

Abstract
Family A G protein-coupled receptors (GPCRs) control diverse biological processes and are of great clinical relevance. Their archetype rhodopsin becomes naturally light sensitive by binding covalently to the photoswitchable tethered ligand (PTL) retinal. Other GPCRs, however, neither bind covalently to ligands nor are light sensitive. We sought to impart the logic of rhodopsin to light-insensitive Family A GPCRs in order to enable their remote control in a receptor-specific, cell-type-specific, and spatiotemporally precise manner. Dopamine receptors (DARs) are of particular interest for their roles in motor coordination, appetitive, and aversive behavior, as well as neuropsychiatric disorders such as Parkinson’s disease, schizophrenia, mood disorders, and addiction. Using an azobenzene derivative of the well-known DAR ligand 2-(N-phenethyl-N-propyl)amino-5-hydroxytetralin (PPHT), we were able to rapidly, reversibly, and selectively block dopamine D1 and D2 receptors (D1R and D2R) when the PTL was conjugated to an engineered cysteine near the dopamine binding site. Depending on the site of tethering, the ligand behaved as either a photoswitchable tethered neutral antagonist or inverse agonist. Our results indicate that DARs can be chemically engineered for selective remote control by light and provide a template for precision control of Family A GPCRs.
Graphical Abstract
INTRODUCTION
Heptahelical G protein-coupled receptors (GPCRs) constitute the largest superfamily of membrane proteins.1 GPCRs are activated by diverse stimuli (e.g., hormones, odorants, or neurotransmitters) and regulate a wide variety of biological processes via heterotrimeric G protein and arrestin signaling proteins. The Family A GPCRs make up the majority of the GPCR superfamily (~85%)1 and represent targets of 25–30% of all available medications.2 Recent genetic and structural advancements have substantially enhanced our understanding of this receptor family.3 However, because of the high degree of similarity between related Family A GPCRs as well as their heterogeneous expression across diverse cell types throughout the body, it has been difficult to selectively target and thus uncover the roles of individual receptors in vivo.
A major challenge in neuroscience has been to elucidate the physiological and behavioral roles of neuromodulator-binding Family A GPCRs that regulate synaptic and circuit function in the central nervous system. Dopamine (DA) and its five receptors (DARs) are particularly notable, as they control critical functions such as locomotion, learning, and memory.4 DARs are divided into two subfamilies based on sequence homology and function. Whereas D1-like receptors (D1/D5) couple to Gs/olf, D2-like receptors (D2/D3/D4) couple to Gi/o/z.5 These G proteins have opposing actions on adenylyl cyclases, which catalyze the synthesis of the secondary messenger cyclic AMP (cAMP). Changes in cAMP directly and indirectly modulate an array of downstream proteins, including kinases,6 ion channels,7–11 and transcription factors.12 DARs can directly control downstream proteins such as G protein-coupled inwardly rectifying potassium channels (GIRKs) via Gβγ subunits.13,14 DARs also recruit arrestins,15,16 which terminate G protein signaling and facilitate G protein-independent signaling.17 Although DARs within a given sub-family couple to similar downstream signaling transducers, they diverge functionally by interacting with unique accessory proteins via their intracellular loops and C-termini (e.g., dopamine receptors 1 and 5, D1R and D5R, can interact directly with NMDA18 and GABAA 19 receptors, respectively) and have distinct cellular localization patterns.20
Deciphering the roles of individual DARs in vivo has implications not only for understanding the synaptic and neural circuit actions of DA but also the mechanisms underlying debilitating disorders such as Parkinson’s disease,21 schizophrenia,22 addiction,23 ADHD,24 obsessive compulsive disorder,25 and Tourette’s syndrome.26 However, current tools that are used to target DARs suffer from substantial limitations. A rich pharmacological toolkit of DAR agonists (full, partial) and antagonists (neutral antagonists, inverse agonists) has been developed over more than half a century.27,28 Nevertheless, there are few ligands that selectively bind individual DARs due to the high degree of similarity in their orthosteric binding sites (OBSs) that bind DA.29 Of note, there are no ligands that selectively bind D1R over D5R or D2R over D3R/D4R.29 In any case, diffusible ligands are not cell-type-specific and thus cannot differentiate between a DAR that has distinct and, in some cases, opposing roles in neighboring neurons or brain regions.20 Therefore, it is impossible to disambiguate the roles of individual DARs using a classical pharmacological approach.
Alternatively, genetic approaches, i.e., the overexpression, knockdown, or knockout of individual proteins, can be used to control individual DARs in specific cell types.30 However, these modifications affect receptor function over long time scales, which limits our understanding of the temporal aspects of DAR activation and can result in confounding compensatory effects on neuronal physiology.
Optogenetic and pharmacogenetic tools, such as optoXRs31–33 and RASSLs34/DREADDs,35 respectively, have been developed to cell-type-specifically interrogate GPCR function in vivo with greater temporal control than traditional genetic approaches. For example, a chimeric receptor, consisting of a partial sequence of D1R as well as the naturally light-sensitive components of rhodopsin (opto-D1R), was used to remotely activate D1R-mediated signaling in vivo.36 However, although such tools represent a powerful means to engage GPCR-dependent processes, they are non-native proteins that lack critical aspects of DARs, including their endogenous ligand binding site that binds DA, and/or receptor elements that control signaling, localization, and trafficking. Furthermore, they cannot be used to block DARs from being activated by DA, which is required for establishing which receptors are necessary for mediating specific DA-dependent processes. Thus, current opto- and pharmacogenetic tools may provide an incomplete, and possibly inaccurate, view of DAR function in vivo.
To address these limitations, we sought to engineer DARs that can be activated or blocked in vivo in a receptor-specific, cell-type-specific, and spatiotemporally precise manner. To develop such tools, we pursued a strategy implemented previously that utilizes azobenzene-containing, photoswitchable tethered ligands (PTLs) to optically control various ion channels.37–42 For example, we developed ionotropic glutamate receptors (iGluRs) that can be controlled with light (LiGluRs) in vitro37,43,44 and in vivo.45 At a cysteine introduced adjacent to the glutamate binding site, LiGluR reacts covalently with the maleimide of maleimide–azobenzene–glutamate (MAG). The azobenzene transitions from a trans-to-cis configuration and vice versa in response to UV and visible light, respectively, altering the position of glutamate with respect to its receptor binding site. The channel can be either activated or blocked depending on where MAG is tethered. Unlike the opto- and pharmacogenetic tools described above,31,35 these light-gated receptors are near-native proteins with only a single point mutation.46,47
We recently extended this approach to metabotropic GluRs (LimGluRs),48 which are Family C GPCRs. In contrast to iGluRs and mGluRs, which have large extracellular “venus flytrap” domains that bind glutamate, DARs bind DA within the upper third of the transmembrane bundle.49 Whether light-insensitive Family A GPCRs, such as DARs, are amenable to optical control using azobenzene-containing PTLs was hitherto unknown, although previous studies indicate that synthetic, non-photoswitchable covalent ligands can bind and activate or block this class of receptors.50–53 Furthermore, we showed recently that untethered azobenzene-containing photochromic ligands (PCLs) can photoswitch Family A GPCRs including opioid54 and muscarinic acetylcholine receptors.55 Moreover, the archetypical Family A GPCR rhodopsin obtains its sensitivity to light by binding covalently to retinal, making retinal a natural PTL.56
In this study, we used a tetherable azobenzene conjugated to the synthetic DAR agonist 2-(N-phenethyl-N-propyl)amino-5-hydroxytetralin (PPHT) to develop light-gated DARs (LiDARs; Figure 1) of D1R and D2R (LiD1R and LiD2R). These tools have the potential to enhance our understanding of DAR function in vivo and inform the development of therapeutics with enhanced efficacy and decreased side effects for DA-associated disorders.
Figure 1.
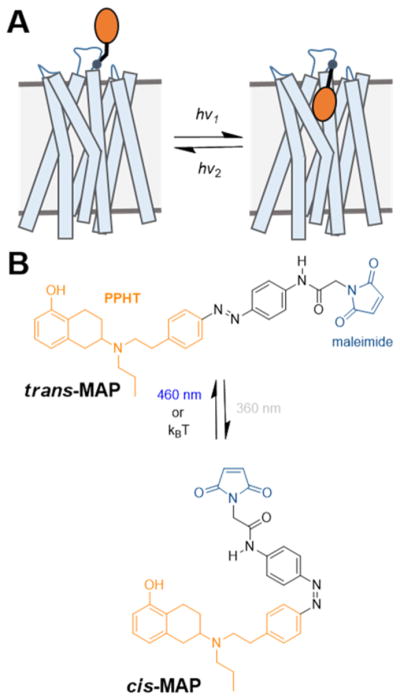
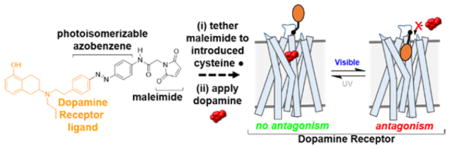
Design of a photoswitchable tethered ligand (PTL) to control DARs with light. (A) Schematic of a dopamine receptor (DAR) bound covalently to a PTL. (B) Azobenzene and maleimide (blue) incorporated into the DAR ligand PPHT (orange). Maleimide–azobenzene–PPHT (MAP) photoisomerizes from its trans-to-cis isomer and vice versa in response to UV and blue light, respectively.
RESULTS
Synthesis of a Photoswitchable Tethered Dopamine Receptor Ligand, MAP
To develop light-gated receptors, we first set out to conjugate a DAR ligand to the cysteine-conjugating photoswitch, maleimide-azobenzene. The catechol of DA (Figure S1) is sensitive to oxidation in aqueous solution56 as well as metabolism in vivo,57 making it undesirable as a parent compound for a DAR PTL. Instead, we used PPHT (Figure S1), a rigidified aminotetralin analogue of DA that contains only one hydroxyl as well as N-propyl and N-phenylethyl groups that enhance metabolic stability and affinity toward DARs.58–63 In addition, the N-phenylethyl group was previously chemically modified for the development of fluorescent tracers of DARs,64 suggesting that PPHT may tolerate the incorporation of azobenzene at this position without impairing its activity at DARs. Thus, we synthesized maleimide–azobenzene–PPHT (MAP; Figure 1B) as well as its non-covalent analogue azobenzene–PPHT (AP).
