

Abstract
Dynamic ensembles hold great promise in advancing RNA-targeted drug discovery. Here, we subjected the transactivation response element (TAR) RNA from human immunodeficiency virus type-1 to experimental high-throughput screening against ~100,000 drug-like small molecules. Results were augmented with 170 known TAR-binding molecules and used to generate sub-libraries optimized for evaluating enrichment when virtually screening (VS) a dynamic ensemble of TAR determined by combining NMR spectroscopy data and molecular dynamics (MD) simulations. Ensemble-based VS scores molecules with an area under the receiver operator characteristic curve of ~0.85-0.94 and with ~40-75% of all hits falling within the top 2% of scored molecules. The enrichment decreased significantly for ensembles generated from the same MD simulations without input NMR data and for other control ensembles. The results demonstrate that experimentally determined RNA ensembles can significantly enrich libraries with true hits, and that the degree of enrichment is dependent on the accuracy of the ensemble.
Keywords: RNA-targeted drug discovery, HIV-1, TAR, computational docking, virtual screening, ensemble, high-throughput screening
INTRODUCTION
The discovery of regulatory non-coding RNAs (ncRNAs) has been accompanied by a growing interest in targeting RNA using small molecules for therapeutics development1–5. Small molecules enjoy favorable pharmacological properties and do not suffer from delivery limitations inherent to oligonucleotide-based therapeutics5. However, targeting RNA with small molecules comes with a unique set of challenges. Most ncRNAs are non-enzymatic, making it difficult to directly screen for inhibitors. High-throughput screening (HTS) assays targeting RNA often yield hits with low specificity, unfavorable pharmacological properties, and/or poor activity in cell-based assays. Additionally, libraries used in HTS are biased to compounds that bind the deep hydrophobic pockets of proteins, not the polar and solvent exposed pockets typical of RNA targets. Rational approaches to identify small molecules that bind specific RNA secondary structures have had some success6, but achieving the desired selectivity and efficacy is difficult given the prevalence of similar secondary structural motifs across the transcriptome.
Structure-based approaches such as computational docking7,8 potentially provide a powerful means to broadly pre-screen compound libraries and generate sub-libraries enriched with diverse compounds that selectively bind the unique pockets of ncRNAs. However, applying virtual screening (VS) to RNA drug targets is complicated by the high flexibility of RNA and its propensity to undergo large conformational changes upon small molecule binding9. Several approaches have been developed to address protein flexibility including ‘soft docking’10, methods that vary side chain rotamers11, and induced-fit docking12. Unfortunately, none of these approaches can treat the large conformational changes accompanying RNA recognition while maintaining the high computational efficiency needed for VS applications. An alternative approach treats the receptor as an ensemble of many conformations each of which is subjected to VS13–15(reviewed in 7,8). However, the force fields used in molecular dynamics (MD) simulations to generate ensembles of conformations remain underdeveloped and poorly tested for RNA16,17. Because of this, and the much higher flexibility of RNA16,17, there is a greater risk of including artifactual conformations in the ensemble that are rarely sampled in solution, leading to false positives in VS18–20. There is also a greater risk of not sampling conformers with favorable binding pockets because of RNA’s more rugged energy landscape and high propensity for kinetic traps21, thus increasing the likelihood of false negatives.
Recent approaches that combine experimental data with computational methods are making it possible to determine ensembles of proteins and nucleic acids at atomic resolution22–26. Interestingly, ensembles of the apo-state determined using these hybrid approaches often include conformations similar to those observed for the biomolecule when bound to cognate partners23–25. Inspired by these discoveries, we9 and others27,28 have carried out ensemble based VS (EBVS) using experimentally informed ensembles. The utility of this approach in targeting RNA was demonstrated in a prospective study9 utilizing an ensemble of the transactivation response element (TAR) RNA from human immunodeficiency virus type-1 (HIV-1) (Fig. 1a). The ensemble was determined using two sets of NMR residual dipolar coupling (RDC) data29,30 to guide selection of conformations from a pool generated using MD simulations23. RDCs depend on the orientation of bond vectors in a biomolecule relative to a molecule-fixed alignment frame and are sensitive to internal motions spanning a broad range of timescales (picosecond-to-millisecond)29,30. The top 57 scoring small molecules out of a screen of 51,000 compounds included six molecules that bind TAR in vitro. These include the first example of a small molecule that binds an RNA apical loop and an aminoglycoside that binds TAR with high selectivity, inhibiting HIV replication (IC50∼20 μM) in an indicator cell line9.
Figure 1.
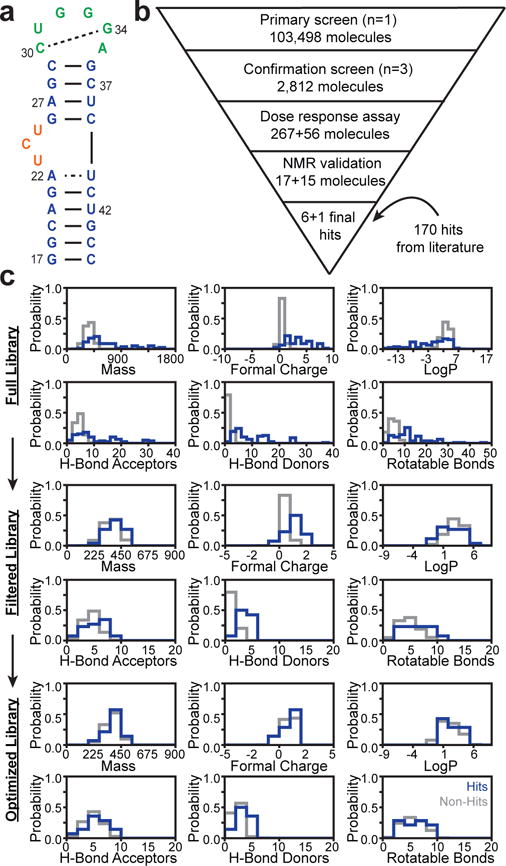
Experimental HTS of HIV-1 TAR RNA to generate libraries for EBVS a. Secondary structure of HIV-1 TAR. b. HTS workflow identifying hits and non-hits. c. Chemical property distributions of hits (blue) and non-hits (gray) for the Full, Filtered and Optimized libraries.
A critical evaluation of VS requires retrospective studies that test the ability of docking to discriminate between known hits and non-hits31. Such studies are routine in protein applications but have been scarce for RNA. Thus far, no study has evaluated the utility of experimentally informed RNA ensembles in enriching true hits using EBVS. In addition, most RNA studies employ non-hits that are not experimentally verified but rather selected using decoy-generation approaches developed for proteins that have not been validated for RNA32–34. Here, we generated a rich dataset by subjecting HIV-1 TAR to experimental HTS against ~100,000 drug-like organic molecules (Fig. 1b). This represents one of the largest RNA-small molecule screens reported to date. After augmenting the ~100,000 compound library with 170 known TAR binders, we generated experimentally validated datasets of hits and non-hits optimized for testing VS, following the general protocol used to generate the database of useful (docking) decoys enhanced (DUD-E) in protein applications31. The results demonstrate that experimentally determined RNA ensembles significantly enrich libraries with true hits and that the degree of enrichment is dependent on the accuracy of the ensemble.
RESULTS
Experimental high-throughput screening to identify TAR hits and non-hits
Using a Tat peptide displacement assay we subjected HIV-1 TAR (Fig. 1a) to experimental HTS against ~100,000 drug-like molecules following the workflow shown in Figure 1b (details in Methods). The library was initially tested in a primary screen employing single point measurements and 260-fold excess small molecule. The 2,812 primary screen hits were subjected to a secondary confirmation screen employing triplicate measurements. The 267 confirmed hits were tested in dose response assays yielding 17 hits with competitive doses to displace 50% of Tat peptide (CD50) values < 100 μM. These compounds were repurchased and re-tested for TAR binding using the displacement assay and NMR chemical shift mapping experiments. This yielded six confirmed hits (Table 1 and Supplementary Fig. 1) and identified three false positives (see Methods and Supplementary Fig. 2). To limit false negatives, we re-tested 56 non-hits with chemical similarity to the hits using dose response assays and NMR experiments. This resulted in the identification of one additional hit (Table 1 and Supplementary Fig. 1) and confirmation of many non-hits with high chemical similarity to our hits (examples in Supplementary Fig. 3). The fact that small structural changes can ablate binding is consistent with the hit molecules making specific interactions with TAR.
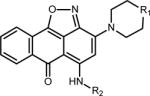
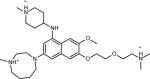
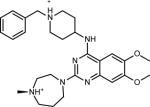
Table 1.
