

We report a new thirdhand smoke exposure route indoors from semivolatile organic compounds partitioning to aerosols.
Abstract
Aerosol composition measurements made in an indoor classroom indicate the uptake of thirdhand smoke (THS) species to indoor particles, a novel exposure route for THS to humans indoors. Chemical speciation of the organic aerosol fraction using mass spectrometric data and factor analysis identified a reduced nitrogen component, predominantly found in the indoor environment, contributing 29% of the indoor submicron aerosol mass. We identify this factor as THS compounds partitioning from interior surfaces to gas phase and then aerosol phase. Partitioning of THS vapors to aerosols requires an aqueous phase for reactive uptake of the reduced nitrogen species (RdNS), leading to seasonal differences in THS concentration indoors. RdNS protonate under the acidic conditions expected for indoor aerosols of outdoor origin. Controlled laboratory measurements performed using cigarette smoke deposited into a Pyrex vessel showed a similar partitioning behavior to aerosol of outdoor origin and mass spectral features comparable to the measured indoor THS factor after 1 week of residence time in the closed vessel. This study reports a new, potentially large THS exposure route from partitioning of surface volatile organic compounds into the aerosol phase and subsequent dispersion in a mechanically ventilated building.
INTRODUCTION
The negative health effects of smoking cigarettes and exposure to secondhand smoke (SHS) are well known (1). Many public areas have restrictions on smoking, including distance from doorways, nonsmoking buildings, and full smoking bans on campus for some universities. These smoking limitations serve to limit the exposure of nonsmoking populations to SHS. Thirdhand smoke (THS), the residue of tobacco smoke that is sorbed to indoor surfaces and clothing, remains the least studied exposure route, with increasing attention given to the various THS processes (2). THS, as an indirect and involuntary exposure route, has been shown to detrimentally affect growth and immunity in mice (3). Surface reactions of THS residue with oxidants, such as nitrous acid (HONO), can produce known carcinogenic compounds (4). Recognized THS exposure routes include dermal uptake from surfaces (2) and clothing (5) exposed to cigarette smoke, volatilization of semivolatile components and subsequent inhalation (4), and exposure to resuspended particles (6). However, no mechanisms for THS exposure from volatilization of THS vapors and subsequent uptake to the aerosol phase have been proposed. Herein, we identify a novel exposure route for reduced nitrogen-containing THS species involving semivolatile partitioning from indoor surfaces to the gas phase and the subsequent reactive uptake via acid-base chemistry of these vapors into the aqueous aerosol phase. We used indoor and outdoor aerosol composition measurements made in a nonsmoking and unoccupied classroom in a mechanically ventilated building to elucidate the mechanism of aerosol uptake and identify it as an important particle mass source and exposure route of THS species for nonsmoking populations. Although the mechanistic understanding and THS concentrations observed in the particle phase are for a single location, the chemical mechanism and behavior are nonspecific to the location, and similar observations are expected when THS residue and aqueous aerosols are present. Controlled laboratory studies of deposited tobacco smoke were able to reproduce the observations, indicating that these results are not limited to the measurement location.