The synthesis of MAP (1; Scheme 1) starts with permethylation of dihydroxy naphthalene (3 to 4) that was followed by birch reduction using sodium in ethanol. The resulting methyl enol ether (5) was cleaved during acidic workup and tautomerized to the corresponding tetralone. Reductive amination with propylamine gave rise to aminotetraline (6). Reductive amination using freshly prepared homobenzylic aldehyde (7) installed the photoswitch into the molecule (8). For further functionalization, the nitro group was then reduced to the corresponding aniline (9). Deprotection of the methyl ether (10) and amide bond formation accomplished the synthesis of MAP. AP (2) was synthesized (Supplementary Scheme 1) with good overall yield through two sequential reductive aminations using amine 11 and propionic aldehyde.
Scheme 1.

Synthesis of Maleimide–Azobenzene–PPHT (MAP)
Structure–Activity Relationship Analysis of MAP at DARs
We evaluated the effects of azobenzene and maleimide–azobenzene addition (AP and MAP, respectively) to a close analogue of PPHT, 4-amino-PPHT64 (Figure S1), at D1R and D2R using previously established bioluminescence resonance energy transfer (BRET)-based, DAR-mediated G protein activation assays. Gs-activation downstream of D1R was measured indirectly using the cAMP sensor YFP-Epac-Rluc (CAMYEL; Figure S2A).65 In response to a Gs-mediated enhancement in cAMP, a conformational change in the cAMP-binding protein Epac results in a decrease in RET between Renilla luciferase (Rluc) and YFP. Gi1-activation downstream of D2R was measured directly by assessing agonist-induced conformational changes within the G protein (Figure S2B).66 Gαi1 was fused with Renilla luciferase 8 at position 91 of the α-helical domain (Gαi1-Rluc8) and Gβ1 and Gγ2 were fused to a split mVenus (V1-Gβ1 and V2-Gγ2) at their N-termini. Receptor activation induces a conformational change and/or the dissociation between Gαi1 and Gβ1γ2, resulting in decreased RET.
In these assays, 4-amino-PPHT was a potent and robust agonist of D1R and D2R relative to DA (Figure 2). We assessed the activity of trans-AP and trans-MAP using preparations of these compounds that were unexposed to light. Both compounds activated D1R and D2R with similar or slightly reduced efficacy compared to 4-amino-PPHT (Figure 2B,E). However, the azobenzene of AP significantly decreased its potency relative to 4-amino-PPHT (248- and 52-fold at D1R and D2R, respectively; Figure 2C,F). The potencies of AP and MAP were not significantly different, indicating that maleimide did not further decrease agonist activity (Figure 2C,F).
Figure 2.

Incorporation of azobenzene but not maleimide into PPHT decreases agonist potency at D1R and D2R. Representative dose–response curves for dopamine (DA), 4-amino-PPHT, trans-AP, and trans-MAP in (A) a D1R-dependent cAMP accumulation assay and (D) a D2R-dependent G protein activation assay. Dose–response curves are representative of three independent experiments performed in triplicate. Emax values for these compounds are summarized for D1R (B) and D2R (E). The potencies of these compounds (pEC50s) are summarized for D1R (C) and D2R (F) (**p < 0.001, ns = not significant, one-way ANOVA, Tukey’s posthoc comparison test). Error bars indicate SEM.
Photochemical Properties of MAP
We characterized the photochemical properties of AP and MAP using UV/vis spectroscopy. Both compounds were efficiently converted from their trans- to cis-configuration by irradiation with 360 nm light and vice versa by 460 nm light (Figure S3A). Both compounds could efficiently be switched over several cycles without any loss of activity (Figure S3B). Furthermore, consistent with the photochemical properties of azobenzenes lacking any strong auxochromic groups, the cis-configuration of these compounds is sufficiently stable toward thermal relaxation (T1/2 > 1 h; Figures S3B), and thus the compounds are considered bistable.
Photoswitching Properties of an Untethered Analogue of MAP, AP
We next evaluated the light sensitivity of AP modulation of DARs. Untethered, azobenzene-containing PCLs have been used to rapidly and reversibly control a variety of biological molecules with light,67,68 albeit with off-target effects associated with untethered ligands and slower kinetics than their tethered counterparts. In iGluRs, the ability of PCLs to photoswitch their receptor targets was predictive of the success of tethered variants that were designed from them.69 We compared G protein activation by AP that was not exposed to light and was therefore in the trans configuration (trans-AP) to G protein activation by AP that had been exposed to 360 nm light and was therefore largely in the cis configuration (cis-AP; Figure 3A). cis-AP was ~1.5- and ~3-fold more potent at D1R and D2R, respectively (two-tailed paired t test, P = 0.005 and 0.003, respectively; Figure 3B,C). Similarly, cis-AP was ~4-fold more potent than trans-AP in a BRET-based, D2R-mediated arrestin recruitment assay66 (Figure S2C; Figure 3D), which measures D2R-mediated translocation of arrestin3 to the plasma membrane. The enhancement of AP’s potency in these functional assays could result from increased affinity and/or efficacy. To address this, we evaluated AP binding to D2R in a homogeneous time-resolved fluorescence resonance energy transfer (HTRF) binding assay70 and found that cis-AP displayed ~3-fold higher affinity than trans-AP for D2R (two-tailed unpaired t test, P = 0.017; Figure 3E). Thus, the cis-isomer of AP is a higher affinity agonist than the trans-isomer.
Figure 3.

Characterization of the photochromic ligand (PCL) azobenzene–PPHT (AP) at D1R and D2R. (A) AP photoisomerizes from its trans- to cis-isomer and vice versa in response to UV and blue light, respectively. (B) In a D1R-dependent cAMP accumulation assay, AP exposed to UV light (cis-AP) was slightly more potent (~1.5-fold) than when not exposed to light (trans-AP). Shown are representative dose–response curves for three independent experiments performed in triplicate. Error bars indicate SEM. (C) In a D2R-dependent G protein activation assay, cis-AP was ~3-fold more potent than trans-AP. Shown are representative dose–response curves for three independent experiments performed in triplicate. Error bars indicate SEM. (D) In a D2R-mediated arrestin recruitment assay, cis-AP was ~4-fold more potent than trans-AP. Shown are representative dose–response curves for three independent experiments performed in triplicate. Error bars indicate SEM. (E) In a D2R binding assay, cis-AP had ~3-fold higher affinity for D2R than trans-AP. Representative binding curves for two independent experiments performed in duplicate. A decrease in the maximal binding indicates the displacement of the fluorescent D2R antagonist NAPS-DY-647 by AP. Error bars indicate SEM.
MAP as a Photoswitchable Tethered Ligand of D1R
Based on the functionality and isomer dependence of action of AP, we pursued MAP as a PTL of D1R. We hypothesized that, while the untethered AP is an agonist, tethered MAP could behave as an agonist or antagonist, depending on where it was tethered and thus its orientation within the D1R orthosteric binding site (OBS). The OBS of D1R, like in all aminergic Family A GPCRs, is formed by TMs 3, 5, 6 and 7, and is “capped” by the second extracellular loop (EL2).71 DA binds to this site via an interaction between its catechol and a cluster of aromatic residues in TM6, as well as a salt bridge between its protonated amine and an aspartate in TM3 (D963.32 according to Ballesteros–Weinstein numbering72). In addition, the two hydroxyls of DA form hydrogen bonds with three TM5 serines (S1985.42, S1995.43, and S2025.46), interactions that promote receptor activation.73 Based on this, an interaction between MAP and the aromatic cluster and/or D963.32, but not the TM5 serines, would be expected to result in antagonism, whereas all three interactions are required for agonism.
Guided by a homology model of D1R based on a crystal structure of the Family A GPCR, β2-adrenergic receptor (β2AR),74 we individually mutated to cysteine 15 residues in D1R, which cluster in TM2, EL1, EL2, or TM7. These sites were chosen because they are predicted to be accessible to solvent and encompass the majority of the surface area surrounding the entryway to the OBS. To evaluate photoswitching, we used D1R-mediated activation of the G protein activated inward rectifier potassium (GIRK) channel. D1R cysteine mutants were individually co-expressed with GIRK1 and GIRK2 subunits (to form the GIRK1/2 heterotetrameric channel) as well as with a chimeric G protein consisting of Gαi that contains the terminal 13 residues of Gαs (Gαis13),75 which enables the Gs-coupled D1R to activate GIRK. Following incubation and washout of MAP, transfected cells were patch-clamped in whole-cell configuration and exposed to alternating 360 and 460 nm light, interconverting MAP between its cis- and trans-isomers, respectively. There was no effect on wildtype (WT) D1R, which does not contain an introduced cysteine anchoring site for MAP (Figure S4B), indicating that our washout conditions efficiently removed untethered MAP.
MAP most robustly photoswitched D1R(I183C), which contains a cysteine in EL2 (Figure 4A). This mutant localized to the plasma membrane like WT (Figure S5A) and was activated by DA with similar potency to WT D1R (pEC50 = 6.2 and 6.4, D1R(I183C) and WT D1R, respectively; Figure S5B). In addition, the synthetic D1-like receptor partial agonist SKF3839376 similarly activated WT D1R and D1R(I183C) (Figures S5C–E). Thus, the introduction of cysteine at position I183 did not substantially alter receptor function. Following incubation and washout of MAP (30 μM for 60 min), transition from 360 to 460 nm light resulted in a reversible reduction (by 41 ± 3%, n = 9) in inward-current evoked by a saturating concentration (10 μM) of DA (Figure 4B), indicating that trans-MAP is a D1R antagonist when tethered to I183C. The magnitude of photoblock at this mutant is comparable to that observed for a previously reported photoblocked metabotropic glutamate receptor 2 (LimGluR2-block).48 Voltage ramps showed that the MAP photoblocked current is inwardly rectifying, consistent with the antagonism of the D1R activation of GIRK channels (Figure 4C). The kinetics of onset and offset of MAP photoblock were similar (τonset= 6.0 s, τoffset = 5.7 s; Figure 4D). Both the photoblock and its reversal persisted in the dark after the light flash, consistent with bistability of MAP (Figure 4E), as has been shown for azobenzene PTLs of glutamate receptors.42,48,77 The magnitude of photoblock decreased with increasing concentrations of DA, in accordance with a competitive interaction between DA and MAP (Figure 4F).
Figure 4.

MAP is a neutral antagonist at D1R(I183C). (A) Ribbon representation of a D1R homology model bound to protonated DA (red). Amino acid I183 in EL2 is shown in green above the OBS that binds DA. (B) When MAP is attached to I183C, photoswitching between 360 nm (gray bars) and 460 nm light (blue bars) had no effect. However, current evoked by DA was photoblocked in response to 460 nm light. (C) Representative trace of currents induced by DA under 360 nm light (gray) or 460 nm light (blue). Ramp currents were subtracted from baseline ramps taken in the absence of DA under 360 nm light. Currents show inward rectification typical of GIRK current. Inset shows close overlap between the normalized traces. (D) The kinetics of the onset and offset of photoblock at D1R(I183C) by MAP were not significantly different (n.s. = not significant, two-tailed, unpaired t test). Error bars represent SEM. (E) Photoblock of D1R(I183C) by MAP is bistable. A brief flash of 460 nm light (blue bar) induced a decrease in dopamine-evoked current that was sustained in the dark for ~30 s until it was reversed by a brief flash of 360 nm light (gray bar). (F) The magnitude of photoblock of D1R(I183C) by MAP decreases with increasing concentrations of DA. Error bars represent SEM for n = 3–9 cells per concentration of DA.
