

Abstract
Genome-wide association studies (GWAS) have identified over 100 risk loci for schizophrenia, but the causal mechanisms remain largely unknown. We performed a transcriptome-wide association study (TWAS) integrating a schizophrenia GWAS of 79,845 individuals from the Psychiatric Genomics Consortium with expression data from brain, blood, and adipose tissues across 3,693 primarily control individuals. We identified 157 TWAS significant genes, of which 35 did not overlap a known GWAS locus. 42/157 genes were associated to specific chromatin features measured in independent samples, highlighting potential regulatory targets for follow-up. Suppression of one identified susceptibility gene, MAPK3, in zebrafish showed a significant effect on neurodevelopmental phenotypes. Expression and splicing from brain captured the majority of the TWAS effect across all genes. This large-scale connection of associations to target genes, tissues, and regulatory features is an essential step in moving towards a mechanistic understanding of GWAS.
INTRODUCTION
Genome-wide association studies (GWAS) have yielded thousands of robustly associated variants for schizophrenia (SCZ) and many other complex traits, but relatively few of these associations have implicated specific biological mechanisms1,2, as GWAS association signals often span many putative target genes, may affect gene expression through regulatory3 or structural elements4, and may affect genes at considerable genomic distances via chromatin looping5,6. A growing body of research has demonstrated the enrichment of SCZ GWAS risk variants and heritability within regulatory elements identified through maps of chromatin modifications and accessibility1,7–13. Since chromatin modifications are themselves under genetic control6,14–19, a plausible causal mechanism for most SCZ loci is genetic variation leading to regulatory changes marked by chromatin, to gene expression, and finally to disease risk. Indeed, QTLs for chromatin (and other molecular phenotypes) are enriched within GWAS associations, further supporting this hypothesis6,18,20,21.
In this work, we leveraged gene expression and splicing data from SCZ and bipolar (BIP) cases and controls in brain, as well as gene expression data from controls in other tissues, to perform a transcriptome-wide association study (TWAS)22–24 in a large SCZ GWAS data set1 and identify genes whose expression is genetically correlated with SCZ. We subsequently performed a TWAS for a diverse set of chromatin phenotypes to connect putative SCZ susceptibility genes with specific regulatory elements. To our knowledge, this is the first TWAS of any disease to integrate analysis of gene expression, splicing, and chromatin variation, moving beyond top SNPs to implicate SCZ-associated molecular phenotypes across the regulatory cascade (Fig. 1).
Figure 1. Schematic of TWAS approach.
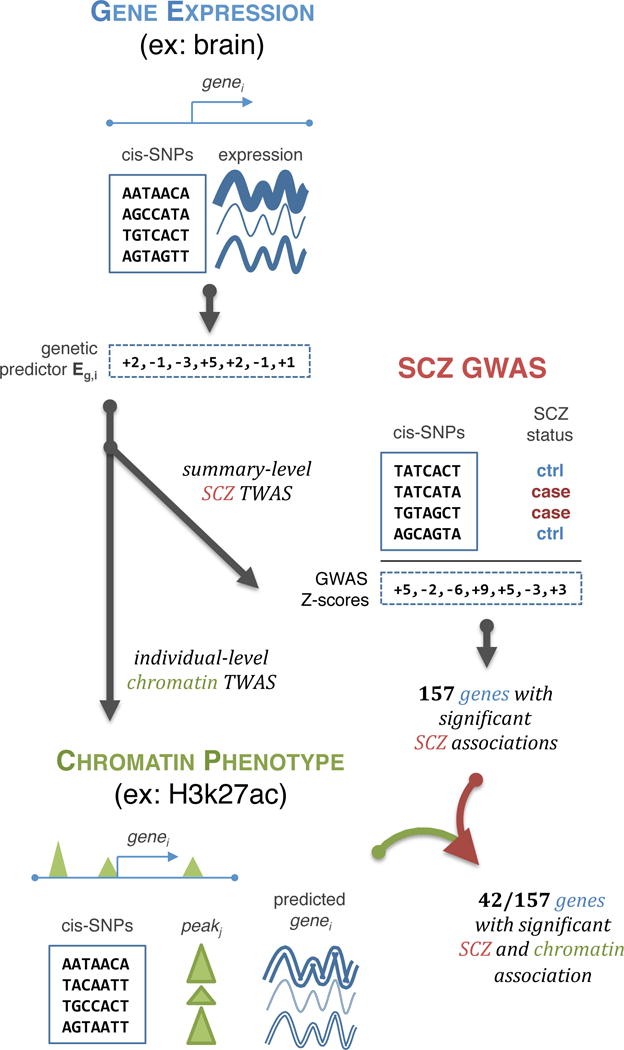
Illustration of the TWAS approach: genetic predictor of gene expression (Eg) is learned in a reference panel (top); integrated with SCZ GWAS association statistics to infer SCZ-Eg association (middle); further integrated with individual-level chromatin phenotypes to infer genes with SCZ and chromatin-Eg associations (bottom). See Supplementary Fig. 1 for detailed analysis flowchart.
RESULTS
TWAS for SCZ identifies new susceptibility genes
We analyzed gene expression and genome-wide SNP array data in 3,693 individuals across four expression reference panels: RNA-seq from the dorsolateral prefrontal cortex (PFC) of 621 individuals – including 283 SCZ cases, 47 BIP cases, and 291 controls – collected by the CommonMind Consortium (CMC)25, expression array data measured in peripheral blood from 1,245 unrelated control individuals from the Netherlands Twin Registry (NTR)26, expression array data measured in blood from 1,264 control individuals from the Young Finns Study (YFS)23, and RNA-seq measured in adipose tissue from 563 control individuals from the Metabolic Syndrome in Men study (METSIM)23. We further characterized splicing events27 in the CMC/brain RNA-seq data (Online Methods). Average cis and trans estimates of SNP-heritability of expression ( , Online Methods) were highly significant in each panel, with nominally significant (P <0.01) for a total of 18,084 genes summed across the four panels (10,819 unique genes; Supplementary Table 1), as well as an additional 9,009 splicing events in brain (in 3,908 unique genes; Supplementary Table 1).
We performed a TWAS using each of the four gene expression reference panels and summary-level data from the PGC SCZ GWAS of 79,845 individuals1 in order to identify genes associated to SCZ (Fig. 1, Supplementary Fig. 1A). Briefly, this approach integrates information from expression reference panels (SNP-expression correlation), GWAS summary statistics (SNP-SCZ correlation), and LD reference panels (SNP-SNP correlation) to assess the association between the cis-genetic component of expression and phenotype (expression-SCZ correlation)23. In practice, the expression reference panel was used as the LD reference panel, and cis SNP-expression effect sizes were estimated using a sparse mixed linear model28 (Online Methods). As SCZ is a highly polygenic trait, we expect these control reference samples to carry disease-affecting regulatory variants. By leveraging genetic predictors of expression our approach is immune to reverse causality (disease → expression), but pleiotropic effects on expression and trait cannot be ruled out without additional analyses (see Discussion)23.
The TWAS identified 247 transcriptome-wide significant gene-SCZ and intron-SCZ associations (summed across expression reference panels) for a total of 157 unique genes, including 49 genes that were significant in more than one expression panel (Fig. 2, Supplementary Fig. 2, Table 1, Supplementary Table 2, 3). We observed no significant differences when performing the TWAS using brain expression from SCZ/BIP cases or controls separately, confirming that the presence of cases in the reference panel did not affect our results (Supplementary Note, Supplementary Table 4). We observed hotspots29 of multiple TWAS-associated genes at 33 loci (defined by genes <500kb apart). However, only 6/33 loci exhibited evidence of statistically independent genetic effects using a summary-based joint test30, suggesting that most of these loci can be explained by a single genetic effect (Online Methods, Supplementary Table 3). Across all TWAS associations, the implicated gene was the nearest gene to the top SNP at the locus in only 56% of instances (using the 10,819 cis-heritable genes as background; decreasing to 24% of instances when using all 26,469 known RefSeq genes) underscoring previous findings23,24,29,31. We confirmed that the summary-based approach was consistent with individual-level predictions using individual-level PGC data and replicated the associations in aggregate using out-of-sample SCZ+BIP phenotypes (Supplementary Note, Supplementary Table 5, 6, Supplementary Fig. 1A, 3, 4, 5).
Figure 2. SCZ TWAS associations and polygenic effects.
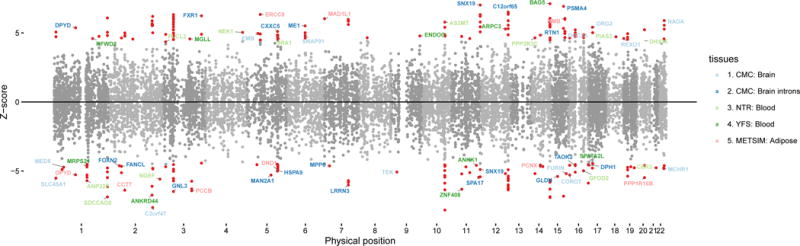
(top) Manhattan plot of all TWAS associations. Each point represents a single gene tested, with physical position plotted on x-axis and Z-score of association between gene and SCZ plotted on y-axis. Transcriptome-wide significant associations are highlighted as red points, with jointly significant independent associations (see Methods) labeled with gene names and color-coded by expression reference (red CMC; blue METSIM, purple YFS, green NTR, black ALL). (bottom) Polygenic TWAS effects across reference tissues. Out of sample SCZ prediction R2 for gene-based polygenic risk scores (GE-PRS) as a function of significance cutoff. Significant correlations (after Bonferroni correction for number of thresholds tested) are indicated with a (*) and the most significant P-value reported. Right-most panel shows prediction from all tissues jointly (black) and from CMC/brain genes + splicing events jointly (red). R2 computed after subtracting ancestry principal components and converting to liability scale with population prevalence of 1%.
Table 1.
