseases linked to cellular senescence
| Condition or disease | Pro-senescence stress | Location of SNCs | Markers used | Senescence intervention method | Effect of SNCs | Refs |
|---|---|---|---|---|---|---|
| Senescence markers only | ||||||
| Alzheimer disease | Human disease state | Astrocytes | • Expression levels of p16INK4A and MMP1 | NA | NA | 90 |
| Aneurysm | Kawasaki disease | Endothelial cells | • SA-β-Gal • Expression levels of p16INK4A and SASP factors |
NA | NA | 184 |
| Cystic fibrosis | Human disease state | Airway epithelial cells | • SA-β-Gal • Expression level of γH2AX • Phospho-CHK2 levels |
NA | NA | 193 |
| Fibrosis in pancreatitis | Mouse disease model | Pancreatic stellate cells | SA-β-Gal | NA | NA | 196 |
| Glaucoma | Human disease state | Outflow tract | SA-β-Gal | NA | NA | 185 |
| Hypertension | Human disease state | Arterial wall | • Expression levels of p53 and p21 • Telomere uncapping |
NA | NA | 197 |
| Idiopathic pulmonary fibrosis | Human disease state | Fibroblasts | • SA-β-Gal • Larger cell size |
NA | NA | 190 |
| Bleomycin-induced model | Alveolar epithelial cells | • SA-β-Gal • Larger cell size • Expression levels of p21, p38 MAPK, γH2AX and SASP factors |
NA | NA | 191, 192 | |
| Inflammatory bowel disease | Human disease state | Crypt cells | Immunostaining for HP1γ and γH2AX | NA | NA | 188 |
| Intervertebral disc degeneration | Human disease state | Nucleus pulposus | • SA-β-Gal • Expression levels of p16INK4A and SASP factors |
NA | NA | 95,96 |
| Macular degeneration | Human disease state | RPE cells | • Expression levels of p53 and p21 • Phospho-p38 MAPK levels |
NA | NA | 186, 187 |
| Osteoarthritis | Human disease state | Articular cartilage | • Reduced proliferation • SA-β-Gal • Telomere length |
NA | NA | 189 |
| Type 2 diabetes mellitus in obesity | Human disease state | White adipose tissue | • Expression levels of HMGA2, p14ARF (p19ARF) and p53 • SA-β-Gal |
NA | NA | 194, 195 |
| Senescence markers and intervention by senescence pathway inactivation | ||||||
| Adipose atrophy | Progeria | Pre-adipocytes | • SA-β-Gal • Expression levels of p16INK4A and SASP factors |
p16INK4A deficiency | • Detrimental • Drives atrophy |
9 |
| Atherosclerosis | Dyslipidemia | Smooth muscle cells | None | Tp53 knockout | • Beneficial • Blocks apoptosis |
198 |
| Unclear | None | Cdn1b knockout | • Beneficial • Restricts proliferation |
199 | ||
| Unclear | None | Cdkn1a knockout | • Beneficial • Restricts proliferation |
168 | ||
| Smooth muscle cells | • SA-β-Gal • Expression levels of p16 INK4A and p19ARF |
9p21 (Cdkn2a locus) deletion | • Beneficial • Restricts proliferation |
169 | ||
| Cataracts | Advanced age | Unclear | Expression levels of p16INK4A and SASP factors | p16INK4A deficiency or Cdkn1a knockout | Detrimental | 9, 200 |
| COPD | LPS-induced model | Clara cells | SA-β-Gal | Clara cell-specific Tp53 knockout | • Detrimental • Drives disease |
89 |
| Idopathic pulmonary fibrosis | Bleomycin-induced model | Alveolar epithelial cells | • SA-β-Gal • Phospho-RB levels |
Cav1 knockout | • Detrimental • Drives fibrosis |
203 |
| Kidney transplant failure | Ischaemia | Tubular epithelial cells | Proliferative arrest | p16INK4A deficiency | Detrimental | 201 |
| Liver fibrosis | Carbon tetrachloride | Hepatic stellate cells | • SA-β-Gal • Expression levels of p16INK4A and p21 • Proliferation |
Tp53/Cdkn2a double knockout | • Beneficial • Restricts fibrosis |
108 |
| Loss of bone mass | Advanced age | Mesenchymal stem cells | p16INK4A expression | p16INK4A deficiency | • Detrimental • Drives bone ageing |
204 |
| Myocardial infarction | Infarct | Cardiac fibroblasts | • SA-β-Gal • Expression levels of CDKi and p53 |
Tp53 knockout | • Beneficial • Restricts fibrosis |
202 |
| Sarcopenia | Advanced age | Satellite cells | • SA-β-Gal • Expression level of p16INK4A |
p38 MAPK inhibition | Satellite cell dysfunction | 97–100 |
| Progeria | Fibroadipogenic progenitors | Expression levels of SASP factors | p16INK4A deficiency | • Detrimental • Drives sarcopenia |
9 | |
| Type 2 diabetes mellitus | Mouse hypercaloric model | White adipose tissue | • SA-β-Gal • Expression levels of γH2AX and SASP factors |
Tp53 knockout | • Detrimental • Drives disease |
195 |
| Wound healing | CCN1 (also known as CYR61) | Wound fibroblasts | • SA-β-Gal • Expression level of p16INK4A |
Cyr61 mutant | • Beneficial • Restricts fibrosis |
163 |
| Senescence markers and intervention by SNC clearance | ||||||
| Adipose atrophy | Advanced age | Pre-adipocytes | • SA-β-Gal • Expression levels of p16INK4A and SASP factors |
INK-ATTAC model | Detrimental | 7 |
| Allopecia | p19ARF overexpression | Skin | Expression level of p19ARF | ABT-737 treatment | Detrimental | 24 |
| Atherosclerosis | Dyslipidemia | Intraplaque foam cells | • SA-β-Gal • Expression levels of p16INK4A and SASP factors |
• INK-ATTAC model • INK-NTR model* • p16-3MR model • Navitoclax treatment |
Detrimental | 11 |
| Cancer | Advanced age | Unclear | None | INK-ATTAC model | Detrimental | 7 |
| Cardiomyocyte hypertrophy | Advanced age | Pericardial epithelium | • SA-β-Gal • Expression levels of p16INK4A and SASP factors |
INK-ATTAC model | Detrimental | 7 |
| Cataracts | Progeria | Unclear | Expression levels of p16INK4A and SASP factors | INK-ATTAC model | Detrimental | 10 |
| Glomerulo- sclerosis | Advanced age | Kidney proximal tubules | • SA-β-Gal • Expression levels of p16INK4A and SASP factors |
INK-ATTAC model | Detrimental | 7 |
| Idiopathic pulmonary fibrosis | Bleomycin-induced model | Multiple | Expression levels of p16INK4A, p21 and SASP factors | • INK-ATTAC model • Dasatinib plus quercetin treatment |
Detrimental | 86 |
| Osteoarthritis | • Joint trauma • Ageing |
Joint articular surface | • SA-β-Gal • Expression levels of p16INK4A and SASP factors |
• p16-3MR model • UBX-101 treatment |
Detrimental | 8 |
| Sarcopenia | Progeria | Fibroadipogenic progenitors | Expression levels of p16INK4A and SASP factors | INK-ATTAC model | Detrimental | 10 |
| Wound healing | Dermal wound | • Myofibroblasts • Endothelium |
Expression levels of p16INK4A and p21 | • p16-3MR model • INK-ATTAC model |
Beneficial | 15 |
γH2AX, phosphorylated histone H2AX; CDKi, inhibitors of cyclin-dependent kinases; CHK2, checkpoint kinase 2; COPD, chronic obstructive pulmonary disease; HMGA2, high-mobility group AT hook protein 2; HP1γ, heterochromatin protein 1 homologue-γ; INK-ATTAC, INK-linked apoptosis through targeted activation of caspase; INK-NTR, INK-linked nitroreductase; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MMP1, matrix metalloproteinase 1; NA, not applicable; RB, retinoblastoma protein; RPE, retinal pigment epithelium; SA-β-Gal, senescence-associated β-galactosidase; SASP, senescence-associated secretory phenotype.
An alternative p16INK4A-positive senescent cell killing system that uses bacterial nitroreductase to reduce metronidazole (a prodrug delivered in the drinking water) to a toxic metabolite.
It is challenging to assess the contribution of senescence to ageing and disease owing to the lack of markers that enable the unequivocal identification of SNCs. The most reliable approach currently available involves the use of a combination of semi-selective markers, including SA-β-Gal, lipofuscin, loss of nuclear high-mobility group box 1 (HMGB1) or lamin B1, increased levels of cell cycle inhibitors — such as p16INK4A, p14ARF or 19ARF as well as p21 — or commonly observed SASP factors35,41,42,91,92. Causality (that is, that SNCs are necessary for a particular disease) can be demonstrated by blocking the establishment of cellular senescence (through inactivation of key components of the senescence programme, such as p16INK4A) or more conclusively by selectively eliminating SNCs from diseased tissues and observing the impact on a disease process7,11. Two similar but non-identical transgenic mouse models have been used for the selective elimination of SNCs in an inducible fashion through the administration of an ‘activator’ molecule: INK-ATTAC mice10 and p16-3MR mice. In the p16-3MR mouse model, a viral thymidine kinase is expressed in p16INK4A-positive SNCs, which phosphorylates the prodrug ganciclovir, thereby converting the compound into a toxic metabolite that results in mitochondrial DNA damage and cell death93,94.
Disorders in which SNCs have been implicated in disease may therefore be categorized according to whether SNC markers alone are used (that is, SNCs correlate with disease), or whether prevention of cellular senescence or senolysis halts or reverses disease (that is, SNCs cause disease) (TABLE 1). In cases such as Alzheimer disease and intervertebral disk degeneration, markers of SNCs are present in dysfunctional tissue, but there is no evidence for a causal role in disease pathogenesis90,95,96. In others, such as COPD and sarcopenia, inactivation of molecular pathways implicated in establishing the SNC fate provided evidence for causality89,97–100. However, this approach does not account for non-SNC roles of these pathways and presumably blocks both SNC cell cycle arrest as well as SNC paracrine effects, such as the SASP. We consider the strongest individual line of evidence for showing SNC causality in disease to be the selective removal of SNCs, an approach that not only demonstrates SNCs are bioactive in disease but mimics the therapeutic goal of senolysis.