Although MAP photoblocked DA-induced activation of D1R(I183C), it had no effect on basal inward-current (Figure 4B). This was in striking contrast to the effects of MAP on D1R(G88C), which contains a cysteine in EL1 (Figure 5A). The transition from 360 to 460 nm light (cis- to trans-MAP) reduced the DA-evoked current, but also reduced the basal inward current (35 ± 4% and 15 ± 4% of maximal DA-evoked current, respectively, n = 5; Figure 6B). The photoblock of basal inward current was repeatable (Figure 5C) and bistable (Figure 5D), consistent with the photochemical properties of MAP. The effect of MAP in its trans-configuration on both basal and agonist-induced current suggested that it may be an inverse agonist that reduces constitutive receptor activity. To address this possibility, we tested whether WT D1R, D1R(I183C), and D1R(G88C) exhibit constitutive activity by measuring changes in basal inward current in response to a saturating concentration of the D1-like receptor inverse agonist LE300.78 LE300 similarly reduced inward current in cells expressing WT D1R, D1R(I183C), and D1R(G88C) (Figure 5E–G), indicating that they each have constitutive activity, consistent with basal current photoblock being of this constitutive activity. If this interpretation is correct and MAP is, indeed, an inverse agonist, its photoeffect on basal current would be reduced by LE300. This prediction was borne out (Figure 5H). However, LE300 did not completely abolish the effect of MAP on basal current of D1R(G88C) (Figure 5H), suggesting that MAP is a more robust inverse agonist than LE300.
Figure 5.

MAP is an inverse agonist at D1R(G88C). (A) Ribbon representation of a D1R homology model bound to protonated DA (red). Amino acid G88 in EL1 (shown in blue and highlighted by an arrow) is adjacent to the OBS that binds DA. (B) When MAP is attached to G88C, photoswitching from 360 and 460 nm light decreases both basal and DA-evoked inward current. (C) Photoswitching of the basal current is repeatable. (D) Photoswitching of the basal current is bistable. (E) Basal inward current is reduced in response to LE300 in cells expressing wildtype D1R. (F) Basal inward current is reduced in response to LE300 in cells expressing D1R(G88C). (G) Summary of the effects of LE300 on basal inward current in cells expressing WT D1R, G88C, and I183C. Error bars represent SEM for n = 3–4 cells per receptor (not significant, one-way ANOVA). (H) LE300 partially attenuated the ability of MAP to photoblock the basal inward current in cells expressing D1R(G88C).
Figure 6.

MAP photoblocks D1R. (A) A surface representation of a D1R homology model. Fifteen residues surrounding the DA binding site of D1R were individually mutated to cysteines. These sites were located on the extracellular face of TM2 (orange), EL1 (blue), EL2 (green), or extracellular face of TM7 (cyan). Protonated DA is shown bound to the OBS of D1R (red). (B) Summary of photoblock at cysteine mutants of D1R by MAP. Error bars represent SEM for n = 3–9 cells per cysteine mutant.
MAP photoswitched mutants of D1R with cysteines in EL1 or EL2, and to a lesser degree TM2 and TM7 (Figure 6). DA-mediated activation of all photoswitchable D1R mutants was blocked in response to 460 nm light, indicating that trans-MAP inhibits D1R when tethered to the receptor.
MAP as a Photoswitchable Tethered Ligand of D2R
Although both D1R and D2R bind and are activated by DA, they are only ~40% homologous in their TM domains79 and have no similarity in their ELs. This dissimilarity suggests that MAP could have distinct effects at these receptors. We first evaluated MAP at WT D2R, which unlike D1R contains a native cysteine deep within its OBS (C1183.36). We previously found this cysteine to be sensitive to covalent modification by selected methyl thiosulfate (MTS) compounds based on their charge.80 We determined the effects of MAP, MTSEA, and MTSES on WT D2R by measuring DA-induced G protein activation following incubation and washout. Consistent with our previous results, whereas negatively charged MTSES had no effect on D2R activation, positively charged MTSEA completely abolished receptor activity (Figure S6A). Notably, MAP had little effect, suggesting lack of accessibility of C118 to this relatively bulky reagent. Consistent with this, following incubation and washout of WT D2R in MAP, exposure to DA still activated GIRK channels (Figure S6B,C), and channel activity was not photoswitched by alternating exposure between 360 and 460 nm light (Figure S6C).
Fourteen D2R residues located near the OBS, based on a homology model that used the crystal structure of D3R49 (Figure 7B) were mutated to cysteine to introduce the MAP conjugation site. The largest photomodulation of D2R was seen when MAP was tethered to I184C in EL2. The transition from 360 to 460 nm light photoblocked (by 49 ± 8%, n = 6) the GIRK current evoked by 10 μM DA (Figure 7A,C). Here too, as seen in D1R above, basal GIRK current was reduced by 460 nm light (Figure 7A), suggesting that trans-MAP is an inverse agonist. Consistent with this, spiperone, a robust inverse agonist of D2R,81 also reduced inward current both in cells expressing WT D2R or D2R(I184C) (Figures S8).
Figure 7.
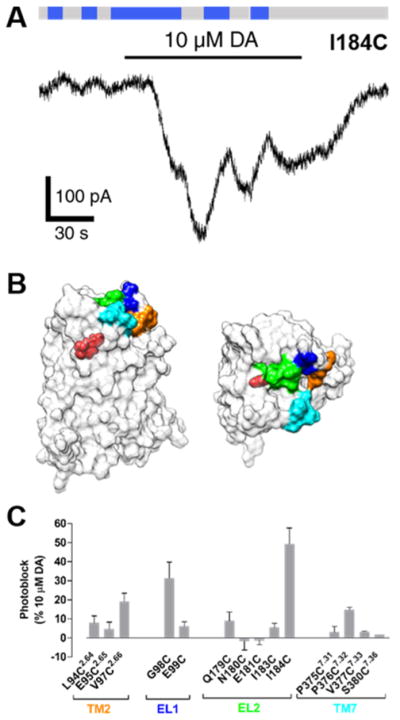
MAP photoblocks D2R. (A) When MAP is attached to I184C, photoswitching from 360 and 460 nm light decreases both basal and DA-evoked current. (B) A surface representation of a D2R homology model. Fourteen residues surrounding the dopamine binding site of D2R were individually mutated to cysteines. These sites were located on the extracellular face of TM2 (orange), EL1 (blue), EL2 (green), or extracellular face of TM7 (cyan). Protonated dopamine is shown bound to the orthosteric binding site (OBS) of D2R (red). (C) Summary of photoblock of cysteine mutants of D2R by MAP. Error bars represent SEM for n = 3–7 cells per cysteine mutant.
Aside from the ability to conjugate to MAP and endow the receptor with photocontrol, the I184-to-cysteine mutation did not alter the function of D2R: localization of D2R to the plasma membrane was normal (Figure S7A), and there was no change in sensitivity to DA (pEC50 = 6.3 and 6.0 for WT and mutant, respectively; Figure S7B) or activation by the synthetic D2-like agonist quinpirole (Figures S7C–E).
Interestingly, MAP operated as a trans-inverse photoagonist when attached to cysteines to either EL1 or EL2 of D2R (Figure 7A). This is in contrast to its effects at D1R, whereby MAP was a neutral antagonist or inverse agonist when tethered to cysteines in EL1 or EL2, respectively (Figures 4 and 5).
DISCUSSION
In this study, we developed a DAR PCL (AP) and PTL (MAP) based on the synthetic agonist PPHT. MAP attached to an introduced cysteine near the OBS yielded photoblock of both D1R (“LiD1R”) and D2R (“LiD2R”), providing what to our knowledge is the first means to remotely control full-length DARs in a receptor subtype-specific, cell-type-specific, and spatiotemporally precise manner. Because MAP can be directly anchored to a given DAR, off-target effects at other DARs and non-DAR proteins are avoided. Thus, MAP is a reversible and selective ligand of individual DARs. Moreover, because these tools are controlled with light, they can be used to rapidly regulate individual DARs on time scales that are relevant to native dopaminergic signaling in the brain.82
A major goal of this study was to use MAP to selectively photoblock DARs. Photoblockade is a powerful means to determine whether a receptor of interest is necessary for specific physiological or behavioral processes in vivo. Photoactivation, on the other hand, can be used to determine whether it is sufficient. Like 4-amino-PPHT and AP, MAP activated D1R and D2R in its untethered form. In contrast, when tethered to a receptor, MAP either had no effect or photoblocked the receptor at the variety of cysteine attachment positions that we tested. This is not a surprise given that photoantagonism could arise from a variety of geometries that improperly position the ligand in the OBS, whereas photoagonism demands proper docking. Indeed, only one out of eight attachment sites for the mGluR2 photoswitch, MAG, produced photoactivation.48
The distinction between photoagonism and photoantagonism could be due to subtle differences in the anchoring site of the PTL from one residue to its neighbor in three-dimensional space, as well as differences in the PTL’s length and bond angles. A non-photoswitchable, covalent agonist of D2R was developed recently,50 suggesting that it may be possible to achieve photoagonism at DARs. Toward this end, further chemical modifications of MAP may result in photoagonism, e.g., by adjusting the linker length between PPHT and azobenzene and/or azobenzene and the maleimide (Figure 1B), thus altering how MAP is positioned within the OBS. Alternatively, the position and number of hydroxyls in aminotetralins are known to dictate efficacy at DARs, and it is possible that MAP is limited as a photoagonist because it has only a single hydroxyl.61,62 Thus, an additional hydroxyl could enhance photoagonism, albeit at the cost of the metabolic stability of the compound.
Interestingly, we found MAP to be either a photoswitchable tethered neutral antagonist (PTNA) or photoswitchable tethered inverse agonist (PTIA) at D1R, depending on where it is tethered. GPCRs like DARs are dynamic ensembles of inactive and active conformations.83 It has been proposed that neutral antagonists occlude the binding of agonists but do not shift the equilibrium between active and inactive states, whereas inverse agonists stabilize inactive states and reduce constitutive activity.84 Crystallographic studies of Family A GPCRs suggest that chemically related neutral antagonists and inverse agonists bind similarly within the OBS and stabilize receptor states with only minor structural differences.51 Similarly, MAP may adopt subtly distinct binding poses within the OBS, depending on where it is tethered, allowing it to be behave as a neutral antagonist or inverse agonist.