Number of TWAS-associated genes across all phenotypes and tissues.
| CMC/brain introns* | CMC/brain | NTR/blood | YFS/blood | MET/adipose | Total** | |
|---|---|---|---|---|---|---|
| Heritable | (9,009) 3,890 | 5,514 | 2,743 | 5,418 | 4,654 | 11,749 |
| SCZ associated | (80) 46 | 44 | 35 | 48 | 39 | 157 |
| SCZ associated (novel†) | (12) 10 | 9 | 6 | 6 | 7 | 35 |
|
| ||||||
| Chromatin associated | (224) 125 | 244 | 182 | 346 | 232 | 806 |
| SCZ and chromatin associated | (10) 8 | 11 | 10 | 13 | 7 | 42 |
Number of splicing events shown in parenthesis.
Total number of unique gene associations.
Novel defined as not overlapping (±500kb) 108 published PGC SCZ GWAS regions.
Of the 108 published PGC GWAS regions1, 47 regions were located near (±500kb) at least one TWAS gene (accounting for 122/157 genes), with the remaining 35/157 genes implicating novel targets. The GWAS association statistics at novel TWAS loci were often well below genome-wide significance (Supplementary Fig. 6), and we hypothesized that some of the new discoveries may be driven by the TWAS aggregating partially independent effects on SCZ that operate through a single gene. As evidence of this, the TWAS association was stronger than the lead SNP for 27% of TWAS associations that did not overlap a genome-wide significant SNP, but only 3% of TWAS associations that did (Fisher’s exact P =8.1×10−07). Across all TWAS associations 21/247 were more significant than the lead GWAS SNP, with the percent of cis expression heritability explained by the top eQTL for these 21 genes significantly lower than for the rest (56% vs. 88%, P =9.6×10−05), indicative of secondary QTL effects. We excluded the MHC region (chr6:28–34MB) from our primary analyses due to its complex haplotype and LD structure. However, as a positive control we specifically tested the C4A gene recently fine-mapped for SCZ4, which lies inside the MHC, and confirmed a highly significant TWAS association between C4A expression in brain tissue and SCZ (P =1.8×10−18).
Splicing events in brain accounted for 46 transcriptome-wide significant gene associations (of which 10 were at novel loci), comparable to the 44 significant gene associations from brain (Table 1, Supplementary Table 3), despite the fact that splicing events accounted for 30% fewer significantly cis-heritable genes than total expression (Supplementary Table 1). Overall, 20/46 associations corresponded to genes that were not tested in the analysis of total gene expression due to non-significant expression heritability, and 19 of the remaining 26 did not have a transcriptome-wide significant association for total gene expression. This is consistent with the recent observation that sQTLs are typically independent of eQTLs at the same gene27. We caution that effect direction for splice events is difficult to interpret because alternatively spliced exons are often negatively correlated (Supplementary Note, Supplementary Fig. 7). Although the largest number of associations came from brain the enrichment was not striking after accounting for the total number of heritable genes (Table 1), suggesting that expression data quality and sample size currently play a more important role than tissue-specificity in finding significant associations.
TWAS associations may be caused by coincidental overlap between eQTLs and non-causal disease variants at a GWAS locus32, which we investigated using formal colocalization and conditional analyses. First, we used the COLOC method33 to estimate the posterior probability of a single shared causal variant for TWAS implicated genes and SCZ using the marginal association statistics. We calibrated a 5% false discovery threshold for considering a gene “colocalized” using randomly selected heritable genes in the same SCZS GWAS regions (Online Methods). Colocalization between eQTLs and SCZ was observed for 55% of the TWAS implicated genes (Supplementary Fig. 8, Supplementary Table 3). We note that COLOCs posterior is highly dependent on the prior probability of a single shared causal variant (Supplementary Fig. 9) and conservative when multiple causal variants mediate the effect on expression and trait23, so that colocalization at the remaining loci may be underestimated. For the 45% genes that did not colocalize significantly, the percent of cis expression heritability explained by the top eQTL was lower than for the rest (79% vs. 89%), suggestive of secondary effects, but the difference was not statistically significant. Second, conditioning on the predicted expression of a TWAS-associated gene (using summary-level data30, Online Methods) reduced the χ2 of the lead GWAS SNP at the locus (including genome-wide significant and non-significant loci) from 42 to 10 on average, and explained more of the association signal than conditioning on the corresponding top expression-QTL (eQTL) (Supplementary Table 7). For the 43 lead GWAS SNPs at genome-wide significant loci that were correlated (r2>0.05) with the predicted expression of at least one TWAS-significant gene (out of 47 overlapping index SNPs), joint conditioning on the predicted expression of all such genes reduced the median SNP P-value from P =1.2×10−10 to P =0.028 (Online Methods, Supplementary Table 8). Given that the expression predictor typically captures only 60-80% of the cis component of gene expression at the expression panel sample sizes used here23, the complete elucidation of the cis component could potentially explain the entire GWAS signal at these loci.
This SCZ GWAS data1 was recently evaluated in a TWAS with gene expression in blood using Summary-based Mendelian Randomization (SMR)24, identifying 16 transcriptome-wide significant associated genes (in contrast to 157 identified here). Of the 16 gene associations identified by SMR, 12 were tested in our study in blood, all replicated at nominal P <0.05 (with consistent sign), and 9 were transcriptome-wide significant – a striking concordance given the different methods and independent expression panels used.
Functional validation of TWAS-associated genes using chromatin interaction data
We leveraged recently published chromatin interaction (Hi-C) data in developing human brain34 to investigate whether TWAS-associated genes are supported by physical chromatin interactions that occur during brain development (Supplementary Fig. 1B). We used the Hi-C data to construct a set of comparison SCZ risk genes based on 3D chromatin interactions between gene transcription start sites (TSS) and SNPs in the fine-mapped 95% causal credible set (Online Methods). This yielded a set of 59 loci with both TWAS and fine-mapped Hi-C data, containing 474 Hi-C predicted SCZ risk genes. The 474 Hi-C predicted genes overlapped with 105/157 TWAS-associated genes (Supplementary Fig. 10; Fisher’s exact test P =1.03×10−18, OR=4.68 compared to random heritable genes at these loci), indicating that most of the TWAS associated genes were supported by 3D chromatin interactions with an SCZ SNP in developing brain. TWAS associations were also significantly correlated with increased expression during mid-fetal developmental in independent samples (P <0.05/19; Supplementary Note, Supplementary Fig. 11, 12) further underscoring the etiological relevance of mechanisms active during brain development.
Polygenic TWAS signal largely explained by expression in brain
To assess the full polygenic architecture of the TWAS associations, we relaxed the transcriptome-wide significance threshold and constructed gene-based polygenic risk scores (GE-PRS) from their predicted expression in the CMC (SCZ+BIP) case-control samples (Supplementary Fig. 1C). For each out-of-sample individual, the GE-PRS is the sum of predicted expression weighted by its signed SCZ TWAS Z-score (Online Methods). The GE-PRS was significantly associated with SCZ status (conditioned on ancestry) across the full spectrum of TWAS association P-values (Fig. 2), as seen with SNP-based polygenic scores1,35,36. Although the prediction was significant in all tissues individually, there was clear evidence of increased effect in brain (in contrast to the transcriptome-wide significant results), with the prediction from brain (genes and splicing events) capturing 92% of the joint prediction from all tissues (Fig. 2, Supplementary Fig. 13). A GE-PRS from actual measured expression and differential splicing in brain was significant but substantially less so than the genetic GE-PRS (Supplementary Fig. 13). Based on polygenic theory37,38, the best TWAS GE-PRS was estimated to account for 26% of the total SCZ SNP-heritability, providing an upper-bound on the amount of trait variance that could be mediated by the steady-state expression in these tissues (Supplementary Note).
Chromatin TWAS identifies specific regulatory features associated with expression
We next sought to identify relationships between the expression of TWAS genes and cis-regulatory elements marked by chromatin activity. We used population-level ChIP-Seq chromatin phenotypes measured in 76 HapMap YRI LCLs for H3k27ac (marking active enhancers), H3k4me1 (enhancers), H3k4me3 (promoters), and DNAse (open chromatin)6, and in 45 HapMap CEU LCLs for H3k27ac, H3k4me1, H3k4me3, PU1 (regulatory transcription factor) and RNA polymerase II (RPB2, associated with active transcription)18. For each of the nine chromatin phenotypes, regions with an excess of ChIP-Seq reads were segmented into local peaks, and the chromatin abundance within each peak treated as a quantitative trait6,18. Both cohorts additionally had gene expression measured by RNA-seq in the same samples, and we confirmed that the genetic correlation was highly significant between expression and each chromatin mark (as well as between different chromatin marks) and persisted as far as 500kb from the TSS (Supplementary Fig. 14, 15, 16, Supplementary Table 9, Online Methods).
We applied individual-level TWAS methods23 to predict expression of the 10,819 significantly heritable genes and 9,009 differentially spliced introns into samples with chromatin phenotypes and searched for expression-chromatin associations (Fig. 1, Supplementary Fig. 1D). Prediction was performed from expression to chromatin phenotype samples (instead of from chromatin phenotype to expression samples) due to increased prediction accuracy in the larger expression panels, but we note that this choice was agnostic to the direction of causality (Supplementary Note). Our approach yielded an average of 2.4× more Bonferroni significant expression-chromatin associations than the conventional approach using in-sample cis-eSNP/cQTL overlap39,40, primarily due to associations >10kb from the TSS (Fig. 3, Supplementary Fig. 17); with similar results when overlapping all cis-eQTLs6,18 and in simulation (Supplementary Note; Supplementary Fig. 18, 19, 20; Supplementary Table 10). Across all tissues, 806 unique genes had a transcriptome-wide significant association (Online Methods) with at least one chromatin phenotype (Supplementary Fig. 18B, Supplementary Table 11), and 4,294 genes were significant at the 10% (per-phenotype) FDR used in previous studies6,18 (Supplementary Table 12). In contrast, only 224 of 9,009 splicing events in the CMC had a transcriptome-wide significant chromatin association, corresponding to 2-3× fewer associations than identified using total CMC gene expression (depending on the chromatin phenotype, Supplementary Table 13). Half of the chromatin associations were distal (10–500kb from TSS) and these were significantly enriched for Hi-C interactions in LCLs6 relative to random (distance-matched) gene-peak pairs (Supplementary Fig. 22, 23, 21, 1G, 24). No other differences in chromatin mark usage or mark-gene distance were observed across the expression reference panels. However, we found that genes with associations to multiple chromatin peaks were more likely to be driven by a single eQTL (Supplementary Table 14), suggesting that multiple chromatin TWAS peaks were typically related by a single genetic mechanism.