RESULTS
Indoor and outdoor aerosol composition
Aerosol mass spectrometry is used extensively in outdoor measurements of aerosol composition; however, its use in the indoor environment is limited (7). Here, we use alternating outdoor and indoor measurements with a 4-min switching frequency to compare the chemical composition of submicron aerosol in a mechanically ventilated university classroom with that of the outdoor environment that supplied it. Results of indoor and outdoor composition comparison indicate that most of the indoor aerosol originates outdoors. Factor analysis of the organic aerosol mass spectral data identified four distinct organic aerosol types, with one of the aerosol types predominantly found in the indoors. This indoor factor has significant mass spectral signal (19%) coming from ions of the general formula CxHyNz+. This indoor organic aerosol component is identified as consisting of THS species based on subsequent laboratory investigations showing mass spectral similarity to deposited cigarette smoke. These investigations are discussed in detail in a later section. The THS factor is responsible for 29% (an average indoor concentration of 0.85 μg m−) of the total submicrometer aerosol measured in the indoor classroom, which has no modern history of smoking. The total aerosol mass composition comparison for all measured components is given in Fig. 1A, with the enhancement of the THS component observed in the indoor aerosol. Figure 1B shows the indoor/outdoor (I/O) concentration ratios for hourly data for the measurements. All components except for the THS factor showed significantly lower concentrations indoors (I/O < 1), as expected, due to mechanical losses from filtration and deposition upon transport to the indoors. For additional discussion of the I/O ratios and factor analysis, see the Supplementary Materials. The indoor enhancement of CxHyNz+ can also be confirmed without factor analysis by dividing the signal from these ions to the total organic aerosol mass spectral signal (fCHN) in the indoor and outdoor environment. Summertime measurements of fCHN show that the indoor aerosol always had significantly higher fractions than the outdoor fCHN (see fig. S1A). Together, the factor analysis and ion family signal show the presence of a unique indoor factor.
Fig. 1. Aerosol chemical composition and I/O ratios.
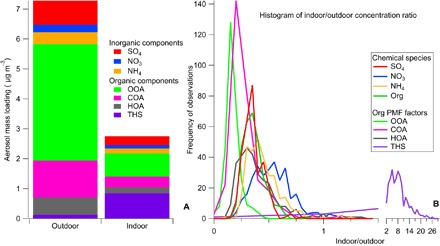
(A and B) Aerosol mass spectrometer (AMS) species average contributions (A) and histogram of hourly I/O ratio (B) for all reported chemical species. Values less than 1 indicate loss from outdoor to indoor transport and weak or absent indoor sources, whereas values greater than 1 indicate a source in the indoor environment. Sulfate is a proxy for nonvolatile particle loss from outdoor to indoor transport. HOA, hydrocarbon-like organic aerosol; OOA, oxygenated organic aerosol; COA, cooking organic aerosol.
THS sources and chemical behavior
Identification of the possible sources of THS in the indoor environment is important to understand the potential ubiquity of THS in other indoor environments. Indoor smoking activity is highly unlikely because of the nonsmoking rules of the building. In addition, aerosol compositional measurements made in the nonsmoking classroom showed a persistent THS aerosol component with no episodic increases (that is, no spikes in aerosol concentration) that would be associated with short-duration smoking activity and/or SHS penetration into the building (see fig. S2). However, the classroom where measurements were performed is 20 m down the hall from an outdoor balcony where illicit smoking activity occurs. Another potential source is the presence of several smokers in shared office space near the study classroom and is on the same heating, ventilation, and air conditioning (HVAC) system zone as the classroom. The balcony and smokers represent the likely sources of the THS species that are sorbed to interior surfaces, providing a reservoir for vapors to partition to the gas phase. Irrespective of the exact source of the THS, the partitioning behavior for this aerosol component is consistent with what has been observed for nicotine and its oxidation products originating from environmental tobacco smoke (ETS). It is important to note that the reduced nitrogen species (RdNS) discussed here are a subset of the THS chemical species originating from ETS. Other relevant RdNS components of THS include but are not limited to 3-ethenylpyridine, 2-picoline, 3-picoline, pyrrole, pyridine, and myosmine (8, 9). Chemical differences between constituents of THS will determine the deposition and subsequent revolatilization of the chemically distinct components, and consequently, not all chemicals associated with THS will behave as the herein observed RdNS (for example, polycyclic aromatic hydrocarbon components of ETS will not have the acid-base activity expected of RdNS). Organic compounds associated with tobacco smoking activities cover a range of vapor pressures and partitioning coefficients (10). In addition, sorption to indoor surfaces and organic films, reactions with other chemicals in the indoor environment, indoor temperature and relative humidity (RH), and ventilation rates play key roles in the lifetimes of THS species indoors (11, 12). The aerosol component determined from factor analysis is named the THS factor to link it to its source.