Below, we highlight case studies on two major human diseases of ageing, atherosclerosis and osteoarthritis, that demonstrate a causal role for SNCs in these diseases. Although there are additional published studies linking SNCs to other diseases and demonstrating the potential therapeutic utility of senolytics21,23,24, we focus on these two diseases because the associated studies provide a detailed mechanistic explanation as to how SNCs might drive these diseases of later life. In both atherosclerosis and osteoarthritis, the presence of SNCs in diseased tissue has been demonstrated using a combination of markers. Furthermore, multiple transgenic and pharmaco logical approaches have been used to eliminate SNCs, and the clearance of SNCs has been experimentally validated.
Case study: Atherosclerosis
Atherosclerosis, which is the leading cause of vascular disease worldwide, is characterized by the gradual accumulation of lipid and protein-filled ‘plaques’ on the inner walls of arteries. Over time, these plaques become unstable and thrombogenic (that is, blood clot- inducing), which results in heart attack, stroke or other severe ischaemic injuries101. The mechanism of plaque initiation and growth, which is the root cause of this devastating illness, is incompletely understood, and currently available therapies slow but do not (or only partially) regress disease.
Atherosclerosis begins when elevated levels of blood lipids, particularly low-density lipoprotein (LDL), results in LDL accumulation in artery walls102. LDL oxidation triggers signalling from the arterial wall, which recruits circulating monocytes to invade, differentiate into macro phages and engulf lipid103. Over time, macrophage- driven inflammation and protease activity promotes plaque growth and evolution of the plaque from an innocuous, small lesion into a clinically dangerous, structurally unstable form. Plaques from a mouse model of atherosclerosis, which is LDL receptor (Ldlr)- negative, accumulate senescent macrophage foam cells in the artery walls from the earliest stages of plaque initiation, that is, with the appearance of fatty-streak lesions11. Additional SNC types resembling vascular smooth muscle cells (VSMCs) and endothelial cells accumulate as the lesion matures. SNCs that are isolated directly from late-stage lesions overexpress several pro-atherogenic secreted factors (IL-1α, monocyte chemotactic protein 1 (MCP1; also known as CCL2), MMP12, and MMP13) compared with non-senescent control cells10. The use of three different genetic SNC elimination strategies, as well as the senolytic molecule navitoclax, have demonstrated that the selective elimination of SNCs from Ldlr−/− mice blocks lesion growth, in part by reducing the levels of factors implicated in plaque formation, such as MCP1, IL-1α, tumour necrosis factor-α (TNFα) and the leukocyte receptor vascular cell adhesion protein 1 (VCAM1). Intriguingly, the selective elimination of SNCs from established atherosclerotic plaques favourably alters plaque structure. Senolysis increases the relative thickness of the fibrous cap, a plaque-stabilizing structure consisting of VSMCs and the ECM they synthesise, that partitions thrombogenic plaque contents from the circulating clotting cascade. A thick fibrous cap is a hallmark of more stable, clinically less dangerous atherosclerotic lesions, which suggests that senolysis could facilitate the remodelling of dangerous atherosclerotic disease into a more stable form.
Case study: Osteoarthritis
Osteoarthritis is a degenerative joint disease, characterized by the gradual loss of joint cartilage, distorted bone growth, joint inflammation and joint pain, which impacts most humans in later life104–106. Standard of care begins with NSAIDS (non-steroidal anti-inflammatory drugs), injection of corticosteroids and — ultimately — mechanical replacement of the affected joint107. The root cause of this debilitating chronic illness has not been elucidated, and no disease-modifying therapies currently exist. Although SNCs have been reported to accumulate in osteoarthritis-affected joints compared with non- osteoarthritis-affected joints, the notion that SNCs have a causal role in the pathogenesis of osteoarthritis has only recently been demonstrated using a mouse model of traumatic osteoarthritis in which senescent chondrocytes accumulate at the articular surface of the arthritic joint8. Microsphere cultures of explanted chondrocytes directly showed a deleterious effect of SNCs on cartilage deposition, which suggests that a factor made by SNCs inhibits cartilage regeneration8. Clearance of SNCs through a transgenic system or intra-articular injection of the senolytic molecule UBX0101 (see below) reduces pain and promotes the repair of damaged cartilage8 (FIG. 1). The observed reduction in levels of the SASP factors MMP13, IL-6 and IL-1β could partially explain this finding. Importantly, SNC clearance has also been demonstrated through use of the luciferase reporter activity present in p16-3MR transgenic mice, SA-β-Gal and immunostaining for loss of nuclear HMGB1. Finally, clearance of naturally occurring SNCs using INK-ATTAC prevents age-related osteoarthritis in naturally aged mice8.
Therapeutic targeting of SNCs
The pivotal finding that SNCs can potentially be eliminated for therapeutic benefit without triggering negative side effects7,8,10 has opened the door for the development of agents and strategies to specifically target SNCs for the prevention and treatment of age-related diseases. Broadly, these strategies consist of the selective elimination of SNCs, referred to as senolysis, immune-mediated SNC clearance and SASP neutralization. Between senolysis and SASP suppression, senolysis holds the most therapeutic promise for two reasons. First, permanent removal of SNCs leads to a durable abolishment of deleterious SASP components. Second, once a SNC is eliminated, there is no risk of tumorigenic ‘escape’ from senescence, which may be possible if SNCs are permitted to linger indefinitely.
Here, we discuss each of the emerging strategies to therapeutically target SNCs, summarize recent progress towards bringing these strategies into the clinic, and highlight their relative advantages and risks. Specific agents that have been tested explicitly as senotherapies, as well as agents that have been developed for other purposes but may have senotherapeutic potential, are summarized in TABLE 2.
Table 2.
Candidate senotherapies
| Agent | Target (or targets) | Target class | Development status | Refs |
|---|---|---|---|---|
| Small molecules | ||||
| ABT-737 | BCL-X and BCL-W L | Pro-survival proteins | Preclinical animal models | 24 |
| Dasatinib | Pan-receptor tyrosine kinases | Receptor tyrosine kinases | Phase II clinical trial (NCT02848131) for chronic kidney disease | 23 |
| Metformin | IKK and/or NF-κB and Dicer | SASP | Approved for T2DM | 206–208 |
| Navitoclax | BCL-2, BCL-X L and BCL-W | Pro-survival proteins | Clinical trials (phase I/II (NCT00445198), phase II (NCT02591095), phase I (NCT02520778) and phase II (NCT02079740)) for various cancers | 11,21,205 |
| Piperlongumine | Unknown | Unknown | Preclinical animal models | 22 |
| Quercetin | Numerous | Numerous | Phase II clinical trial (NCT02848131) for chronic kidney disease | 23 |
| Rapamycin | mTOR | SASP | Approved for immunosuppression | 20,209 |
| Antibodies | ||||
| Anakinra | IL-1R | Cytokine receptor | Approved for rheumatoid arthritis | 214 |
| Canakinumab | IL-1β | Cytokine | Approved for cyropyrin-associated periodic syndromes | 211 |
| Etanercept | TNFα | Cytokine | Approved for autoimmune diseases | 213 |
| Infliximab or Adalimumab | TNFα | Cytokine | Approved for autoimmune diseases | 210 |
| Rilonacept | IL-1α and IL-1β | Cytokine | Approved for cyropyrin-associated periodic syndromes | 212 |
| Siltuximab | IL-6 | SASP factor | Approved for multicentric Castleman disease | 147 |
| Tocilizumab | IL-6R | Cytokine receptor | Approved for autoimmune diseases | 148 |
B cell lymphoma 2, BCL-2; IL, interleukin; IL-6R, interleukin-6 receptor; IKK, inhibitor of NF-κB kinase; mTOR, mechanistic target of rapamycin; NF-κB, nuclear factor-κB; SASP, senescence-associated secretory phenotype; T2DM, type 2 diabetes mellitus; TNFα, tumour necrosis factor-α.
Senolysis: eliminating SNCs
SNCs themselves are viable therapeutic targets for several key reasons. First, despite arising from different tissues, diseases and cell types, SNCs may share a similar biochemistry, which potentially allows reuse of therapeutic strategies across multiple diseases in which SNCs are causal. Second, a pharmaceutical development programme targeting SNCs could synergize well with other well-established programmes. For example, senolysis has been shown to retard cancer progression in mice7, and — given that DNA-damaging anticancer therapy also produces collateral senescence — senolytics could become a key component of the chemotherapeutic arsenal (BOX 2). Third, SNCs have mostly been described to have negative effects on ageing and disease, which lowers the possibility of off-target effects of senolysis. However, SNCs do have some beneficial effects: they arise in wounds to enhance healing7,15 and may also direct tissue regeneration71, restrict fibrosis108 and promote neoplastic cell clearance via the SASP64 (BOX 1). Notably, these effects seem largely transient and modulatory, as constitutive transgenic removal of SNCs has no overt negative health effects in mice7. One exception may be the potential role of cellular senescence in promoting insulin secretion by pancreatic β-cells during ageing18 (BOX 1). Surprisingly, senescent pancreatic β-cells from aged mice show enhanced glucose-stimulated insulin secretion, which suggests that senescence in this cell type may be a homeostatic mechanism. Whether this applies to other secretory cell types or whether this is of physiological importance is currently unclear but should be kept in mind. Fourth, SNC clearance extends healthspan — a global measure of ageing — in mice7,10, which suggests that senotherapy might retard ageing in general, thereby positively impacting numerous age-related illnesses.
Box 2. Cancer and therapy-induced senescence.
Senescence is a tumour-suppressive cell fate, but senescent cells (SNCs) that arise in aged tissue7 or in tumour stroma69,88,91 can promote cancer progression, potentially through effects of the senescence-associated secretory phenotype (SASP). The elimination of these naturally arising tumorigenic SNCs is a promising strategy for cancer prevention and treatment. However, SNCs generated incidentally91 or deliberately171 by anticancer therapy are also disease-modifying. A new dimension to cancer treatment may be added by defining how to produce, delete and control these therapy-induced SNCs.
