

Abstract
Despite the identification of a β-hydroxyhexaene produced by the enediyne polyketide synthases (PKSs), the post-PKS biosynthetic steps to the individual members of this antitumor and antibiotic family remain largely unknown. The massive biosynthetic gene clusters (BGCs) that direct the formation of each product caution that many steps could be required. It was recently demonstrated that the enediyne PKS in the dynemicin A BGC from Micromonospora chersina gives rise to both the anthraquinone and enediyne halves of the molecule. We now present the first evidence for a mid-pathway intermediate in dynemicin A biosynthesis, an iodoanthracene bearing a fused thiolactone, which was shown to be incorporated selectively into the final product. This unusual precursor reflects just how little is understood about these biosynthetic pathways, yet constrains the mechanisms that can act to achieve the key heterodimerization to the anthraquinone-containing subclass of enediynes.
Keywords: anthraquinones, biosynthesis, enediynes, iodine, natural products
Graphical abstract
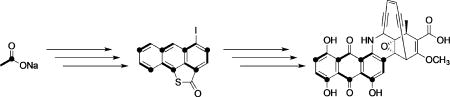
Hiding in plain sight: The first mid-pathway intermediate in the biosynthesis of an enediyne antitumor antibiotic has been characterized. A thiolactone-fused iodoanthracene was identified as an advanced precursor to the anthraquinone half of dynemicin A, revealing unexpected biosynthetic complexity and providing mechanistic insight into the construction of this family of natural products.
The enediyne antitumor antibiotics are a striking family of architecturally complex small molecules that combine a helix-binding element with an enediyne core, which initiates DNA cleavage.[1] Commensurate with their structural complexity, large biosynthetic gene clusters (BGCs) encoding up to 75 open reading frames have been sequenced and linked to enediyne production.[2–4] Considerable effort in the past 15 years has been devoted to defining the function of the highly reducing PKS that anchors all enediyne BGCs.[5, 6] It is now generally accepted that several, if not all, of these multidomain iterative enzymes initiate their biosynthetic pathways by assembling β-hydroxyhexaene octaketide 1 (Figure 1) bound to their acyl-carrier protein domain.[7,8] This labile intermediate can be cleaved by a separately encoded thioesterase (TE), accompanied by decarboxylation and dehydration to afford heptaene 2 (Figure 1), a universally observed enediyne shunt product.[9, 10] In competition with this pathway, it has been proposed that 1 can be directed to each of the enediyne final products by processes that remain unknown.[7–9]
Figure 1.

The enediyne PKS, DynE8, biosynthesizes β-hydroxyhexaene 1, which can be cleaved by a thioesterase in trans to produce heptaene 2. Enzyme-bound 1 is also believed to be an intermediate towards the anthraquinone and enediyne halves of dynemicin A (3). Black dots indicate carbon atoms originating from the carbonyl group of acetate, and bold lines represent intact acetate units.[7, 24]
We recently uncovered a dual role for the enediyne PKS DynE8 in the biosynthesis of dynemicin A (3; Figure 1).[11] DynE8 was found to provide the carbon skeletons for both the enediyne and anthraquinone halves of dynemicin, presumably from 1, which represents two remarkably different molecular fates for this simple precursor. Guided by the hypothesis that partitioning of this precursor would not be perfectly 1:1, and that the cyclization and oxidation cascades to each compound are different, we sought to test whether intermediates to one, or even both, could be detected in the dynemicin producer Micromonospora chersina. Herein, we report the first isolation and characterization of a mid-pathway intermediate in the biosynthesis of an enediyne natural product.