ETS contains myriad compounds, including heterocyclic compounds such as nicotine and other alkaloids. These species contain significant amounts of reduced nitrogen and are known to be semivolatile in their neutral, free-base form (13, 14). However, protonated versions are strongly nonvolatile under acidic conditions. The acid-base and partitioning chemistry of nicotine from mainstream smoke has been described in detail by other researchers (13). Applying this framework in the context of THS and partitioning to indoor aerosols is straightforward. The activity of the free-base and protonated form of nicotine can be assumed to be close to 1, and for the case of indoor aerosols in this study, we assume that the pH will be similar to the outdoor aerosol because the unoccupied classroom has no other major sources of aerosol other than outdoor aerosol introduced through the mechanical ventilation of the room. Recent work has estimated the outdoor aerosol pH to range from 0 to 2, even with decreasing sulfur emissions (15). Following the framework presented by Pankow (13), we calculate that the ratio of the protonated form of nicotine to the free-base form in aqueous aerosol at pH 1 is 107:1. At pH 2, the ratio is 106:1. Reactive uptake of free-base nicotine into the particle phase will be dominant until pH 7 when the ratio of protonated to nonprotonated nicotine will be 10:1. The acidic nature of ambient outdoor-originated aerosols therefore provides a sink for any gas-phase nicotine, nicotine by-products, and other alkaloid species that participate in acid-base chemistry. The mass spectral signature of the THS factor, shown in fig. S2, is consistent with RdNS commonly found in cigarette smoke (8, 9). However, mass spectral similarity to pure nicotine is poor, indicating chemical modification of the deposited ETS. The chemical signature of THS therefore changes with time.
Unique mass spectral characteristics of the THS factor include CH4N+, C2H4N+, and C3H8N+ at mass/charge ratios (m/z) of 30, 42, and 58, respectively. Fresh ETS has been identified using C5H10N+ at an m/z of 84 as a proposed tracer (16) and is the dominant ion in the standard National Institute of Standards and Technology mass spectrum of nicotine. The ion C5H10N+ is due to the methyl-pyrrolidine ring in nicotine and related compounds. In our THS factor, the reduced signal associated with this ion is indicative of secondary chemistry of ETS occurring after deposition to indoor surfaces, consistent with laboratory studies (4).
Laboratory investigation of THS partitioning and composition
Laboratory experiments performed with deposited ETS support this conclusion of chemical processing. Figure 2 shows the results of the laboratory experiments in which cigarette smoke was introduced into a Pyrex vessel such that ETS deposited to the walls of the container (see Materials and Methods for a full description). Following the deposition of ETS, inlet-filtered air drawn through the container showed no measurable aerosol mass loading, indicating that the uptake mechanism of THS species into aerosol is purely through a gas-to-particle mechanism and is not due to resuspension of deposited particles (see Fig. 2A). One day following the deposition of ETS, unfiltered outdoor air was pulled through the vessel, and aerosol organic mass increased as compared to sampling outdoor aerosol not drawn through the vessel (see Fig. 2B). This increase is attributed to the partitioning of THS species onto the existing aerosol. In addition, the composition was altered, with the fractional contribution of ions from RdNS in the organic aerosol (fCHN) increasing from 7 to 20% for the outdoor air only and the outdoor air sampled after pulling through the vessel, respectively. Figure 2D shows the residual mass spectrum of added THS species into the aerosol from the deposited ETS in the Pyrex vessel 1 day after deposition. The residual mass spectrum generated from the subtraction of outdoor only and the outdoor aerosol passed through the jar shows that C5H10N+ ion is the dominant peak observed from THS partitioning into outdoor aerosol after 1 day of residence time in the container. Sealing the vessel and performing identical measurements 1 week later showed that THS partitioning to outdoor aerosol was still occurring, although the contribution to aerosol mass was smaller after 1 week of aging in the Pyrex vessel (see Fig. 2C). Similar to observations from 1 day after deposition of ETS, fCHN showed a considerable increase when outdoor air was pulled through the container, indicating continued partitioning of THS species to the aerosol phase. In addition, there were chemical changes to the THS-associated mass spectrum. The C5H10N+ ion was virtually gone, and ion signals for CH4N+, C2H4N+, and C3H8N+ increased and became the dominant contributors of reduced nitrogen ions measured in the organic aerosol mass spectrum (see Fig. 2E). For the final mass spectrum, Fig. 2F shows the THS factor mass spectrum as a point of comparison. The CxHyNz ions show similarity with the 1-week-aged mass spectrum in Fig. 2E. Differences in the CxHyOz ion concentration are likely due to the differences in chemical processing between the jar (no added oxidants) and the THS factor from the indoor measurements.