Although cytotoxic anticancer therapy now includes more selective drugs, most protocols still employ chemical or radiation-induced DNA damage to disproportionately affect tissue with a high proliferative index, including tumours172. This paradigm has off-target sequelae in other tissues with rapid turnover and produces acute adverse effects on the gastrointestinal tract, bone marrow, integument and reproductive system. In addition to short-term consequences, DNA-damaging protocols produce collateral senescence that may underpin long-lasting side effects seen in cancer survivors173–175. Such therapy-induced senescence may cause tissue dysfunction, both by removal of progenitor cells from the cycling pool as well as through the SASP. Indeed, cancer survivors have a higher incidence of age-related diseases linked to senescence, including cardiovascular disease176,177, neurodegeneration178, sarcopenia179 and secondary neoplasia180. Given the large patient population receiving DNA-damaging anticancer therapy and the emergence of chemotherapeutic agents aimed at arresting tumour growth by deliberately inducing senescence, managing the consequences of therapy-associated senescence through senotherapy should be considered as part of cancer management91.
Although the SASP promotes immune cell recruitment and cancer surveillance under normal circumstances46, this may not be the case in a tumour microenvironment that is challenged with pro-senescent chemotherapies. For example, the pro-senescent drug palbociclib (a cyclin-dependent kinase 4 (CDK4) and CDK6 inhibitor) is initially tumour-suppressive but — in the long-term — results in collateral induction of SASP factors in the tumour stroma that are tumour-promoting171. This phenomenon is somewhat unexpected, as overexpression of p16INK4A, which is a potent CDK4 and CDK6 inhibitor, does not result in a SASP in normal diploid cells181. Similarly, SRC homology phosphatase 2 (SHP2; also known as PTPN11) inhibition by GS-493 can both prevent and arrest mammary tumours in mice through tumour cell senescence182, but the process of activating signal transducer and activator of transcription 3 (STAT3) causes secretion of SASP factors that suppress tumour immune surveillance10. Preservation of maximal tumour immune response with SHP2 inhibition could be achievable by co-administration of a STAT3 inhibitor183. Alternatively, follow-up treatment with a senolytic drug could prevent these chronic therapy-induced SNCs from causing secondary neoplasia, cancer relapse and accelerated ageing in patients receiving chemotherapy.
Many important unresolved questions remain in the emerging space of senotherapy in cancer treatment. For instance, it is unclear how pro-senescence therapy will compare with DNA-damaging protocols in their side effects on non-tumour tissue. The properties of SNCs arising in aged tissue, tumour stroma or tumours challenged by chemotherapy are under-characterized despite their importance in defining the senescence–cancer relationship. The identification of markers that permit isolation and molecular characterization of in vivo SNCs is therefore a high priority.
Existing senolytics
Senolytics were the first potential senotherapy to be successfully tested in preclinical in vivo models, and several senolytic agents currently exist. Many of these agents target upregulated anti- apoptosis systems within SNCs, such as signalling through the BCL-2 family of proteins (BCL-2, BCL-XL, and BCL-W)21,24. These family member proteins bind to and functionally neutralize pro-apoptotic BCL-2 family members. Pro-apoptotic BCL-2 proteins activate the BAX and BCL-2 homologous antagonist/killer (BAK) proteins to trigger MOMP, thereby causing cytochrome c release to drive programmed cell death109. The senolytic molecules navitoclax and ABT-737 occupy the inhibitory binding grooves of BCL-2, BCL-XL and BCL-W, which counteracts their anti-apoptotic functions and permits SNCs to initiate apoptosis110. Administration of navitoclax to sublethally irradiated mice reduces the SNC burden in muscle satellite cells111 and the bone marrow, in which haematopoietic function was partially restored21. Navitoclax has also been shown to eliminate senescent foam cell macrophages in early atherosclerotic lesions and block SNC-dependent progression of atherosclerosis11. ABT-737, which is an earlier-generation analogue of navitoclax, was similarly able to drive apoptosis of irradiation-induced lung epithelial SNCs and p19ARF- induced dermal cells in vivo24. Finally, piperlongumine has recently been shown to be a pro-apoptotic senolytic that displays good selectivity and potency in vitro21. Piperlongumine acts synergistically with navitoclax, which suggests a novel mechanism of action.
Other anti-apoptotic pathways could also be inhibited by small molecules to eliminate SNCs112. For example, the senolytic compound UBX0101 induces apoptosis of SNCs following intra-articular injection into arthritic joints, thereby reducing expression of SASP factors and improving joint function8. In both 2D and 3D cell cultures, senolytic activity of UBX0101 has been demonstrated on primary chondrocytes that were freshly isolated from osteoarthritic human knee cartilage with concordant reduction in MMP13, IL-6 and IL-1β expression levels. UBX0101 also improves functional measures (increases in aggrecan and collagen II levels, and weight-bearing ability) in a mouse model of osteoarthritis, which indicates that this molecule is a promising candidate for future advancement into clinical trials (FIG. 3).
Figure 3. Preclinical testing of senolytic candidates.

Preclinical testing of senolytic efficacy should be designed to test whether the candidate drug kills senescent cells (SNCs), modulates senescence-associated secretory phenotype (SASP) factors linked to the disease state and results in a therapeutically relevant improvement in tissue function. These properties should be shown in human cell types, diseased or aged human tissue explants and in an in vivo animal model of the disease state. The preclinical testing of UBX0101 for osteoarthritis is an informative example of this workflow in action. The compound was shown to cause SNC elimination (reduced p16INK4A levels and senescence-associated β-galactosidase (SA-β-Gal) activity), attenuate SASP factors (matrix metalloproteinase 13 (MMP13), interleukin-6 (IL-6) and IL-1β) and improve functional measures (increased aggrecan, collagen II and weight-bearing ability). UBX0101-mediated SNC elimination was demonstrated using three model systems with different types of relevance to the human disease state: in vitro culture of human chondrocytes and synoviocytes, 3D culture of human cells from arthritic joints and a mouse model of osteoarthritis (anterior cruciate ligament transection (ACLT)).
Another senolytic regimen, the combined use of dasatinib plus quercetin, targets less well-established anti-apoptotic signalling in SNCs. The scientific basis for exploring the senolytic properties of these molecules originated from the observation that survival of irradiated pre-adipocytes depends on six pro-survival signalling proteins: the ephrins EFNB1 and EFNB3, the kinase phosphatidylinositol 3-kinase-δ (PI3Kδ), the serpin PAI1, BCL-XL and the CDKi p21 (REF. 23). In combination, dasatinib (a pan-tyrosine kinase inhibitor with the ability to block ephrin-dependent receptor signalling113) and quercetin (a plant flavonoid with a myriad of presumed biological effects114,115, including partial inhibition of serpins) demonstrate senolytic activity in cultured pre-adipocytes and fat tissue of aged mice23 but not in IMR90 cells116. Dasatinib plus quercetin treatment was also tested in a mouse model of atherogenesis but did not replicate the anti- atherogenic effect of SNC elimination that had been achieved with multiple transgenic systems or navitoclax117. In addition, dasatinib plus quercetin treatment was shown to kill profibrotic senescent primary human fibroblasts in vitro and clear SNCs in the widely used bleomycin-induced pulmonary fibrosis mouse model112. However, although early dasatinib plus quercetin treatment positively impacts exercise capacity and lung compliance, it does not alter fibrosis, the key pathological feature of human idiopathic pulmonary fibrosis86. Overall, the fact that dasatinib and quercetin target such a broad spectrum of biological pathways and mechanisms makes it difficult to ascribe any observed biological effect specifically to senolysis.
SNC killing can also be induced by interfering with the p53–forkhead box protein O4 (FOXO4) interaction116. FOXO4 has been shown to target p53 to the nucleus of IMR90 cells that had been induced to undergo senescence upon exposure to ionizing radiation. Disruption of the p53–FOXO4 interaction using a d-retro-inverso peptide (DRI-FOXO4) that corresponds to the reverse sequence of the FOXO4–p53-binding domain causes nuclear exclusion of p53, which allows it to induce apoptosis by catalysing cytochrome c release into the cytoplasm from mitochondria. In progeroid and aged mice, short-term DRI-FOXO4 treatment has been shown to restore hair growth, kidney function and running wheel activity. However, it remains to be established how these healthspan improvements were obtained, particularly because earlier studies documented that clearance of SNCs in progeroid mice with pre- established ageing phenotypes had no rejuvenating effects.
Immune-mediated SNC clearance
Immune evasion by SNCs could arise from the intrinsic decline of immune system efficiency with ageing, immune-impairing factors that are present in the diseased tissue microenvironment or SNC ‘camouflage’ (REF. 46). Strategies to overcome these potential problems are being developed in cancer therapy. Recent efforts have progressed to engineer patient-derived cytotoxic T cells with chimeric antigen receptors (CAR T cells) that are targeted to cancer epitopes. Indeed, initial trials using CAR T cells against the B cell antigen CD19 are encouraging118, which suggests that similar cytotoxicity could be achieved if a SNC-specific cell surface marker could be identified.
Immune surveillance seems to be divided into an initial cytotoxic T cell infiltration followed by clearance of cellular debris (efferocytosis), but the details differ between models. In a mouse model of hepatocellular carcinoma (HCC), re-expression of p53 in Tp53−/− liver tumours drives cellular senescence, which is accompanied by a strong neutrophil, natural killer and macrophage response in the liver119. Natural killer cell- mediated SNC clearance also restricts fibrosis in chronic liver injury108. By contrast, hepatocytes overexpressing NRAS-H12V undergo oncogene-induced senescence and are cleared by a combination of innate and adaptive immunity87. In this scenario, monocyte-derived macrophages, but not liver-resident Kupffer cells, prime a T helper 1 (TH1)-type response from CD4+ T cells to trigger SNC killing.