Wild-type M. chersina was fermented alongside the ΔDynE8 mutant,[11] and their metabolite profiles were compared by high performance liquid chromatography (HPLC). This analysis revealed two late-eluting species (4 and 5) that accumulate concurrently with dynemicin A in the wild-type strain (Figure 2a), with UV/Vis spectra resembling neither those of the dynemicins previously isolated from M. chersina[12, 13] nor off-pathway enediyne PKS products (Figure 2b).[14–16] Both 4 and 5 were purified from a fermentation of wild-type M. chersina and examined by ultra-performance liquid chromatography coupled to electrospray ionization mass spectrometry (UPLC-ESI-MS). The earlier-eluting 4 produced a [M+H]+ ion at m/z 237.0370 (see the Supporting Information, Figure S1), which fits to a molecular formula of C15H8OS within 1.7 ppm. Although the presence of sulfur was met with skepticism on biosynthetic grounds, the fifteen carbon count was consistent with an octaketide precursor, such as 1.[7,8] Poor ionization of 5 precluded its analysis by UPLC-ESI-MS, but electron-impact mass spectrometry (EI-MS) produced a clear nominal mass of 361.9 Da, with a prominent fragmentation ion at m/z = 235 Da (Figure S2). We reasoned that the clean loss of 126.9 Da could correspond to iodine, a minor component of the dynemicin fermentation medium.[17] Exact mass analysis of 5 by EI-MS subsequently revealed an [M]+ ion at m/z = 361.9268 (Figure S3). This mass correlated to a molecular formula of C15H7IOS within 1.6 ppm, supporting the unexpected presence of iodine and indicating that 4 and 5 likely share a common sulfur-containing scaffold.
Figure 2.

a) HPLC comparison of fermentation extracts of wild-type M. chersina and the ΔDynE8 mutant. Production of 3 in comparison to 4 and 5 is underrepresented at 280 nm. b) UV/Vis spectral overlay of 4 and 5. c) Structures of anthracene derivatives 4 and 5. d) ORTEP representation of 5, with ellipsoids set at 50% probability.
The iodine atom in 5 heightened our curiosity, as a mere 0.5 mgL−1 of sodium iodide (NaI) in the dynemicin fermentation medium has been reported to boost the yields of the enediyne by a factor of more than ten.[1] In our hands, NaI supplementation elevated dynemicin production more than 300-fold, and removing it from the fermentation completely abolished detectable production of 4 and 5 (Figure S4). By contrast, sodium chloride and sodium bromide were found to be ineffective substitutes for iodide, even when supplied at a tenfold larger molar concentration (Figure S4). These results suggested a specific role for iodide (and perhaps 5) in dynemicin A biosynthesis, leading us to fully elucidate its structure.
A suite of 1D and 2D NMR experiments (Figures S5–S9) was performed on 5 isolated from a fermentation of wild-type M. chersina supplemented with [1-13C]- and [2-13C]sodium acetate to enhance its 13C NMR signals. Three isolated aromatic spin systems were readily identified by 1H NMR (Figure S5) and correlation spectroscopy (COSY; Figure S7): 1) four coupled hydrogen atoms reminiscent of the unhydroxylated A ring of dynemicin generated by cytochrome P450 knockouts,[11] 2) two coupled hydrogen atoms split into sharp doublets with chemical shifts differing by about 0.5 ppm, and 3) a slightly broadened singlet. As our molecular formula eliminated the possibility of an anthraquinone, we reasoned from the 1H NMR data that 5 was an anthracene derivative with an unmodified A ring. We further deduced that the singlet could correspond to a lone B ring hydrogen atom, which was supported by chemical shift comparison to the corresponding similarly broadened singlet of anthracene.[18] Finally, we assigned the isolated doublets to either an ortho- or para-disubstituted C ring. With an anthracene core structure proposed, we were left with just one carbon atom from C15H7IOS unassigned, as well as two of twelve degrees of unsaturation (DoU). Analysis of the 13C NMR spectrum (Figure S6) revealed a carbonyl group at δ = 192.1 ppm, which we positioned in the C ring para to the iodine atom based on a heteronuclear multiple bond correlation (HMBC) experiment that was optimized for two to three bond couplings (Figure S9). In particular, prominent cross-peaks were detected between the upfield hydrogen atom of the C ring doublets and the carbonyl group, and the B ring hydrogen atom and the C–I bond (distinguished by its large upfield shift owing to the heavy atom effect).[19] Finally, to account for the presence of sulfur and the remaining DoU, we proposed a five-membered thiolactone ring with the sulfur attached to the B ring, and thus arrived at structure 5 (Figure 2c).