Chemical structure and CD50 values (with and without 100-fold excess tRNA) for TAR hits identified through HTS. Reported values represent the mean and s.d. from n=3 independent experiments.
| Chemical Structure | Molecule Name | CD50 (μM) | 100X tRNA CD50 (μM) | |
|---|---|---|---|---|
|
|
CCG-133994 | 12 ± 4 | 16 ± 5 |
|
|
CCG-133895 | 17 ± 1 | 29 ± 17 | |
|
|
CCG-133868 | 31 ± 7 | 21 ± 10 | |
|
|
CCG-133905 | 53 ± 30 | 12 ± 1 | |
|
|
CCG-133879 | 29 ± 8 | 24 ± 2 | |
|
CCG-208662 | 41 ± 14 | NA | |
|
CCG-208677 | 55 ± 13 | NA | |
To test for false negatives, we re-tested 10 non-hits that score in the top 5% of EBVS using NMR. Four molecules were identified that bind TAR, including an aminoglycoside which was missed in HTS due to insolubility in DMSO, a weak binder that does not satisfy our hit criteria, and two compounds whose binding affinities could not be verified due to fluorescence interference effects (see Methods and Supplementary Fig. 4). These compounds were removed from the VS libraries to avoid biasing results. These results highlight potential weaknesses in experimental HTS and provide a blind test for EBVS to identify TAR binders.
Overall, HTS yielded seven hits, which represent two novel classes of RNA binding small molecules (Table 1). Five of the hit molecules share an anthraquinone scaffold while the other two have napthyl and quinazoline cores. The anthraquinone molecules show selectivity relative to tRNA (Table 1) and insignificant activity in a microRNA screen (Dr. A.L. Garner University of Michigan, personal communication). Of particular relevance to this study, the HTS yielded 103,349 experimentally verified non-hits that can be used as decoys to test the performance of EBVS.
Building small molecule libraries for EBVS evaluation
The HTS library was augmented with 170 diverse small molecules reported in the literature to bind TAR with dissociation or inhibition constants that satisfy our hit criteria (See Supplementary Note 1 and Supplementary Table 1). The hits include derivatives of beta-carboline, quinolone, diphenylfuran, nucleosides, aminoglycosides, and many others as well as 36 molecules with demonstrated activity in cell (or cell-extract) based assays. To avoid bias and maximize chemical diversity, the 177 hits were clustered based on Bemis-Murcko atomic frameworks and the compound with highest affinity selected as a representative of each scaffold. This resulted in the “Full” library consisting of 78 hits (19 with cell-based activity) and 103,349 non-hits.
The chemical properties of hits and non-hits in the Full library are markedly different (Fig. 1c). On average, the hits, which include several aminoglycosides, have larger molecular weight, charge, number of rotatable bonds, hydrogen bond donors and acceptors as well as lower LogP values. Similar differences between RNA binders and compound libraries used in HTS have been noted previously35. Such differences can lead to artificial enrichment in VS by biasing docking scores for hits versus non-hits based solely on differences in 1D chemical properties and not 3D structure complimentarity31,36. We therefore generated two additional property-matched libraries that provide a more stringent test for docking-based enrichment (see Supplementary Note 1). A “Filtered” library containing 26 hits (8 with cell-based activity) and 102,307 non-hits was generated by omitting small molecules with outlier chemical properties (Fig. 1c and Supplementary Fig. 5). An “Optimized” library containing 14 hits (5 with cell-based activity) and 637 non-hits was generated following the general protocol for decoy generation used in the DUD-E31 for protein applications where a set number of property-matched and topologically distinct non-hits are selected for each hit (Fig. 1c, Supplementary Fig. 5, and Supplementary Fig. 6a-b). Together, the three small molecule libraries provide the means to robustly evaluate the performance of VS against TAR RNA.
Ensemble based virtual screening
EBVS was carried out against a recently reported RDC informed dynamic ensemble (E0,4rdc) of HIV-1 TAR RNA (Fig. 2a)24. The ensemble contains twenty unique and equally populated (5% each) conformations24. Compared to the previous TAR ensemble used in VS9,23 (see E1,2rdc below), this ensemble was determined using four rather than two sets of RDCs17 and a longer MD simulation (8.2 μs versus 80 ns) to generate the starting pool of TAR conformations24. The TAR ensemble displays a high degree of flexibility; the pairwise RMSD between any two conformations is >1.9 Å and on average 5.9 Å. This substantially exceeds the flexibility of most protein targets, presenting a significant challenge to docking based approaches.
Figure 2.
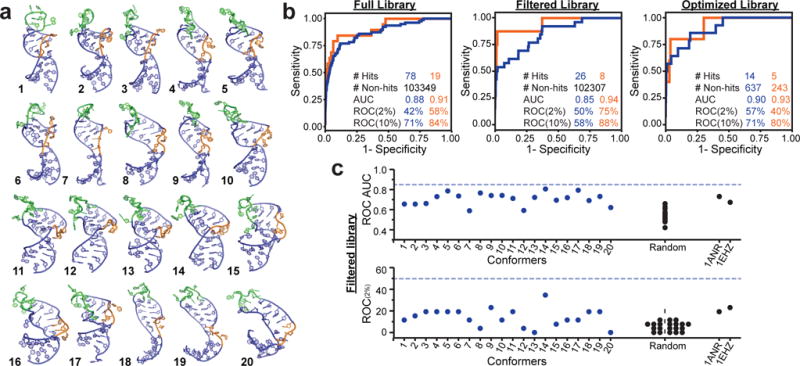
Evaluating EBVS against the RDC TAR dynamic ensemble a. The twenty conformers of the of TAR dynamic ensemble (E0,4rdc). b. ROC curve analysis showing EBVS enrichment of all hits (blue) and cell-active hits (orange) for all three libraries. c. ROC AUC and ROC(2%) scores for docking against individual conformers of the E0,4rdc ensemble, a randomly selected MD ensemble (E0,ran), and the lowest energy NOE-based structures for apo-TAR (PDB 1ANR) and tRNA (PDB 1EHZ) for the Filtered library. Dashed lines indicate the values for the full N=20 E0,4rdc ensemble. Results for the Full and Optimized libraries are shown in Supplementary Fig. 7. ROC plots were generated from one run of docking all molecules to all receptors.
Each small molecule was docked against every TAR conformer using Internal Coordinate Mechanics (ICM)37. Each small molecule was assigned a docking score corresponding to the best score across the 20 conformers, a Boltzmann-weighted average score, or an arithmetic average score (see Methods). The global enrichment of true binders was assessed based on the area under the curve (AUC) of a receiver operator characteristic (ROC) curve, with AUC=1.0 representing perfect enrichment and AUC=0.5 representing random selection of hits and non-hits. For the Full library, optimal enrichment was obtained using the Boltzmann average or best score, whereas the arithmetic average yielded slightly better enrichment for the Filtered and Optimized libraries (Supplementary Fig. 6c). The Full library had larger variation in enrichment across scoring approaches because it contains molecules with highly varied docking scores across conformers. In what follows, we use the Boltzmann average score for the Full library and arithmetic average score for both Filtered and Optimized libraries. Results for all scoring approaches and for including all hits without clustering are presented in Figure 2b and Supplementary Figure 6c.
EBVS globally enriches the Full library with ROC AUC=0.88 and 42% of hits are identified after screening only 2% of non-hits (ROC(2%)=42%) (Fig. 2b). This corresponds to a hit rate of 1.6% as compared to 0.075% when screening the entire library, and an enrichment factor EF(2%)=21. Similar levels of enrichment were obtained for the Filtered (ROC AUC= 0.85 and ROC(2%)= 50%) and Optimized (ROC AUC=0.90 and ROC(2%)=57%) libraries (Fig. 2b). EBVS significantly enriches hits with cell-based activity with ROC AUC=0.91-0.94 and ROC(2%)=40-75% (Fig. 2b). This performance is comparable to best-case results when docking to known bound structures of proteins38–40.
The enrichment was lower for individual TAR conformers derived from the ensemble and decreased further for single conformers randomly selected from the MD pool (Fig. 2c and Supplementary Fig. 7a). Docking against the lowest energy NOE-based structure of free TAR (PDB 1ANR)41 generally performed better than other single conformers, but consistently worse than EBVS (Fig. 2c and Supplementary Fig. 7a). Enrichment was also lower for an NMR structure of tRNA (PDB 1EHZ)42 compared to the TAR ensemble (Fig. 2c and Supplementary Fig. 7a). The TAR binders, including those with cell activity, had higher scores on average for docking against tRNA compared to TAR suggesting that VS would have identified these as selective TAR binders (Supplementary Table 2). The similar level of enrichment observed across the three libraries when VS the ensemble, single conformers, or tRNA argues against significant artificial enrichment in the Full library.
Enrichment depends on ensemble size
On average, enrichment decreased when using smaller sub-ensembles derived from the full N=20 ensemble, reaching a minimum at N=1 (Fig. 3a and Supplementary Fig. 7b). This is despite the fact that increasing the ensemble size increases the risk of including artifactual conformations that can lead to false positives18–20. The N=20 TAR ensemble represents the smallest ensemble that satisfies the RDC data, with smaller ensembles failing to reproduce the RDCs to within experimental uncertainty24. Accordingly, the sub-ensembles have diminishing accuracy as measured based on their agreement with the four RDC datasets (RDC RMSD) (Fig. 3b). Consequently, enrichment decreases on average with increasing RDC RMSD (Fig. 3c). Similar trends were observed for all libraries and for other ensembles (See below and Supplementary Fig. 7c-d). These results show that all 20 conformations contribute to the high enrichment observed for TAR and suggest a correlation between enrichment and ensemble accuracy.