Cancer cells escape programmed immune clearance through a combination of decoy receptor presentation, immunomodulatory cytokines and checkpoint ligands. For example, decoy receptor 2 (DCR2; also known as TNFRSF10D) and DCR3 (also known as TNFRSF6B), which are expressed widely on cancer cells, titrate away FAS ligand (FASL; also known as TNFSF6) and TNF-related apoptosis-inducing ligand (TRAIL; also known as TNFSF10) that are presented by cytotoxic T cells, thereby blocking apoptosis120–124. Similarly, hepatic SNCs produced by carbon tetrachloride (CCl4)-induced liver injury have upregulated DCR2 levels, which neutralizes natural killer cell activation of the FAS-mediated extrinsic apoptosis pathway. Instead, natural killer cells target these hepatic SNCs using perforin and granzyme granule exocytosis, and — accordingly — perforin-knockout mice show an increased number of liver SNCs and greater fibrosis when challenged with CCl4 compared with wild-type mice125. One possibility is that the preference for granule- mediated death signalling may prevent autoimmunity during short-term tissue repair. Alternatively, SNC- recruited granzymes may carry out extracellular matrix degradation through their serine protease activity to restrict fibrosis126. DCR2 is overexpressed in SNCs that result from other stimuli127, but it is unclear whether this is true for age-associated SNCs or whether this mechanism significantly impairs the efficiency of natural killer cell-mediated SNC clearance.
Another major immunomodulator is the programmed cell death ligand 1 (PDL1), which permits cancer cell immune evasion by suppressing apoptosis in regulatory T cells and promoting death in effector T cells128. CD4+ T cells, which are involved in SNC clearance, express the PDL1 receptor (PD1), but to what extent this ligand is used by SNCs for immune evasion is unknown129.
Together, these findings indicate that blockade of decoy receptors or immune checkpoint inhibitors could, in theory, restore immune surveillance of chronic SNCs and therefore serve as indirect senolytics by harnessing the immune system.
SASP neutralization
An additional approach to senotherapy would be to disrupt the SNC secretome, which contains pro- inflammatory cytokines, chemokines, growth factors, matrix metalloproteinases and other bioactive molecules that are implicated in diseases of ageing3,5,8,11. Activation of several stress response signalling cascades results in the secretion of SASP factors, with control exerted at both the transcriptional and translational level. Although there seem to be several common canonical SASP factors, the ultimate composition of the secretome depends on the mode of senescence induction, the cell type, the duration of senescence and which signalling cascades are active. Insights into the composition and properties of the SASP were mainly obtained from cell culture experiments and need to be validated in vivo41. For SASP-neutralizing senotherapies in development, three broad approaches can be considered: blocking pro-SASP signalling cascades within SNCs, disrupting secretion of the SASP or inhibiting the activity of individual components of the SASP.
One potential problem posed by suppressing intracellular pro-SASP signalling is how to disrupt expression of SASP factors without elevating cancer risk. The blunt disruption of pro-SASP signalling can be clearly tumorigenic in some cases, and several SASP factors have been reported to maintain or transmit the SNC state64,66,130,131. For example, SASP suppression via NF-κB inhibition in a mouse lymphoma model blunts immune surveillance following therapy-induced senescence and synergizes with p53 insufficiency for senescence escape, which drives treatment resistance and relapse132. Similarly, mechanistic target of rapamycin complex 1 (mTORC1) inhibition with rapamycin abolishes cytostatic p53 translation in prostate epithelial cells lacking PTEN, which is a common tumour suppressor lost in prostate cancer47. mTORC1 inhibition, which inhibits the SASP20, by rapamycin or analogues also has the side effect of broad immunosuppression133, which would limit its usefulness as part of a chronic senotherapy regimen.
Surprisingly, epigenetic disruption of the SASP may not influence SNC growth arrest. Knockdown of the epigenetic regulators mixed-lineage leukaemia protein 1 (MLL1; also known as KMT2A)134, bromodomain- containing protein 4 (BRD4)135 and HMGB2 (REF. 136) seem to prevent SASP emergence while leaving the cell cycle arrest unaffected. It is currently difficult to understand how SASP suppression via alteration of these epigenetic regulators does not permit cell cycle re-entry, given the known roles of SASP factors such as IL-6 in suppressing the cell cycle59,137. It will be important to determine if these findings hold true for animal models in which cancer risk and immunosuppression can be assessed, as well as to test the durability of the epigenetic reprogramming of SNCs. Intriguingly, a small- molecule inhibitor (inflachromene) of HMGB2 as well as the pro-inflammatory SASP factor HMGB1 has been identified and, although not yet tested specifically against SNCs, is effective in blocking microglial inflammation138. Further understanding of the SASP and maintenance of senescence versus senescence escape in vivo is needed.
A second approach involves blocking the secretion or maturation of the SASP. Although the signalling cascades initiating and maintaining the SASP are fairly well understood, less emphasis has been placed on understanding the nature of SASP secretion and maturation. What is collectively referred to as a ‘secretome’ is in reality a mix of classically secreted proteins42, cargoes packaged into exosomes139, ions (including reactive oxygen species)140, shed extracellular domains of transmembrane proteins141 and (likely) poorly characterized metabolites of other classes. Proteases are probably central to modulating the activity of the SASP, given their high activity in the SNC microenvironment (due to their inclusion in the secretome), activation at the SNC membrane and import through SNC-surveilling immune cells. For example, SASP interleukins of the IL-1 superfamily — including IL-1α, IL-1β, IL-18 (REF. 142) and IL-33 (REF. 143) — are matured through partial protease digestion to achieve maximal immune-stimulatory activity144. IL-1α, for example, can be cleaved by granzyme B — which is highly expressed by natural killer cells and cytotoxic T cells — mast cell chymase, neutrophil elastase and calpains, which are activated by elevated Ca2+ levels in SNCs145. In addition, high cell surface activity of a disintegrin and metalloproteinase 17 (ADAM17) in SNCs drives ectodomain shedding of amphiregulin and TNF receptor 1 (REF. 141). These findings suggest that agents targeted against SASP proteases may not only be effective in preventing aberrant ECM degradation caused by SNCs but may also impair protease-mediated activation of other SASP components. However, the proteases MMP1 and MMP3, which are present in the SASP, have been reported to cleave MCP1, which reduces its chemoattractant activity in vitro146. Therefore, not all proteases that are present in the SASP are necessarily good therapeutic targets and some — such as MMP1 and MMP3 — may act to restrict activity of other SASP factors.
Last, the blockade of specific SASP factors or their receptors, such as IL-6 or its receptor, has the benefit of a well-defined target and therefore potentially a low risk of off-target effects. A monoclonal antibody (mAb)- mediated approach, particularly one that is reusing existing drugs, could therefore potentially be applied here. For example, siltuximab (a mAb against IL-6) or tocilizumab (a mAb against the IL-6 receptor) are approved anti-inflammatory drugs147,148. However, a downside of this approach is that only one of many components can be targeted at a time. To date, no study of such agents in ageing or healthspan has been conducted.
Therapeutic considerations and challenges
Strategies to eliminate SNCs or disrupt their impact appear within reach. However, there are several challenges and important considerations that must be kept in mind for the successful development of senotherapies.
In vitro and in vivo models
Several important insights have emerged from early efforts to develop senolytic compounds. Different cell lineages react differently to a given stress and, indeed, SNCs derived from different cell types have overlapping but distinct molecular signatures41,42. As a consequence, SNCs in cell culture display different vulnerabilities to senolytics from different mechanistic classes. Furthermore, the method of senescence induction contributes to this heterogeneity, that is, making a cell senescent through overexpression of an oncogene renders it differentially sensitive to various senolytic molecules as compared to a cell made senescent via irradiation. It remains to be established what type of senescence induction method in vitro is most relevant to ageing or to any given disease. Identification of disease-relevant cellular systems that translate into preclinical in vivo models is necessary for the discovery of new senolytic molecules.
A senolytic molecule must selectively eliminate SNCs from disease-relevant tissues, attenuate the SASP and demonstrate disease-modifying characteristics in a relevant preclinical in vivo model in which SNCs drive the disease state (FIG. 3). Three categories of preclinical in vivo models are generally used to develop senolytic molecules. First, administration of known stressors (for example, chemotherapeutics, ionizing radiation or bleomycin) induces senescence in a selected tissue or throughout an animal. Second, surgical models are used where injury can induce SNC accumulation in the tissue of interest. Third, aged animals can be used. The combination of either of the first two types of models with the third (ageing) is useful, as human disease often presents in the context of old age. Given the significant differences between humans and mice, validation of senolytic activity in human clinical specimens, such as ex vivo cultures of chondrocytes from human arthritic knee joints, is a crucial component of preclinical testing.
Optimal diseases for senotherapy
To develop clinically meaningful senotherapeutics, the right disease must be matched with the right SNC targeting strategy. The optimal disease for initial senotherapeutic development should meet several criteria. A causal link between SNCs and disease progression should be well established. Furthermore, the mechanism by which SNCs drive disease, such as through the SASP, should be mechanistically understood and targeted using the correct senotherapy. If SNCs contribute both beneficial and detrimental effects, the risk– benefit trade-off should be assessed in preclinical models. In addition, the current standard of care for this disease should have significant drawbacks (for example, weakly effective, tolerability and adherence concerns, provide only temporary and symptomatic relief) that could be improved upon by a replacement with or the incorporation of senotherapy. Through consideration of the diseases to pursue, it is essential to establish credible, testable hypotheses by which a senotherapeutic agent would address the specific set of identified needs. Based on these criteria, osteoarthritis is a prime candidate SNC-driven disease for clinical testing of senotherapy. While preclinical studies have also identified patients with atherosclerosis as potential beneficiaries of senotherapy, osteoarthritis has the major advantage that senolytics can be administered locally, thus reducing the risk of undesirable side effects. Other age-related conditions amendable to local senolysis are glaucoma and idiopathic pulmonary fibrosis, but their clinical advancement is dependent on the outcome of ongoing preclinical research.
Biomarkers and patient selection
Diseases associated with ageing are likely to have a diverse aetiology. For example, the prevalence of senescence- associated osteoarthritis is currently unknown, as risk factors beyond that of age include obesity, metabolic disease, biomechanical instability and acute injury. It is unknown whether osteoarthritis that results from these varied risk factors involves SNCs. Biomarkers for senescence could both aid in selecting patients that are most likely to benefit from senotherapy and also provide a tool for assessing treatment response.