To ensure the accuracy of our NMR interpretation and remove any ambiguity about the location and identity of the three heteroatoms, the structure of 5 was also determined by X-ray crystallography (Figure 2d and Table S1), which fully confirmed it as 5-iodo-2H-anthra[9,1-bc]thiophen-2-one.[20] Then, with the structure of 5 as a guide, NMR analysis (Figures S10–S14) and characterization of 4 proceeded without complication. The 1H NMR (Figure S10) and COSY (Figure S12) spectra of 4 revealed an additional hydrogen atom in the C ring, at the position taken up by iodine in 5. Therefore, 4 was determined to be 2H-anthra[9,1-bc]thiophen-2-one, as depicted in Figure 2c.
Recognizing the resemblance of both 4 and 5 to the dynemicin anthraquinone, and the potential utility of iodide as a leaving group to install the C–N bond between the two halves of dynemicin, we proceeded to investigate the possible biosynthetic role of 5. Accordingly, the iodoanthracene was purified from a fermentation of wild-type M. chersina without carbon labeling and resupplied to a fermentation lacking NaI. When both NaI and 5 were excluded from the fermentation, dynemicin A was barely detected by HPLC analysis; conversely, when only about 5 µm 5 was included, production was restored to nearly wild-type levels fermented with NaI (Figure 3a). Consistent with the observation that 4 does not accumulate in M. chersina in the absence of NaI, 5 was found to be partially degraded to 4 during this fermentation (Figure 3b). This result suggested that iodide release could be responsible for stimulating enediyne production instead of 5 itself. Therefore, the fermentation was repeated with 5 labeled from [1-13C]- and [2-13C]sodium acetate. Dynemicin A produced by feeding of 13C-enriched 5 showed clear incorporation of the carbon label by UPLC-ESI-MS (Figure 4c). In contrast, dynemicin A produced by supplementation with both NaI and 13C-enriched 4 saw no carbon enrichment (Figure 4b), indicating that 4 is not iodinated to 5, and that 5 is specifically incorporated into dynemicin A.
Figure 3.

a) Dynemicin A production by wild-type M. chersina fermented with or without NaI and 5. The y axis scale bar represents 25 milli-absorbance units at 570 nm. b) Production of 4 and 5 in the same fermentations as in (a). The “standard” of 5 is the purified material that was added to the “no NaI + 5” fermentation. The y axis scale bar represents 200 milli-absorbance units at 280 nm.
Figure 4.

Mass spectra of dynemicin A from fermentations a) with NaI, b) with NaI and 13C-enriched 4, and c) without NaI and with 13C-enriched 5.
To finally establish that 5 is an on-pathway precursor to the anthraquinone—but not the enediyne—the iodoanthracene was isolated from a fermentation supplemented with 2 mm [1-13C]sodium acetate to label every other carbon atom (seven in total), and re-fed to a fermentation of the higher-producing dynemicin A mutant ΔPKS5[11] without NaI. Comparison of the 13C NMR spectrum of derivatized triacetyldynemicin A (for improved solubility)[12] from this fermentation to a reference of unlabeled triacetyldynemicin A showed enrichment (ca. 2% per site) at the expected seven anthraquinone carbon atoms (Figure 5). Four of the carbon atoms (C11, C13, C15, and C21) aligned approximately with their well-separated counterparts in the unlabeled spectrum. A fifth carbon atom (C19) also aligned, but appeared to have been misassigned as C14,[12] the carbon atom related by bilateral symmetry across the horizontal axis of the anthraquinone. It is also plausible that these closely spaced signals simply switched positions under the conditions of our NMR experiment. Unexpectedly, migrations of observed chemical shifts were seen throughout the sample and reference spectra when compared to each other, and to other spectra of the same compound acquired previously.[11] Deviations were particularly pronounced in the cluster of resonances correlating to C9, C10, C16, and C17 containing two signals of enhanced intensity, which we assigned to C9 and C17. The unlabeled carbon atoms C12, C25, and C26 in Figure 5 further exemplify this behavior. We attribute these small chemical shift differences to sample concentration, water content, and pH (triacetyldynemicin A has two ionizable groups).