Figure 3.
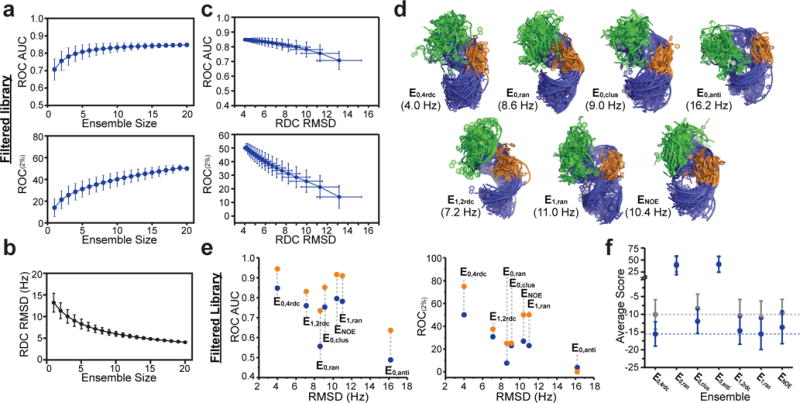
Dependence of EBVS enrichment on ensemble size and ensemble accuracy a. Dependence of the ROC AUC and ROC(2%) scores on ensemble size for the Filtered library. b. Dependence of the RDC RMSD on the ensemble size. c. Dependence of the ROC AUC and ROC(2%) scores on the RDC RMSD for the Filtered library. For a, b, and c the mean and s.d. values over all possible sub-ensembles of each ensemble size are plotted. d. Distinct ensembles of apo-TAR with variable accuracy as assessed based on RDC RMSD (shown in parentheses). e. Dependence of the ROC AUC and ROC(2%) on RDC RMSD for all hits (blue) and cell-active hits (orange) of the Filtered library. f. Mean and s.d. of EBVS scores for hits (blue) and non-hits (gray) of the Filtered library for all ensembles. Dashed lines represent the values for the E0,4rdc ensemble. Results for the Full and Optimized libraries are shown in Supplementary Fig. 7 and 9. All ROC values were generated from one run of docking all molecules to all receptors.
Although all conformations contribute to enrichment, some conformers are predicted to be more or less preferentially bound across the Full library (Supplementary Fig. 8a) and the preferences are different for hit molecules relative to the Full library (Supplementary Fig 8b). Interestingly, conformer 5, which most resembles a known ligand-bound TAR conformation, yields the lowest docking score for many molecules across the Full library but is favored by a smaller percentage of hit molecules, suggesting a favorable but non-selective binding pocket (Supplementary Fig. 8b). Conformers 8, 10, and 17, which also most resemble known ligand-bound TAR conformations, yield the best docking score for more hits than non-hits although conformer 17 is also often selected by false positive hits (top 2% scored non-hits) (Supplementary Fig.8b).
Hyper-enriching sub-ensembles, which exhibit higher enrichment than the N=20 parent ensemble, tend to be enriched in conformers that score highly for hits compared to the Full library, such as conformer 2 (Supplementary Fig. 8c). On the other hand, conformers, such as conformers 5 and 15, that are not favored by hit molecules relative to the library are found in fewer hyper-enriching ensembles. Despite small variations, most conformers are not significantly under- (<20%) or overrepresented (>80%) in hyper-enriching ensembles, supporting that all conformers contribute to enrichment. Taken together, these results highlight how a given conformer can contribute positively to enrichment when placed within an appropriate sub-ensemble even though it may have poor enrichment when considered in isolation or in a different ensemble context.
Enrichment depends on ensemble accuracy
We carried out EBVS on six additional N=20 TAR ensembles with varying degrees of accuracy as assessed by RDC RMSD (Fig. 3d). E0,4rdc, determined using four sets of RDCs and an MD generated pool (MD0), predicts the four RDC data sets with an optimal RMSD=4.0 Hz. Three additional ensembles were generated from the same MD pool by randomly selecting conformations (E0,ran), clustering the MD pool by heavy atom RMSD (E0,clus), or by selecting an ensemble that poorly satisfies the RDCs (E0,anti). These ensembles predict RDCs with less favorable RMSDs of 10.4 Hz, 9.0 Hz, and 16.2 Hz, respectively. Additionally, we examined a previously reported TAR ensemble (E1,2rdc; RMSD=7.2 Hz) determined using two sets of RDCs and a different MD pool (MD1)23 as well as a corresponding control ensemble (E1,ran; RMSD=11.0 Hz) obtained by randomly selecting 20 conformations from the MD1 pool. Finally, we examined the NOE-based bundle of HIV-1 TAR structures (ENOE; RMSD=8.6 Hz)41. Note that the high RDC RMSD observed for ENOE is not surprising considering that NOE-derived distance restraints are orthogonal to RDC-derived orientational restraints and that the bundle of structures is not a statistical ensemble but rather a collection of single structures that satisfy the experimental constraints.
As shown in Figure 3e, Supplementary Fig. 9a and Table 2, E0,4rdc, which best satisfies the RDCs, robustly yields the highest enrichment across the three libraries whereas E0,anti, which least satisfies the RDCs, generally yields the lowest enrichment. The enrichment observed for the remaining ensembles falls between these extremes, and is generally better for the two experimentally informed ensembles (E1,2rdc and ENOE). The three purely computational ensembles (E0,ran, E0,clus and E1,ran) show significant variations in enrichment, highlighting the risks of generating ensembles without experimental input. E0,ran and E0,clus have very similar RDC RMSDs but E0,clus consistently yields higher enrichment, showing that RDC RMSD is not the only predictor of enrichment. This is not surprising considering that RDCs are insensitive to translational aspects of RNA structure that are likely important for predicting binding and that multiple degenerate ensembles can satisfy a given set of RDCs43.
Differences in docking scores and binding pockets help explain the different enrichment levels observed across different ensembles (Fig. 3f and Supplementary Fig. 9b). The difference in scores between hits and non-hits is greater for E0,4rdc relative to other ensembles. The average scores of hits for E0,4rdc are lower than most other ensembles, consistent with formation of optimal pockets. E1,2rdc and E1,ran have comparatively lower average scores for non-hits increasing the likelihood of false positives. Conformers in these ensembles tend to have larger binding pockets relative to other ensembles (Supplementary Fig. 9c). The average scores for E0,ran and E0,anti are significantly elevated for both hits and non-hits and correspondingly they have smaller binding pockets on average (Fig. 3f and Supplementary Fig. 9b-c). All other ensembles had similar binding pocket sizes and accessibility, indicating that enrichment is not determined only by these gross binding pocket features (Supplementary Fig. 9c).
Enrichment correlates to overlap with ligand-bound conformations
We examined how well the different ensembles encompass six previously determined NMR structures of ligand-bound TAR (acetylpromazine (1LVJ)44, rbt550 (1UTS)45, rbt203 (1UUD)46, rbt205 (1UUI)46, neomycin B (1QD3)47, and arginine (1ARJ)48). First, we focused on the relative orientation of TAR helices, which is an important determinant of RNA binding pockets49 and is the least well modeled aspect of TAR in MD simulations16. The average inter-helical orientation has also been independently validated for three ligand-bound TAR conformations based on order tensor analysis of RDCs for arginine50, acetylpromazine51, and neomycin B51. However, these RDC studies also highlighted uncertainty in the NOE-based structures due to deviations in the local geometry and/or unaccounted flexibility (Supplementary Note 2 and Supplementary Fig. 10a).
Overall, ensembles that best overlap with the ligand-bound conformations showed the best EBVS enrichment (Fig. 4a). As noted previously24, E0,4rdc encompasses the six ligand-bound TAR inter-helical conformations despite a very broad MD0 pool. In contrast, E0,anti, which shows the weakest enrichment, shows the poorest overlap with the ligand-bound conformations. Interestingly, E0,clus, which shows better enrichment than either E0,anti or E0,ran, shows the most significant overlap among the three computational ensembles. The MD1 pool has a different spread of inter-helical angles than MD0 that does not overlap as well with the ligand-bound conformations. The ensembles derived from MD1 (E1,2rdc, E1,ran, and ENOE) all show intermediate overlap with the ligand-bound conformations.
Figure 4.