Figure 3. Chromatin TWAS associations compared to top eSNP/cQTL associations.
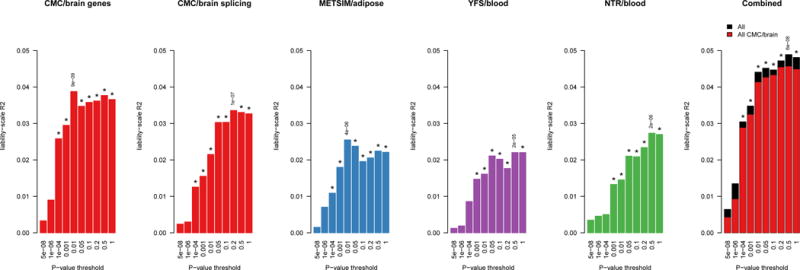
Number of unique genes significantly associated with a chromatin peak after Bonferroni correction for a given distance from the gene (x-axis): (left) using top eSNP in chromatin cohort; (right) using chromatin TWAS from all reference panels. Results from CEU (YRI) populations shown in top (bottom) panels.
We used the measured RNA-seq expression in the chromatin individuals to confirm these associations. Across the 806 chromatin TWAS-associated genes, the correlation between measured expression and an associated chromatin phenotype was highly significant when compared against a distance-matched background null (Supplementary Fig. 1E, 14B), with the average TWAS-associated chromatin peak explaining a striking 20% of the variance in expression of its target gene in CEU (Supplementary Fig. 25, 26, 27, 28, Supplementary Table 16). For the three chromatin phenotypes that were measured in both CEU and YRI, chromatin TWAS peaks implicated in one population were predictive of correlation with measured expression in the other (Supplementary Fig. 1F, 29, 30, Supplementary Table 17), supporting our use of chromatin phenotypes from multiple populations.
Putative regulatory mechanisms for SCZ-associated genes
Focusing on the 157 transcriptome-wide significant genes from the SCZ TWAS, we identified 42 genes (including 7 genes at novel loci) that also had Bonferroni significant chromatin TWAS associations (to a total of 78 individual chromatin peaks) in analyses using the same expression reference panel (Supplementary Fig. 1H, Table 1, Supplementary Table 2, 3, 18, 19). Only 8 of the 78 chromatin peaks underlying joint SCZ TWAS and chromatin TWAS associations were within the promoter (±2kb of the TSS) of their associated gene, suggesting that most regulatory elements affecting SCZ are distally located, as previously observed in other traits6,8,20. SCZ TWAS genes were nominally enriched for chromatin TWAS associations (OR=1.53, P =4×10−4) but the effect was largely dampened after matching on cis-genetic properties of genes (P =0.01; Supplementary Table 20) and could potentially be explained by other unknown properties.
Significant evidence of chromatin-SCZ association and colocalization was observed for the majority of the identified peaks using independent statistical methods (Supplementary Fig. 1H). We analyzed the subset of SCZ TWAS loci with expression-chromatin associations by applying COLOC to (1) SNP-expression and SNP-chromatin association data to investigate expression-chromatin colocalization and (2) SNP-chromatin and SNP-SCZ association data to investigate chromatin-SCZ colocalization. Colocalization was observed for 100% of the expression-chromatin associations and 97% of the chromatin-SCZ associations in CEU (Supplementary Fig. 8, Supplementary Table 19). The chromatin associations in YRI pose a model violation for COLOC due to differences in LD structure between populations, but colocalization still remained much higher than background, with 70% (43%) of expression-chromatin (chromatin-SCZ) associations colocalized (Supplementary Fig. 8). Predicting chromatin into SCZ using SMR24 (which tests the best cQTL) or a TWAS-like test (testing all SNPs in the BSLMM predictor) replicated >60% of the associations at Bonferroni significance and >90% at P <0.05 (Supplementary Note, Supplementary Table 21, 3, 19). However, the chromatin sample size was insufficient to robustly estimate genetic predictors of chromatin and carry out a full chromatin-wide association study.
Examples of SCZ and chromatin TWAS loci
We highlight three examples of TWAS associations to both SCZ and chromatin phenotypes. We visualize these loci using a “TWAS scatterplot” of the relationship between each marginal GWAS/QTL association (Z-score, y axis) and the correlation (x axis) between TWAS predicted expression (GEpred) and the marginal GWAS/QTL association. This relationship is expected to be linear and without outliers under the TWAS model (Supplementary Note, Fig. 4, 5, 32, 33, 34).
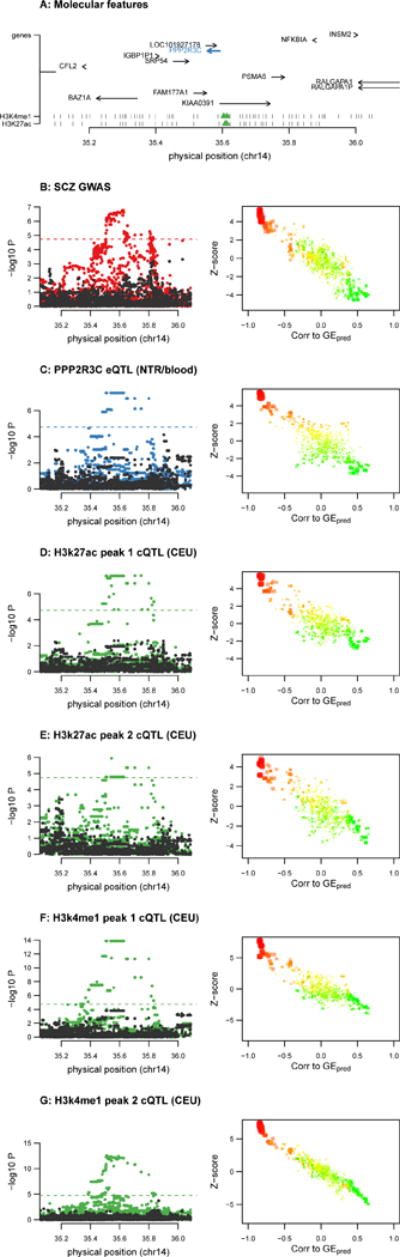
First, total expression of PPP2R3C in NTR/blood was associated with SCZ (P =3.4×10−6) – despite no genome-wide significant SNPs at the locus – as well as four distal chromatin peaks (minimum P =1.0×10−9; Fig. 4). Conditioning each GWAS SNP on the predicted expression of PPP2R3C explained all significant marginal associations for the implicated phenotypes and formal colocalization was supported between all features and SCZ (average posterior=92%; Supplementary Table 24). PPP2R3C was the nearest gene to the most significantly associated SNP at the locus and to the implicated chromatin peaks. However, because the locus was not genome-wide significant, this association would not have been identified in a conventional analysis of known GWAS loci. PPP2R3C was recently identified by SMR analysis of SCZ in an independent expression panel24 and our findings pinpoint specific regulatory elements for experimental follow-up.
Figure 4. Chromatin and SCZ TWAS association at PPP2R3C.
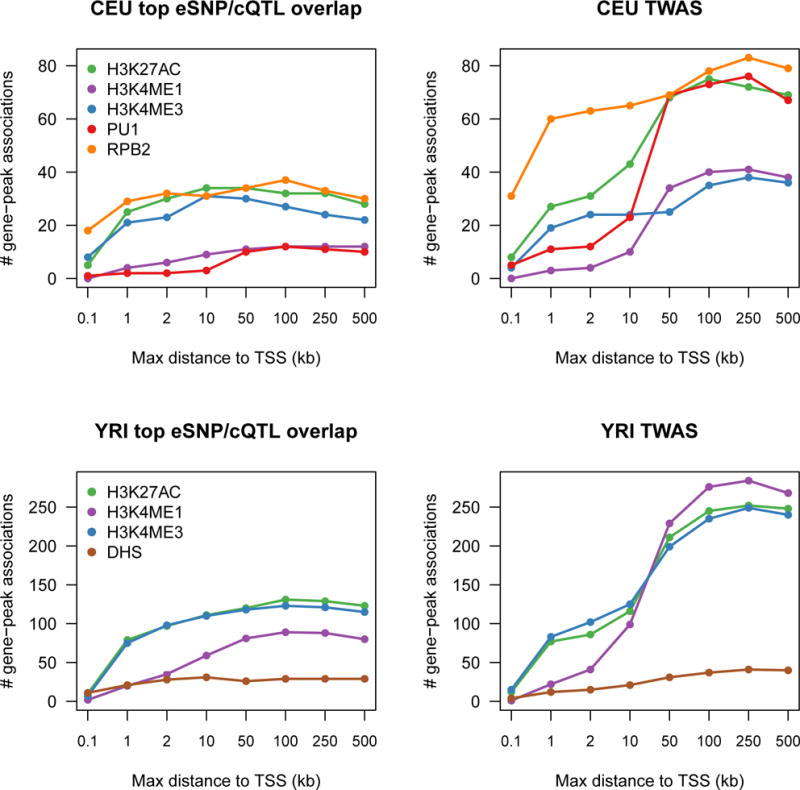
Example association of PPP2R3C gene expression and SCZ and four nearby chromatin peaks. (A) locus schematic with all nearby genes and chromatin peaks; TWAS associated features highlighted in blue and green. (B–G left) Manhattan plots of marginal association statistics before and after conditioning on the TWAS predicted expression (colored/dark dots, respectively). Dashed line shows local significance threshold after Bonferroni correction for number of SNPs. (B–G right) Relationship between marginal GWAS/QTL association (y axis) and the correlation (x-axis) between TWAS predicted expression (GEpred estimated in the 1000 Genomes reference) and marginal GWAS/QTL association. The color of each point reflects the eQTL effect size of the expression used for GEpred and size of each point reflects absolute significance of the eQTL. (B) SCZ GWAS association; (C) PPP2R3C expression phenotype used for TWAS prediction and associated with SCZ/chromatin; (D) 1st TWAS associated H3k27ac peak in CEU; (E) 2nd TWAS associated H3k27ac peak in CEU; (F) 1st TWAS associated H3k4me1 peak in CEU; (G) 2nd TWAS associated H3k4me1 peak in CEU. See Supplementary Note, Supplementary Fig. 32, 33, 34 for additional examples and simulations.