Development of biomarkers for use in clinical trials, particularly in an area of rapidly evolving biology such as senescence, is non-trivial. The path to biomarker discovery and development depends heavily on how these biomarkers will be used. For the development of a senolytic agent, markers of cellular senescence are most relevant. These include predictive markers, target occupancy markers and pharmacodynamic or target engagement markers. Although predictive biomarkers may be of greatest use in a clinical trial, prognostic biomarkers will also play a part, and much of the work done to date by others has been in the prognostic biomarker category149. For example, in regard to osteoarthritis, many inflammatory biomarkers have been associated with disease progression. It is likely that a significant proportion of these are related to the inflammatory nature of senescence150–153.
The identification of a pharmacodynamic biomarker is important, as it can be used for early insight into a treatment effect on a potential clinical end point, including safety. For senescence associated-disease, pharmacodynamic biomarkers are especially crucial given the novel therapeutic approach (that is, senolysis), the potential variability in response and the lack of other ways to determine dose escalation, dosing frequency and dose duration.
In a clinically heterogeneous human patient population with varied comorbidities, finding the biomarker signal for senescence-associated disease (whether predictive, prognostic or pharmacodynamic) among the noise of ageing, multiple disease states and chronic inflammation will be difficult. However, newer approaches, such as detection of circulating tumour-derived nucleic acids154, could potentially be repurposed for detection of SNC-derived RNA and therefore hold promise to provide specificity for senescence-associated disease biomarkers.
Safety and selectivity
For a senolytic to be safe, it must have minimal off- target effects in non-SNCs. In addition, biologically relevant killing of beneficial SNCs should be minimized. Notably, SNCs enhance wound healing, direct tissue patterning in embryogenesis, control vascularization of the placenta, potentially enhance tissue regeneration and promote insulin secretion by pancreatic β-cells during ageing12,14–18,152 (BOX 1). Interestingly, in mice, SNCs promote wound closure but are ultimately not essential, as wounds without SNCs still close, albeit more slowly7,15.
There are several strategies for optimizing selectivity. For example, selectivity for SNCs over non-SNCs could be achieved by targeting cellular signalling pathways — such as apoptotic pathways — that are specifically altered in SNCs, which makes them more vulnerable to elimination by modulation of apoptosis control proteins. Another strategy to achieve cellular selectivity may be to synthesize inactive prodrug molecules that can be converted into an active senolytic molecule within the cytoplasm of a SNC by an enzymatic activity that is specifically upregulated in SNCs. This approach has been employed to activate mesoporous silica nanoparticles coated with galacto-oligosaccharide, which is a substrate of the senescent biomarker SA-β-Gal. In senescent X-linked diskeratosis congenita cells that express SA-β-Gal, the galacto-oligosaccharide is cleaved, which releases rhodamine packaged within the silica nanoparticles155. Similar activation of a prodrug could be achieved extracellularly by enzymes that are part of the SASP. For example, one potential approach could be to create prodrugs that are cleaved by SASP-enriched MMPs.
Another approach to achieving selectivity could be to construct a targeted conjugate that combines a senolytic moiety with an epitope that selectively recognizes SNCs156. For example, oxidized vimentin157, DCR2 (REF. 158), density-enhanced phosphatase 1 (DEP1; also known as PTPRJ)159 and β2 microglobulin (B2MG)159 are overexpressed at the SNC cell surface and could be used for directing local accumulation of senolytics. However, these and other ‘SNC-specific’ epitopes may be broadly expressed in vivo or generally upregulated in aged tissue; controls are not typically found in the literature but important to designing therapies deployed against age-related disease.
Tissue selectivity could also be achieved by using local routes of administration. Based on reported findings in clinical studies with BCL-2 family inhibitors, these molecules are unlikely to have the required safety assessment profile for systemic use owing to thrombocytopenia and neutropenia160. Therefore, their use in the therapeutic setting could be initially achieved by local administration to diseased tissue. Alternatively, given that SNCs are non-dividing, elimination of SNCs in disease should not require chronic treatment or prolonged exposure but should instead only require acute treatment with shorter exposure. Acute dosing of molecules administered directly to the site where they are required to eliminate SNCs provides the opportunity to use molecules with short systemic half-lives, thereby minimizing the exposure of non-SNCs to these molecules.
Finally, intermittent, infrequent dosing of a senolytic may potentially limit off-target effects and avoids the unwanted reduction of acute, beneficial SNCs. Indeed, SNC killing twice per week in mice seems to lack side effects7.
Conclusions and outlook
Although our understanding of how SNCs may actively contribute to ageing and age-related diseases is in its infancy, the potential of therapeutically targeting SNCs is beginning to emerge. Through in vitro testing, several senolytic molecules have now been identified that show promising potency and selectivity. Furthermore, utilization of these ‘first-generation’ senolytic strategies in preclinical models of osteoarthritis and atherosclerosis are profoundly disease-modifying, perhaps through attenuation of the SASP8,11. This indicates that diseases driven by SNCs may be amenable to localized senotherapy clinical trials in the very near future.
However, whereas strategies to eliminate SNCs or disrupt their impact appear within reach, important considerations must be kept in mind. First, senotherapy has not yet been attempted in humans, and the demonstration that strategies targeting SNCs are both safe and effective is therefore paramount. In particular, drugs targeting SNCs, especially SASP suppressors, should be investigated carefully to ensure that they maintain permanent cell cycle arrest, as bypassing this process could drive tumorigenesis85,161. In addition, little is known about the actual senescence burden in various diseases, as our current methodologies require invasive approaches to acquire tissue samples. Without robust methods to retrieve and quantify SNCs in diseased tissue, we have limited molecular knowledge about these cells and their SASP profiles. As SNCs also have a variety of beneficial biological functions, identification of the beneficial components of the SASP could lead to strategies that eliminate SNCs while simultaneously supplementing or preserving SNC-derived crucial factors.
Ageing has not classically been considered a disease per se, and no studies to date have focused on how treatments impact the ageing process in general. However, in recent years, there has been interest in examining how long-term treatment of patients with metformin impacts healthspan, defined as time from emergence of a first feature of ageing to a second, independent feature162. For drugs designed to target ageing or age- related diseases, the patient population will be aged and potentially facing a myriad of different pathologies, which will be an inherent difficulty in assessing how these drugs affect a single isolated disease. The first clinical trials of senotherapies should therefore focus on short-term interventions with surrogate biomarkers and clear end points, such as delivery of senolytic molecules into an arthritic joint, where pain and mobility are rapidly assessed. A well-designed clinical trial has the potential to take a first-generation senolytic into clinical development in the near future.
Acknowledgments
The authors thank N.David and Y.Poon of Unity Biotechnology for invaluable intellectual contributions to this Review and for thoroughly editing the text, and C.Yohn for feedback on the manuscript. The writing of this Review was supported by a grant from the Paul F.Glenn Foundation (J.M.v.D. and D.J.B.) and US National Institutes of Health (NIH) grants R01CA96985 and CA168709 (J.M.v.D.).
Glossary
- Senolysis
The therapeutic killing of senescent cells using small molecules (also known as senolytics)
- Senescence
A tumour-suppressive cell fate undertaken in response to irreparable damage. It is characterized by permanent withdrawl from the cell cycle and acquisition of a pro-inflammatory, proteolytic secretome
- Senotherapies
Therapeutic strategies that aim to neutralize the deleterious effects of senescent cells as a treatment for age-related diseases
- Senescence-associated β-galactosidase
(SA-β-Gal). A lysosomal hydrolase with optimal activity at pH 6.0 in senescent cells (SNCs). Detection of SA-β-Gal enzymatic activity is frequently used to stain SNCs in vitro and in vivo
- Cyclin-dependent kinase inhibitor
(CDKi). A protein that arrests the cell cycle by binding to and deactivating cyclin-dependent kinases. p16INK4A, p19ARF, and p21 belong to this class of proteins and are commonly used as senescence markers
- Senescence-associated secretory phenotype
(SASP). The suite of secreted factors produced by senescent cells, including metalloproteinases, cytokines, chemokines, and growth factors, as well as non-protein metabolites
- Cytokines
Secreted protein factors that act as ligands for receptor- mediated cell signalling
- Chemokines
Cytokines, such as monocyte chemotactic protein 1 (MCP1), promote cellular migration (also known as chemotaxis)
- BCL-2 family members
A protein class that consists of 25 members that share B cell lymphoma 2 (BCL-2) homology domains. These proteins either inhibit or promote mitochondrion-mediated apoptosis. BCL-2, BCL-W, and BCL-XL are three anti-apoptotic members that are inhibited by the senolytic and chemotherapeutic compound navitoclax
- High-mobility group box 1
(HMGB1). A member of a class of non-histone, chromatin proteins that modify transcription by binding to and distorting DNA. HMGB1 is released from senescent cells as an inflammatory ‘alarmin’. Another member, HMGB2, is a positive regulator of senescence-associated secretory phenotype (SASP) factor transcription
- Metalloproteinases
A group of enzymes that cleave a peptide bond through the catalytic action of a coordinated metal ion (often zinc)
Footnotes
Competing interests statement
J.M.v.D. is a cofounder of Unity Biotechnology, which is a company developing senolytic medicines, including small molecules that selectively eliminate senescent cells. R.M.L., J.M.v.D., D.J.B. and B.G.C. are co-inventors on patent applications licensed to or filed by Unity Biotechnology. R.M.L., J.D. and D.M. are employed by Unity Biotechnology.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Weismann A. In: Collected Essays upon Heredity and Kindred Biological Problems. Poulton EB, editor. Clarendon; 1881. [Google Scholar]
- 2.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. First detection of cellular senescence in vitro. [DOI] [PubMed] [Google Scholar]
- 3.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 5.van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408. doi: 10.1038/nrc3960. This review comprehensively examined the question of SNC detection, quantification and identification in vivo, and is an excellent resource for new investigators to the field. [DOI] [PubMed] [Google Scholar]
