

Abstract
Cyclic amines are ubiquitous core structures of bioactive natural products and pharmaceutical drugs. While the site-selective abstraction of C–H bonds is an attractive strategy for preparing valuable functionalized amines from their readily available parent heterocycles, this approach has largely been limited to substrates that require protection of the amine nitrogen atom. In addition, most methods rely on transition metals and are incompatible with the presence of amine N–H bonds. Here we introduce a protecting group free approach for the α-functionalization of cyclic secondary amines. An operationally simple one-pot procedure generates products via a process involving intermolecular hydride transfer to generate an imine intermediate that is subsequently captured by a nucleophile such as an alkyl or aryl lithium compound. Reactions are regioselective and stereospecific and enable the rapid preparation of bioactive amines, as exemplified by the facile synthesis of anabasine and (−)-solenopsin A.
Graphical Abstract
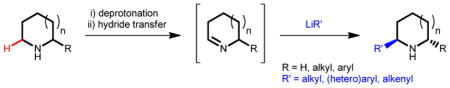
Fully or partially saturated nitrogen heterocycles are key constituents of numerous pharmaceutical drugs.1 However, of the many diverse methods that enable the synthesis of functionalized derivatives of such heterocycles, the vast majority require multistep sequences.2 A particularly attractive entry to substituted, saturated nitrogen heterocycles is the modification of readily available amines via the transformation of ubiquitous but poorly reactive C–H bonds.3,4 Significant challenges notwithstanding, major efforts by the synthetic community continue to focus on the development of methods for the direct functionalization of C–H bonds, including C–H bonds in α-position of the amine nitrogen atom.3,4 Although highly desirable for further processing, current protocols are largely incompatible with the presence of a free amine N–H bond which interferes with C–H bond functionalization. Thus, progress in amine C–H functionalization has largely been limited to methods that rely on protected or tertiary amines. Furthermore, the use of directing groups is a strict requirement of many approaches. These directing groups typically double as amine protecting groups and not infrequently are hard to remove, significantly limiting the applicability of the underlying methods. Here we report a conceptually new strategy that addresses several important limitations.
Outlined in Fig.1 are the most common approaches for amine α-C–H bond functionalization. A widely applied method utilizes N-Boc protected pyrrolidine and related heterocycles as starting materials.5–11 As an example, α-deprotonation of N-Boc protected pyrrolidine with a strong base in presence of a chiral ligand, followed by transmetalation and, finally, palladium/phosphine ligand catalyzed coupling with an aryl halide, results in the formation of α-arylated N-Boc pyrrolidine in highly enantioenriched form (Fig. 1a).12 Other directing group based approaches utilize a transition metal catalyst in the absence of exogenous base (Fig. 1b).13–15 The undoubtedly most widely explored method involves oxidative C–H functionalization and capture of intermediate iminium ions with various nucleophiles such as alkynes (Fig 1c).16–18 While highly useful for certain substrates, the scope of most reactions in this category is often limited to moderately activated N-aryl amines. Somewhat related although mechanistically distinct are photoredox-based approaches (Fig. 1d).19–21 Other strategies for the α-C–H bond functionalization of tertiary amines not depicted in Fig. 1 include hydrogen atom22 or hydride transfer23 and metal-carbenoid insertion.24 Isolated examples of other amine C–H bond functionalizations have been reported, including the functionalization of more remote sites.8,25,26 None of the above mentioned methods are applicable to the C–H bond functionalization of secondary amines.
Figure 1. Methods for amine α-C–H bond functionalization and new concept for secondary amines.

This Figure summarizes some of the most widely used methods for amine α-C–H bond functionalization: a, Deprotonative/cross-coupling approach. b, Directing group-based approach involving oxidative C–H insertion by a metal catalyst. c, Oxidative approach (Cross-dehydrogenative coupling). d, Photoredox-based method. e, Hydroaminoalkylation. f, Redox-neutral condensation. g, This work: Intermolecular hydride transfer based approach.
Methods for the direct α-C–H bond functionalization of secondary amines are rare and essentially limited to hydroaminoalkylation27 (Fig. 1e). While impressive results have been achieved with this approach, due to mechanistic constraints, the method cannot be used to introduce aryl, alkenyl or alkynyl groups or non-α-branched alkyl groups. Our group has developed a mechanistically unique method for the α-C–H bond functionalization of secondary amines that features azomethine ylides as intermediates (Fig. 1f).28,29 However, reactions invariably involve N-alkylation; thus the products of these reaction are themselves tertiary amines.