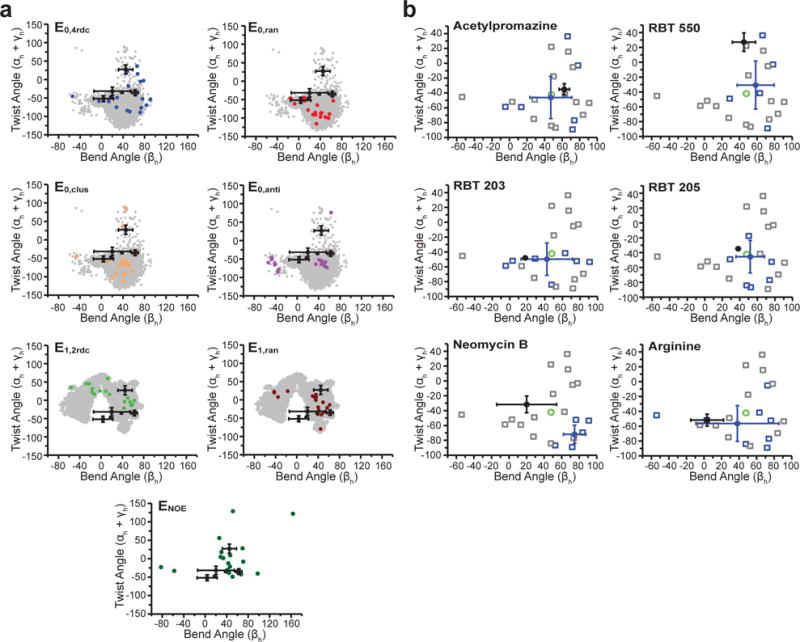
Assessing EBVS-predicted small molecule bound TAR conformations. a. Inter-helical bend (βh) and twist (αh+γh) angles59 (negative and positive twist angles correspond to over- and under-twisting, respectively) for each TAR ensemble (colored) compared to its respective parent MD pool (gray) and all ligand-bound TAR NOE-based NMR structures (black, mean and s.d. values over all deposited structures). b. For each small molecule, the inter-helical angles of the ligand-bound NMR structures (black, mean and s.d. values over all deposited structures) are compared to the conformers of the E0,4rdc ensemble (open squares), the average values over all conformers (green), the Boltzmann-weighted EBVS-predicted structures (blue circles, mean and s.d. values over n=20 independent docking runs), and all conformers predicted to be > 25% populated over n=20 independent docking runs (blue open squares).
Excluding neomycin B, the average inter-helical conformations predicted by EBVS against E0,4rdc is within error of the NMR structures for four out of five molecules and all five bend angles are within error (Fig. 4b). In contrast, only two structures are within error for EBVS against E0,ran (Supplementary Fig. 10b). In the case of neomycin B, docking prefers a conformer that differs considerably from the NMR structure. Here, the larger size of neomycin B likely contributes to greater uncertainty in the docking predictions as is observed in benchmark studies (Fig. 5 and Ref 52).
Figure 5.
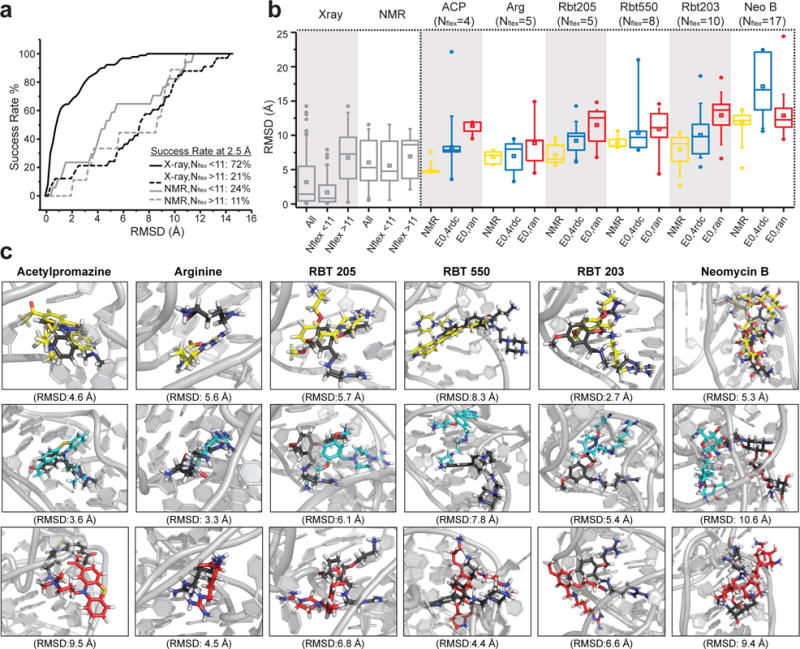
Evaluating ligand-bound poses predicted using EBVS a. Success rates for a benchmark re-docking X-ray (black) or NMR (gray) structures of RNA bound to ligands (see Supplementary Table 3). Data shown for molecules with number of rotatable bonds Nflex< 11 (solid line) and Nflex> 11 (dashed line). RMSD values correspond to the best scoring pose over n=20 independent docking runs. b. Benchmark RMSDs when re-docking ligands to their X-ray (123 structures) or NMR (26 structures) RNA structure and for molecules with Nflex< 11 (17 NMR structures and 90 X-ray structures) and Nflex> 11 (9 NMR structures and 33 X-ray structures). Also shown are the RMSDs over n=20 independent docking runs for each ligand-bound TAR NMR structure after re-docking to the NMR structure (yellow) or when carrying out EBVS against E0,4rdc (blue) and E0,ran (red). (center line, median; center square, mean; box limits, 25th and 75th percentiles; whiskers, 5th and 95th percentiles; points, outliers) c. Lowest RMSD bound poses over n=20 independent docking runs based on re-docking the NMR structure (yellow) or when carrying out EBVS against E0,4rdc (blue) or E0,ran (red). All poses are superimposed onto the NMR structure (black) using the binding pocket and ligand.
Comparison of ligand-bound poses reveals that, with the exception of neomycin B, EBVS correctly places the ligands within or near the RNA binding pocket defined by the NOE-based NMR structure. A more quantitative comparison is complicated by many factors, including the fact that EBVS predicts an ensemble of bound conformations not a single structure, differences in NMR and EBVS predicted RNA structures that complicate alignment, and by evidence for uncertainty in local aspects of the NOE-based NMR structure50 which may arise from the dynamic nature of these complexes (Supplementary Note 2 and Supplementary Fig. 10a). Notwithstanding the above complications, we compared the EBVS predicted ligand poses with the NMR structures.
We first carried out benchmark studies by re-docking known RNA ligand-bound structures (Supplementary Table 3) and computing the ligand RMSD between the re-docked pose and original NMR structure. For X-ray structures of RNA bound to ligands with less than 11 rotatable bonds (Nflex<11), we obtained a success rate of 72% for an RMSD cutoff of 2.5 Å (Fig. 5a). However, the success rate dropped significantly for NMR structures or molecules with Nflex>11 (Fig. 5a). These results highlight the fact that our docking protocol is able to recapitulate bound poses when the structure is well-defined and the molecule is not highly flexible.
To compare EBVS predicted ligand poses to the NMR structures, we computed the ligand RMSD after superimposing structures using both the RNA binding pocket and ligand (see Methods). On average, EBVS predicts the ligand-bound poses (RMSD= 8.3 ± 3.5 Å (acetylpromazine), 7.0 ± 2.0 Å (arginine), 9.2 ± 1.8 Å (rbt205), 10.3 ±3.4 Å (rbt550), 10.1 ± 3.3 Å (rbt203) and 17.1 ± 4.5 Å (neomycin B)) with an accuracy that is comparable, albeit consistently slightly poorer, than those obtained when re-docking the ligands against their NMR structure (RMSD= 4.9 ± 0.6 Å (acetylpromazine), 6.8 ± 0.9 Å (arginine), 7.1 ± 1.3 Å (rbt205), 8.9 ± 0.9 Å (rbt550), 8.0 ± 2.2 Å (rbt203) and 12.0 ± 1.8 Å (neomycin B)) (Fig. 5b). These RMSDs are on the high end for re-docking NMR structures (Fig. 5b). This could be because the apo-ensemble does not perfectly reproduce the ligand-bound TAR conformations and/or because of uncertainty in the NOE-based NMR structures due to lack of RDC restraints and/or unaccounted flexibility. When only considering the lowest RMSD pose over 20 docking runs, EBVS E1,4rdc agrees better with the NMR structure than re-docking the NMR structure itself for three of the six ligands and EBVS against E0,ran yields the poorest agreement for all ligands except neomycin B (Fig. 5c). In light of our benchmark study, the poor pose prediction of neomycin B may be attributed in part to its large number of rotatable bonds.
Discussion
Advances in hybrid experimental-computational methods are enabling the determination of dynamic ensembles with ever increasing accuracy. One of the emerging themes from studies thus far is that bound conformations of biomolecules are often significantly populated in the apo-state ensemble. Even though ensemble-based docking is becoming a popular method for treating flexibility during VS7,8,13–15, only three studies have subjected experimentally informed ensembles to VS9,27,28. Rather, static structures or purely computational ensembles are typically subjected to VS. Here, we present the first perspective study evaluating the enrichment performance of VS experimentally informed ensembles and comparing it to that of computational ensembles.