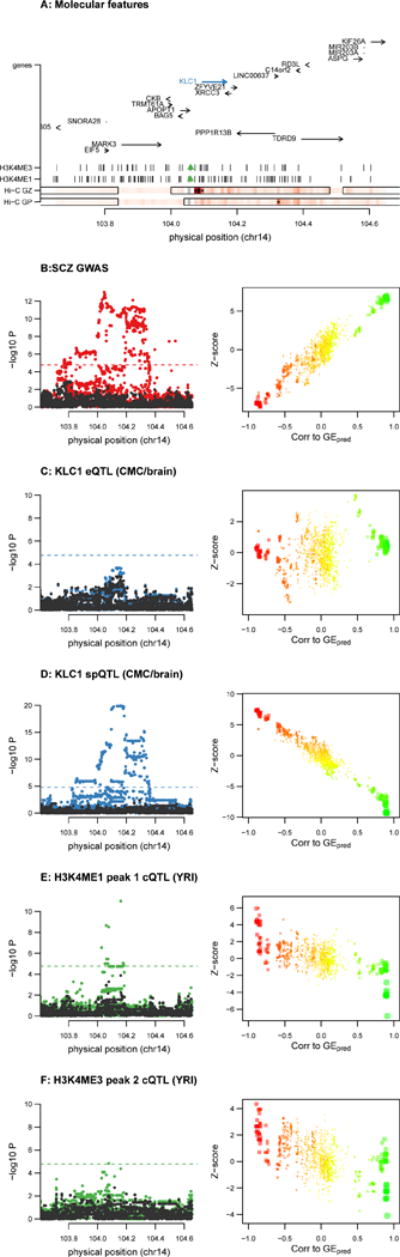
Second, a splicing event at KLC1 in CMC had an SCZ TWAS P =6.7×10−12 and overlapping H3k4me1/me3 chromatin TWAS associations (minimum P =2.5×10−07) (Fig. 5). Conditioning on the top sQTL explained all significant SCZ GWAS signal at the locus whereas conditioning on the most significant eQTL had a negligible effect, highlighting an effect on SCZ explained by splicing independent of total expression. Notably, both chromatin TWAS associations were supported by Hi-C interactions to the KLC1 promoter in developing brain34 (FDR 0.01 significant and the most significant interaction in the locus), serving as a functional validation of coordinated activity (Fig. 5, Supplementary Fig. 35). We performed a TWAS-like test for chromatin-SCZ association, which was highly significant for both peaks (best P =2.6×10−13; Supplementary Table 3). Evidence for colocalization was high for KLC1 splicing and SCZ (posterior=58%) as well as for the chromatin phenotypes and both KLC1 splicing and SCZ (posterior >80%), in spite of the fact that the chromatin phenotypes were identified in YRI and may exhibit LD differences across populations (Supplementary Table 24). Differential DNA methylation41 and expression at KLC1 in SCZ cases versus controls was recently identified in two independent analyses of brain tissue, further supporting a cis-regulatory effect on SCZ.
Figure 5. Chromatin and SCZ TWAS association at KLC1.
Example association of KLC1 splice event to SCZ with evidence of chromatin interaction in Hi-C from developing brain. (A) locus schematic with all nearby genes and chromatin peaks; TWAS associated features highlighted in blue and green. Hi-C GZ/CP rows show significance of Hi-C chromatin interaction between the 10kb block containing the associated chromatin peaks (gray, with neighboring white blocks not tested) and every other 10kb block in the region; 10kb being the highest resolution for this Hi-C data. Dark red shading indicates more significant and interactions significant at 0.01 FDR are labeled with stars. The most significant interaction in the locus overlaps the KLC1 promoter. The interactions are shown for fetal brain data from CP (cortical and subcortical plate) and GZ (germinal zone) and corresponding topological domains (TADs) are outlined with solid black lines. (B–F left) Manhattan plots of marginal association statistics before and after conditioning on the TWAS predicted expression (colored/dark dots, respectively). Dashed line shows local significance threshold after Bonferroni correction for number of SNPs. (B–F right) Relationship between marginal GWAS/QTL association (y axis) and the correlation (x-axis) between TWAS predicted expression (GEpred estimated in the 1000 Genomes reference) and marginal GWAS/QTL association. The color of each point reflects the eQTL effect size of the expression used for GEpred and size of each point reflects absolute significance of the eQTL.(B) SCZ GWAS association; (C) KLC1 total expression, both panels show independence from the TWAS predicted expression; (D) KLC1 splicing event phenotype used for TWAS prediction and associated with SCZ/chromatin; (E) TWAS associated H3k4me1 chromatin peak in YRI; (F) TWAS associated H3k4me3 chromatin peak in YRI. See Supplementary Note, Supplementary Fig. 32, 33, 34 for additional examples and simulations.
Third, total expression of MAPK3 in CMC/brain was associated with SCZ (P =1.3×10−6) as well as two chromatin peaks near the TSS: H3k27ac (P =7×10−6) and RPB2 (P =1×10−11). In the CEU chromatin phenotype samples, where MAPK3 expression was also measured in LCLs, the H3k27ac and RPB2 peaks explained 36% (P =7×10−6) and 23% (P =5×10−4) of the variance in measured expression, respectively, with only the H3k27ac peak significant in a joint model. Formal colocalization analysis supported a single shared causal variant across all combinations of eQTL/cQTL/GWAS for the implicated features (posterior probabilities 54–97%; Supplementary Table 24). We confirmed that the associated peaks were observed in epigenetic data from H3k27ac, H3k4me3 and ATAC-seq measured in brain tissues42 and contained two SNPs with significant allele-specific effect43 on MAPK3 (Supplementary Note, Supplementary Fig. 36, 37, 38, 39). Strikingly, these peaks overlapped two recently identified human-gained neuro-developmental enhancers in independent fetal cortex tissues44 (Supplementary Fig. 36). This class of enhancers clusters with genes important to cortical development and neuronal differentiation, and has been hypothesized to play a key role in human cortical evolution.
Functional interrogation of MAPK3 in zebrafish
MAPK3 maps within the 16p11.2 600kb copy number variant (CNV) that has been associated with both SCZ and autism45–49. Previous studies have shown that dosage perturbation of another transcript in that region KCTD13 can induce reciprocal head size and neuro-proliferative defects consistent with the anatomical pathology in patients45. Critically, pairwise dosage analyses showed a genetic interaction of KCTD13 with MAPK3 (as well as a third locus, MVP)45, while independent transcriptional studies in human cells and mouse models highlighted a functional ”cassette“ composed of KCTD13, MVP and MAPK3, a set of coregulated genes associated with the head size phenotype48. Together with our TWAS observations, these data implicate a transcriptional relationship between these genes in the 16p11.2 region and suggest that MAPK3 (and its expression thereof) might be a functional trigger. If so, suppression of MAPK3 should rescue the pathology induced by increased expression of KCTD13. To test this hypothesis, we performed an experimental assay in zebrafish embryos (Online Methods). Consistent with prior studies, overexpression of human KCTD13 (associated with microcephaly in humans) induced both a decrease in head size and a concomitant decrease in the number of cycling cells in the brain (Fig. 6). However, suppression of endogenous mapk3 in KCTD13 over-expressants was able to rescue both phenotypes reproducibly (Fig. 6; n=37-69 embryos per injection, replicated, scored blind).
Figure 6. Suppression of endogenous mapk3 rescues the microcephaly and neuronal proliferation phenotypes induced by overexpression of wild-type KCTD13.
Dorsal view of 4 days post fertilization (dpf) control larvae (A) and embryos injected with either morpholino (MO) against endogenous mapk3 (B), human capped wild-type (WT) KCTD13 mRNA (C) or combinatorial administration of mapk3 MO and WT human KCTD13 mRNA (D). Quantification of the headsize phenotype across the four conditions (E). Dorsal view of 3dpf embryos stained with an antibody against phospho-histone 3 (PH3), a marker of neuronal proliferation of control larvae (F) or embryos injected with either MO against mapk3 (G), human capped wild-type (WT) KCTD13 mRNA (H) or combinatorial administration (I). Graph showing quantification of the proliferating neuronal count across the four conditions (J). Student’s t-test was used to determine statistical significance.
DISCUSSION
The landmark PGC SCZ GWAS paper concluded that “if most risk variants are regulatory, available eQTL catalogues do not yet provide power, cellular specificity, or developmental diversity to provide clear mechanistic hypotheses for follow-up experiments”1. In this work, we integrate data from GWAS, expression, splicing, and chromatin activity to identify mechanistic hypotheses. We found 157 unique genes with transcriptome-wide significant associations to SCZ, which were significantly supported by chromatin contact measured during brain development. Genes below the transcriptome-wide significance threshold continued to be strongly associated with SCZ and exhibited enrichment for expression and splicing in the brain (though this can also reflect expression data quality). Associations for splicing events that were independent of total expression highlighted an important source of disease-relevant variation27 with potential therapeutic implications50,51. 42/157 SCZ-associated genes were significantly associated with nearby chromatin phenotypes, implicating specific regulatory features for functional follow-up. We interrogated one TWAS association, MAPK3, in zebrafish embryos and observed a significant effect on neurodevelopmental phenotypes with consistent direction, prioritizing this as a candidate for further follow-up.