A new concept for the α-C–H bond functionalization of secondary amines is depicted in Fig. 1g, using pyrrolidine as a prototypical example. Lithiated pyrrolidine (1), readily obtained by in situ deprotonation of pyrrolidine, engages a sacrificial hydride acceptor (a simple carbonyl compound) to provide cyclic imine 4 (via proposed transition state 2) in addition to lithium alkoxide 3. Capture of imine 4 by an organolithium reagent furnishes functionalized amine 5. The ability of lithium amides such as 1 to serve as hydride donors is well documented.30 Likewise, there is precedent for the nucleophilic addition of organolithium species to imines.31 However, these steps apparently have never been applied in sequence. In addition, there seem to have been no concerted efforts to utilize this unique reactivity for amine α-functionalization. This is perhaps not surprising, as there are significant challenges associated with this proposal. Early work by Wittig and coworkers, who reported on the ability of lithiated amines to serve as hydride donors in reactions with carbonyl compounds such as benzophenone, established that imines such as 4 are rapidly deprotonated by the corresponding lithium amide starting material (e.g., 1) to form a 1-azaallyl anion (e.g., 6) and free amine, in this case pyrrolidine.32 The results by Wittig et al. appear to suggest that deprotonation is a more rapid process than hydride transfer and the outcome of their experiments were dominated by 1-azaallyl anion reactivity.32 Another well-appreciated challenge is the known propensity of imines such as 4 to undergo trimerization.33 Several conditions must be met in order to successfully execute the proposed amine α-functionalization. First, reaction conditions must be identified under which the rate of hydride transfer significantly exceeds the rates of both imine deprotonation (by lithium amide 1) or trimerization. In addition, the rate of nucleophilic addition has to be greater than both the rates of imine deprotonation (by the nucleophile) or trimerization.
Following significant experimentation (see Supplementary Table 1), we developed a simple procedure for the α-C–H bond functionalization of a broad range of cyclic N–H amines (Table 1). Deprotonation of the amine, followed by addition of a hydride acceptor and, finally, an organolithium nucleophile, allowed for the formation of a range of products. This method is applicable to amines with various ring sizes. Aryl lithiums bearing different functional groups and heteroaryl lithium reagents were found to be competent nucleophiles. While simple n-alkyl lithium reagents such as n-BuLi (commercial solution in hexanes) were viable nucleophiles, s-BuLi (commercial solution in cyclohexane) and MeLi (commercial solution in ether) did not participate in the title reaction. Organolithium compounds outperformed other organometallic nucleophiles in all cases. For instance, replacement of PhLi for PhMgBr or Ph2CuLi resulted in the formation of 8 in 21% and 41% yield, respectively. Benzophenone was identified as the hydride acceptor of choice in the case of pyrrolidine. Remarkably, hydride transfer was found to occur rapidly at −78° C. In some cases, tert-butyl phenyl ketone outperformed benzophenone as the hydride acceptor. For substrates with attenuated reactivities, trifluoroacetophenone was identified as an efficient hydride acceptor. Notably, a high level of trans-diastereoselectivity was observed for products 23, 24, and 29. The relative stereochemistry of 29 is complementary to that observed in the α-functionalization of N-Boc-4-substituted-piperidine through deprotonative α-lithiation, where the corresponding cis-diastereomers are formed with excellent selectivity.8
Table 1.
α-C–H bond functionalization of cyclic amines.

|
Benzophenone was used as the hydride acceptor (10 min reaction time).
Tert-butyl phenyl ketone was used as the hydride acceptor (30–90 min reaction time).
Trifluoroacetophenone was used as the hydride acceptor (60 min reaction time).