While an ensemble of structures can often be identified that outperforms single X-ray or NMR structures in retrospective enrichment studies53,54, identifying the successful ensemble in advance of VS can prove difficult19,55. This is a significant problem for RNA given that a handful of conformers have to be selected from thousands of conformations as representatives of a broad conformational landscape. Our results emphasize the potential importance of conformational penalties27 when developing and testing scoring functions against highly flexible RNA targets9,56. In the case of TAR, the performance varies significantly when drawing N=20 ensembles from the same MD pool without guidance from experimental data (Fig. 3 and Table 2). Data from NMR, X-ray, or other methods can guard against artifactual conformations and guide identification of the most populated conformations, which carry the least conformational penalties for ligand binding22–26,57. Experimentally informed conformer populations can also be directly translated into scoring penalties during EBVS27. Additionally, experimental data can define an optimally small ensemble for VS applications, whereas there is no general recipe for selecting ensemble size without experimental input53,55. In the case of RDCs, it has been shown that the minimum sized ensembles that satisfy the data represent a data driven clustering of the real ensemble43. Here, the ensemble size is naturally tuned to the level of dynamics with greater flexibility calling for larger ensembles to satisfy the RDCs43.
Table 2.
Enrichment scores for all TAR ensembles. ROC values were generated from one run of docking all molecules to all receptors.
| RDC RMSD (Hz) | Full library Boltzmann weighting | Filtered library arithmetic average | Optimized library arithmetic average | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| AUC | ROC 2% | AUC | ROC 2% | AUC | ROC 2% | ||
| E0,4rdc | 4.0 | 0.88 | 42% | 0.85 | 50% | 0.90 | 57% |
| E0,ran | 10.4 | 0.47 | 21% | 0.56 | 8% | 0.51 | 0% |
| E0,clus | 9.0 | 0.79 | 29% | 0.75 | 23% | 0.82 | 36% |
| E0,anti | 16.2 | 0.51 | 14% | 0.49 | 4% | 0.47 | 0% |
| E1,2rdc | 7.2 | 0.87 | 50% | 0.76 | 31% | 0.86 | 36% |
| E1,ran | 11.0 | 0.81 | 29% | 0.78 | 23% | 0.86 | 50% |
| ENOE | 8.6 | 0.73 | 31% | 0.80 | 27% | 0.76 | 36% |
Our study also highlights future challenges and opportunities in RNA VS applications. First, while our results indicate that EBVS significantly enriches compounds with activity in cell-based (or cell extract based) assays, there is a need to more directly assess the RNA binding selectivity of hits and to assess the ability of EBVS to enrich for selective RNA binders. Second, rigorous evaluation of pose predictions from EBVS against flexible ncRNA targets will require more high-resolution structures of RNA-small molecule complexes by X-ray or NMR, so long as RDCs and other experimental restraints are used to improve the accuracy of NMR structures. Finally, there is room to further refine ensemble determination approaches by including low-populated conformational states that may have optimal binding pockets. For example, as noted previously23,24, the experimentally informed TAR ensemble does not contain conformers with the U23-A27-U38 base triple which forms on ARG recognition50,58. The integration of conformational penalties from experimentally informed ensembles may help identify pitfalls in docking scoring functions that are currently obscured by treatment of RNA receptors as static structures. Notwithstanding the above future challenges, our results indicate that EBVS can immediately be applied to significantly enrich compound libraries with RNA binders.
ONLINE METHODS
HTS library composition
The small molecule library used in experimental HTS consisted of 103,498 drug-like small molecules available at the Center for Chemical Genomics (CCG), University of Michigan, Ann Arbor. 100,000 molecules were synthetic organic molecules with drug-like properties (ChemDiv). The other 3,498 compounds consisted of 2,000 bioactive molecules (MicroSource Discovery Systems Inc.), 446 molecules (National Institute of Health clinical collection), and 1052 molecules that the CCG had previously found to be active against other targets. The library was stored as 2-5 mM stock solutions in DMSO for ~3 years for initial screens. Repurchased molecules were stored as 3-20 mM stock solutions in DMSO for ~1 year, except for CCG-39701 which was stored as a powder and dissolved in water before use.
Preparation of HIV-1 TAR RNA and Tat peptide
HIV-1 TAR for NMR and binding assays was prepared by in vitro transcription using DNA template containing the T7 promoter (Integrated DNA Technologies). DNA template was annealed at 50 μM DNA in 3 mM MgCl2 by heating to 95°C for 5 min and cooling on ice for 30 min. The transcription reaction was carried out at 37°C for 12 hours with T7 RNA polymerase (New England BioLabs) in the presence of 13C/15N labeled or unlabeled nucleotide triphosphates (Cambridge Isotope Laboratories, Inc). RNA was purified using 20% (w/v) denaturing polyacrylamide gel electrophoresis with 8 M urea and 1X TBE. Purified RNA was extracted from the gel by electroelution in 1X TAE buffer and purified by ethanol precipitation. Purified RNA was dissolved in water to 50 μM RNA, heated to 95°C for 5 min and cooled on ice for 1 hour to anneal. For NMR experiments, 13C/15N labeled RNA was exchanged into NMR buffer [15 mM NaH2PO4/Na2HPO4, 25 mM NaCl, 0.1 mM EDTA, 10% (v/v) D2O at pH 6.4]. For in vitro assays, unlabeled RNA was diluted to 150 nM in Tris-HCl assay buffer [50 mM Tris-HCl, 50 mM KCl, 0.01% (v/v) Triton X-100 at pH 7.4].
The Tat peptide used in HTS, (5-FAM)-AAARKKRRQRRRAAA-Lys(TAMRA), was purchased (LifeTein) with purity > 95% as assessed by Electrospray Ionization Mass Spectrometry. The peptide was stored at −20°C as a 100 μM stock solution in Tris-HCl assay buffer and diluted to 60 nM with assay buffer for use in HTS.
High-throughput screening
Assay
HTS utilized a previously described TAR-Tat displacement assay60. The Tat peptide is highly flexible when free in solution and becomes structured upon binding to TAR61–63. When the Tat peptide is flexible, its two terminal fluorophores, fluorescein and TAMRA, interact and their fluorescence is quenched. Alternatively, in its extended form bound to TAR, the fluorophores are held at a distance allowing fluorescence resonance energy transfer (FRET) from fluorescein to TAMRA. Thus, as inhibitor displaces Tat, there is a decrease in fluorescence signal (excitation: 485 nm, emission: 590 nm). For these assays, we used 50 nM TAR and 20 nM Tat because this ratio gave the maximal fluorescence signal. In the literature, this assay commonly uses a 1:1 ratio of TAR to Tat, so the excess TAR in our assay results in higher CD50 values and a relatively more stringent test of binding. Using neomycin B as a control, we found that the CD50 obtained using our assay (CD50 = 0.96 ± 0.42 μM) is slightly higher than the same assay with a 1:1 ratio of TAR to Tat (CD50 = 0.32 ± 0.10 μM).
The library was tested in a primary screen using a single point measurement (n=1) and 260-fold excess molecule [50 nM TAR, 20 nM Tat, and 13 μM molecule] followed by a confirmation screen of triplicate measurements (n=3) for the 2812 molecules that showed activity, defined as a change in fluorescence signal three standard deviations above the negative control (Tat alone). Molecules were pin-tooled (200 nL) into opaque 384-well microplates by Biomek FX 384-well nanoliter HDR (Beckman) and Mosquito X1 (TTP Labtech). TAR and Tat were dispensed with Multidrop reagent dispenser (Thermo Scientific). Assay mixtures were incubated at room temperature for 10–15 minutes prior to fluorescence measurements using a Pherastar plate reader (BMG Labtech). Each plate during HTS contained 16 wells of TAR and Tat without molecule (negative control) and 16 wells of Tat only (positive control). The Z-factor64 was calculated for each microplate; the average Z-factor throughout the screening campaign was 0.71.
Dose response assays
A total of 267 molecules with reproducible activity were tested in a dose response assay and those with CD50 < 100 μM were considered hits. Dose-response assays were performed such that the final assay concentrations were 50 nM TAR, 20 nM Tat, and 1-1000 μM molecule in assay buffer. Assays were performed in parallel with and without 100-fold excess bulk yeast tRNA to test specificity and in the absence of RNA (Tat only) to measure background signal. There were 137 molecules that caused fluorescence intensity change with Tat alone, suggesting they bound Tat; these were removed from further analysis. Assays were performed in opaque 384-well microplates and read with a Clariostar plate reader (BMG Labtech). Fluorescence signal was normalized to the highest intensity after subtracting background signal. Dose response curves were fit to Equation 1 with OriginPro (OrginLab) using the instrumental weighting method. Equation 2 was used to obtain CD50 values,
| (Equation 1) |
| (Equation 2) |
where A1 and A2 are the lowest and highest signals, respectively; p is the hill slope; and logx0 is the logarithm to base 10 of the concentration at half response. All variables were allowed to float during the fit. Assays were measured in triplicate and the mean and standard deviation (s.d.) is reported.
Validation of hits
The 17 small molecule hits from the dose response assays were re-purchased and re-tested for activity in addition to 56 molecules with chemical similarity to these hits, defined as having >80% similarity based on sphere exclusion clustering performed with JKlustor package (ChemAxon). Next, 32 molecules, including all 17 hits and 15 chemically similar molecules with possible activity in the assay, were tested for TAR binding by NMR chemical shift titrations employing [13C-1H] SOFAST-HMQC NMR experiments65 performed at 298 K on 600 MHz and 800 MHz Agilent spectrometers equipped with triple-resonance HCN cryogenic probes. 13C/15N-labeled TAR was exchanged into NMR buffer. Concentrated stocks of molecule in DMSO were added to TAR such that no more than 10% (v/v) DMSO was added to the buffer. Free TAR controls had equivalent volumes of DMSO to compensate for minor changes that may be induced by DMSO. Spectra were processed using nmrPipe66 and SPARKY67.