We conclude with several limitations and future directions of this study. First, although TWAS is not confounded by reverse-causality (disease → expression independent of SNP), instances of pleiotropy (where a SNP or linked SNPs influence SCZ and expression independently) are statistically indistinguishable from truly causal susceptibility genes. As molecular studies grow and the chance of incidental QTL/GWAS overlap increases, experimental causal inference is necessary to validate these findings. Second, the chromatin phenotypes analyzed here were measured in LCLs (as population-level chromatin data from other tissues is currently unavailable), preventing us from identifying expression-chromatin associations that are brain-specific. Third, the use of summary-based data necessitates linear predictors of expression, which may misinterpret relationships between expression and disease/chromatin where, for example, the strongest eQTLs/cQTLs have weak effect on the trait due to context specificity. Lastly, although we did not observe significant pathway/ontology enrichment for the identified susceptibility genes, we posit that these genes and chromatin features can serve as anchors for network-based analyses of genome-wide co-expression and co-regulation; we view this as an intriguing future direction.
As tissue acquisition may be the biggest hurdle for producing larger data sets, methods that do not depend on measurements from the same samples will remain critical. Beyond specific mechanistic findings for SCZ, this work outlines a systematic approach to identify functional mediators of complex disease.
ONLINE METHODS
Data and quality control
Genotypes and expression from the NTR26, YFS23, and METSIM23 were processed as described in ref23 (and below) and the corresponding expression weights were downloaded directly from the TWAS web-site (see Web Resources). Genotypes and expression data from the CMC25 were processed using the GTEx Consortium guidelines for eQTL analysis of RNA-seq data. Specifically, RNA-seq RPKM was quantile normalized across samples; genes having >10 individuals with zero reads were removed; each gene was rank-normalized; 15 PEER factors were computed; and the residual expression used.
For alternative splice variants in brain, we used the LeafCutter algorithm27,52 to quantify de novo intron excision in the CMC RNA-seq data by clustering reads that spanned intron junctions. These clusters correspond to individual isoforms and enable an estimate of differential intron splicing computed from the ratio of reads spanning an intron relative to the total isoform read count. Splice variants were quantified using default parameters: a minimum of 50 reads per cluster, and a maximum intron length of 500kb. Based on the guidelines in ref.52, the following quality controls were applied to the inferred isoform clusters: clusters having >10 individuals with zero reads were removed; clusters with <100 individuals having >20 reads were removed; and introns with <5 individuals having non-zero counts were removed. The inferred per-sample abundance for each intron was then treated as a molecular phenotype, normalized, and PEER-corrected as with total expression above. This process identified 123,480 splicing events, of which 99,562 mapped to canonical gene introns. We treated the differential splicing of these 99,562 splicing events as quantitative traits in the same manner as total expression.
For genotype data in the above studies, individuals failing a sex check or having 5% missing SNPs were removed. Additionally, SNPs were removed if they had >5% missing calls; P <0.05 case-control missing association; P <5×10−6 Hardy-Weinberg disequilibrium; P <5×10−3 association to batch; P <5×10−8 missing haplotype association; or frequency <1%. Principal components (PCs) were computed using ×all samples for the NTR, YFS, and METSIM data directly and using SNPweights (v2.1)53 for the CMC data, outliers were removed (samples >6 standard deviations away the mean along any top component), and PCs included as fixed-effects in estimating .
For all datasets, related individuals with GRM values >0.05 were also removed prior to estimating .
For chromatin data, we used population-level ChIP-Seq chromatin phenotypes measured in 76 HapMap YRI LCLs for H3k27ac (marking active enhancers), H3k4me1 (enhancers), H3k4me3 (promoters), and DNAse (open chromatin)6, and in 45 HapMap CEU LCLs for H3k27ac, H3k4me1, H3k4me3, PU1 (regulatory transcription factor) and RNA polymerase II (RPB2, associated with active transcription)18. We did not perform any additional QC of the functional features, which were previously adjusted for PEER/covariates and normalized6,18.
estimation
Cis and trans were estimated using variance-components, modeling the phenotype as a multi-variante Normal where K are the standard genetic relatedness matrices from SNPs in the cis locus (Kcis) and in the rest of the genome (Ktrans). The σ2 parameters were fit for each gene using AI-REML as implemented in the GCTA software54, with principal components and sex included as fixed effects. For of splicing events, the intron ratios condition out isoform abundance but total gene expression was also included as a covariate to account for any residual correlation. As in previous studies26, individual estimates outside the plausible 0-1 range were allowed in order to achieve unbiased mean estimates. The standard error of each estimate was approximated as the standard deviation divided by the square root of the number of genes tested; however, significant differences were confirmed by permutation tests (see below).
To evaluate the contribution of low-frequency variants, we imputed the NTR data to the Haplotype Reference Consortium reference, yielding high-quality imputed SNPs down to MAF of 0.001. On average, we did not observe a significantly non-zero contribution of imputed rare variants to , nor did we see a significant change in common due to denser imputation relative to array SNPs (Supplementary Table 1). Though recent work has identified biases in estimates of from rare variants55, we expect these biases to be small in the cis region and largely mitigated by the two-component model. We did not further evaluate the contribution of rare variants to . No difference was observed when using dosages to construct the cis GRM.
In the CMC data, where SCZ/BIP and control status was also available, the average cis-genetic correlation of expression between (SCZ/BIP) cases and controls was 1.00 (s.e. 0.02), indicating consistent direction of eQTL effect sizes between cases and controls and motivating us to use the full cohort as a TWAS reference panel (Supplementary Table 25).
Schizophrenia TWAS
We performed a Transcriptome Wide Association Study (TWAS) using publicly available summary statistics from the PGC GWAS of 79,845 individuals1 and four gene expression reference panels in independent samples (Web Resources). For a given gene, SNP-expression weights in the 1MB cis locus were first computed using the Bayesian Sparse Linear Mixed Model (BSLMM)28, which models effects on expression as a mixture of normal distributions to account for the sparse expression architecture. Given weights w, SCZ Z-scores Z, and SNP correlation (LD) matrix D; the association between predicted expression and SCZ (i.e. the TWAS statistic) is estimated as (see ref23 for methodological details). We computed TWAS statistics using either the SNPs genotyped in each expression reference panel, or imputed HapMap3 SNPs (which typically represent well-imputed SNPs). To account for multiple hypotheses, we applied Bonferroni correction within each expression panel that was used. This threshold was chosen so as to maximize consistency with previous published results and not penalize for additional (and often highly correlated) expression panels tested. Specifically, we report “transcriptome-wide” significance after correcting for the number of genes tested within each of the five reference panels (CMC, CMC-splicing, NTR, YFS, METSIM; 5,419 tests on average). This is consistent with the correction applied in previous TWAS results of multiple expression references23.
Summary-based joint/conditional tests and figures
Conditional and joint analysis was performed using the summary statistic-based method described in ref.30, adapted to genes instead of SNPs. This joint test aims to distinguish genes with independent genetic predictors (that are also SCZ associated) from those that are merely co-expressed with a shared genetic predictor. This requires marginal association statistics (i.e. the main TWAS results) and a correlation/LD matrix to evaluate the joint/conditional model. The correlation matrix was estimated by predicting the cis-genetic component of expression for each TWAS gene into the 1000 Genomes genotypes and computing Pearson correlations across all pairs of genes as well as between all gene-SNP pairs (with correlations below 0.01 set to zero due to sampling noise). The 247 transcriptome-wide significant TWAS associations across four reference panels were then added to the model one at a time in decreasing order of significance and retained if their conditional TWAS association remained significant after Bonferroni correction for 247 tests. To quantify strongly independent gene associations at hotspot loci, this procedure was repeated with the additional constraint that genes were only added to the model at each step if they had r2<0.30 with all genes already in the model. To assess how much of the SCZ GWAS association signal remained after the TWAS signal was removed, each GWAS SNP association was conditioned on the joint gene model one SNP at a time. For Manhattan plots in Fig. 4, 5 each GWAS SNP was conditioned on the predicted expression of the single target TWAS gene. For TWAS scatterplots in Fig.s 4, 5 the correlation of each SNP to the TWAS predicted phenotype was computed by predicting expression into the 1000 Genomes reference.
Colocalization analyses
We used the COLOC software33 to estimate the posterior probability of two phenotypes sharing a causal variant (which we refer to as “colocalization”). For a locus and pair of traits (e.g. chromatin and SCZ) the corresponding SNP-trait QTL effect sizes (and standard errors) were tested with the coloc.abf function, with molecular phenotypes treated as quantitative traits and SCZ input treated as a case-control trait with 43% cases. The posterior probability of one shared causal variant (H4) was reported. The default prior on sharing (P =1×10−5) was used for all primary analyses, with priors P =1×10−4 and P =1×10−3 evaluated separately with all other parameters unchanged.
Functional validation of TWAS-associated genes using chromatin interaction data
The TWAS genes were validated by presence of chromosome conformation capture (Hi-C) interaction with finemapped SCZ GWAS SNPs in the locus.
SCZ GWAS loci were fine-mapped as described34. First, independent, genome-wide significant SNPs and all nearby SNPs in LD (r2>0.6) having P <1×10−5 were selected. The CAVIAR fine-mapping algorithm56 was then applied in each locus (allowing for a maximum of 2 causal variants) to identify the 95% credible set of causal SNPs. Functional SNPs (SNPs that cause nonsense and missense variation as well as that reside within gene promoters) were directly assigned to their target genes, while non-annotated SNPs were assigned to the genes of action based on chromatin interactions in developing human cortices34.
Hi-C interactions were defined as described34. We fit background interaction profiles as a function of distance from all 9,444,230 imputed PGC GWAS SNPs using a Weibull distribution. The significance for a given Hi-C contact was then measured by calculating the probability of observing a stronger contact under this null. Fine-mapped GWAS SNPs were assigned to 10kb bins (which is the highest resolution available for the fetal brain Hi-C data), and the significance of interactions for every bin within a 1Mb flanking region (500kb upstream to downstream) was calculated. Significant Hi-C interacting regions (FDR<0.01) are then overlapped with Gencode v19 gene coordinates to identify the potential gene targets, and only genes with significant SNP heritability estimates ( P <0.01) were evaluated, resulting in 474 genes. These Hi-C defined schizophrenia risk genes were then overlapped with the genes and splice variants identified by TWAS. Overrepresentation analysis was performed by Fisher’s exact test with a background gene list of 1,392 genes that reside within a ±500kb window to any credible SNP and have nominally significant (P <0.01).