A highly attractive feature of the new α-C–H bond functionalization strategy is its applicability to the regioselective functionalization of amines with an existing α-substituent (Table 2). Reactions are highly regioselective and lead to the replacement of the electronically less activated and sterically more accessible C–H bond, allowing for the synthesis of α,α′-disubstituted amines. The majority of the reactions are highly diastereoselective and lead to the preferential formation of the trans-configured products, consistent with the nucleophile attacking the less hindered face of the intermediate imine. An exception is product 36, which is formed with a slight preference for the cis-isomer, possibly due to the silyl ether acting as a weak directing group. Importantly, reactions are highly stereospecific. For instance, amine (R)-8 was converted to product (R,R)-39 without loss of enantiomeric excess. This finding was exploited in a one-step synthesis of the natural product (−)-solenopsin A34 from readily available amine (R)-48. The natural product anabasine (26),35 itself obtained in one step (see Table 1), was readily converted into derivative 49. Further demonstrating the utility of this method for drug discovery, late-stage intermediates to the commercial drugs varenicline36 and risperidone37 readily underwent α-C–H bond functionalization with high levels of diastereoselectivity.
Table 2.
Regioselective and stereospecific α-C–H bond functionalization of cyclic amines.

|
Benzophenone was used as the hydride acceptor (10 min reaction time).
Tert-butyl phenyl ketone was used as the hydride acceptor (60 min reaction time).
Trifluoroacetophenone was used as the hydride acceptor (60 min reaction time).
Control experiments were performed to test for the potential involvement of the above-mentioned 1-pyrroline trimer 7 (Fig. 2). Apparently, this compound is not an intermediate in this process, as judged by the fact that 7 does not undergo formation of 2-phenyl pyrrolidine (8) when exposed to the standard reaction conditions (Fig. 2a). The same is true when the reaction is performed in the presence of in-situ-generated lithium alkoxide (corresponding to compound 3 in Fig 1). To shed light on the relative rates of the different reaction steps and to potentially simplify the setup conditions, a number of experiments were performed (Fig. 2b). Addition of n-BuLi to a preformed mixture of pyrrolidine and benzophenone, followed by addition of phenyl lithium, resulted in the formation of 8 with similar efficiency as compared to conditions of Table 1, where addition of n-BuLi preceded the addition of benzophenone. This indicates that the deprotonation of pyrrolidine by n-BuLi is significantly faster than any reaction of n-BuLi with benzophenone. The procedure for the formation of 8 could be simplified further by employing phenyl lithium as the base in the first step. Simply adding an excess of phenyl lithium to a mixture of pyrrolidine and benzophenone led to the formation of 8 in nearly identical yield as observed before. This experiment establishes, rather unexpectedly, that the rates of both pyrrolidine deprotonation and hydride transfer vastly exceed the rate of the potentially competing addition of phenyl lithium to benzophenone, a known process.38 We also evaluated this simplified reaction setup in the α-n-butylation of azacyclotridecane. While the yield of 35 was substantially lower than in the stepwise process (32% vs. 64%), this result is quite remarkable as it indicates that the reduction of benzophenone by the intermediate lithium amide can compete with the known reduction of benzophenone by n-BuLi.39
Figure 2. Control studies and simplification of setup conditions.

a, The 1-pyrroline trimer 7 is not involved as an intermediate in pyrrolidine α-C–H bond functionalization. b, Experiments that involve pre-mixing of amine and hydride acceptor.
In conclusion, we have developed an operationally convenient method for the α-C–H bond functionalization of cyclic N–H amines. This approach complements existing methodology, is protecting group free, and is uniquely effective in the stereospecific functionalization of cyclic amines with preexisting substituents. In contrast to the vast majority of current approaches for amine C–H functionalization, no transition metals are required in this process.
Supplementary Material
Acknowledgments
Financial support from the NIH–NIGMS (R01GM101389) is gratefully acknowledged. We thank Dr. Tom Emge (Rutgers University) for crystallographic analysis.