Nine molecules were inactive in both the displacement assay and NMR when retested with fresh molecule, suggesting that the original activity was due to contamination or degradation. One of the 56 molecules with chemical similarity to the hits, CCG-133994, was active in both the displacement assay and NMR, despite not being identified as a hit in the primary screen. Three molecules had activity in the assay, but did not bind based on NMR chemical shift titrations. Inspection of the Tat-only control for these molecules suggest that they likely bind Tat rather than TAR in the displacement assay (Supplementary Fig. 2). These should have been identified earlier in the workflow, but the fluorescence change in the presence of Tat may not have been large enough. Overall, seven molecules were confirmed to bind TAR RNA based on their activity in the TAR-Tat displacement assay and their ability to induce chemical shift perturbations in the TAR NMR spectra (Table 1 and Supplementary Fig. 1).
Hit molecules
The anthraquinone hits and chemically similar molecules exhibited a color change from orange to blue when diluted from 100% DMSO to an aqueous solution, likely due to DMSO reacting with the anthraquinone to form DMSO-anthraquinone, as described previously68. All experiments were performed with the derivatives in the blue state. The addition of the small molecule hits to TAR resulted in large chemical shift perturbations or line broadening in 2D NMR spectra for several residues throughout TAR (Supplementary Fig. 1b). As expected, hits with similar chemical structures induce similar chemical shift perturbations indicating that they interact with TAR via similar binding modes (Supplementary Fig. 1b). There are however two interesting exceptions. One of the five anthraquinone molecules, CCG-133905, induces significantly more broadening consistent with tighter binding and/or partial aggregation (Supplementary Fig. 1). CCG-133994, which contains an ester and an amine, induces chemical shift perturbations that are distinct from the other anthraquinone molecules, suggesting a distinct binding mode for this molecule (Supplementary Fig. 1). Furthermore, NMR reveals that CCG-133994 is in slow exchange, which is in agreement with the fact that it is the tightest binder in the TAR-Tat displacement assays.
Identification of false negatives
To investigate possible false negatives in the HTS, we selected ten molecules in the top 5% of docking scores and tested them for TAR binding using NMR. Four of the ten molecules did in fact bind TAR under NMR conditions (Supplementary Fig. 4). Closer analysis revealed that different factors led to the exclusion of these small molecules from HTS during the primary screen. One aminoglycoside molecule, CCG-39701, was insoluble in DMSO but was active in the assay when dissolved in water (Supplementary Fig. 4). CCG-174885, does not displace the Tat-peptide strongly enough to be a hit in our assay, but NMR clearly shows that it does bind TAR. The other two molecules, CCG-208298 and CCG-100975, had fluorescence interference at high concentration preventing determination of an accurate CD50 (Supplementary Fig. 4). To avoid biasing results, these molecules were not included in EBVS. Although these results demonstrate sources of uncertainty in our HTS results, our database is still based on more experimental data than the current standard of docking decoys and our Optimized library should limit the number of false negatives by removing molecules topologically similar to hit molecules (see below). These results also provide a blind test of EBVS since we were able to identify TAR binders.
Virtual Screening
VS was performed using the docking program Internal Coordinate Mechanics (ICM, Molsoft)37. The protocol allowed full ligand flexibility and rigid receptors. Docking was set up as described previously19. Briefly, each of the 20 conformers of the TAR dynamic ensemble34 was uploaded to ICM in PDB format and converted to ICM objects using the default options (waters deleted and hydrogens optimized). Binding pockets were identified with the ICM PocketFinder Module using a tolerance value of 4.6. The volume and buriedness of the binding pocket are given by ICM. Receptor maps were generated to include all atoms within 5 Å of the predicted binding pockets with atom occupancy weighted. Docking was run with a thoroughness value of 1, flexible ring sampling level 2, and covalent geometry relaxed. Protonation states of the small molecules were assigned in ICM at pH 7 with the exception of neomycin B which was manually assigned a charge of +5 based on previous reports69. The full library was docked to each ensemble a single time for the enrichment studies. Docking against the parent E0,4rdc ensemble was replicated and shown to give similar scores/enrichment (ROC AUC/ROC2%= 0.88/42%, 0.81/35%, 0.87/50% for Full, Filtered and Optimized libraries respectively).
Ensemble-Based Docking Scores
The docking scores provided by ICM represent predicted binding energies in kcal/mol. For each molecule, a composite score across all conformers was assigned as the arithmetic average, the top score, or the Boltzmann-weighted average. To calculate the Boltzmann-weighted average, the fractional population of all 20 TAR conformers was calculated using the Boltzmann distribution (Eq. 3). The population of each conformer was multiplied by its docking score and these values were summed over all conformers to calculate the population-weighted score of each molecule (Eq. 4).
| (Equation 3) |
| (Equation 4) |
Where pi is the population of conformer i, εi is the docking score of conformer i, R is the gas constant (1.987×10−3 kcal K−1 mol−1), T is temperature (298 K), and M is the number of conformers in the ensemble.
Receiver Operator Characteristic Curves
An in-house python script was used to generate the ROC plots using Equations 5 and 6 and to calculate the ROC scores (ROC AUC, ROC(2%)),
| (Equation 5) |
| (Equation 6) |
where n is the number of true negatives (TN), true positives (TP), false negatives (FN) or false positives (FP) at every possible score threshold.
Generating TAR ensembles
The RDC-derived TAR ensembles (E0,4rdc and E1,2rdc) were determined as reported previously23,24. Note that no RDCs were measured in the TAR apical loop and this structure is not directly informed by experimental data. The NOE-based NMR ensemble (ENOE) consists of all 20 models of apo-TAR downloaded from the PDB (1ANR)41. The randomly selected ensembles (E0,ran and E1,ran) were constructed by using a random number generator to randomly select 20 structures from the two pools of TAR conformations generated using MD simulations23,24 containing 10,000 (MD0) and 80,000 (MD1) conformations, respectively. Another ensemble was generated by clustering MD0 into 20 clusters by heavy-atom RMSD of all non-terminal nucleotides and taking representative structures from each cluster (E0,clus). Finally, an ensemble that poorly agrees with all four RDC data sets (E0,anti) was generated using a sample and select (SAS) Monte Carlo selection scheme to maximize the χ2 function assessing the agreement between measured and predicted RDCs (Eq. 7)23,
| (Equation 7) |
where i runs over all the RDCs measured for the different constructs j and δ is the weight used to normalize different RDC data sets, and is set at one tenth of the range of RDCs measured for each TAR construct24. Dexp are the experimentally measured RDCs and Dcalc are the predicted RDCs that were calculate by PALES70,71 as described below.
The quality of the various TAR ensembles used in this study was determined by evaluating how well they agree with four sets of RDC data measured on variably elongated TAR RNA molecules as described previously24. Briefly, the program PALES70,71 was used to calculate predicted RDCs based on the structures in the ensemble, after in silico elongation as described previously24. A scaling factor was used to account for variations in experimental conditions. The predicted RDCs are averaged for all structures of the ensemble assuming equal probabilities (Eq. 8),
| (Equation 8) |
where k runs over the N conformers of the ensemble, λj is the scaling factor for the jth TAR construct and Di,j is the ith coupling in the jth construct. These calculated RDCs were then compared to measured RDCs and the RMSD (Hz) was calculated.
Fitting RDCs to ligand-bound NMR structures
Previously published one bond C-H RDCs49–51 were used to assess the quality of NOE-based NMR structures of TAR in complex with arginine (1ARJ)48, acetylpromazine (1LVJ)44 and neomycin B (1QD3)47. Specifically, we computed RMSD between the measured RDCs and values calculated when using the best-fit order tensor determined using RAMAH72.
Benchmarking docking predicted poses
Using an updated set of ligand-bound RNA structures from the PDB that include 123 X-ray structures (90 with Nflex ≤ 11) and 26 NMR (17 with Nflex ≤ 11) structures (Supplementary Table 3), we re-docked each structure 20 times using the same docking procedure as described above. The binding pockets in the NMR structures were defined as any residue within 5 Å of the small molecule. Complexes with metal interactions near the binding site were not included in this benchmark. The RMSD between the re-docked structure and the original pose was calculated using the heavy atoms of the ligand for the best scoring pose over twenty runs.