Fig. 2. Laboratory investigation of THS partitioning.
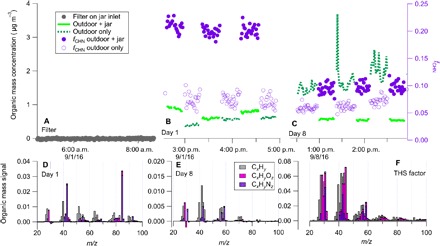
(A) AMS-measured organic aerosol concentration when a filter was placed on the inlet to the Pyrex vessel and particle-free air passed through the jar. (B and C) Concentration of outdoor organic aerosol and outdoor organic aerosol sampled through the Pyrex vessel with deposited smoke and the mass fraction of the organic aerosol from CxHyNz ions in the mass spectrum on days 1 and 8 after smoke deposition. (D and E) Mass spectra associated with the added THS species on days 1 and 8, respectively. (F) THS mass spectrum from positive matrix factorization (PMF) analysis for comparison of CxHyNz ion signals.
As previously mentioned, the THS partitioning to the outdoor aerosol mass was significantly less as compared to the first day after deposition. This reduction and corresponding mass spectral changes suggest chemical modification of the deposited ETS in a sealed container over a 1-week time scale. The reduction in signal intensity for the pyrrolidine ring–associated ion at an m/z of 84 (C5H10N+) is not surprising, given the literature on various oxidation pathways for nicotine. For oxidation by OH, the pyrrolidine ring has the most energetically favorable reaction sites for hydrogen abstraction and lifetime of hours (17). Nicotine lifetime with ozone (O3) exposure, which is more common in the indoor environment, is approximately 6 days, and proposed reaction products all show modification of the pyrrolidine ring (18). Surface oxidation of nicotine by HONO also preferentially modifies the pyrrolidine ring, forming carcinogenic nitrosamine products (4). Oxidation with OH, HONO, and ozone shows chemical modification of the pyrrolidine ring on relatively short time scales. Consequently, the decrease in the C5H10N+ ion associated with the ring is expected and consistent with the loss of this ion in both our laboratory experiments and in the THS factor measured in the classroom.