Footnotes
Author contributions: W.C. and L.M. developed the amine α-functionalization and contributed equally to this work. A.P. further developed the reaction and expanded the scope. D.S. conceived and supervised the project and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Data availability statement
Supplementary information including experimental procedures and compound characterization data are available in the online version of the paper.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Vitaku E, Smith DT, Njardarson JT. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J Med Chem. 2014;57:10257–10274. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 2.Vo CVT, Bode JW. Synthesis of Saturated N-Heterocycles. J Org Chem. 2014;79:2809–2815. doi: 10.1021/jo5001252. [DOI] [PubMed] [Google Scholar]
- 3.Campos KR. Direct sp(3) C-H bond activation adjacent to nitrogen in heterocycles. Chem Soc Rev. 2007;36:1069–1084. doi: 10.1039/b607547a. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell EA, Peschiulli A, Lefevre N, Meerpoel L, Maes BUW. Direct alpha-Functionalization of Saturated Cyclic Amines. Chem Eur J. 2012;18:10092–10142. doi: 10.1002/chem.201201539. [DOI] [PubMed] [Google Scholar]
- 5.Beak P, Lee WK. α-Lithioamine synthetic equivalents from dipole-stabilized carbanions: The t-Boc group as an activator for α-lithiation of carbamates. Tetrahedron Lett. 1989;30:1197–1200. [Google Scholar]
- 6.Beak P, Kerrick ST, Wu S, Chu J. Complex Induced Proximity Effects: Enantioselective Syntheses Based on Asymmetric Deprotonations of N-Boc-pyrrolidines. J Am Chem Soc. 1994;116:3231–3239. [Google Scholar]
- 7.McGrath MJ, O’Brien P. Catalytic Asymmetric Deprotonation Using a Ligand Exchange Approach. J Am Chem Soc. 2005;127:16378–16379. doi: 10.1021/ja056026d. [DOI] [PubMed] [Google Scholar]
- 8.Seel S, et al. Highly Diastereoselective Arylations of Substituted Piperidines. J Am Chem Soc. 2011;133:4774–4777. doi: 10.1021/ja201008e. [DOI] [PubMed] [Google Scholar]
- 9.Beng TK, Woo JS, Gawley RE. Synthetic Applications and Inversion Dynamics of Configurationally Stable 2-Lithio-2-arylpyrrolidines and -piperidines. J Am Chem Soc. 2012;134:14764–14771. doi: 10.1021/ja306276w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cordier CJ, Lundgren RJ, Fu GC. Enantioconvergent Cross-Couplings of Racemic Alkylmetal Reagents with Unactivated Secondary Alkyl Electrophiles: Catalytic Asymmetric Negishi α-Alkylations of N-Boc-pyrrolidine. J Am Chem Soc. 2013;135:10946–10949. doi: 10.1021/ja4054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Coldham I. Synthesis of 1,1-Disubstituted Tetrahydroisoquinolines by Lithiation and Substitution, with in Situ IR Spectroscopy and Configurational Stability Studies. J Am Chem Soc. 2014;136:5551–5554. doi: 10.1021/ja500485f. [DOI] [PubMed] [Google Scholar]
- 12.Campos KR, Klapars A, Waldman JH, Dormer PG, Chen C-y. Enantioselective, Palladium-Catalyzed α-Arylation of N-Boc-pyrrolidine. J Am Chem Soc. 2006;128:3538–3539. doi: 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]
- 13.Pastine SJ, Gribkov DV, Sames D. sp(3) C-H bond arylation directed by amidine protecting group: alpha-arylation of pyrrolidines and piperidines. J Am Chem Soc. 2006;128:14220–14221. doi: 10.1021/ja064481j. [DOI] [PubMed] [Google Scholar]
- 14.Spangler JE, Kobayashi Y, Verma P, Wang DH, Yu JQ. α-Arylation of Saturated Azacycles and N-Methylamines via Palladium(II)-Catalyzed C(sp3)–H Coupling. J Am Chem Soc. 2015;137:11876–11879. doi: 10.1021/jacs.5b06740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jain P, Verma P, Xia G, Yu JQ. Enantioselective amine α-functionalization via palladium-catalysed C–H arylation of thioamides. Nat Chem. 2017;9:140–144. doi: 10.1038/nchem.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shono T, Matsumura Y, Tsubata K. Electroorganic chemistry. 46 A new carbon-carbon bond forming reaction at the .alpha.-position of amines utilizing anodic oxidation as a key step. J Am Chem Soc. 1981;103:1172–1176. [Google Scholar]
- 17.Li ZP, Li CJ. CuBr-catalyzed efficient alkynylation of sp(3) C-H bonds adjacent to a nitrogen atom. J Am Chem Soc. 2004;126:11810–11811. doi: 10.1021/ja0460763. [DOI] [PubMed] [Google Scholar]