Remarkably, despite the low overall degree of homology between D1R and D2R, MAP most robustly photoblocked these receptors when tethered to cysteines in EL2 (I183C and I184C, respectively), and to a lesser degree EL1. Although EL2 is highly divergent in sequence between D1R and D2R, it commonly serves as a “lid” over the OBS. This combined with the fact that EL2 is relatively conformationally dynamic85 may allow MAP greater freedom to access the OBS than if tethered to the TM domain. EL2 acts as a lid over the OBS of a variety of Family A GPCRs,71 suggesting that it may represent a common target for controlling other receptors in this family using PTLs.
The ability to photoblock DARs is of great clinical interest. Notably, all antipsychotics are blockers of D2R-like receptors. 86,87 However, because these medications are nonselective for D2R-like receptors and are diffusible, it has been challenging to identify which of these D2-like receptors (D2R, D3R, or D4R) is responsible or to determine which cells expressing the receptor(s) play a role in efficacy and side effects. LiD2R offers the possibility of blocking D2R in a cell-type-specific manner, allowing us to mimic selected effects of antipsychotic medications in isolation. Moreover, antipsychotics have varying degrees of inverse agonism at D2R, and it is unclear if and where in the brain this is relevant to clinical efficacy.88 LiD2R variants that operate as inverse photoagonists may help to resolve this issue.
Several approaches have developed recently to interrogate GPCR function in vivo, including DREADDs35 and opto-XRs.31–33 Opto-XRs are of particular interest because, like the light-gated receptors developed here, they can be precisely controlled with light. Although opto-XRs cannot be used to block receptors from being activated by their endogenous ligands, they can be targeted to specific cell types and activated in an inducible-manner. Interestingly, Siuda et al. developed biased mutants of opto-β2AR that selectively activate only a subset of signaling proteins downstream of this receptor (i.e., G proteins versus arrestins).33 Considerable effort has gone toward uncovering the roles of distinct signaling pathways downstream of DARs,5 and a number of biased DAR mutants have been developed.89,90 Given that mutations in these receptors are not involved in ligand binding, they can likely be integrated into LiDARs, which would enable signaling-pathway-specific and spatiotemporally precise control of near-native DARs.
CONCLUSION
Family A GPCRs are subdivided into groups of structurally related receptors, making it difficult in particular cases to target individual receptor subtypes. Considerable medicinal chemistry efforts have gone toward developing subtype-selective drugs, with varying degrees of success.29 However, our understanding of the physiological and clinical roles of a given receptor will require not only receptor-specific but also cell-type-specific and spatiotemporally precise control over its function. The success of LiDARs indicates that Family A GPCRs are amenable to rapid, cell-type- and receptor-specific control using PTLs. Thus, we anticipate that our approach will also work for difficult-to-target Family A GPCRs, including but not limited to the neuromodulator-binding opioid, serotonin, and cannabinoid receptors.
EXPERIMENTAL SECTION
General Experimental Details
All reactions were carried out with magnetic stirring and, if moisture- or air-sensitive, under nitrogen or argon atmosphere using standard Schlenk techniques in oven-dried glassware (140 °C oven temperature). External bath temperatures were used to record all reaction temperatures. Low temperature reactions were carried out in a Dewar vessel filled with acetone/dry ice (−78 °C) or distilled water/ice (0 °C). High temperature reactions were conducted using a heated silicon oil bath in reaction vessels equipped with a reflux condenser or in a sealed flask. Tetrahydrofuran (THF) was distilled over sodium and benzophenone prior to use. Dichloromethane (CH2Cl2), triethylamine (Et3N), and diisopropylethylamine (DIPEA) were distilled over calcium hydride under a nitrogen atmosphere. All other solvents were purchased from Acros Organics as “extra dry” reagents. All other reagents with a purity >95% were obtained from commercial sources (Sigma-Aldrich, Acros, Alfa Aesar, and others) and used without further purification.
Flash column chromatography was carried out with Merck silica gel 60 (0.040–0.063 mm). Analytical thin layer chromatography (TLC) was carried out using Merck silica gel 60 F254 glass-backed plates and visualized under UV light at 254 nm. Staining was performed with ceric ammonium molybdate (CAM) or by oxidative staining with an aqueous basic potassium permanganate (KMnO4) solution and subsequent heating.
NMR spectra (1H NMR and 13C NMR) were recorded in deuterated chloroform (CDCl3) or deuterated methanol (d4-MeOH) on a Bruker Avance III HD 400 MHz spectrometer equipped with a CryoProbe, a Varian VXR400 S spectrometer, a Bruker AMX600 spectrometer, or a Bruker Avance III HD 800 MHz spectrometer equipped with a CryoProbe and are reported as follows: chemical shift δ in ppm (multiplicity, coupling constant J in Hz, number of protons) for 1H NMR spectra and chemical shift δ in ppm for 13C NMR spectra. Multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, br = broad, m = multiplet, or combinations thereof. Residual solvent peaks of CDCl3 (δH = 7.26 ppm, δC = 77.16 ppm) and d4-MeOH (δH = 3.31 ppm, δC = 49.00 ppm) were used as an internal reference. NMR spectra were assigned using information ascertained from COSY, HMBC, and HSQC experiments.
High-resolution mass spectra (HRMS) were recorded on a Varian MAT CH7A, or a Varian MAT 711 MS instrument by electron impact (EI) or electrospray ionization (ESI) techniques at the Department of Chemistry, Ludwig-Maximilians-Universität Munich.
Infrared (IR) spectra were recorded from 4000 to 600 cm−1 on a Perkin Elmer Spectrum BX II instrument. For detection a Smiths DetectioN DuraSamplIR II Diamond ATR sensor was used. Samples were prepared as a neat film or a thin powder layer. IR data in frequency of absorption (cm−1) is reported as follows: w = weak, m = medium, s = strong, br = broad, or combinations thereof.
All yields are isolated, unless otherwise specified.

Dimethylsulfate (43.3 mL, 468 mmol) was added to a suspension of potassium carbonate (75.5 g, 548 mmol) and 1,6-dihydroxynaphthalene (25 g, 156 mmol) in acetone (250 mL). The mixture was heated at 70 °C for 3.5 h, cooled to room temperature, and filtered. The solvent was evaporated under reduced pressure, and the resulting residue was redissolved in ethyl acetate. The solution was washed with a solution of sodium hydroxide (1 M) and brine and dried over magnesium sulfate. Evaporation of the solvent in vacuo and purification of the resulting residue by flash column chromatography (EtOAc:-pentane = 0:100–20:80, Rf = 0.7 (EtOAc:pentane 20:80)) gave 1,6-dimethoxynaphthalene 4 (27.6 g, 147 mmol, 94%) as a colorless solid.
Data for 4: 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.17 (dt, J = 8.8, 0.8 Hz, 1H), 7.38–7.31 (m, 2H), 7.15–7.10 (m, 2H), 6.70 (dd, J = 6.9, 1.7 Hz, 1H), 3.99 (s, 3H), 3.92 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) = 158.2, 155.8, 136.0, 126.8, 123.8, 120.9, 119.4, 117.6, 105.8, 102.1, 55.6, 55.4. IR (ATR): νmax (cm−1) = 2997 (w), 2966 (w), 2836 (w), 1929 (w), 1625(m), 1599 (m), 1581 (s), 1510 (w), 1468 (w), 1451 (s), 1430 (s), 1372 (s), 1386 (w), 1372 (s), 1254 (m), 1220 (vs), 1199 (m), 1165 (m), 1143 (m), 1098 (m), 1070 (m), 1026 (vs), 988 (m), 935 (m), 871 (m), 839 (m), 828 (s), 782 (vs), 748 (s), 727 (m), 695 (m). HRMS (EI): calcd for C12H12O2 [M]+ 188.0837, found 188.0819.
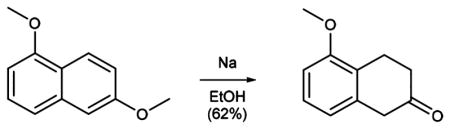
Sodium (33.69 g, 1.47 mol) was added portionwise to a solution of 1,6-dimethoxynaphthalene 4 (27.6 g, 147 mmol) in ethanol (400 mL) at 50 °C, and the mixture was then stirred at 80 °C for 2.5 h. After cooling down to room temperature the mixture was acidified with concentrated hydrochloric acid to pH = 1, heated at 80 °C for 30 min, and stirred at room temperature overnight. The solution was diluted with water (230 mL) and extracted with CH2Cl2. The organic phase was washed with a solution of sodium chloride (10%) and brine and dried over magnesium sulfate. Evaporation of the solvent in vacuo and purification of the resulting residue by flash column chromatography (EtOAc:pentane = 0:100–10:90, Rf = 0.5 (EtOAc:pentane 10:90) gave 3,4-dihydronaphthalenone 5 (15.94 g, 90.47 mmol, 62%) as a yellow oil.
Data for 5: 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.16 (t, J = 7.9 Hz, 1H), 6.76 (dd, J = 8.2, 1.0 Hz, 1H), 6.70 (dd, J = 7.6, 1.0 Hz, 1H), 3.83 (s, 3H), 3.52 (s, 2H), 3.06 (t, J = 6.8 Hz, 2H), 2.48 (dd, J = 7.4, 6.2 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ (ppm) = 210.4, 156.2, 134.8, 127.3, 124.8, 120.2, 108.2, 55.2, 44.4, 44.4, 37.6, 37.6, 20.8. IR (ATR): νmax (cm−1) = 2957 (w), 2904 (w), 2838 (w), 1713 (vs), 1587 (s), 1471 (s), 1441 (m), 1403 (w), 1344(w), 1299 (w), 1263 (vs), 1237 (m), 1197 (w), 1172 (w), 1087 (vs), 1076 (s), 1025 (w), 977 (w), 963 (m), 849 (m), 778 (s), 745 (m), 724 (m), 695 (m). MS (EI): calcd for C11H12O2 [M]+ 176.08, found 176.03.