Acid-base behavior of RdNS
Nicotine and related ETS species will protonate under acidic conditions. Both pyrrolidine and pyridine rings of the nicotine molecule serve as weak bases with pKa1 value of 8.02 for the pyrrolidine ring of nicotine and pKa2 of 3.12 for the pyridine nitrogen. Proposed reaction products of nicotine surface oxidation by ozone include cotinine and myosmine with pKa values of 8.8 and 7.81, respectively (19). These molecules are also preferentially protonated at neutral and acidic pH values. Similar to nicotine, these molecules will not participate in semivolatile partitioning in their protonated form but will participate in their neutralized, free-base form. Surfaces exposed to ETS will retain protonated species, and they will persist until neutralized with a stronger base such as ammonia. This behavior may explain why these RdNS have been observed to persist in chamber studies beyond their expected ventilation lifetime based only on their nonprotonated volatility (8). Once in the free-base form, these molecules can participate in semivolatile equilibrium partitioning and will volatilize into the gas phase to reach equilibrium. Experimentally, it has been shown that exposure to ammonia will drive bound amines (20), including nicotine (21, 22), off a surface and into the gas phase, and similar behavior is expected for other weak bases present in ETS. Recent work has shown human emissions of ammonia at roughly 5.9 mg/hour per person, providing a significant source in the indoor environment (23). Once free-base nicotine and related molecules are in the gas phase, they can repartition to surfaces or to existing aerosol particles. Partitioning to aerosols results in reactive uptake via protonation of these weak bases. This uptake is a nonstandard semivolatile organic compound (SVOC) partitioning mechanism due to the irreversible aerosol uptake process under acidic conditions, which concentrates these species within the aqueous particle phase. Previous work supports this mechanism, with particle partitioning coefficients higher by several orders of magnitude than an alkane or polycyclic aromatic hydrocarbon with similar volatility (24).
RdNS partitioning to aqueous phase
Two additional observations from the measurements support an aqueous phase partitioning mechanism rather than organic phase partitioning. Traditional SVOC partitioning into the aerosol phase is dependent on the concentration of organic species in the aerosol (12, 25). Partitioning to the aqueous phase will be dependent on both the presence of an aqueous phase (that is, the particles have deliquesced or taken up water) and the total concentration of all aerosol species. Figure 3 shows the relationship between the measured concentration of the RdNS versus the concentration of the total indoor aerosol mass minus the THS factor and the total indoor organic aerosol mass minus the THS factor. The strong correlation with the total aerosol (R = 0.81) and the lack of correlation with the remaining organic aerosol (R = 0.05) support the conclusion that the indoor THS factor preferentially partitions to the aqueous phase and not the organic phase of the indoor aerosol particles. The robust relationship observed with non-THS aerosol concentration indicates that the total mass uptake is limited by available aerosol in the indoor environment because THS contribution does not level off at higher indoor aerosol concentrations. This correlation also suggests that we would not have observed aerosol contributions by THS exceeding ~29% because increases in THS-associated mass are accompanied by proportional increases in total indoor aerosol mass.
Fig. 3. Correlation of THS with indoor aerosol components.
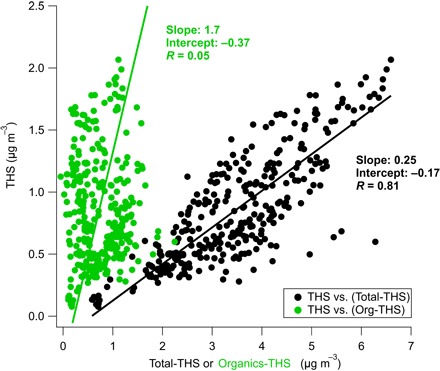
The black dots show the scatter plot of the concentration of the THS factor versus the total indoor aerosol minus THS factor. The green dots show the scatter plot of THS factor concentration versus the indoor organic aerosol minus the THS factor.