- 7.Baker DJ, et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. This is a first demonstration that SNCs reduce lifespan and negatively impact healthspan in naturally-aged, non-progeroid mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeon OH, et al. Clearance of senescent cells prevents the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23:775–781. doi: 10.1038/nm.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker DJ, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10:825–836. doi: 10.1038/ncb1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. Proof-of-concept study for transgene-mediated SNC killing and first demonstration that removing SNCsonce they arise extends healthspan in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Childs BG, et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–477. doi: 10.1126/science.aaf6659. This study links the pro-inflammatory, proteolytic properties of plaque SNCs to progression and destabilization of atherosclerosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munoz-Espin D, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–1118. doi: 10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 13.Mosteiro L, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016;354:aaf4445. doi: 10.1126/science.aaf4445. [DOI] [PubMed] [Google Scholar]
- 14.Storer M, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–1130. doi: 10.1016/j.cell.2013.10.041. References 12 and 14 show that cellular senescence contributes to tissue patterning during mammalian embryogenesis. [DOI] [PubMed] [Google Scholar]
- 15.Demaria M, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chuprin A, et al. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013;27:2356–2366. doi: 10.1101/gad.227512.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li T, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helman A, et al. p16Ink4a-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med. 2016;22:412–420. doi: 10.1038/nm.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudin CM, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laberge RM, et al. MTOR regulates the pro- tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17:1049–1061. doi: 10.1038/ncb3195. This study provides proof of concept that the inflammatory SNC secretory phenotype could be pharmacologically inhibited using an approved drug (rapamycin). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83. doi: 10.1038/nm.4010. First demonstration that pharmacological inhibition of anti-apoptotic BCL-2 family members shows senescent cell killing (senolytic) activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging. 2016;8:2915–2926. doi: 10.18632/aging.101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14:644–658. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yosef R, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016;7:11190. doi: 10.1038/ncomms11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karatza C, Stein WD, Shall S. Kinetics of in vitro ageing of mouse embryo fibroblasts. J Cell Sci. 1984;65:163–175. doi: 10.1242/jcs.65.1.163. [DOI] [PubMed] [Google Scholar]
- 26.Ponten J, Stein WD, Shall S. A quantitative analysis of the aging of human glial cells in culture. J Cell Physiol. 1983;117:342–352. doi: 10.1002/jcp.1041170309. [DOI] [PubMed] [Google Scholar]
- 27.Cristofalo VJ, Sharf BB. Cellular senescence and DNA synthesis. Thymidine incorporation as a measure of population age in human diploid cells. Exp Cell Res. 1973;76:419–427. doi: 10.1016/0014-4827(73)90394-7. [DOI] [PubMed] [Google Scholar]
- 28.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 29.Dimri GP, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. This study reports the development of the SA-β-Gal stain, which is a widely used biomarker for detecting SNCs in vitro and in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 31.Alcorta DA, et al. Involvement of the cyclin- dependent kinase inhibitor p16INK4a in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Serrano M, et al. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 33.Beausejour CM, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Z, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. This is a first example of a loss of tumour suppressor function (PTEN insufficiency), which results in cellular senescence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evangelou K, et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell. 2017;16:192–197. doi: 10.1111/acel.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minamino T, et al. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544. doi: 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 37.Krishnamurthy J, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 39.Aguilera A, Garcia-Muse T. Causes of genome instability. Annu Rev Genet. 2013;47:1–32. doi: 10.1146/annurev-genet-111212-133232. [DOI] [PubMed] [Google Scholar]
- 40.Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell. 2010;141:81–93. doi: 10.1016/j.cell.2010.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coppe JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coppe JP, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE. 2010;5:e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodier F, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodier F, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124:68–81. doi: 10.1242/jcs.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sturmlechner I, Durik M, Sieben CJ, Baker DJ, van Deursen JM. Cellular senescence in renal ageing and disease. Nat Rev Nephrol. 2017;13:77–89. doi: 10.1038/nrneph.2016.183. [DOI] [PubMed] [Google Scholar]
- 46.Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age- related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–1435. doi: 10.1038/nm.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alimonti A, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest. 2010;120:681–693. doi: 10.1172/JCI40535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bartkova J, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 49.Pluquet O, Pourtier A, Abbadie C. The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am J Physiol Cell Physiol. 2015;308:C415–C425. doi: 10.1152/ajpcell.00334.2014. [DOI] [PubMed] [Google Scholar]
- 50.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- 51.Eischen CM, Lozano G. The Mdm network and its regulation of p53 activities: a rheostat of cancer risk. Hum Mutat. 2014;35:728–737. doi: 10.1002/humu.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herbig U, Wei W, Dutriaux A, Jobling WA, Sedivy JM. Real-time imaging of transcriptional activation in live cells reveals rapid up-regulation of the cyclin-dependent kinase inhibitor gene CDKN1A in replicative cellular senescence. Aging Cell. 2003;2:295–304. doi: 10.1046/j.1474-9728.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- 53.Wong ES, et al. p38MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Dev Cell. 2009;17:142–149. doi: 10.1016/j.devcel.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 54.Ohtani N, et al. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–1070. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- 55.Takahashi A, et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8:1291–1297. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- 56.Johmura Y, et al. Necessary and sufficient role for a mitosis skip in senescence induction. Mol Cell. 2014;55:73–84. doi: 10.1016/j.molcel.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 57.Le ON, et al. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell. 2010;9:398–409. doi: 10.1111/j.1474-9726.2010.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Acosta JC, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 59.Kuilman T, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. References 41, 58 and 59 established that the secretory profile of SNCs is a hallmark of this cell type. [DOI] [PubMed] [Google Scholar]
- 60.Acosta JC, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–990. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 62.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 63.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoare M, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol. 2016;18:979–992. doi: 10.1038/ncb3397. This study demonstrated that the SNC secretory profile changes composition over time to coordinate distinct paracrine signalling events to immune cells and surrounding tissue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/ IL-8 cytokine network. Proc Natl Acad Sci USA. 2009;106:17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eyman D, Damodarasamy M, Plymate SR, Reed MJ. CCL5 secreted by senescent aged fibroblasts induces proliferation of prostate epithelial cells and expression of genes that modulate angiogenesis. J Cell Physiol. 2009;220:376–381. doi: 10.1002/jcp.21776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo X, et al. Stromal-initiated changes in the bone promote metastatic niche development. Cell Rep. 2016;14:82–92. doi: 10.1016/j.celrep.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Angelini PD, et al. Constitutive HER2 signaling promotes breast cancer metastasis through cellular senescence. Cancer Res. 2013;73:6095–6095. doi: 10.1158/0008-5472.CAN-12-2301. [DOI] [PubMed] [Google Scholar]
- 69.Ruhland MK, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762. doi: 10.1038/ncomms11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eggert T, et al. Distinct functions of senescence- associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30:533–547. doi: 10.1016/j.ccell.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ritschka B, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31:172–183. doi: 10.1101/gad.290635.116. This work implicated signals arising from the SNC secretome to stemness during tissue regeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Inoue K, et al. Serial coronary CT angiography-verified changes in plaque characteristics as an end point: evaluation of effect of statin intervention. JACC Cardiovasc Imaging. 2010;3:691–698. doi: 10.1016/j.jcmg.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 73.Eren M, et al. PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc Natl Acad Sci USA. 2014;111:7090–7095. doi: 10.1073/pnas.1321942111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117–3126. doi: 10.1158/0008-5472.CAN-06-3452. [DOI] [PubMed] [Google Scholar]
- 75.Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014;15:1139–1153. doi: 10.15252/embr.201439245. This review gives a broad overview of the possible advantages of senescence compared with apoptosis as a fate for damaged cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burton DG, Faragher RG. Cellular senescence: from growth arrest to immunogenic conversion. Age (Dordr) 2015;37:27. doi: 10.1007/s11357-015-9764-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tavana O, et al. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle. 2010;9:3328–3336. doi: 10.4161/cc.9.16.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Webley K, et al. Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Mol Cell Biol. 2000;20:2803–2808. doi: 10.1128/mcb.20.8.2803-2808.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Timofeev O, et al. p53 DNA binding cooperativity is essential for apoptosis and tumor suppression in vivo. Cell Rep. 2013;3:1512–1525. doi: 10.1016/j.celrep.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y, et al. DNMT3a plays a role in switches between doxorubicin-induced senescence and apoptosis of colorectal cancer cells. Int J Cancer. 2011;128:551–561. doi: 10.1002/ijc.25365. [DOI] [PubMed] [Google Scholar]
- 81.Hayward RL, et al. Antisense Bcl-xl down-regulation switches the response to topoisomerase I inhibition from senescence to apoptosis in colorectal cancer cells, enhancing global cytotoxicity. Clin Cancer Res. 2003;9:2856–2865. [PubMed] [Google Scholar]
- 82.Tang JJ, Shen C, Lu YJ. Requirement for pre- existing of p21 to prevent doxorubicin-induced apoptosis through inhibition of caspase-3 activation. Mol Cell Biochem. 2006;291:139–144. doi: 10.1007/s11010-006-9206-7. [DOI] [PubMed] [Google Scholar]