Polygenic TWAS signal from gene-based polygenic risk scores (GE-PRS)
We extend to genes the SNP-based polygenic risk score35 to evaluate TWAS predictive accuracy and validation. Given a 1-by-M vector z of signed association statistics in the discovery study (e.g. PGC) and an N-by-M matrix X of predicted expression for the corresponding M genes in the replication study, we constructed a GE-PRS S=Xz. The M genes were either all transcriptome-wide significant genes (Supplementary Note) or all genes passing relaxed p-value thresholds. This risk score was then tested with ancestry against case/control status by a standard linear model y~S+P +e where S is the risk score and P is a matrix of principal components accounting for ancestry. Risk-score performance was measured as the linear R2 from the above model less the R2 from the model y~P +e to account for ancestry, and converted to the liability scale assuming a prevalence of 1%.
For the TWAS using METSIM, YFS, and NTR expression reference panels, the cis-genetic component of expression was predicted in CMC samples. For the TWAS using the CMC expression panel, either the total expression was used (Supplementary Fig. 13) or the cis-genetic component of expression was estimated directly using BSLMM (equivalent up to a scaling factor to estimating genetic values by dropping each individual in turn). We stress that the case/control label from the CMC data was never used to identify the TWAS associations, and that the GePRS from the CMC expression panel were thus evaluated against an independent CMC case/control phenotype. Ascertaining cases in the CMC expression panel may increase the frequency of causal variants and make the prediction more accurate than using a randomly ascertained expression panel, however, we observed little difference when performing the TWAS using an expression panel consisting of CMC controls only (Supplementary Table 4).
Individual-level chromatin TWAS
We used cis SNP-expression effect sizes computed by BSLMM scores in the four expression reference panels (including splicing events) to predict individual-level expression into the 45 CEU18 and 766 YRI individuals with measured chromatin phenotypes. We retained only post-QC SNPs that were typed in both studies and removed strand-ambiguous SNPs. We note that even though the YRI target samples are of different ethnicity, this prediction does not require an LD-reference panel and is therefore only expected to suffer loss in power (but not increased type I error) due to the differences in LD. For each predicted gene, we identified all chromatin peaks within a given window of the TSS (primary results used ±500kb) and tested each mark for association to predicted expression by linear regression.
Top eSNP/cQTL overlap analysis
We compared the chromatin TWAS to a traditional approach of identifying SNPs that are significant both as cQTLs and eQTLs in real data (Fig. 18B). For each population and given distance to TSS, we performed this analysis in two stages. Stage 1: We used fastQTL40 to identify the most significantly associated eSNP for each gene by permuting the expression and re-testing the cis-locus, and restricted to those genes with eSNP P <0.01 (for consistency with the TWAS gene selection). Stage 2: Each significant eSNP from Stage 1 was then tested for an association to all nearby chromatin peaks by standard linear regression, and those passing Bonferroni correction for all gene-peak pairs for each chromatin phenotype (e.g. H3K27ac in CEU) were reported. This analysis was compared to the chromatin TWAS analysis where each gene was tested against any peak within the given distance (by standard linear regression), and number of significant results reported after Bonferroni correction for total number of gene-peak pairs tested in that phenotype. We separately considered an approach where all significant eQTLs in a gene are evaluated for overlap (as in refs.6,18), which underperformed the permuted top eSNP/cQTL approach here (Supplementary Note).
Multiple hypothesis correction for chromatin TWAS
The large number of correlated phenotypes analyzed – expression from five experiments and chromatin from nine experiments in two populations – allows for several approaches to multiple testing correction. For the chromatin TWAS, we corrected for the number of gene-peak pairs tested within a single expression reference and chromatin phenotype experiment (for example, number of gene-peak pairs when evaluating predicted CMC expression with the CEU:H3k27ac chromatin phenotype). This is directly comparable to the experiment-wide corrections applied in previous eQTL/cQTL analyses6,18. The same correction was applied for the SCZ/chromatin TWAS overlap: for example, the 44 SCZ TWAS genes identified using CMC expression were within 500kb of 1,528 total peaks in the CEU:H3k27ac experiment and “overlap” was reported for any peak that had a chromatin TWAS association P <0.05/1,528.
For comparison, we separately calculated the number of associations that were significant at 5% FDR across all molecular experiments. This yielded approximately 3.5× more chromatin TWAS associations and 1.2× more SCZ and chromatin TWAS associations (Supplementary Table 2), demonstrating that the above experiment-wide Bonferroni correction strategy corresponds to a conservative study-wide FDR.
Estimating support for mediation by expression/chromatin
We sought to evaluate the evidence in support of two models of mediation: MCH, where SNP → chromatin → expression → disease; and MEX, where SNP → expression → chromatin → disease. Under the assumption of linear, additive variance across molecular phenotypes, this can be estimated via the ratio of genetic covariance (covg) between chromatin-SCZ and expression-SCZ. Conceptually, the genetic effect of a given molecular phenotype on SCZ will be attenuated by environmental noise, which will manifest itself as lower covg to SCZ for phenotypes further along the molecular cascade. The fraction of environmental variance on expression (envEX) under each model of mediation can then computed from the following equation (see Supplementary Note for derivation):
We inferred these quantities from both CEU and YRI using covg estimates from cross-trait LD score regression57 and computed significance by comparing to randomly sampled gene-peak pairs within 500kb of the TSS. We separately considered a partial correlation approach using residuals of expression and chromatin in turn (Supplementary Note). We caution that the estimate of env in the above equation was computed from an average across all loci, and could also be consistent with confounding from different levels of measurement error for ChIP-seq and RNA-seq, a mixture of models MCH and MEX that favors model MCH, or mediation by other unobserved molecular phenotypes.
In vivo complementation in zebrafish embryos and whole-mount immunostaining
The human wild-type (WT) mRNA of KCTD13 (NM 178863) was cloned into the pCS2+ vector and transcribed in vitro using the SP6 Message Machine kit (Ambion), as described45. To suppress endogenous mapk3 we identified ENSDART00000103746.5 with 93% similarity and 88% identity to human MAPK3 (NM_002746) as the sole zebrafish ortholog, against which we designed a splice blocking morpholino (MO) targeting exon 2 (mapk3 sb: CTGTGAGTGTTTAAGGATACACATC). We injected 10ng of mapk3 MO and 150pg of WT KCTD13 RNA alone and in combination into wild-type zebrafish embryos at the 1-to 4-cell stage. For the evaluation of neuronal proliferation, experiments were performed as described58. Proliferating neurons were quantified by counting all positive cells on a dorsal view of 3 days post fertilization (dpf) embryos, excluding the eyes from the scored area, using the ITCN ImageJ plugin that considers cells with 20 pixel width and 5 pixel minimum distance between them in order to be considered as separate cells. Statistical significance for this assay was established using Student’s t-test. For the head size assay, injected larvae were grown to 4 dpf and imaged live on dorsal view. The area of the head was traced excluding the eyes from the measurements and statistical significance was calculated using Student’s t-test. The sample size for the head size assay consisted of control=67, mapk3_MO=59, KCTD13_WT=61 and mapk3_MO+KCTD13_WT=60; for PH3 it consisted of control=37, mapk3_MO=40, KCTD13_WT=39 and mapk3_MO+KCTD13_WT=40. All experiments were repeated in duplicate and scored blind to injection cocktail.
Supplementary Material
Table 2. TWAS genes with association to schizophrenia and chromatin phenotypes.
42 genes (including 7 genes at novel loci, highlighted with a [*]) had a significant TWAS association with SCZ and chromatin phenotypes. For each significant TWAS association with SCZ, the number of significant genechromatin associations (FWER 5% among TWAS gene-mark associations, by Bonforroni correction) are reported. In the middle columns ‘.’ represents genes that were not heritable in the study and therefore not TWAS-associated. In the right columns ‘.’ represents no identified association; genes with no chromatin associations are not shown. Top panel shows results from genes, with TSS listed as position; bottom panel shows results from splicing events in CMC with exon-exon junction listed as position (details in Supplementary Table 18). The minority of loci without subsequent evidence of colocalziation of molecular phenotype with SCZ are indicated with † (see Supplementary Table 3 for full numerical results).