Computing inter-helical angles
EBVS was used to predict inter-helical angles for six TAR-ligand complexes and values were compared to inter-helical angles in the NOE-based NMR structures of the complexes (acetylpromazine (1LVJ)44, rbt550 (1UTS)45, rbt203 (1UUD)46, rbt205 (1UUI)46, neomycin B (1QD3)47, and arginine (1ARJ)48). For each of these molecules, docking against a TAR ensemble was repeated twenty times using the protocol described above. The inter-helical angles (αh, βh, γh) were computed for each conformer of all ensembles as well as for each model of the bound TAR NMR structures using an in-house software as described previously49. For this calculation, the lower helix was defined by base pairs C19-G43, A20-U42 and G21-C41 and the upper helix was defined by base pairs G26-C39, A27-U38 and G28-C37. For each docking run, the inter-helical angles were population-weighted based on the Boltzmann-weighted docking scores and averaged over all twenty replicates. The inter-helical angles for the NOE-based NMR bundles were averaged over all models assuming equal populations.
Analysis of ligand-bound poses
Ligand poses predicted by EBVS were compared to the NOE-based NMR structures for six-ligand TAR complexes by computing the heavy-atom RMSD between ligands after superimposing structures by both the ligand and RNA binding pocket (defined as any residue within 5 Å of the ligand in the NMR structure). As a control, we first re-docked all ligands to the lowest energy NMR structure twenty times using the same docking protocol as above, defining the binding pocket as all resides within 5 Å of the ligand. The RMSD values were calculated for the best scoring pose over all twenty runs. Next, each ligand was docked to E0,4rdc or E0,ran ensembles twenty times using the same docking protocol. For each run, the ligand RMSD was calculated for the best scoring pose(s) from EBVS (some runs resulted in two significantly (>25%) populated poses) to all structures in the NMR bundle and the best-fit RMSDs over all 20 runs were averaged.
Data Availability
Results from the high-throughput screen have been deposited on PubChem (AID: 1259389). The SDF files for the Full, Filtered and Optimized libraries have been made available at https://sites.duke.edu/alhashimilab/resources/. All other data can be made available upon request.
Code Availability
All custom scripts have been made available at https://sites.duke.edu/alhashimilab/resources/ or can be provided upon request.
Supplementary Material
Acknowledgments
We would like to thank M. Larsen and S. Vander Roest (University of Michigan Center of Chemical Genomics) for their help and input in carrying out high-throughput screening. We also thank the Duke Magnetic Resonance Spectroscopy Center for NMR resources and assistance in carrying out experiments and the Duke Compute Cluster for computational resources and support. This work was supported by the US National Institutes of Health (P50 GM103297, R01 AI066975 and P01 GM0066275 to H.M.A.-H; T32 GM08487 and F31 GM119306 to L.R.G).
Footnotes
AUTHOR CONTRIBUTIONS
Experiments were designed by L.R.G, J.L. and H.M.A.-H., performed by L.R.G. J.L., A.R., D.K.M, M.L.K, A.D.K., and B.S. and analyzed by L.R.G. and H.M.A-H. L.R.G. and H.M.A.-H. wrote the manuscript with input from all other co-authors.
COMPETING FINANCIAL INTERESTS
H.M.A.–H is an advisor to and holds an ownership interest in Nymirum Inc, an RNA-based drug discovery company. Some of the technology used in this paper has been licensed to Nymirum.
References
- 1.Howe JA, et al. Selective small-molecule inhibition of an RNA structural element. Nature. 2015;526:672–7. doi: 10.1038/nature15542. [DOI] [PubMed] [Google Scholar]
- 2.Connelly CM, Moon MH, Schneekloth JS. The emerging role of RNA as a therapeutic target for small molecules. Cell Chem Biol. 2016;23:1077–90. doi: 10.1016/j.chembiol.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shortridge MD, Varani G. Structure based approaches for targeting non-coding RNAs with small molecules. Curr Opin Struct Biol. 2015;30:79–88. doi: 10.1016/j.sbi.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hermann T. Small molecules targeting viral RNA. Wiley Interdiscip Rev RNA. 2016;7:726–43. doi: 10.1002/wrna.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2016;16:167–79. doi: 10.1038/nrd.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Disney MD, Yildirim I, Childs-Disney JL. Methods to enable the design of bioactive small molecules targeting RNA. Org Biomol Chem. 2014;12:1029–39. doi: 10.1039/c3ob42023j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feixas F, Lindert S, Sinko W, Mccammon JA. Exploring the role of receptor flexibility in structure-based drug discovery. Biophys Chem. 2014;186:31–45. doi: 10.1016/j.bpc.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amaro RE, Li WW. Emerging methods for ensemble-based virtual screening. Curr Top Med Chem. 2010;10:3–13. doi: 10.2174/156802610790232279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stelzer AC, et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat Chem Biol. 2011;7:553–9. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrari AM, Wei BQ, Costantino L, Shoichet BK. Soft docking and multiple receptor conformations in virtual screening. J Med Chem. 2004;47:5076–84. doi: 10.1021/jm049756p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerqueira NMFSA, Bras NF, Fernandes PA, Ramos MJ. MADAMM: A multistaged docking with an automated molecular modeling protocol. Proteins. 2009;74:192–206. doi: 10.1002/prot.22146. [DOI] [PubMed] [Google Scholar]
- 12.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–53. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 13.Knegtel RMA, Kuntz ID, Oshiro CM. Molecular docking to ensembles of protein structures. J Mol Biol. 1997;266:424–40. doi: 10.1006/jmbi.1996.0776. [DOI] [PubMed] [Google Scholar]
- 14.Carlson HA, et al. Developing a dynamic pharmacophore model for HIV-1 integrase. J Med Chem. 2000;43:2100–14. doi: 10.1021/jm990322h. [DOI] [PubMed] [Google Scholar]
- 15.Lin JH, Perryman AL, Schames JR, McCammon JA. Computational drug design accommodating receptor flexibility: The relaxed complex scheme. J Am Chem Soc. 2002;124:5632–33. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- 16.Yang S, Salmon L, Al-Hashimi HM. Measuring similarity between dynamic ensembles of biomolecules. Nat Methods. 2014;11:552–4. doi: 10.1038/nmeth.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salmon L, et al. Modulating RNA alignment using directional dynamic kinks: Application in determining an atomic-resolution ensemble for a hairpin using NMR residual dipolar couplings. J Am Chem Soc. 2015;137:12954–65. doi: 10.1021/jacs.5b07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barril X, Morley SD. Unveiling the full potential of flexible receptor docking using multiple crystallographic structures. J Med Chem. 2005;48:4432–43. doi: 10.1021/jm048972v. [DOI] [PubMed] [Google Scholar]
- 19.Craig IR, Essex JW, Spiegel K. Ensemble docking into multiple crystallographically derived protein structures: An evaluation based on the statistical analysis of enrichments. J Chem Inf Model. 2010;50:511–24. doi: 10.1021/ci900407c. [DOI] [PubMed] [Google Scholar]
- 20.Tian S, et al. Assessing an ensemble docking-based virtual screening strategy for kinase targets by considering protein flexibility. J Chem Inf Model. 2014;54:2664–79. doi: 10.1021/ci500414b. [DOI] [PubMed] [Google Scholar]
- 21.Treiber DK, Williamson JR. Beyond kinetic traps in RNA folding. Curr Opin Struct Biol. 2001;11:309–14. doi: 10.1016/s0959-440x(00)00206-2. [DOI] [PubMed] [Google Scholar]
- 22.Blackledge M. Recent progress in the study of biomolecular structure and dynamics in solution from residual dipolar couplings. Prog Nucl Magn Reson Spectrosc. 2005;46:23–61. [Google Scholar]
- 23.Frank AT, Stelzer AC, Al-Hashimi HM, Andricioaei I. Constructing RNA dynamical ensembles by combining MD and motionally decoupled NMR RDCs: new insights into RNA dynamics and adaptive ligand recognition. Nucleic Acids Res. 2009;37:3670–9. doi: 10.1093/nar/gkp156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salmon L, Bascom G, Andricioaei I, Al-Hashimi HM. A general method for constructing atomic-resolution RNA ensembles using NMR residual dipolar couplings : the basis for interhelical motions revealed. J Am Chem Soc. 2013;135:5457–66. doi: 10.1021/ja400920w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lange OF, et al. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–5. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 26.Salmon L, Yang S, Al-Hashimi HM. Advances in the determination of nucleic acid conformational ensembles. Annu Rev Phys Chem. 2014;65:293–316. doi: 10.1146/annurev-physchem-040412-110059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer M, Coleman RG, Fraser JS, Shoichet BK. The incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat Chem. 2014;6:575–83. doi: 10.1038/nchem.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tóth G, et al. Targeting the intrinsically disordered structural ensemble of α -Synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PLoS ONE. 2014;9:e87133. doi: 10.1371/journal.pone.0087133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH. Nuclear magnetic dipole interactions in field-oriented proteins: information for structure determination in solution. Proc Natl Acad Sci USA. 1995;92:9279–83. doi: 10.1073/pnas.92.20.9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–4. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 31.Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J Med Chem. 2012;55:6582–94. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Calin GA, Zhang S. Novel insights of structure-based modeling for RNA-targeted drug discovery. J Chem Inf Model. 2012;52:2741–53. doi: 10.1021/ci300320t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, et al. Accuracy assessment of protein-based docking programs against RNA targets. J Chem Inf Model. 2010;50:1134–46. doi: 10.1021/ci9004157. [DOI] [PubMed] [Google Scholar]