- 83.Hanks S, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159–1161. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 84.Donehower LA. The p53-deficient mouse: a model for basic and applied cancer studies. Semin Cancer Biol. 1996;7:269–278. doi: 10.1006/scbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- 85.Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413:83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- 86.Schafer MJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kang TW, et al. Senescence surveillance of pre- malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. This work demonstrates that the anticancer effects of cellular senescence extend beyond cell cycle arrest, in particular that SNCs may promote recruitment of tumour-suppressive immune effectors. [DOI] [PubMed] [Google Scholar]
- 88.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. This study provided early support for the hypothesis that SNCs promote carcinogenesis in a cell non-autonomous fashion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bar-Shai A, Sagiv A, Alon R, Krizhanovsky V. The role of Clara cell senescence in the pathogenesis of COPD. Eur Respir J. 2014;44:3245. [Google Scholar]
- 90.Bhat R, et al. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE. 2012;7:e45069. doi: 10.1371/journal.pone.0045069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Demaria M, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–176. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23:2066–2075. doi: 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Laberge RM, et al. Mitochondrial DNA damage induces apoptosis in senescent cells. Cell Death Dis. 2013;4:e727. doi: 10.1038/cddis.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Demaria M, Desprez PY, Campisi J, Velarde MC. Cell autonomous and non-autonomous effects of senescent cells in the skin. J Invest Dermatol. 2015;135:1722–1726. doi: 10.1038/jid.2015.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roberts S, Evans EH, Kletsas D, Jaffray DC, Eisenstein SM. Senescence in human intervertebral discs. Eur Spine J. 2006;15(Suppl. 3):S312–S316. doi: 10.1007/s00586-006-0126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Le Maitre CL, Freemont AJ, Hoyland JA. Accelerated cellular senescence in degenerate intervertebral discs: a possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Res Ther. 2007;9:R45. doi: 10.1186/ar2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sousa-Victor P, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- 98.Cosgrove BD, et al. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med. 2014;20:255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bernet JD, et al. p38 MAPK signaling underlies a cell- autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med. 2014;20:265–271. doi: 10.1038/nm.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Du J, et al. Aging increases CCN1 expression leading to muscle senescence. Am J Physiol Cell Physiol. 2014;306:C28–C36. doi: 10.1152/ajpcell.00066.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30:1282–1292. doi: 10.1161/ATVBAHA.108.179739. [DOI] [PubMed] [Google Scholar]
- 102.Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. doi: 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 104.Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!) Osteoarthritis Cartilage. 2013;21:16–21. doi: 10.1016/j.joca.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 105.Wieland HA, Michaelis M, Kirschbaum BJ, Rudolphi KA. Osteoarthritis — an untreatable disease? Nat Rev Drug Discov. 2005;4:331–344. doi: 10.1038/nrd1693. [DOI] [PubMed] [Google Scholar]
- 106.Lawrence RC, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58:26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.March L, Amatya B, Osborne RH, Brand C. Developing a minimum standard of care for treating people with osteoarthritis of the hip and knee. Best Pract Res Clin Rheumatol. 2010;24:121–145. doi: 10.1016/j.berh.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 108.Krizhanovsky V, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. This work shows a beneficial function (fibrosis restriction) for cellular senescence beyond tumour suppression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Luna-Vargas MP, Chipuk JE. Physiological and pharmacological control of BAK, BAX, and beyond. Trends Cell Biol. 2016;26:906–917. doi: 10.1016/j.tcb.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tse C, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 111.Zhu Y, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15:428–435. doi: 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bai L, Wang S. Targeting apoptosis pathways for new cancer therapeutics. Annu Rev Med. 2014;65:139–155. doi: 10.1146/annurev-med-010713-141310. [DOI] [PubMed] [Google Scholar]
- 113.Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib- resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007;7:345–356. doi: 10.1038/nrc2126. [DOI] [PubMed] [Google Scholar]
- 114.Boots AW, Haenen GR, Bast A. Health effects of quercetin: from antioxidant to nutraceutical. Eur J Pharmacol. 2008;585:325–337. doi: 10.1016/j.ejphar.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 115.Chondrogianni N, et al. Anti-ageing and rejuvenating effects of quercetin. Exp Gerontol. 2010;45:763–771. doi: 10.1016/j.exger.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 116.Baar MP, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017;169:132–147.e16. doi: 10.1016/j.cell.2017.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roos CM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15:973–977. doi: 10.1111/acel.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. This work tested and validated the hypothesis that SNCs can be removed by the immune system and provided support for the idea that the SNC secretome drives this immune surveillance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vindrieux D, et al. Down-regulation of DcR2 sensitizes androgen-dependent prostate cancer LNCaP cells to TRAIL-induced apoptosis. Cancer Cell Int. 2011;11:42. doi: 10.1186/1475-2867-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang Z, Rosen DG, Yao JL, Huang J, Liu J. Expression of p14ARF, p15INK4b, p16INK4a, and DCR2 increases during prostate cancer progression. Mod Pathol. 2006;19:1339–1343. doi: 10.1038/modpathol.3800655. [DOI] [PubMed] [Google Scholar]
- 122.Aydin C, et al. Decoy receptor-2 small interfering RNA (siRNA) strategy employing three different siRNA constructs in combination defeats adenovirus- transferred tumor necrosis factor-related apoptosis- inducing ligand resistance in lung cancer cells. Hum Gene Ther. 2007;18:39–50. doi: 10.1089/hum.2006.111. [DOI] [PubMed] [Google Scholar]
- 123.Wu Q, et al. Aberrant expression of decoy receptor 3 in human breast cancer: relevance to lymphangiogenesis. J Surg Res. 2014;188:459–465. doi: 10.1016/j.jss.2014.01.058. [DOI] [PubMed] [Google Scholar]
- 124.Ge Z, Sanders AJ, Ye L, Jiang WG. Aberrant expression and function of death receptor-3 and death decoy receptor-3 in human cancer. Exp Ther Med. 2011;2:167–172. doi: 10.3892/etm.2011.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sagiv A, et al. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene. 2013;32:1971–1977. doi: 10.1038/onc.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab Invest. 2009;89:1195–1220. doi: 10.1038/labinvest.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.He J, Hu Y, Hu M, Li B. Development of PD-1/ PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci Rep. 2015;5:13110. doi: 10.1038/srep13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Konkel JE, et al. PD-1 signalling in CD4+ T cells restrains their clonal expansion to an immunogenic stimulus, but is not critically required for peptide- induced tolerance. Immunology. 2010;130:92–102. doi: 10.1111/j.1365-2567.2009.03216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang G, et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci USA. 2006;103:16472–16477. doi: 10.1073/pnas.0605752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Elzi DJ, et al. Plasminogen activator inhibitor 1— insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc Natl Acad Sci USA. 2012;109:12052–12057. doi: 10.1073/pnas.1120437109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chien Y, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Baroja-Mazo A, Revilla-Nuin B, Ramirez P, Pons JA. Immunosuppressive potency of mechanistic target of rapamycin inhibitors in solid-organ transplantation. World J Transplant. 2016;6:183–192. doi: 10.5500/wjt.v6.i1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Capell BC, et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 2016;30:321–336. doi: 10.1101/gad.271882.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tasdemir N, et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 2016;6:612–629. doi: 10.1158/2159-8290.CD-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aird KM, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol. 2016;215:325–334. doi: 10.1083/jcb.201608026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kojima H, Inoue T, Kunimoto H, Nakajima K. IL-6-STAT3 signaling and premature senescence. JAK- STAT. 2013;2:e25763. doi: 10.4161/jkst.25763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee S, et al. A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nat Chem Biol. 2014;10:1055–1060. doi: 10.1038/nchembio.1669. [DOI] [PubMed] [Google Scholar]
- 139.Lehmann BD, et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008;68:7864–7871. doi: 10.1158/0008-5472.CAN-07-6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xin MG, Zhang J, Block ER, Patel JM. Senescence-enhanced oxidative stress is associated with deficiency of mitochondrial cytochrome c oxidase in vascular endothelial cells. Mech Ageing Dev. 2003;124:911–919. doi: 10.1016/s0047-6374(03)00163-5. [DOI] [PubMed] [Google Scholar]
- 141.Effenberger T, et al. Senescence-associated release of transmembrane proteins involves proteolytic processing by ADAM17 and microvesicle shedding. FASEB J. 2014;28:4847–4856. doi: 10.1096/fj.14-254565. [DOI] [PubMed] [Google Scholar]
- 142.Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet D. H p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. 2013;210:2057–2069. doi: 10.1084/jem.20130783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rovillain E, et al. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene. 2011;30:2356–2366. doi: 10.1038/onc.2010.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. 2015;42:991–1004. doi: 10.1016/j.immuni.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 145.McCarthy DA, Clark RR, Bartling TR, Trebak M, Melendez JA. Redox control of the senescence regulator interleukin-1alpha and the secretory phenotype. J Biol Chem. 2013;288:32149–32159. doi: 10.1074/jbc.M113.493841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McQuibban GA, et al. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160–1167. [PubMed] [Google Scholar]
- 147.van Rhee F, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15:966–974. doi: 10.1016/S1470-2045(14)70319-5. [DOI] [PubMed] [Google Scholar]
- 148.Emery P, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67:1516–1523. doi: 10.1136/ard.2008.092932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mobasheri A, Bay-Jensen AC, van Spil WE, Larkin J, Levesque MC. Osteoarthritis year in review 2016: biomarkers (biochemical markers) Osteoarthritis Cartilage. 2017;25:199–208. doi: 10.1016/j.joca.2016.12.016. [DOI] [PubMed] [Google Scholar]
- 150.Fernandez-Puente P, et al. Identification of a panel of novel serum osteoarthritis biomarkers. J Proteome Res. 2011;10:5095–5101. doi: 10.1021/pr200695p. [DOI] [PubMed] [Google Scholar]
- 151.Kraus VB, et al. OARSI clinical trials recommendations: soluble biomarker assessments in clinical trials in osteoarthritis. Osteoarthritis Cartilage. 2015;23:686–697. doi: 10.1016/j.joca.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bay-Jensen AC, et al. Osteoarthritis year in review 2015: soluble biomarkers and the BIPED criteria. Osteoarthritis Cartilage. 2016;24:9–20. doi: 10.1016/j.joca.2015.10.014. [DOI] [PubMed] [Google Scholar]