| Gene | Chr | Position | TWAS P-value
|
Associated marks
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| YFS/blood | MET/adipose | NTR/blood | CMC/brain | DHS | H3K27AC | H3K4ME1 | H3K4ME3 | PU1 | RPB2 | |||
| RERE | 1 | 8,483,747 | 4e-07 | 2e-06 | 2e-06 | . | . | . | . | . | . | 3 |
| SLC45A1 | 1 | 8,378,144 | . | . | . | 4e-08 | . | . | . | . | . | 1 |
| MAP7D1* | 1 | 36,621,565 | 6e-04 | . | 1e-06 | . | . | . | . | . | . | 1 |
| MED8 | 1 | 43,855,483 | 5e-01 | . | . | 2e-06 | . | . | 1 | . | . | . |
| ANP32E | 1 | 150,207,026 | . | . | 1e-08 | . | . | . | . | 1 | . | . |
| MRPS21 | 1 | 150,266,261 | 3e-06 | 3e-03 | 6e-03 | 2e-02 | . | . | 1 | . | . | . |
| RFWD2*† | 1 | 176,176,380 | 4e-06 | . | . | . | . | . | 1 | . | . | . |
| C2orf69 | 2 | 200,775,978 | . | 6e-10 | . | . | . | . | . | 1 | . | . |
| GLT8D1† | 3 | 52,737,714 | . | . | 5e-08 | 3e-08 | . | 1 | . | . | . | . |
| GLYCTK | 3 | 52,321,835 | 2e-08 | . | . | . | . | 1 | . | . | . | . |
| GNL3 | 3 | 52,719,935 | 7e-09 | 6e-07 | . | 5e-02 | . | . | . | 1 | . | . |
| NEK4† | 3 | 52,804,965 | . | . | . | 2e-09 | . | . | . | 1 | . | . |
| NT5DC2† | 3 | 52,567,793 | 6e-06 | 6e-06 | . | 7e-01 | . | 1 | . | 1 | . | . |
| PPM1M | 3 | 52,279,808 | 2e-07 | 2e-07 | . | 2e-03 | . | 1 | . | . | . | . |
| TMEM110 | 3 | 52,931,597 | 1e-02 | 4e-01 | 1e-08 | 6e-06 | . | 1 | 1 | 2 | . | . |
| PCCB | 3 | 135,969,166 | 1e-08 | 1e-10 | . | 3e-10 | 1 | . | 3 | . | . | . |
| RP11-53O19.3 | 5 | 44,826,178 | . | 6e-06 | . | . | . | . | 1 | . | . | . |
| DND1 | 5 | 140,053,171 | . | 8e-07 | 1e-02 | . | . | 1 | . | 1 | . | . |
| IK† | 5 | 140,027,383 | 4e-06 | 1e-06 | . | 5e-05 | . | 1 | . | 1 | . | . |
| NDUFA2 | 5 | 140,027,370 | 2e-06 | . | . | 4e-06 | . | 1 | . | 2 | . | . |
| PCDHA2 | 5 | 140,174,443 | . | . | . | 7e-06 | . | 1 | . | 1 | . | . |
| ZMAT2 | 5 | 140,080,031 | 5e-06 | 1e-03 | . | 3e-06 | . | . | . | 1 | . | . |
| AS3MT | 10 | 104,629,209 | . | 6e-08 | 7e-09 | 1e-05 | . | 1 | . | . | . | . |
| MPHOSPH9† | 12 | 123,717,785 | 4e-09 | 1e-05 | . | 2e-08 | . | 1 | . | . | 1 | . |
| KIAA0391* | 14 | 35,591,526 | 7e-01 | 2e-07 | 5e-01 | . | . | 2 | 1 | . | . | . |
| PPP2R3C* | 14 | 35,591,748 | 6e-05 | 1e-01 | 3e-06 | 2e-02 | . | 2 | 2 | . | . | . |
| MAPK3 | 16 | 30,134,630 | 5e-05 | . | . | 1e-06 | . | 1 | . | . | . | 1 |
| GFOD2 | 16 | 67,753,273 | . | . | 6e-07 | 2e-05 | . | 1 | . | 2 | . | . |
| TSNAXIP1 | 16 | 67,840,780 | . | . | . | 2e-06 | . | . | 1 | 2 | . | . |
| DUS2L | 16 | 68,038,024 | 1e-06 | . | 3e-06 | 4e-04 | . | . | . | 2 | . | . |
| PRMT7† | 16 | 68,344,876 | 1e-05 | 8e-04 | . | 8e-06 | . | . | 1 | 1 | . | . |
| GRAP* | 17 | 18,950,336 | . | . | 5e-07 | . | . | . | . | . | . | 1 |
| RNF112* | 17 | 19,314,490 | 8e-06 | . | . | . | . | . | . | . | . | 1 |
| ACTR5† | 20 | 37,377,096 | 2e-07 | 2e-04 | . | 7e-01 | 1 | . | 1 | . | . | . |
| CBR3 | 21 | 37,507,262 | 6e-03 | 2e-03 | 2e-06 | 5e-04 | 1 | . | 2 | . | . | . |
|
| ||||||||||||
| CMC/brain splicing | ||||||||||||
| TBC1D5 | 3 | 17,255,862 - 17,279,655 | . | . | . | 3e-06 | . | . | 1 | . | . | . |
| NEK4† | 3 | 52,800,010 - 52,800,194 | . | . | . | 1e-06 | . | . | 1 | . | . | . |
| CCDC90B | 11 | 82,985,783 - 82,991,184 | . | . | . | 3e-07 | . | 1 | . | . | . | . |
| SBNO1† | 12 | 123,821,038 - 123,825,535 | . | . | . | 4e-10 | . | . | . | . | 1 | . |
| KLC1 | 14 | 104,145,855 - 104,151,323 | . | . | . | 7e-12 | . | . | 1 | 1 | . | . |
| RTN1* | 14 | 60,074,210 - 60,193,637 | . | . | . | 1e-06 | . | . | 1 | . | . | . |
| TAOK2† | 16 | 29,997,825 - 29,998,165 | . | . | . | 4e-06 | . | . | . | . | . | †1 |
| PPP4C† | 16 | 30,094,168 - 30,094,715 | . | . | . | 2e-06 | . | . | . | . | . | †1 |
Novel, not overlapping 108 PGC SCZ GWAS loci.
No significant SCZ colocalization posterior in any reference (exlucding chromatin features in YRI).
Acknowledgments
We would like to acknowledge Michael Gandal, Bryce van de Geijn, Arthur Ko, Po-Ru Loh, Luke O’Connor, Paivi Pajukanta, and Noah Zaitlen for helpful discussions. This research was funded by NIH grants F32GM106584, R01GM105857, R01MH109978, R01MH107649, R01MH105472, R01HG009120. HKF was supported by the Fannie and John Hertz Foundation. The project described was also supported by award Number T32GM007753 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health
We are grateful to the CommonMind Consortium and the PsychENCODE Consortium for making data publicly and readily available. Data were generated as part of the CommonMind Consortium supported by funding from Takeda Pharmaceuticals Company Limited, F. Hoffman-La Roche Ltd and NIH grants R01MH085542, R01MH093725, P50MH066392, P50MH080405, R01MH097276, RO1-MH-075916, P50M096891, P50MH084053S1, R37MH057881 and R37MH057881S1, HHSN271201300031C, AG02219, AG05138 and MH06692. Brain tissue for the study was obtained from the following brain bank collections: the Mount Sinai NIH Brain and Tissue Repository, the University of Pennsylvania Alzheimers Disease Core Center, the University of Pittsburgh NeuroBioBank and Brain and Tissue Repositories and the NIMH Human Brain Collection Core. CMC Leadership: Pamela Sklar, Joseph Buxbaum (Icahn School of Medicine at Mount Sinai), Bernie Devlin, David Lewis (University of Pittsburgh), Raquel Gur, Chang-Gyu Hahn (University of Pennsylvania), Keisuke Hirai, Hiroyoshi Toyoshiba (Takeda Pharmaceuticals Company Limited), Enrico Domenici, Laurent Essioux (F. Hoffman-La Roche Ltd), Lara Mangravite, Mette Peters (Sage Bionetworks), Thomas Lehner, Barbara Lipska (NIMH). Data were generated as part of the PsychENCODE Consortium, supported by: U01MH103339, U01MH103365, U01MH103392, U01MH103340, U01MH103346, R01MH105472, R01MH094714, R01MH105898, R21MH102791, R21MH105881, R21MH103877, and P50MH106934 awarded to: Schahram Akbarian (Icahn School of Medicine at Mount Sinai), Gregory Crawford (Duke), Stella Dracheva (Icahn School of Medicine at Mount Sinai), Peggy Farnham (USC), Mark Gerstein (Yale), Daniel Geschwind (UCLA), Thomas M. Hyde (LIBD), Andrew Jaffe (LIBD), James A. Knowles (USC), Chunyu Liu (UIC), Dalila Pinto (Icahn School of Medicine at Mount Sinai), Nenad Sestan (Yale), Pamela Sklar (Icahn School of Medicine at Mount Sinai), Matthew State (UCSF), Patrick Sullivan (UNC), Flora Vaccarino (Yale), Sherman Weissman (Yale), Kevin White (UChicago) and Peter Zandi (JHU).
Footnotes
The authors declare no competing financial interests.
WEB RESOURCES
BRAINSPAN transcriptomes:
http://www.brainspan.org/static/download.html(/p)(p)CommonMind consortium:
https://www.synapse.org/cmc(/p)(p)YRI chromatin data:
http://chromovar3d.stanford.edu(/p)(p)PGC summary data:
https://www.med.unc.edu/pgc/downloads(/p)(p)PLINK:
https://www.cog-genomics.org/plink2(/p)(p)PsychENCODE knowledge portal:
https://www.synapse.org/#!Synapse:syn4921369/wiki/235539(/p)(p)SNPWeights for principal component analysis:
http://www.hsph.harvard.edu/alkes-price/software/(/p)(p)CEU chromatin data (provided by the authors):
http://gardeux-vincent.eu/Cell2015/description.peaks.zip,
http://gardeux-vincent.eu/Cell2015/quantified.peaks.zip,
http://gardeux-vincent.eu/Cell2015/quantified.peaks.PEER.centered.zip
AUTHOR CONTRIBUTIONS
AG, BP, ALP designed this study. AG, NM, HW, HKF, YR conducted analyses. MK, LS, AS, GEC, DHG, NK, PFS conducted and supervized experiments. Psychiatric Genomics Consortium, SM, BN, RAO, MCO, PFS collected the data. AG, BP, ALP wrote the paper.