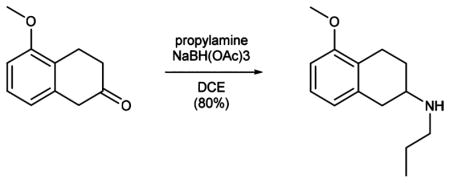
Propylamine (2.1 mL, 25.54 mmol) and NaBH(OAc)3 (6.50 g, 30.66 mmol) were sequentially added to a solution of 3,4-dihydronaphthalenone 5 (3.00 g, 17.02 mmol) in DCE (100 mL), and the reaction was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the resulting residue was portioned between EtOAc and aqueous NaHCO3 solution. The organic phase was washed with brine, dried over magnesium sulfate, and concentrated under reduced pressure to yield secondary amine 6 (2.99 g, 13.62 mmol, 80%) as a colorless oil.
Data for 6: 1H NMR (400 MHz, d4-MeOH): δ (ppm) = 7.07 (t, J = 7.9 Hz, 1H), 6.69 (dd, J = 13.6, 7.9 Hz, 2H), 3.79 (s, 3H), 2.98 (ddddd, J = 27.8, 20.8, 17.7, 5.4, 2.6 Hz, 3H), 2.81–2.69 (m, 2H), 2.57 (ddd, J = 30.0, 16.8, 8.0 Hz, 2H), 2.16 (ddt, J = 11.9, 5.7, 2.9 Hz, 1H), 1.69–1.48 (m, 3H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, d4-MeOH): δ (ppm) = 158.6, 136.7, 127.6, 125.5, 122.4, 108.3, 55.6, 54.9, 49.4, 36.2, 29.1, 23.3, 23.2, 12.0. IR (ATR): νmax (cm−1) = 2936 (m), 2875 (w), 2835 (m), 2792 (m), 2734 (m), 2537 (w), 2445 (w), 1587 (s), 1468 (vs), 1438 (s), 1395 (w), 1382 (w), 1346 (w), 1312 (w), 1284 (w), 1258 (vs), 1094 (s), 1068 (m), 1036 (m), 987 (m), 954 (m), 914 (w), 892 (w), 876 (w), 832 (w), 765 (vs), 709 (m), 694 (m). HRMS (EI): calcd for C14H22NO+ [M+H]+ 220.1696, found 220.1696.
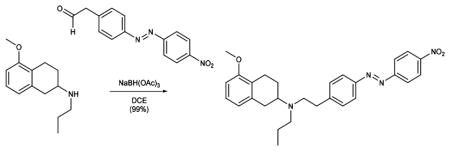
DMP (8.33 g, 21.06 mmol) was added to a solution of (E)-2-(4-((4-nitrophenyl)diazenyl)phenyl) ethan-1-ol (1.90 g, 7.02 mmol) in CH2Cl2, and the reaction was stirred at room temperature for 3 h. The mixture was washed with aqueous Na2S2O3 solution, water, and brine, dried over magnesium sulfate, and evaporated to dryness. The resulting residue was redissolved in DCE (100 mL), secondary amine 6 (700 mg, 3.19 mmol) and NaBH(OAc)3 (2.23 g, 10.53 mmol) were sequentially added, and the reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was portioned between EtOAc and aqueous NaHCO3 solution. The organic phase was washed with brine, dried over magnesium sulfate, and concentrated under reduced pressure. Purification of the resultant residue by flash column chromatography (MeOH:CH2Cl2 = 3:97, Rf = 0.3) gave azobenzene 8 (1.49 g, 3.16 mmol, 99%) as a red solid.
Data for 8: 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.42–8.38 (m, 2H), 8.06–8.02 (m, 2H), 7.94–7.90 (m, 2H), 7.43–7.38 (m, 2H), 7.11 (t, J = 7.9 Hz, 1H), 6.72 (dd, J = 7.6, 1.0 Hz, 1H), 6.67 (d, J = 8.1 Hz, 1H), 3.83 (s, 3H), 3.01 (dddd, J = 10.8, 8.6, 5.5, 2.7 Hz, 2H), 2.87 (s, 5H), 2.81–2.71 (m, 1H), 2.62–2.49 (m, 3H), 2.07 (ddd, J = 14.5, 4.6, 2.0 Hz, 1H), 1.63–1.48 (m, 3H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) = 157.4, 156.0, 151.0, 148.7, 146.6, 138.1, 130.0, 126.3, 125.4, 124.9, 123.6, 123.5, 121.8, 107.1, 56.7, 55.4, 52.8, 52.6, 36.3, 32.5, 25.9, 24.0, 22.4, 12.1. IR (ATR): νmax (cm−1) = 2931 (m), 2836 (w), 1738 (vw), 1602 (w), 1586 (m), 1521 (s), 1499 (w), 1468 (m), 1438 (w), 1416 (vw), 1360 (vs), 1259 (m), 1218 (w), 1143 (m), 1106 (m), 1093 (m), 1071 (m), 1008 (w), 963 (w), 908 (w), 858 (s), 766 (w), 754 (w), 729 (s), 710 (w), 688 (w). HRMS (EI): calcd for C28H33N4O3 + [M+H]+ 473.2547, found 473.2543.
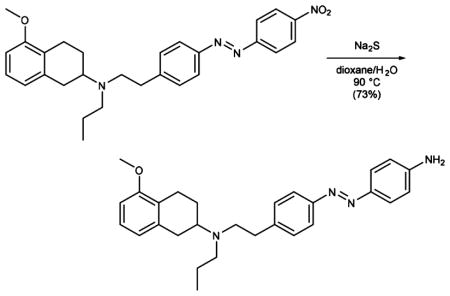
A suspension of azobenzene 8 (500 mg, 1.06 mmol) and sodium sulfide (2.33 mg, 2.12 mmol) in 1,4-dioxane (50 mL) and water (50 mL) was stirred at 90 °C overnight. The reaction was cooled to room temperature, diluted with aqueous NaOH (1 M), and extracted with EtOAc. The organic phase was washed with water and brine, dried over magnesium sulfate, and concentrated under reduced pressure. Purification of the resultant residue by flash column chromatography (MeOH:CH2Cl2 = 2:98, Rf = 0.2) gave aniline 9 (341 mg, 0.770 mmol, 73%) as a red solid.
Data for 9: 1H NMR (400 MHz, CDCl3): δ (ppm) 7.80 (t, J = 8.3 Hz, 4H), 7.32 (d, J = 8.2 Hz, 2H), 7.10 (t, J = 7.8 Hz, 1H), 6.73 (dd, J = 8.3, 6.5 Hz, 3H), 6.66 (d, J = 8.1 Hz, 1H), 4.03 (s, 2H), 3.81 (s, 3H), 3.12–2.94 (m, 2H), 2.94–2.71 (m, 6H), 2.68–2.44 (m, 3H), 2.08 (dt, J = 13.7, 4.2 Hz, 1H), 1.68–1.46 (m, 3H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) = 157.3, 151.4, 149.5, 145.7, 143.2, 138.1, 129.5, 126.3, 125.3, 125.1, 122.4, 121.8, 114.8, 107.0, 67.2, 56.8, 55.3, 52.8, 35.9, 32.4, 25.9, 24.0, 22.3, 12.1. IR (ATR): νmax (cm−1) = 3336 (w), 2933 (m), 2837 (vw), 2508 (w), 2246)vw), 2212 (vw), 1623 (w), 1586 (m), 1516 (m), 1468 (m), 1438 (w), 1378 (vw), 1346 (vw), 1260 (m), 1216)(vw), 1180 (vw), 1128 (vw), 1094 (m), 1071 (m), 96.2 (vw), 907 (s), 826 (m), 768 (m), 700 (vs). HRMS (EI): calcd for C28H35N4O+ [M+H]+ 443.2805, found 443.2811.
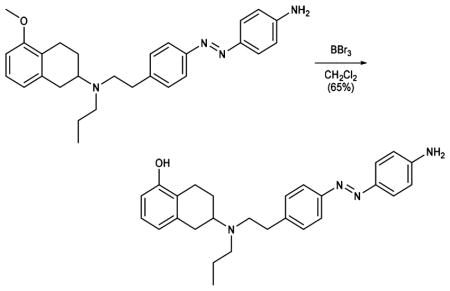
BBr3 (1 M in CH2Cl2, 3.18 mL, 3.18 mmol) was added to a solution of aniline 9 (704 mg, 1.59 mmol) in CH2Cl2 (30 mL) at −78 °C, and the reaction was stirred 1 h at the same temperature and 1 h at room temperature. The reaction mixture was diluted with MeOH (5 mL) and aqueous NaHCO3 solution and extracted with EtOAc. The organic phase was washed with brine, dried over magnesium sulfate, and concentrated under reduced pressure. Purification by flash column chromatography (MeOH:CH2Cl2 = 2:98 to 6:94, Rf = 0.6 (MeOH:CH2Cl2 = 6:94)) gave phenol 10 (440 mg, 1.03 mmol, 65%) as a red solid.
Data for 10: 1H NMR (400 MHz, CDCl3): δ (ppm) 7.81 (t, J = 7.6 Hz, 4H), 7.31 (d, J = 8.0 Hz, 2H), 6.99 (t, J = 7.8 Hz, 1H), 6.73 (d, J = 8.3 Hz, 2H), 6.68 (d, J = 7.6 Hz, 1H), 6.58 (d, J = 7.9 Hz, 1H), 4.04 (s, 2H), 3.09–2.73 (m, 8H), 2.59 (ddd, J = 23.4, 10.2, 5.8 Hz, 3H), 2.12 (dd, J = 12.5, 5.5 Hz, 1H), 1.70–1.47 (m, 3H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) = 153.7, 151.4, 149.5, 145.6, 143.0, 138.9, 129.5, 126.5, 125.1, 123.3, 122.5, 121.6, 114.8, 112.1, 56.8, 52.7, 52.6, 35.5, 32.2, 25.8, 23.8, 21.9, 12.1. IR (ATR): νmax (cm−1) = 3370 (w), 2313 (w), 3027 (w), 2930 (m), 2870 (w), 1907 (vw), 1726 (vw), 1619 (m), 1598 (vs), 1505 (m), 1463 (s), 1428 (m), 1404 (w), 1374 (w), 1341 (w), 1297 (m), 1275 (s), 1138 (vs), 1083 (m), 1065 (m), 1012 (m), 949 (w), 836 (s), 751 (vs), 711 (m), 666(w). HRMS (EI): calcd for C27H33N4O+ [M+H]+ 429.2649, found 429.2645.
Oxalyl chloride (0.05 mL, 0.610 mmol) and DMF (1 drop) were added sequentially to a solution of 2-maleimidoacetic acid (73 mg, 0.469 mmol) in CH2Cl2 (5 mL), and the reaction mixture was stirred for 2 h at room temperature. The solvent was evaporated in vacuo, and the resulting residue was redissolved in THF (3 mL). The solution was added dropwise to an ice-cooled solution of phenol 10 (67 mg, 0.156 mmol) and DIPEA (0.10 mL, 0.610 mmol) in THF (3 mL), and the reaction was stirred for 30 min at room temperature. The solvent was evaporated under reduced pressure, and the resulting residue was purified by flash column chromatography (MeOH:CH2Cl2 = 3:97, Rf = 0.25) to give Mal–Azo–PPHT (41 mg, 0.072 mmol, 46%) as a red solid.