Figure 5.

13C NMR comparison of unlabeled triacetyldynemicin A (bottom) to triacetyldynemicin A produced by supplementing 5 labeled from [1-13C]sodium acetate to a fermentation without NaI (top). Numbers correspond to the dynemicin carbon labeling depicted in Figure 1.
Demonstration of the intermediacy of 5 is an important step forward to unraveling the biosynthesis of dynemicin. It uncovers unknown biochemistry that creates an iodoanthracene bearing a thiolactone, and reveals that the biosynthetic complexity of this enediyne pathway is far greater than previously appreciated.[11] Bioinformatics analysis of the dynemicin BGC provides few clues as to how 5 might be generated, as the cluster lacks both halogenases and sulfur insertion enzymes, and many genes in the cluster have no predicted function.[4] Its characterization provides insight, however, into the timing of various dynemicin biosynthetic events, and provides us with an opportunity to consider how the two halves of dynemicin might be joined.
We envision a coupling of the subunits of dynemicin by replacing the iodine atom of 5 with a presumed nitrogen-containing enediyne precursor (Figure 6). One path to achieve this heterodimerization (forming an anthracene–enediyne precursor such as 7) is nucleophilic aromatic substitution (SNAr), although iodine is not the optimal halogen for this chemistry. The rate-determining step of SNAr reactions is breaking the aromaticity of the ring by addition of the nucleophile, and as a result, the order of reactivity of the halogens is typically F>Cl>Br>I to stabilize the negative charge that develops in the intermediate.[21, 22] Notwithstanding, the weaker resonance stabilization of anthracene in comparison to benzene, in combination with the electron-withdrawing ability of the rigidly coplanar carbonyl group of the thiolactone, may lower the energy barrier of such a reaction enough to allow for this substitution path to prevail. Alternatively, the coupling could follow a unimolecular radical nucleophilic substitution (SRN1) route, which would require a single electron donor to form a carbon-based radical such as hypothetical 6 with the loss of iodide. Lending credence to this path, the halogen reactivity for SRN1 reactions follows the trend of I>Br>Cl>F,[23] and unproductive reaction of radical 6 by hydrogen atom abstraction would form the reduced product, 4. Building on the discovery of 5 and the role of iodide in its generation, efforts are underway to identify potential enediyne partners to which it or a more highly oxidized derivative could couple to produce the dynemicin carbon core.
Figure 6.