- 18.Girard SA, Knauber T, Li CJ. The Cross-Dehydrogenative Coupling of Csp3-H Bonds: A Versatile Strategy for C-C Bond Formations. Angew Chem Int Ed. 2014;53:74–100. doi: 10.1002/anie.201304268. [DOI] [PubMed] [Google Scholar]
- 19.McNally A, Prier CK, MacMillan DWC. Discovery of an alpha-Amino C-H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science. 2011;334:1114–1117. doi: 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science. 2016;352:1304–1308. doi: 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beatty JW, Stephenson CRJ. Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc Chem Res. 2015;48:1474–1484. doi: 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshikai N, Mieczkowski A, Matsumoto A, Ilies L, Nakamura E. Iron-Catalyzed C-C Bond Formation at alpha-Position of Aliphatic Amines via C-H Bond Activation through 1,5-Hydrogen Transfer. J Am Chem Soc. 2010;132:5568–5569. doi: 10.1021/ja100651t. [DOI] [PubMed] [Google Scholar]
- 23.Haibach MC, Seidel D. C-H Bond Functionalization through Intramolecular Hydride Transfer. Angew Chem Int Ed. 2014;53:5010–5036. doi: 10.1002/anie.201306489. [DOI] [PubMed] [Google Scholar]
- 24.Davies HML, Manning JR. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature. 2008;451:417–424. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Millet A, Larini P, Clot E, Baudoin O. Ligand-controlled beta-selective C(sp3)-H arylation of N-Boc-piperidines. Chem Sci. 2013;4:2241–2247. [Google Scholar]
- 26.Topczewski JJ, Cabrera PJ, Saper NI, Sanford MS. Palladium-catalysed transannular C–H functionalization of alicyclic amines. Nature. 2016;531:220–224. doi: 10.1038/nature16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Payne PR, Garcia P, Eisenberger P, Yim JCH, Schafer LL. Tantalum Catalyzed Hydroaminoalkylation for the Synthesis of α- and β-Substituted N-Heterocycles. Org Lett. 2013;15:2182–2185. doi: 10.1021/ol400729v. [DOI] [PubMed] [Google Scholar]
- 28.Zhang C, De CK, Mal R, Seidel D. alpha-Amination of Nitrogen Heterocycles: Ring-Fused Aminals. J Am Chem Soc. 2008;130:416–417. doi: 10.1021/ja077473r. [DOI] [PubMed] [Google Scholar]
- 29.Seidel D. The Azomethine Ylide Route to Amine C–H Functionalization: Redox-Versions of Classic Reactions and a Pathway to New Transformations. Acc Chem Res. 2015;48:317–328. doi: 10.1021/ar5003768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majewski M, Gleave DM. Reduction with lithium dialkylamides. J Organomet Chem. 1994;470:1–16. [Google Scholar]
- 31.Scully FE. Regioselective 2-alkylation and 2-arylation of piperidine and pyrrolidine via organolithiation of cyclic imines. J Org Chem. 1980;45:1515–1517. [Google Scholar]
- 32.Wittig G, Hesse A. Zur Reaktionsweise N-metallierter acyclischer und cyclischer sekundärer Amine. Liebigs Ann Chem. 1971;746:149–173. [Google Scholar]
- 33.Yujiro N, Keiichiro O, Yoshito T, Shuji T. One-step synthesis and structural confirmation of 1-pyrroline trimer. Chem Lett. 1977;6:693–696. [Google Scholar]
- 34.MacConnell JG, Blum MS, Fales HM. Alkaloid from Fire Ant Venom: Identification and Synthesis. Science. 1970;168:840–841. doi: 10.1126/science.168.3933.840. [DOI] [PubMed] [Google Scholar]
- 35.Crooks PA. Analytical Determination of Nicotine and Related Compounds and Their Metabolites. 1999:69–147. [Google Scholar]
- 36.Coe JW, et al. Varenicline: An α4β2 Nicotinic Receptor Partial Agonist for Smoking Cessation. J Med Chem. 2005;48:3474–3477. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- 37.Colpaert FC. Discovering risperidone: the LSD model of psychopathology. Nat Rev Drug Discov. 2003;2:315–320. doi: 10.1038/nrd1062. [DOI] [PubMed] [Google Scholar]
- 38.Yamataka H, Kawafuji Y, Nagareda K, Miyano N, Hanafusa T. Electron transfer in the additions of organolithium reagents to benzophenone and benzaldehyde. J Org Chem. 1989;54:4706–4708. [Google Scholar]
- 39.Yamataka H, Miyano N, Hanafusa T. Comparative mechanistic study of the reactions of benzophenone with n-butylmagnesium bromide and n-butyllithium. J Org Chem. 1991;56:2573–2575. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.