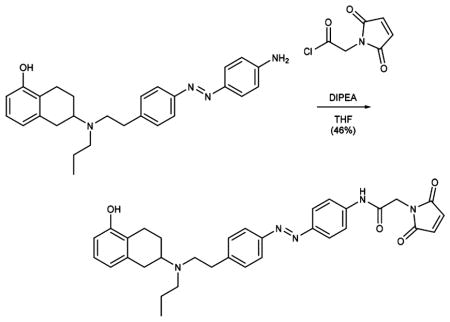
Data for 1: 1H NMR (400 MHz, CDCl3): δ (ppm) 8.00 (s, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.31 (d, J = 8.0 Hz, 2H), 6.98 (t, J = 7.7 Hz, 1H), 6.79 (s, 2H), 6.67 (d, J = 7.6 Hz, 1H), 6.58 (d, J = 7.9 Hz, 1H), 5.56 (s, 1H), 4.37 (s, 2H), 3.04–2.70 (m, 8H), 2.62–2.50 (m, 3H), 1.61 (tt, J = 12.1, 6.2 Hz, 1H), 1.50 (p, J = 7.4 Hz, 2H), 0.89 (t, J = 7.3 Hz, 3H).13C NMR (100 MHz, CDCl3): δ (ppm) = 170.3, 164.5, 153.6, 151.1, 138.5, 134.7, 129.7, 126.5, 124.3, 124.0, 123.1, 122.9, 122.7, 121.8, 121.7, 120.1, 112.1, 56.7, 52.7, 52.6, 41.4, 35.9, 32.3, 25.8, 23.8, 22.2, 12.1. IR (ATR): νmax (cm−1) = 2933(m), 1741(s), 1595 (vs), 1560 (s), 1501 (m), 1464 (m), 1408 (s), 1301 (s), 1253 (s), 1156 (s), 1012 (w), 846 (s), 771 (m), 676 (w). HRMS (ESI): calcd for C33H36N5O4+ [M+H]+ 566.2762, found 566.2763.
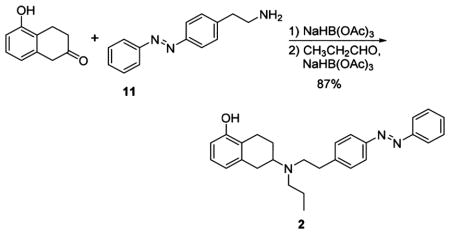
NaHB(OAc)3 (103 mg, 0.46 mmol) was added to a solution of amine 11 (73 mg, 0.32 mmol) and 5-hydroxytetralone (50 mg, 0.31 mmol) in DCE (2 mL), and the resulting suspension was stirred at room temperature overnight. The reaction mixture was portioned between EtOAc and aqueous NaHCO3 solution, and the organic phase was further washed with aqueous NaHCO3 solution, a solution of sodium chloride (10%), and brine. The solution was dried over Na2SO4 and concentrated under reduced pressure, yielding the crude alkylation product as an orange oil, which was immediately transferred to the next step.
The freshly prepared secondary amine (described above) was redissolved in DCE (1.5 mL) and treated with propionaldehyde (25 μL, 0.34 mmol) followed by NaHB(OAc)3 (103 mg, 0.46 mmol). After 5 h, the reaction was quenched and worked up as described above, yielding the crude product tertiary amine. Medium-pressure chromatography (Biotage, 25 g SiO2 column, EtOAc:hexanes = 10:90–100:0, Rf= 0.6 EtOAc:hexanes = 1:1) afforded AP 2 as an orange oil (110 mg, 0.27 mmol, 87% over 2 steps).
Data for 2: 1H NMR (600 MHz, CDCl3): δ = 7.94–7.89 (m, 2H), 7.89–7.83 (m, 2H), 7.54–7.49 (m, 2H), 7.49–7.44 (m, 1H), 7.37–7.32 (m, 2H), 6.99 (t, J = 7.8, 1H), 6.68 (d, J = 7.7, 1H), 6.60 (d, J = 7.9, 1H), 5.40 (s, 1H), 3.06–2.87 (m, 3H), 2.85 (s, 4H), 2.82–2.66 (m, J = 28.9, 15.2, 9.4, 1H), 2.63–2.53 (m, 3H), 2.13–2.07 (m, 1H), 1.68–1.57 (m, 1H), 1.57–1.47 (m, 2H), 0.91 (t, J = 7.3, 3H). 13C NMR (100 MHz, CDCl3): (151 MHz, CDCl3) δ = 153.46, 152.72, 151.07, 144.48, 138.37, 130.72, 129.52, 129.04, 126.40, 122.94, 122.83, 122.73, 121.68, 111.97, 56.58, 52.62, 52.54, 35.81, 32.24, 25.70, 23.60, 22.08, 11.93. IR (ATR): νmax (cm−1) = 3043 (w), 2931 (m), 1585 (s), 1499 (w), 1484 (w), 1464 (s), 1373 (m), 1340 (m), 1277 (s), 1221 (w), 1154 (m), 1104 (w), 1070 (w), 1012 (m), 919 (w), 836 (m), 808 (w), 766 (vs), 712 (w), 687 (vs). HRMS (ESI): calcd for C27H32N3O+ [M+H]+ 414.2545, found 414.2537.
Homology Modeling and Docking
DAR homology models were generated using Bioluminate (Schrodinger, Inc.).91 Models of D1R and D2R were based on β2-adrenergic receptor (β2AR) and D3R, respectively, because of the high relative degree of homology between these receptors. The amino acid sequence of D1R was first aligned to β2AR in BLAST,92 and the TM segments were then structurally aligned with the crystal structure of β2AR bound to the inverse agonist carazolol (PDB: 2RH1). The sequence of D2R was aligned to D3R as described previously,49 and the homology model was generated based on a crystal structure of D3R bound to the antagonist eticlopride (PDB: 3PBL). The three extracellular loops of either D1R or D2R were refined using extended sampling. For docking DA into either D1R or D2R, DA was first prepared using LigPrep (Schrodinger, Inc.) and docked with extra precision (XP) using Glide.93 The hydroxyl groups of the three TM5 serines (Ser5.42, Ser5.43, and Ser5.46) in D1R or D2R that contribute to the OBS were allowed to rotate during the docking procedure. As expected, the protonated amine and hydroxyls of DA were oriented toward Asp3.32 in TM3 and the TM5 serines, respectively. All molecular representations of D1R or D2R were prepared using Chimera, which was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Molecular Biology and Heterologous Expression
For the BRET-based assays used in this study, HEK293T cells were seeded onto 10 cm plates and transfected with a 1:1 ratio of DNA:polyethylenimine (linear, MW 25 000; PolySciences, Inc.). Human D1R and the short isoform of human D2R were cloned into the expression vector pcDNA3.1 with a signal peptide to enhance receptor expression.94 For the BRET-based D1R-mediated cAMP accumulation assay, we transfected pcDNA3.1 plasmids encoding D1R (5 μg) and CAMYEL65 (6 μg; ATCC). For the BRET-based D2R-mediated G protein activation assay, we transfected pcDNA3.1 plasmids encoding D2R (2 μg) and human Gαi1 with Renilla luciferase 8 (Rluc8) inserted at position 91 (Gαi1-91-Rluc8; 0.2 μg)95 and mVenus fragments V1 and V2 fused to human Gβ1 and human Gγ2, respectively (V1-β1 and V2-γ2; 1 and 7 μg, respectively).96 For the D2R-mediated arrestin recruitment assay, we transfected D2R (2 μg), Rluc8-arrestin3-Sp1 (0.25 μg), mem-linker-citrine-SH3 (5 μg), and GRK2 (5 μg).97
For the HTRF-based D2R binding assay, D2R was cloned into pcDNA5/FRT/TO (Invitrogen) with a signal peptide and SNAP-tag added to its N-terminus.70 HEK293 cells were stably transfected with this plasmid using the Flp-in T-Rex system (Invitrogen).
For electrophysiological studies, cysteine mutations were introduced into D1R or D2R using the QuickChange mutagenesis kit (Agilent). Human Gαi1, human chimeric Gαis13,75 human GIRK1, human GIRK2, or tdTomato was cloned into pcDNA3.1. HEK293T cells, sparsely seeded onto 18 mm coverslips, were transiently transfected overnight with GIRK1, GIRK2, and tdTomato (as a marker of transfection) with Lipofectamine 2000 (Invitrogen) at a ratio of 7:7:1 (total DNA of 1.5 μg per coverslip). Each coverslip was also transfected with either 0.7 μg of D1R and 0.35 μg Gαis13 or 0.1 μg D2R and 0.1 μg Gαi1.
Resonance Energy Transfer (RET) Assays
All RET studies were performed in HEK293 or HEK293T cells that were maintained in DMEM (Invitrogen) with 10% fetal bovine serum at 37 °C under 5% CO2. For the BRET studies, HEK293T cells were transiently transfected with the plasmids described above. Cells used in the G protein activation and arrestin recruitment assays were prepared and assayed as described previously in detail.96,97 Briefly, cells were washed, harvested, and resuspended in PBS containing 5 mM glucose at room temperature. Cells (~40 μg of protein per well according to a BCA protein assay kit, ThermoScientific) were distributed into a 96-well microplate (Wallac, PerkinElmer Life, and Analytical Sciences). After incubation with coelenterazine H (5 μM) (Dalton Pharma Services) for 8 min, different ligands were injected and incubated for 2–10 min. Just prior its addition to cells, AP was switched to the cis configuration (cis-AP) by illumination with a hand-held UVGL-15 UV-lamp (power output: ~ 1 mW/mm2; irradiance: 365 nm; UVP, LLC) for 1 min. Using a Pherastar FS plate reader (BMG Labtech), BRET1 signal was determined by quantifying and calculating the ratio of the light emitted by mVenus (510–540 nm) over that emitted by Rluc8 (485 nm).
For experiments used to determine whether MAP binds covalently to WT D2R, MTS reagents or MAP were incubated for 1 h at room temperature with transfected HEK293T cells expressing D2R, Gαi1-91-Rluc8, V1-β1, and V2-γ2. To avoid inactivation of the Gαi1 by either the MTS reagents or MAP, residue C351 was mutated to an isoleucine.