- 153.Ahmed U, et al. Biomarkers of early stage osteoarthritis, rheumatoid arthritis and musculoskeletal health. Sci Rep. 2015;5:9259. doi: 10.1038/srep09259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schwarzenbach H, Hoon DSB, Pantel K. Cell- free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–437. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 155.Agostini A, et al. Targeted cargo delivery in senescent cells using capped mesoporous silica nanoparticles. Angew Chem Int Ed. 2012;51:10556–10560. doi: 10.1002/anie.201204663. [DOI] [PubMed] [Google Scholar]
- 156.Polakis P. Antibody drug conjugates for cancer therapy. Pharmacol Rev. 2016;68:3–19. doi: 10.1124/pr.114.009373. [DOI] [PubMed] [Google Scholar]
- 157.Frescas D, et al. Senescent cells expose and secrete an oxidized form of membrane-bound vimentin as revealed by a natural polyreactive antibody. Proc Natl Acad Sci USA. 2017;114:E1668–E1677. doi: 10.1073/pnas.1614661114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Collado M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 159.Althubiti M, et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014;5:e1528. doi: 10.1038/cddis.2014.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kaefer A, et al. Mechanism-based pharmacokinetic/ pharmacodynamic meta-analysis of navitoclax (ABT-263) induced thrombocytopenia. Cancer Chemother Pharmacol. 2014;74:593–602. doi: 10.1007/s00280-014-2530-9. [DOI] [PubMed] [Google Scholar]
- 161.Sharpless NE, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. This is an important demonstration of the tumour-suppressive properties of a key senescence effector, p16INK4A. [DOI] [PubMed] [Google Scholar]
- 162.Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a tool to target aging. Cell Metab. 2016;23:1060–1065. doi: 10.1016/j.cmet.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH, DePinho RA. The differential impact of p16INK4a or p19ARF deficiency on cell growth and tumorigenesis. Oncogene. 2004;23:379–385. doi: 10.1038/sj.onc.1207074. [DOI] [PubMed] [Google Scholar]
- 165.Cairns P, et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet. 1995;11:210–212. doi: 10.1038/ng1095-210. [DOI] [PubMed] [Google Scholar]
- 166.Herman JG, et al. Inactivation of the CDKN2/p16/ MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–4530. [PubMed] [Google Scholar]
- 167.Merlo A, et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 168.Khanna AK. Enhanced susceptibility of cyclin kinase inhibitor p21 knockout mice to high fat diet induced atherosclerosis. J Biomed Sci. 2009;16:66. doi: 10.1186/1423-0127-16-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Visel A, et al. Targeted deletion of the 9p21 non- coding coronary artery disease risk interval in mice. Nature. 2010;464:409–412. doi: 10.1038/nature08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chiche A, et al. Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell. 2017;20:407–414.e4. doi: 10.1016/j.stem.2016.11.020. [DOI] [PubMed] [Google Scholar]
- 171.Guan X, et al. Stromal senescence by prolonged CDK4/6 inhibition potentiates tumor growth. Mol Cancer Res. 2017;15:237–249. doi: 10.1158/1541-7786.MCR-16-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cheung-Ong K, Giaever G, Nislow C. DNA- damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013;20:648–659. doi: 10.1016/j.chembiol.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 173.Yeh CK. Cellular senescence and aging. Oral Dis. 2016;22:587–590. doi: 10.1111/odi.12483. [DOI] [PubMed] [Google Scholar]
- 174.Oeffinger KC, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355:1572–1582. doi: 10.1056/NEJMsa060185. [DOI] [PubMed] [Google Scholar]
- 175.Schuitema I, et al. Accelerated aging, decreased white matter integrity, and associated neuropsychological dysfunction 25 years after pediatric lymphoid malignancies. J Clin Oncol. 2013;31:3378–3388. doi: 10.1200/JCO.2012.46.7050. [DOI] [PubMed] [Google Scholar]
- 176.Dorth JA, Patel PR, Broadwater G, Brizel DM. Incidence and risk factors of significant carotid artery stenosis in asymptomatic survivors of head and neck cancer after radiotherapy. Head Neck. 2014;36:215–219. doi: 10.1002/hed.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Anzidei M, et al. Longitudinal assessment of carotid atherosclerosis after radiation therapy using computed tomography: a case control Study. Eur Radiol. 2016;26:72–78. doi: 10.1007/s00330-015-3753-9. [DOI] [PubMed] [Google Scholar]
- 178.Wildiers H, et al. International Society of Geriatric Oncology consensus on geriatric assessment in older patients with cancer. J Clin Oncol. 2014;32:2595–2603. doi: 10.1200/JCO.2013.54.8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yip C, et al. Assessment of sarcopenia and changes in body composition after neoadjuvant chemotherapy and associations with clinical outcomes in oesophageal cancer. Eur Radiol. 2014;24:998–1005. doi: 10.1007/s00330-014-3110-4. [DOI] [PubMed] [Google Scholar]
- 180.Armstrong GT, et al. Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol. 2014;32:1218–1227. doi: 10.1200/JCO.2013.51.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Coppe JP, et al. Tumor suppressor and aging biomarker p16I NK4a induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–36403. doi: 10.1074/jbc.M111.257071. This work clearly demonstrated the separability of the SNC secretome from cell cycle arrest. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lan L, et al. Shp2 signaling suppresses senescence in PyMT-induced mammary gland cancer in mice. EMBO J. 2015;34:2383. doi: 10.15252/embj.201592508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Serrano M. SHP2: a new target for pro-senescence cancer therapies. EMBO J. 2015;34:1439–1441. doi: 10.15252/embj.201591616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fukazawa R, et al. Coronary artery aneurysm induced by Kawasaki disease in children show features typical senescence. Circ J. 2007;71:709–715. doi: 10.1253/circj.71.709. [DOI] [PubMed] [Google Scholar]
- 185.Liton PB, et al. Cellular senescence in the glaucomatous outflow pathway. Exp Gerontol. 2005;40:745–748. doi: 10.1016/j.exger.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhu D, Wu J, Spee C, Ryan SJ, Hinton DR. BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration. J Biol Chem. 2009;284:9529–9539. doi: 10.1074/jbc.M809393200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mishima K, et al. Senescence-associated beta- galactosidase histochemistry for the primate eye. Invest Ophthalmol Vis Sci. 1999;40:1590–1593. [PubMed] [Google Scholar]
- 188.Sohn JJ, et al. Macrophages, nitric oxide and microRNAs are associated with DNA damage response pathway and senescence in inflammatory bowel disease. PLoS ONE. 2012;7:e44156. doi: 10.1371/journal.pone.0044156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Martin JA, Brown TD, Heiner AD, Buckwalter JA. Chondrocyte senescence, joint loading and osteoarthritis. Clin Orthop Relat Res. 2004;427(Suppl):S96–S103. doi: 10.1097/01.blo.0000143818.74887.b1. [DOI] [PubMed] [Google Scholar]
- 190.Yanai H, et al. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging. 2015;7:664–672. doi: 10.18632/aging.100807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Aoshiba K, Tsuji T, Nagai A. Bleomycin induces cellular senescence in alveolar epithelial cells. Eur Respir J. 2003;22:436–443. doi: 10.1183/09031936.03.00011903. [DOI] [PubMed] [Google Scholar]
- 192.Aoshiba K, et al. Senescence-associated secretory phenotype in a mouse model of bleomycin-induced lung injury. Exp Toxicol Pathol. 2013;65:1053–1062. doi: 10.1016/j.etp.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 193.Fischer BM, et al. Increased expression of senescence markers in cystic fibrosis airways. Am J Physiol Lung Cell Mol Physiol. 2013;304:L394–L400. doi: 10.1152/ajplung.00091.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Markowski DN, et al. HMGA2 expression in white adipose tissue linking cellular senescence with diabetes. Genes Nutr. 2013;8:449–456. doi: 10.1007/s12263-013-0354-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Minamino T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15:1082–1087. doi: 10.1038/nm.2014. [DOI] [PubMed] [Google Scholar]
- 196.Fitzner B, et al. Senescence determines the fate of activated rat pancreatic stellate cells. J Cell Mol Med. 2012;16:2620–2630. doi: 10.1111/j.1582-4934.2012.01573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Morgan RG, et al. Role of arterial telomere dysfunction in hypertension: relative contributions of telomere shortening and telomere uncapping. J Hypertension. 2014;32:1293–1299. doi: 10.1097/HJH.0000000000000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mercer J, Figg N, Stoneman V, Braganza D, Bennett MR. Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ Res. 2005;96:667–674. doi: 10.1161/01.RES.0000161069.15577.ca. [DOI] [PubMed] [Google Scholar]
- 199.Diez-Juan A, Andres V. The growth suppressor p27(Kip1) protects against diet-induced atherosclerosis. FASEB J. 2001;15:1989–1995. doi: 10.1096/fj.01-0130com. [DOI] [PubMed] [Google Scholar]
- 200.Baker DJ, Weaver RL, van Deursen J. M p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep. 2013;3:1164–1174. doi: 10.1016/j.celrep.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Braun H, et al. Cellular senescence limits regenerative capacity and allograft survival. J Am Soc Nephrol. 2012;23:1467–1473. doi: 10.1681/ASN.2011100967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhu F, et al. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS ONE. 2013;8:e74535. doi: 10.1371/journal.pone.0074535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shivshankar P, et al. Caveolin-1 deficiency protects from pulmonary fibrosis by modulating epithelial cell senescence in mice. Am J Respir Cell Mol Biol. 2012;47:28–36. doi: 10.1165/rcmb.2011-0349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Li H, et al. FOXP1 controls mesenchymal stem cell commitment and senescence during skeletal aging. J Clin Invest. 2017;127:1241–1253. doi: 10.1172/JCI89511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Green DRA. BH3 mimetic for killing cancer cells. Cell. 2016;165:1560. doi: 10.1016/j.cell.2016.05.080. [DOI] [PubMed] [Google Scholar]
- 206.Maruthur NM, et al. Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med. 2016;164:740–751. doi: 10.7326/M15-2650. [DOI] [PubMed] [Google Scholar]
- 207.Moiseeva O, et al. Metformin inhibits the senescence- associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell. 2013;12:489–498. doi: 10.1111/acel.12075. [DOI] [PubMed] [Google Scholar]
- 208.Noren Hooten N, et al. Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell. 2016;15:572–581. doi: 10.1111/acel.12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Webster AC, Lee VW, Chapman JR, Craig JC. Target of rapamycin inhibitors (sirolimus and everolimus) for primary immunosuppression of kidney transplant recipients: a systematic review and meta-analysis of randomized trials. Transplantation. 2006;81:1234–1248. doi: 10.1097/01.tp.0000219703.39149.85. [DOI] [PubMed] [Google Scholar]
- 210.Jobanputra P, Barton P, Bryan S, Burls A. The effectiveness of infliximab and etanercept for the treatment of rheumatoid arthritis: a systematic review and economic evaluation. Health Technol Assess. 2002;6:1–110. doi: 10.3310/hta6210. [DOI] [PubMed] [Google Scholar]
- 211.Kuemmerle-Deschner JB, et al. Canakinumab (ACZ885, a fully human IgG1 anti-IL-1beta mAb) induces sustained remission in pediatric patients with cryopyrin-associated periodic syndrome (CAPS) Arthritis Res Ther. 2011;13:R34. doi: 10.1186/ar3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hoffman HM, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin- associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58:2443–2452. doi: 10.1002/art.23687. [DOI] [PubMed] [Google Scholar]
- 213.Klareskog L, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363:675–681. doi: 10.1016/S0140-6736(04)15640-7. [DOI] [PubMed] [Google Scholar]
- 214.Cohen S, et al. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:614–624. doi: 10.1002/art.10141. [DOI] [PubMed] [Google Scholar]