Two possible routes (SNAr and SRN1) to the dynemicin anthraquinone from iodoanthracene 5.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr. A. Majumdar, Dr. M. A. Siegler, and Dr. I. P. Mortimer for assistance with NMR spectroscopy, X-ray crystallography, and mass spectrometry, respectively. We also extend our deepest thanks to P. Pal for invaluable conversations about data analysis and her chemical insight into this problem. This work was supported by National Institutes of Health grants R01 ES001670 and T32 GM080189.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201802036.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Borders DB, Doyle TW, editors. Enediyne Antibiotics as Antitumor Agents. Marcel Dekker; New York: 1995. pp. 1–15. [Google Scholar]; Borders DB, Doyle TW, editors. Enediyne Antibiotics as Antitumor Agents. Marcel Dekker; New York: 1995. p. 303. [Google Scholar]
- 2.Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Staffa A, Bachmann BO, Huang K, Fonstein L, Czisny A, Whitwam RE, Farnet CM, Thorson JS. Science. 2002;297:1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 3.Liu W, Christenson SD, Standage S, Shen B. Science. 2002;297:1170–1173. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]
- 4.Gao Q, Thorson JS. FEMS Microbiol. Lett. 2008;282:105–114. doi: 10.1111/j.1574-6968.2008.01112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zazopoulos E, Huang K, Staffa A, Liu W, Bachmann BO, Nonaka K, Ahlert J, Thorson JS, Shen B, Farnet CM. Nat. Biotechnol. 2003;21:187–190. doi: 10.1038/nbt784. [DOI] [PubMed] [Google Scholar]
- 6.Rudolf JD, Yan X, Shen B. J. Ind. Microbiol. Biotechnol. 2015:1–16. doi: 10.1007/s10295-015-1671-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belecki K, Townsend CA. Angew. Chem. Int. Ed. 2012;51:11316–11319. doi: 10.1002/anie.201206462. Angew. Chem.2012, 124, 11478–11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belecki K, Townsend CA. J. Am. Chem. Soc. 2013;135:14339–14348. doi: 10.1021/ja406697t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horsman GP, Yihua C, Thorson JS, Shen B. Proc. Natl. Acad. Sci. USA. 2010;107:11331–11335. doi: 10.1073/pnas.1003442107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belecki K, Crawford JM, Townsend CA. J. Am. Chem. Soc. 2009;131:12564–12566. doi: 10.1021/ja904391r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen DR, Townsend CA. Nat. Chem. 2018;10:231–236. doi: 10.1038/nchem.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konishi M, Ohkuma H, Tsuno T, Oki T, VanDuyne GD, Clardy J. J. Am. Chem. Soc. 1990;112:3715–3716. [Google Scholar]
- 13.Miyoshi-Saitoh M, Morisaki N, Tokiwa Y, Iwasaki S, Konishi M, Saitoh K, Oki T. J. Antibiot. 1991;44:1037–1044. doi: 10.7164/antibiotics.44.1037. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, Guo Z-F, Lai PM, Sze KH, Guo Z. Angew. Chem. Int. Ed. 2010;49:7926–7928. doi: 10.1002/anie.201003369. Angew. Chem.2010, 122, 8098–8100. [DOI] [PubMed] [Google Scholar]
- 15.Kong R, Goh LP, Liew CW, Ho QS, Murugan E, Li B, Tang K, Liang Z-X. J. Am. Chem. Soc. 2008;130:8142–8143. doi: 10.1021/ja8019643. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Van Lanen SG, Ju J, Liu W, Dorrestein PC, Li W, Kelleher NL, Shen B. Proc. Natl. Acad. Sci. USA. 2008;105:1460–1465. doi: 10.1073/pnas.0711625105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lam KS, Veitch JA, Lowe SE, Forenza S. J. Ind. Microbiol. 1995;15:453–456. [Google Scholar]
- 18.Tobisu M, Nakamura R, Kita Y, Chatani N. J. Am. Chem. Soc. 2009;131:3174–3175. doi: 10.1021/ja810142v. [DOI] [PubMed] [Google Scholar]
- 19.Spiesecke H, Schneider WG. J. Chem. Phys. 1961;35:731–738. [Google Scholar]
- 20.CCDC 1823652 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 21.Bartoli G, Todesco PE. Acc. Chem. Res. 1977;10:125–132. [Google Scholar]
- 22.Carey FA, Sundberg RJ. Advanced Organic Chemistry, Part A: Structure and Mechanisms. 5. Springer; New York: 2007. pp. 816–821. [Google Scholar]
- 23.Bunnett JF. Acc. Chem. Res. 1978;11:413–420. [Google Scholar]
- 24.Tokiwa Y, Miyoshi-Saitoh M, Kobayashi H, Sunaga R, Konishi M, Oki T, Iwasaki S. J. Am. Chem. Soc. 1992;114:4107–4110. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.