The HTRF-based binding assay was described previously in detail.70 Briefly, 10 000 cells per well were seeded into black 96-well plates (Greiner) pretreated with 50 μg/mL poly-D-lysine. Cells were induced with 1 μg/mL tetracycline in growth medium 24 h after seeding. Forty-eight hours after seeding, cells were incubated with 100 nM Tag-Lite Lumi4 (CisBio) in growth medium for 1 h and washed three times with Tris-Krebs buffer (20 mM Tris, pH 7.4; 118 mM NaCl; 1.2 mM KH2PO4; 1.2 mM MgSO4; 4.7 mM KCl; 1.8 mM CaCl2). Twenty nanomoles of NAPS (N-azidophenethylspiperone)–DY-647 (Cisbio), along with various concentrations of test ligand in a total volume of 200 μL Tris-Krebs/0.1% bovine serum albumin buffer, were added to each well, and plates were incubated for 2 h at room temperature. cis-AP-containing wells were illuminated every 5 min for 30 s with a hand-held UVGL-15 UV-lamp (1 mW/mm2 at 365 nm) to maintain the compound in the cis configuration. Following this incubation period, preparations were excited at 337 nm (excitation of Tag-Lite Lumi4), and emission was measured at 620 nm (emission for Tag-Lite Lumi4) and 665 nm (emission for DY-647) on a Pherastar FS plate reader. We measured 400 μs readings after a 50 μs delay to avoid short-life fluorescence background from the signal.
Electrophysiology
HEK293T cells were maintained in DMEM (Invitrogen) with 10% fetal bovine serum on poly-L-lysine-coated coverslips. Cells were patch-clamped in whole-cell configuration 16–24 h after transfection in high potassium solution containing 120 mM KCl, 25 mM NaCl, 10 mM HEPES, 2 mM CaCl2, and 1 mM MgCl2, pH 7.4. Glass pipettes with a resistance of 3–7 MΩ were filled with intracellular solution containing 140 mM KCl, 10 mM HEPES, 3 mM Na2ATP, 0.2 mM Na2GTP, 5 mM EGTA, and 3 mM MgCl2, pH 7.4. Cells were voltage clamped to −60 or −80 mV using an Axopatch 200A (Molecular Devices) amplifier.
All pharmacological compounds were applied using a gravity-driven perfusion system. Illumination was applied to the entire field of view using a Polychrome V monochromator (TILL Photonics) through a 20× objective (4 mW/mm2 at 460 nm or 0.5 mW/mm2 at 360 nm). pClamp software was used for both data acquisition and control of illumination. For bistable switching, the shutter was manually turned on or off at specific time points. To conjugate MAP to WT and cysteines mutants of D1R or D2R, cells were incubated with 30 μM MAP for 60 min in the dark at 23–27 °C in standard extracellular cell buffers. These conditions are comparable to those used to label other engineered light-gated receptors with maleimide-azobenzene photoswitches. 48,77
Statistics and Data Analysis
Data were analyzed using GraphPad Prism (GraphPad), Clampfit (Axon instruments), or Origin (OriginLab) software. For dose–response curves, data were normalized to vehicle (0%) and dopamine (100%), and nonlinear regression analysis was performed using the sigmoidal dose–response function in GraphPad Prism. To calculate the percentage photoblock of DA by MAP, the DA-induced current under 460 nm light (trans-MAP+DA) was divided by DA-induced current under 360 light (cis-MAP+DA), subtracted from one, and multiplied by 100. Basal current under 360 nm light (cis-MAP−DA) was used as the baseline. This is described below as eq 1:
| (1) |
To calculate inverse photoagonism as a percentage of maximal DA activation, the current under 460 nm light (trans-MAP−DA) was subtracted from cis-MAP−DA, divided by cis-MAP+DA, and multiplied by 100. This is described below as eq 2:
| (2) |
Statistical analyses were performed using Graphpad Prism. All values reported are mean ± SEM.
Supplementary Material
Acknowledgments
The work was supported by National Institutes of Health Nanomedicine Development Center for the Optical Control of Biological Function (2PN2EY018241), Instrumentation Awards S10RR028971, U01NS090527, U01MH109069, and R01MH54137, as well as by the Deutsche Forschungsgemeinschaft (SFB 749 and CIPSM). We thank C. Habrian and Y. Yu for helpful discussions.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b07659. Experimental procedures and characterization of compounds (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Lagerstrom MC, Schioth HB. Nat Rev Drug Discovery. 2008;7:339. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 2.Rask-Andersen M, Almen MS, Schioth HB. Nat Rev Drug Discovery. 2011;10:579. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 3.Kobilka BK. Biochim Biophys Acta, Biomembr. 2007;1768:794. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beninger RJ. Brain Res Rev. 1983;6:173. doi: 10.1016/0165-0173(83)90038-3. [DOI] [PubMed] [Google Scholar]
- 5.Beaulieu JM, Gainetdinov RR. Pharm Rev. 2011;63(1):182. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 6.Greengard P, Allen PB, Nairn AC. Neuron. 1999;23:435. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- 7.Surmeier DJ, Eberwine J, Wilson CJ, Cao Y, Stefani A, Kitai ST. Proc Natl Acad Sci U S A. 1992;89:10178. doi: 10.1073/pnas.89.21.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Neuron. 1995;14:385. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 9.Cepeda C, Chandler SH, Shumate LW, Levine MS. J Neurophysiol. 1995;74:1343. doi: 10.1152/jn.1995.74.3.1343. [DOI] [PubMed] [Google Scholar]
- 10.Hernandez-Lopez S, Bargas J, Surmeier DJ, Reyes A, Galarraga E. J Neurosci. 1997:3334. doi: 10.1523/JNEUROSCI.17-09-03334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schiffmann SN, Lledo PM, Vincent JD. J Physiol. 1995;483:95. doi: 10.1113/jphysiol.1995.sp020570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersson M, Konradi C, Cenci MA. J Neurosci. 2001;21:9930. doi: 10.1523/JNEUROSCI.21-24-09930.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pillai G, Brown NA, McAllister G, Milligan G, Seabrook GR. Neuropharmacology. 1998;37:983. doi: 10.1016/s0028-3908(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 14.Kuzhikandathil EV, Yu W, Oxford GS. Mol Cell Neurosci. 1998;12:390. doi: 10.1006/mcne.1998.0722. [DOI] [PubMed] [Google Scholar]
- 15.Conroy JL, Free RB, Sibley DR. ACS Chem Neurosci. 2015;6:681. doi: 10.1021/acschemneuro.5b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG. J Biol Chem. 2001;276:37409. doi: 10.1074/jbc.M106728200. [DOI] [PubMed] [Google Scholar]
- 17.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Annu Rev Physiol. 2007;69:483. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 18.Lee FJ, Xue S, Pei L, Vukusic B, Chery N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F. Cell. 2002;111:219. doi: 10.1016/s0092-8674(02)00962-5. [DOI] [PubMed] [Google Scholar]
- 19.Liu F, Wan Q, Pristupa ZB, Yu XM, Wang YT, Niznik HB. Nature. 2000;403:274. doi: 10.1038/35002014. [DOI] [PubMed] [Google Scholar]
- 20.Tritsch NX, Sabatini BL. Neuron. 2012;76:33. doi: 10.1016/j.neuron.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dauer W, Przedborski S. Neuron. 2003;39:889. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 22.Howes OD, Kapur S. Schizophr Bull. 2009;35:549. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volkow ND, Fowler JS, Wang GJ, Swanson JM, Telang F. Arch Neurol. 2007;64:1575. doi: 10.1001/archneur.64.11.1575. [DOI] [PubMed] [Google Scholar]
- 24.DiMaio S, Grizenko N, Joober R. J Psychiatry Neurosci. 2003;28:27. [PMC free article] [PubMed] [Google Scholar]
- 25.Denys D, Zohar J, Westenberg HG. J Clin Psychiatry. 2004;65:11. [PubMed] [Google Scholar]
- 26.Buse J, Schoenefeld K, Munchau A, Roessner V. Neurosci Biobehav Rev. 2013;37:1069. doi: 10.1016/j.neubiorev.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Seeman P. Pharmacol Rev. 1980;32:229. [PubMed] [Google Scholar]
- 28.Mailman RB, Huang X. Handbook of Clinical Neurology. 2007;83:77. doi: 10.1016/S0072-9752(07)83004-1. [DOI] [PubMed] [Google Scholar]
- 29.Michino M, Beuming T, Donthamsetti P, Newman AH, Javitch JA, Shi L. Pharmacol Rev. 2015;67:198. doi: 10.1124/pr.114.009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kreitzer AC. Annu Rev Neurosci. 2009;32:127. doi: 10.1146/annurev.neuro.051508.135422. [DOI] [PubMed] [Google Scholar]
- 31.Airan RD, Thompson KR, Fenno LE, Bernstein H, Deisseroth K. Nature. 2009;458:1025. doi: 10.1038/nature07926. [DOI] [PubMed] [Google Scholar]
- 32.Kim JM, Hwa J, Garriga P, Reeves PJ, RajBhandary UL, Khorana HG. Biochemistry. 2005;44:2284. doi: 10.1021/bi048328i. [DOI] [PubMed] [Google Scholar]
- 33.Siuda ER, McCall JG, Al-Hasani R, Shin G, Il Park S, Schmidt MJ, Anderson SL, Planer WJ, Rogers JA, Bruchas MR. Nat Commun. 2015;6:8480. doi: 10.1038/ncomms9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conklin BR, Hsiao EC, Claeysen S, Dumuis A, Srinivasan S, Forsayeth JR, Guettier JM, Chang WC, Pei Y, McCarthy KD, Nissenson RA, Wess J, Bockaert J, Roth BL. Nat Methods. 2008;5:673. doi: 10.1038/nmeth.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urban DJ, Roth BL. Annu Rev Pharmacol Toxicol. 2015;55:399. doi: 10.1146/annurev-pharmtox-010814-124803. [DOI] [PubMed] [Google Scholar]