- 34.Morley SD, Afshar M. Validation of an empirical RNA-ligand scoring function for fast flexible docking using RiboDock®. J Comput Aided Mol Des. 2004;18:189–208. doi: 10.1023/b:jcam.0000035199.48747.1e. [DOI] [PubMed] [Google Scholar]
- 35.Aboul-ela F. Strategies for the design of RNA-binding small molecules. Future Med Chem. 2010;2:93–119. doi: 10.4155/fmc.09.149. [DOI] [PubMed] [Google Scholar]
- 36.Verdonk ML, et al. Virtual screening using protein - ligand docking : avoiding artificial enrichment. J Chem Inf Comput Sci. 2004;44:793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 37.Abagyan R, Totrov M, Kuznetsov D. ICM - A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. [Google Scholar]
- 38.Neves MAC, Totrov M, Ruben A. Docking and scoring with ICM: the benchmarking results and strategies for improvement. J Comput Aided Mol Des. 2012;26:675–86. doi: 10.1007/s10822-012-9547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brozell SR, et al. Evaluation of DOCK 6 as a pose generation and database enrichment tool. J Comput Aided Mol Des. 2012;26:749–773. doi: 10.1007/s10822-012-9565-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaudreault F, Najmanovich RJ. FlexAID: Revisiting docking on non-native-complex structures. J Chem Inf Model. 2015;27:1323–36. doi: 10.1021/acs.jcim.5b00078. [DOI] [PubMed] [Google Scholar]
- 41.Aboul-ela F, Karn J, Varani G. Structure of HIV-1 TAR RNA in the absence of ligands reveals a novel conformation of the trinucleotide bulge. Nucleic Acids Res. 1996;24:3974–81. doi: 10.1093/nar/24.20.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi H, Moore PB. The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: a classic structure revisited. RNA. 2000;6:1091–105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang S, Al-Hashimi HM. Unveiling inherent degeneracies in determining population- weighted ensembles of interdomain orientational distributions using NMR residual dipolar couplings: application to RNA helix junction helix motifs. J Phys Chem B. 2015;119:9614–26. doi: 10.1021/acs.jpcb.5b03859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du Z, Lind KE, James TL. Structure of TAR RNA complexed with a Tat-TAR interaction nanomolar inhibitor that was identified by computational screening. Chem Biol. 2002;9:707–12. doi: 10.1016/s1074-5521(02)00151-5. [DOI] [PubMed] [Google Scholar]
- 45.Murchie AIH, et al. Structure-based drug design targeting an inactive RNA conformation: exploiting the flexibility of HIV-1 TAR RNA. J Mol Biol. 2004;336:625–38. doi: 10.1016/j.jmb.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 46.Davis B, et al. Rational design of inhibitors of HIV-1 TAR RNA through the stabilisation of electrostatic ‘Hot Spots’. J Mol Biol. 2004;336:343–56. doi: 10.1016/j.jmb.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 47.Faber C, Sticht H, Schweimer K, Rösch P. Structural rearrangements of HIV-1 Tat-responsive RNA upon binding of neomycin B. J Biol Chem. 2000;275:20660–6. doi: 10.1074/jbc.M000920200. [DOI] [PubMed] [Google Scholar]
- 48.Aboul-ela F, Karn J, Varani G. The structure of the human immunodeficiency virus type-1 TAR RNA reveals principles of RNA recognition by Tat protein. J Mol Biol. 1995;253:313–32. doi: 10.1006/jmbi.1995.0555. [DOI] [PubMed] [Google Scholar]
- 49.Bailor MH, Sun X, Al-Hashimi HM. Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science. 2010;327:202–6. doi: 10.1126/science.1181085. [DOI] [PubMed] [Google Scholar]
- 50.Pitt SW, Majumdar A, Serganov A, Patel DJ, Al-Hashimi HM. Argininamide binding arrests global motions in HIV-1 TAR RNA: comparison with Mg2+-induced conformational stabilization. J Mol Biol. 2004;338:7–16. doi: 10.1016/j.jmb.2004.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pitt SW, Zhang Q, Patel DJ, Al-Hashimi HM. Evidence that electrostatic interactions dictate the ligand- induced arrest of RNA global flexibility. Angew Chem Int Ed Engl. 2005;44:3412–5. doi: 10.1002/anie.200500075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lang PT, et al. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15:1219–30. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rueda M, Bottegoni G, Abagyan R. Recipes for the selection of experimental protein conformations for virtual screening. J Chem Inf Model. 2010;50:186–193. doi: 10.1021/ci9003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nichols SE, Baron R, Ivetac A, McCammon JA. Predictive power of molecular dynamics receptor structures in virtual screening. J Chem Inf Model. 2011;51:1439–46. doi: 10.1021/ci200117n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korb O, et al. Potential and limitations of ensemble docking. J Chem Inf Model. 2012;52:1262–74. doi: 10.1021/ci2005934. [DOI] [PubMed] [Google Scholar]
- 56.Lind KE, Du Z, Fujinaga K, Peterlin BM, James TL. Structure-based computational database screening, in vitro assay, and NMR assessment of compounds that target TAR RNA. Chem Biol. 2002;9:185–93. doi: 10.1016/s1074-5521(02)00106-0. [DOI] [PubMed] [Google Scholar]
- 57.Yoon S, Welsh WJ. Identification of a mnimal subset of receptor conformations for improved multiple conformation docking and two-step scoring. J Chem Inf Comput Sci. 2004;44:88–96. doi: 10.1021/ci0341619. [DOI] [PubMed] [Google Scholar]
- 58.Puglisi JD, Tan R, Calnan BJ, Frankel AD, Williamson JR. Conformation of the TAR RNA-arginine complex by NMR spectroscopy. Science. 1992;257:76–80. doi: 10.1126/science.1621097. [DOI] [PubMed] [Google Scholar]
- 59.Bailor MH, Mustoe AM, Brooks CL, Al-Hashimi HM. 3D maps of RNA interhelical junctions. Nat Protoc. 2011;6:1536–45. doi: 10.1038/nprot.2011.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matsumoto C, Hamasaki K, Mihara H, Ueno A. A high-throughput screening utilizing intramolecular fluorescence resonance energy transfer for the discovery of the molecules that bind HIV-1 TAR RNA specifically. Bioorg Med Chem Lett. 2000;10:1857–61. doi: 10.1016/s0960-894x(00)00359-0. [DOI] [PubMed] [Google Scholar]
- 61.Calnan BJ, Biancalana S, Hudson D, Frankel AD. Analysis of arginine-rich peptides from the HIV Tat protein reveals unusual features of RNA-protein recognition. Genes Dev. 1991;5:201–10. doi: 10.1101/gad.5.2.201. [DOI] [PubMed] [Google Scholar]
- 62.Loret EP, Georgel P, Johnson WC, Jr, Ho PS. Circular dichroism and molecular modeling yield a structure for the complex of human immunodeficiency virus type 1 trans-activation response RNA and the binding region of Tat, the trans-acting transcriptional activator. Proc Natl Acad Sci USA. 1992;89:9734–8. doi: 10.1073/pnas.89.20.9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shojania S, O’Neil JD. HIV-1 Tat is a natively unfolded protein: the solution conformation and dynamics of reduced HIV-1 Tat-(1-72) by NMR spectroscopy. J Biol Chem. 2006;281:8347–56. doi: 10.1074/jbc.M510748200. [DOI] [PubMed] [Google Scholar]
- 64.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 65.Sathyamoorthy B, Lee J, Kimsey I, Ganser LR, Al-Hashimi H. Development and application of aromatic [13C, 1H] SOFAST-HMQC NMR experiment for nucleic acids. J Biomol NMR. 2014;60:77–83. doi: 10.1007/s10858-014-9856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 67.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- 68.Sutter P, Weis CD. Ring opening reactions of 6H-anthra[1,9-cd]isoxazol-6-ones and related compounds. J Heterocycl Chem. 1982;19:997–1011. [Google Scholar]
- 69.Walter F, Vicens Q, Westhof E. Aminoglycoside – RNA interactions. Curr Opin Chem Biol. 1999;3:694–704. doi: 10.1016/s1367-5931(99)00028-9. [DOI] [PubMed] [Google Scholar]
- 70.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: Aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–2. [Google Scholar]
- 71.Zweckstetter M. NMR: prediction of molecular alignment from structure using the PALES software. Nat Protoc. 2008;3:679–90. doi: 10.1038/nprot.2008.36. [DOI] [PubMed] [Google Scholar]
- 72.Hansen AL, Al-Hashimi HM. Insight into the CSA tensors of nucleobase carbons in RNA polynucleotides from solution measurements of residual CSA: Towards new long-range orientational constraints. J Magn Reson. 2006;179:299–307. doi: 10.1016/j.jmr.2005.12.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Results from the high-throughput screen have been deposited on PubChem (AID: 1259389). The SDF files for the Full, Filtered and Optimized libraries have been made available at https://sites.duke.edu/alhashimilab/resources/. All other data can be made available upon request.