DATA AVAILABILITY AND ACCESSION CODE AVAILABILITY STATEMENTS
The data that support the findings of this study are available at the following:
TWAS results:
http://www.gusevlab.org/projects/chromatinTWAS/(/p)(p)TWAS methods:
Bibliography
- 1.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Price AL, Spencer CC, Donnelly P. Proc R Soc B. Vol. 282. The Royal Society; 2015. Progress and promise in understanding the genetic basis of common diseases; p. 20151684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature. 2016;533:95–99. doi: 10.1038/nature17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–183. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claussnitzer M, Dankel SN, Kim K-H, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, et al. Fto obesity variant circuitry and adipocyte browning in humans. New England Journal of Medicine. 2015;373:895–907. doi: 10.1056/NEJMoa1502214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grubert F, Zaugg J, Kasowski M, Ursu O, Spacek D, Martin A, Greenside P, Srivas R, Phanstiel D, Pekowska A, et al. Genetic control of chromatin states in humans involves local and distal chromosomal interactions. Cell. 2015;162:1051–1065. doi: 10.1016/j.cell.2015.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J. Systematic localization of common disease-associated variation in regulatory dna. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trynka G, Sandor C, Han B, Xu H, Stranger BE, Liu XS, Raychaudhuri S. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nature Genetics. 2013;45:124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pickrell JK. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. The American Journal of Human Genetics. 2014;94:559–573. doi: 10.1016/j.ajhg.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gusev A, Lee SH, Trynka G, Finucane H, Vilhjálmsson BJ, Xu H, Zang C, Ripke S, Bulik-Sullivan B, Stahl E, et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. The American Journal of Human Genetics. 2014;95:535–552. doi: 10.1016/j.ajhg.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kichaev G, Yang WY, Lindstrom S, Hormozdiari F, Eskin E, Price AL, Kraft P, Pasaniuc B. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genetics. 2014;10:e1004722. doi: 10.1371/journal.pgen.1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Won HH, Natarajan P, Dobbyn A, Jordan DM, Roussos P, Lage K, Raychaudhuri S, Stahl E, Do R. Disproportionate contributions of select genomic compartments and cell types to genetic risk for coronary artery disease. PLoS Genetics. 2015;11:e1005622. doi: 10.1371/journal.pgen.1005622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, Anttila V, Xu H, Zang C, Farh K, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nature Genetics. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. Dnase [thinsp] i sensitivity qtls are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McVicker G, van de Geijn B, Degner JF, Cain CE, Banovich NE, Raj A, Lewellen N, Myrthil M, Gilad Y, Pritchard JK. Identification of genetic variants that affect histone modifications in human cells. Science. 2013;342:747–749. doi: 10.1126/science.1242429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasowski M, Kyriazopoulou-Panagiotopoulou S, Grubert F, Zaugg JB, Kundaje A, Liu Y, Boyle AP, Zhang QC, Zakharia F, Spacek DV, et al. Extensive variation in chromatin states across humans. Science. 2013;342:750–752. doi: 10.1126/science.1242510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kilpinen H, Waszak SM, Gschwind AR, Raghav SK, Witwicki RM, Orioli A, Migliavacca E, Wiederkehr M, Gutierrez-Arcelus M, Panousis NI, et al. Coordinated effects of sequence variation on dna binding, chromatin structure, and transcription. Science. 2013;342:744–747. doi: 10.1126/science.1242463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waszak S, Delaneau O, Gschwind A, Kilpinen H, Raghav S, Witwicki R, Orioli A, Wiederkehr M, Panousis N, Yurovsky A, et al. Population variation and genetic control of modular chromatin architecture in humans. Cell. 2015;162:1039–1050. doi: 10.1016/j.cell.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Taudt A, Colome-Tatche M, Johannes F. Genetic sources of population epigenomic variation. Nat Rev Genet. 2016 doi: 10.1038/nrg.2016.45. advance online publication, –. [DOI] [PubMed] [Google Scholar]
- 20.Farh KKH, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moyerbrailean GA, Kalita CA, Harvey CT, Wen X, Luca F, Pique-Regi R. Which genetics variants in dnase-seq footprints are more likely to alter binding? PLoS Genetics. 2016;12:e1005875. doi: 10.1371/journal.pgen.1005875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, Eyler AE, Denny JC, Nicolae DL, Cox NJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nature Genetics. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx B, Jansen R, de Geus E, Boomsma D, Wright F, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nature Genetics. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson M, Powell J, Montgomery G, Goddard M, Wray N, Visscher P, et al. Integration of summary data from gwas and eqtl studies predicts complex trait gene targets. Nature Genetics. 2016 doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 25.Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, Ruderfer DM, Oh EC, Topol A, Shah HR, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nature Neuroscience. 2016;19:1442. doi: 10.1038/nn.4399. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright F, Sullivan P, Brooks A, Zou F, Sun W, Xia K, Madar V, Jansen R, Chung W, Zhou YH, et al. Heritability and genomics of gene expression in peripheral blood. Nature Genetics. 2014;46:430–437. doi: 10.1038/ng.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, van+de+Geijn B, Raj A, Knowles D, Petti A, Golan D, Gilad Y, Pritchard J. Rna splicing is a primary link between genetic variation and disease. Science. 2016;352:600–604. doi: 10.1126/science.aad9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou X, Carbonetto P, Stephens M. Polygenic modeling with bayesian sparse linear mixed models. PLoS Genetics. 2013;9:e1003264. doi: 10.1371/journal.pgen.1003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated snps are more likely to be eqtls: annotation to enhance discovery from gwas. PLoS Genetics. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Ferreira T, Morris AP, Medland SE, Madden PA, Heath AC, Martin NG, Montgomery GW, Weedon MN, Loos RJ, et al. Conditional and joint multiple-snp analysis of gwas summary statistics identifies additional variants influencing complex traits. Nature Genetics. 2012;44:369–375. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nica AC, Montgomery SB, Dimas AS, Stranger BE, Beazley C, Barroso I, Dermitzakis ET. Candidate causal regulatory effects by integration of expression qtls with complex trait genetic associations. PLoS Genetics. 2010;6:e1000895. doi: 10.1371/journal.pgen.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chun S, Casparino A, Patsopoulos NA, Croteau-Chonka DC, Raby BA, De Jager PL, Sunyaev SR, Cotsapas C. Limited statistical evidence for shared genetic effects of eqtls and autoimmune-disease-associated loci in three major immune-cell types. Nat Genet. 2017;49:600–605. doi: 10.1038/ng.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLOS Genetics. 2014;10:1–15. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016;538:523–527. doi: 10.1038/nature19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P, Ruderfer DM, McQuillin A, Morris DW, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, Ripke S, Genovese G, Loh PR, Bhatia G, Do R, et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. The American Journal of Human Genetics. 2015;97:576–592. doi: 10.1016/j.ajhg.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palla L, Dudbridge F. A fast method that uses polygenic scores to estimate the variance explained by genome-wide marker panels and the proportion of variants affecting a trait. The American Journal of Human Genetics. 2015;97:250–259. doi: 10.1016/j.ajhg.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daetwyler HD, Villanueva B, Woolliams JA. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS one. 2008;3:e3395. doi: 10.1371/journal.pone.0003395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The GTEx Consortium. The genotype-tissue expression (gtex) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O. Fast and efficient qtl mapper for thousands of molecular phenotypes. Bioinformatics. 2016;32:1479. doi: 10.1093/bioinformatics/btv722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryan J, Saffery R. Crucial timing in schizophrenia: role of dna methylation in early neurodevelopment. Genome Biology. 2014;15:495. doi: 10.1186/s13059-014-0495-y. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, Crawford GE, Jaffe AE, Pinto D, Dracheva S, Geschwind DH, et al. The psychencode project. Nature neuroscience. 2015;18:1707–1712. doi: 10.1038/nn.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van de Geijn B, McVicker G, Gilad Y, Pritchard JK. Wasp: allele-specific software for robust molecular quantitative trait locus discovery. Nat Meth. 2015;12:1061–1063. doi: 10.1038/nmeth.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reilly SK, Yin J, Ayoub AE, Emera D, Leng J, Cotney J, Sarro R, Rakic P, Noonan JP. Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science. 2015;347:1155–1159. doi: 10.1126/science.1260943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF, et al. Kctd13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11. 2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maillard A, Ruef A, Pizzagalli F, Migliavacca E, Hippolyte L, Adaszewski S, Dukart J, Ferrari C, Conus P, Männik K, et al. The 16p11. 2 locus modulates brain structures common to autism, schizophrenia and obesity. Molecular psychiatry. 2015;20:140–147. doi: 10.1038/mp.2014.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, et al. Microduplications of 16p11. 2 are associated with schizophrenia. Nature Genetics. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Migliavacca E, Golzio C, Männik K, Blumenthal I, Oh EC, Harewood L, Kosmicki JA, Loviglio MN, Giannuzzi G, Hippolyte L, et al. A potential contributory role for ciliary dysfunction in the 16p11. 2 600 kb bp4-bp5 pathology. The American Journal of Human Genetics. 2015;96:784–796. doi: 10.1016/j.ajhg.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Föcking M, Lopez L, English J, Dicker P, Wolff A, Brindley E, Wynne K, Cagney G, Cotter D. Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia. Molecular psychiatry. 2015;20:424–432. doi: 10.1038/mp.2014.63. [DOI] [PubMed] [Google Scholar]
- 50.Sibley CR, Blazquez L, Ule J. Lessons from non-canonical splicing. Nature Reviews Genetics. 2016 doi: 10.1038/nrg.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Rivera RMC, Madhavan S, Pan X, Ran FA, Yan WX, et al. In vivo genome editing improves muscle function in a mouse model of duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li YI, Knowles DA, Pritchard JK. Leafcutter: Annotation-free quantification of rna splicing. bioRxiv. 2016:044107. doi: 10.1038/s41588-017-0004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen CY, Pollack S, Hunter DJ, Hirschhorn JN, Kraft P, Price AL. Improved ancestry inference using weights from external reference panels. Bioinformatics. 2013:btt144. doi: 10.1093/bioinformatics/btt144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang J, Lee SH, Goddard ME, Visscher PM. Gcta: a tool for genome-wide complex trait analysis. The American Journal of Human Genetics. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AA, Lee SH, Robinson MR, Perry JR, Nolte IM, van Vliet-Ostaptchouk JV, et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nature Genetics. 2015 doi: 10.1038/ng.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hormozdiari F, Kostem E, Kang EY, Pasaniuc B, Eskin E. Identifying causal variants at loci with multiple signals of association. Genetics. 2014;198:497–508. doi: 10.1534/genetics.114.167908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, Duncan L, Perry JR, Patterson N, Robinson EB, et al. An atlas of genetic correlations across human diseases and traits. Nature Genetics. 2015 doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jordan DM, Frangakis SG, Golzio C, Cassa CA, Kurtzberg J, Davis EE, Sunyaev SR, Katsanis N. Identification of cis-suppression of human disease mutations by comparative genomics. Nature. 2015;524:225–9. doi: 10.1038/nature14497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.