- 36.Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, Lammel S, Mirzabekov JJ, Airan RD, Zalocusky KA, Tye KM, Anikeeva P, Malenka RC, Deisseroth K. Cell. 2014;157:1535. doi: 10.1016/j.cell.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volgraf M, Gorostiza P, Numano R, Kramer RH, Isacoff EY, Trauner D. Nat Chem Biol. 2006;2:47. doi: 10.1038/nchembio756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin WC, Tsai MC, Davenport CM, Smith CM, Veit J, Wilson NM, Adesnik H, Kramer RH. Neuron. 2015;88:879. doi: 10.1016/j.neuron.2015.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin WC, Davenport CM, Mourot A, Vytla D, Smith CM, Medeiros KA, Chambers JJ, Kramer RH. ACS Chem Biol. 2014;9:1414. doi: 10.1021/cb500167u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sandoz G, Levitz J, Kramer RH, Isacoff EY. Neuron. 2012;74:1005. doi: 10.1016/j.neuron.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chambers JJ, Banghart MR, Trauner D, Kramer RH. J Neurophysiol. 2006;96:2792. doi: 10.1152/jn.00318.2006. [DOI] [PubMed] [Google Scholar]
- 42.Berlin S, Szobota S, Reiner A, Carroll EC, Kienzler MA, Guyon A, Xiao T, Trauner D, Isacoff EY. eLife. 2016;5:12040. doi: 10.7554/eLife.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szobota S, Gorostiza P, Del Bene F, Wyart C, Fortin DL, Kolstad KD, Tulyathan O, Volgraf M, Numano R, Aaron HL, Scott EK, Kramer RH, Flannery J, Baier H, Trauner D, Isacoff EY. Neuron. 2007;54:535. doi: 10.1016/j.neuron.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 44.Janovjak H, Szobota S, Wyart C, Trauner D, Isacoff EY. Nat Neurosci. 2010;13:1027. doi: 10.1038/nn.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaub BM, Berry MH, Holt AE, Reiner A, Kienzler MA, Dolgova N, Nikonov S, Aguirre GD, Beltran WA, Flannery JG, Isacoff EY. Proc Natl Acad Sci U S A. 2014;111:E5574. doi: 10.1073/pnas.1414162111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fehrentz T, Schönberger M, Trauner D. Angew Chem, Int Ed. 2011;50:12156. doi: 10.1002/anie.201103236. [DOI] [PubMed] [Google Scholar]
- 47.Broichhagen J, Frank JA, Trauner D. Acc Chem Res. 2015;48:1947. doi: 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]
- 48.Levitz J, Pantoja C, Gaub B, Janovjak H, Reiner A, Hoagland A, Schoppik D, Kane B, Stawski P, Schier AF, Trauner D, Isacoff EY. Nat Neurosci. 2013;16:507. doi: 10.1038/nn.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Science. 2010;330:1091. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weichert D, Kruse AC, Manglik A, Hiller C, Zhang C, Hubner H, Kobilka BK, Gmeiner P. Proc Natl Acad Sci U S A. 2014;111:10744. doi: 10.1073/pnas.1410415111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosenbaum DM, Rasmussen SG, Kobilka BK. Nature. 2009;459:356. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Portoghese PS, Larson DL, Sayre LM, Fries DS, Takemori AE. J Med Chem. 1980;23:233. doi: 10.1021/jm00177a002. [DOI] [PubMed] [Google Scholar]
- 53.Takemori AE, Portoghese PS. Annu Rev Pharmacol Toxicol. 1985;25:193. doi: 10.1146/annurev.pa.25.040185.001205. [DOI] [PubMed] [Google Scholar]
- 54.Schönberger M, Trauner D. Angew Chem, Int Ed. 2014;53:3264. doi: 10.1002/anie.201309633. [DOI] [PubMed] [Google Scholar]
- 55.Agnetta L, Kauk M, Canizal MCA, Messerer R, Holzgrabe U, Hoffmann C, Decker M. Angew Chem, Int Ed. 2017;56:7282. doi: 10.1002/anie.201701524. [DOI] [PubMed] [Google Scholar]
- 56.Richter HW, Waddell WH. J Am Chem Soc. 1983;105:5434. [Google Scholar]
- 57.Meiser J, Weindl D, Hiller K. Cell Commun Signaling. 2013;11:34. doi: 10.1186/1478-811X-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van der Weide J, De Vries JB, Tepper PG, Horn AS. Eur J Pharmacol. 1986;125:273. doi: 10.1016/0014-2999(86)90037-3. [DOI] [PubMed] [Google Scholar]
- 59.Horn AS, Tepper P, Van der Weide J, Watanabe M, Grigoriadis D, Seeman P. Pharm Weekbl. 1985;7:208. doi: 10.1007/BF02307578. [DOI] [PubMed] [Google Scholar]
- 60.Horn AS, Tepper P, Kebabian JW, Beart PM. Eur J Pharmacol. 1984;99:125. doi: 10.1016/0014-2999(84)90444-8. [DOI] [PubMed] [Google Scholar]
- 61.Seiler MP, Markstein R. Mol Pharmacol. 1984;26:452. [PubMed] [Google Scholar]
- 62.Seiler MP, Markstein R. Mol Pharmacol. 1982;22:281. [PubMed] [Google Scholar]
- 63.Sumners C, Dijkstra D, de Vries JB, Horn AS. Naunyn-Schmiedeberg’s Arch Pharmacol. 1981;316:304. doi: 10.1007/BF00501362. [DOI] [PubMed] [Google Scholar]
- 64.Madras BK, Canfield DR, Pfaelzer C, Vittimberga FJ, Jr, Difiglia M, Aronin N, Bakthavachalam V, Baindur N, Neumeyer JL. Mol Pharmacol. 1990;37:833. [PubMed] [Google Scholar]
- 65.Jiang LI, Collins J, Davis R, Lin KM, DeCamp D, Roach T, Hsueh R, Rebres RA, Ross EM, Taussig R, Fraser I, Sternweis PC. J Biol Chem. 2007;282:10576. doi: 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clayton CC, Donthamsetti P, Lambert NA, Javitch JA, Neve KA. J Biol Chem. 2014;289:33663. doi: 10.1074/jbc.M114.605378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McKenzie CK, Sanchez-Romero I, Janovjak H. Adv Exp Med Biol. 2015;869:101. doi: 10.1007/978-1-4939-2845-3_6. [DOI] [PubMed] [Google Scholar]
- 68.Reiner A, Levitz J, Isacoff EY. Curr Opin Pharmacol. 2015;20:135. doi: 10.1016/j.coph.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Volgraf M, Gorostiza P, Szobota S, Helix MR, Isacoff EY, Trauner D. J Am Chem Soc. 2007;129:260. doi: 10.1021/ja067269o. [DOI] [PubMed] [Google Scholar]
- 70.Michino M, Donthamsetti P, Beuming T, Banala A, Duan L, Roux T, Han Y, Trinquet E, Newman AH, Javitch JA, Shi L. Mol Pharmacol. 2013;84:854. doi: 10.1124/mol.113.087833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shi L, Javitch JA. Annu Rev Pharmacol Toxicol. 2002;42:437. doi: 10.1146/annurev.pharmtox.42.091101.144224. [DOI] [PubMed] [Google Scholar]
- 72.Ballesteros A, Weinstein H. Methods Neurosci. 1995;25:366. [Google Scholar]
- 73.Pollock NJ, Manelli AM, Hutchins CW, Steffey ME, MacKenzie RG, Frail DE. J Biol Chem. 1992;267:17780. [PubMed] [Google Scholar]
- 74.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leaney JL, Milligan G, Tinker A. J Biol Chem. 2000;275:921. doi: 10.1074/jbc.275.2.921. [DOI] [PubMed] [Google Scholar]
- 76.Salmi P, Isacson R, Kull B. CNS Drug Rev. 2004;10:230. doi: 10.1111/j.1527-3458.2004.tb00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gorostiza P, Volgraf M, Numano R, Szobota S, Trauner D, Isacoff EY. Proc Natl Acad Sci U S A. 2007;104:10865. doi: 10.1073/pnas.0701274104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kassack MU, Hofgen B, Decker M, Eckstein N, Lehmann J. Naunyn-Schmiedeberg’s Arch Pharmacol. 2002;366:543. doi: 10.1007/s00210-002-0641-z. [DOI] [PubMed] [Google Scholar]
- 79.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. Mol Pharmacol. 2003;63:1256. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 80.Javitch JA, Li X, Kaback J, Karlin A. Proc Natl Acad Sci U S A. 1994;91:10355. doi: 10.1073/pnas.91.22.10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roberts DJ, Strange PG. Br J Pharmacol. 2005;145:34. doi: 10.1038/sj.bjp.0706073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schultz W. Annu Rev Neurosci. 2007;30:259. doi: 10.1146/annurev.neuro.28.061604.135722. [DOI] [PubMed] [Google Scholar]
- 83.Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, Thian FS, Kobilka TS, Shaw DE, Mueller L, Prosser RS, Kobilka BK. Cell. 2013;152:532. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strange PG. Trends Pharmacol Sci. 2002;23(2):89. doi: 10.1016/s0165-6147(02)01993-4. [DOI] [PubMed] [Google Scholar]
- 85.Kmiecik S, Jamroz M, Kolinski M. Biophys J. 2014;106:2408. doi: 10.1016/j.bpj.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Creese I, Burt DR, Snyder SH. Science. 1976;192:481. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- 87.Seeman P, Lee T, Chau-Wong M, Wong K. Nature. 1976;261:717. doi: 10.1038/261717a0. [DOI] [PubMed] [Google Scholar]
- 88.Strange PG. Trends Pharmacol Sci. 2008;29:314. doi: 10.1016/j.tips.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 89.Lan H, Liu Y, Bell MI, Gurevich VV, Neve KA. Mol Pharmacol. 2009;75:113. doi: 10.1124/mol.108.050534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peterson SM, Pack TF, Wilkins AD, Urs NM, Urban DJ, Bass CE, Lichtarge O, Caron MG. Proc Natl Acad Sci U S A. 2015;112:7097. doi: 10.1073/pnas.1502742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu K, Day T, Warshaviak D, Murrett C, Friesner R, Pearlman D. Proteins: Struct, Funct, Genet. 2014;82:1646. doi: 10.1002/prot.24551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.NCBI Ressource Coordinators. Nucleic Acids Res. 2017;45:D12–D17. doi: 10.1093/nar/gkw1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PSJ. Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 94.Guo W, Shi L, Javitch JA. J Biol Chem. 2003;278:4385. doi: 10.1074/jbc.C200679200. [DOI] [PubMed] [Google Scholar]
- 95.Sauliere A, Bellot M, Paris H, Denis C, Finana F, Hansen JT, Altie MF, Seguelas MH, Pathak A, Hansen JL, Senard JM, Gales C. Nat Chem Biol. 2012;8:622. doi: 10.1038/nchembio.961. [DOI] [PubMed] [Google Scholar]
- 96.Newman AH, Beuming T, Banala AK, Donthamsetti P, Pongetti K, LaBounty A, Levy B, Cao J, Michino M, Luedtke RR, Javitch JA, Shi L. J Med Chem. 2012;55:6689. doi: 10.1021/jm300482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Donthamsetti P, Quejada JR, Javitch JA, Gurevich VV, Lambert NA. Curr Protoc Pharmacol. 2015;70:2.14.1–2.14.14. doi: 10.1002/0471141755.ph0214s70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.