The presence of an aqueous phase is the other necessary factor for this partitioning mechanism because protonation of the free-base semivolatile RdNS will only occur in an aqueous medium. For a mechanically ventilated building, the operation of the HVAC system in different seasons leads to differences in water content of the indoor aerosol. In the summertime, warm air with varying amounts of water content is brought into the building, mixed with recirculated air, and conditioned to cooler temperatures (down to approximately 12.5°C or 55°F by the cooling coil) for the supply airstream. This process leads to deliquescence and significant uptake of water by aerosol particles, as RH values will increase to above 90% in the supply air for all of the temperature and RH combinations observed in this measurement data. Even with the subsequent decrease in RH of the rooms, all of the indoor aerosol will maintain the aqueous phase because the indoor RH does not drop low enough to drive off the water. In the HVAC system at >90% RH, the water content is 66% or greater of the total indoor aerosol volume; at 50% RH, as is observed in the room air, the water volume is 20% of the total volume. This continuous summertime presence of aerosol water allowed the RdNS to partition into the aerosol phase. In the wintertime, the temperature gradient is reversed with colder, drier outdoor air drawn into the HVAC system mixed with recirculating air and heated to temperatures approaching 38°C or 100°F. This process effectively effloresces the aerosol particles, drying them and resulting in the loss of the aqueous phase in the aerosol. Under these conditions, there is little to no available aerosol water for reactive uptake, and consequently, the THS factor will not be efficiently added to indoor aerosols that have been through a heating cycle in the HVAC system. This is consistent with no change in indoor fCHN compared to outdoor (see fig. S3), as well as our finding that there is no THS factor in PMF factor analysis of organic aerosol from an analogous wintertime measurement campaign using the same instrumentation. The combination of this lack of observable RdNS in the wintertime (even when there is still active smoking activity outdoors and in general) and the correlation with the total indoor aerosol minus the THS factor supports an aqueous phase partitioning mechanism.
DISCUSSION
Combining the observational evidence with previous work by others in the scientific literature allows us to propose a mechanistic understanding of the partitioning of THS species to indoor aerosols. Figure 4 presents the proposed mechanism of this novel THS partitioning to indoor aerosols and potential routes of exposure for occupants in a shared space. ETS elements can enter the indoor environment from off-gassing of semivolatile compounds from a smoker or person who has been exposed to cigarette smoke (Fig. 4A) and/or from indoor smoking or penetration of smoke through a window or the building envelope (Fig. 4B). Once in the indoor environment, components of ETS, including RdNS, partition to surfaces, hence becoming THS (Fig. 4C). On surfaces, RdNS can undergo oxidation reactions with a variety of indoor oxidants (Fig. 4D). These deposited RdNS will protonate under acidic to neutral conditions and will remain on surfaces until they are cleaned, transferred to another surface by contact, or deprotonated by reaction with a stronger base such as ammonia (Fig. 4E). In the neutral free-base form, RdNS and reaction products of deposited RdNS will participate in semivolatile partitioning based on their vapor pressure, leading to remission into the gas phase (Fig. 4F). Once in the gas phase, these species can either repartition to an indoor surface or undergo reactive uptake into the aqueous phase of indoor aerosols (Fig. 4G). If the species partition to the aerosol phase, then they will protonate under neutral to acidic conditions and will be effectively locked in the aerosol. The building’s air distribution system can then disperse the aerosols throughout any space served by the HVAC system, leading to involuntary exposures of THS species to other occupants.
Fig. 4. Proposed mechanism of partitioning to aerosol of RdNS from THS.
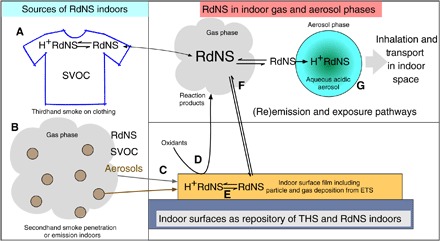
(A and B) Cigarette smoke components (SHS and THS) are introduced into the indoor environment from volatilization off of clothing (A) or SHS emission or penetration (B). (C) Once indoors, smoke vapors can partition and aerosol particles deposit to surfaces in the indoor environment. (D to F) Chemical processing and reactions with indoor oxidants (D) can modify the deposited chemical species. Strong bases such as ammonia can deprotonate deposited species (E), leading to volatilization of semivolatile compounds to the gas phase (F). These species can repartition to surfaces or undergo reactive uptake into the acidic aqueous phase of aerosols. (G) Once in the particle phase, these species are able to be transported through a building via natural or forced convection.
The HVAC system serves not only to condition the aerosols to wet or dry states but also to move air throughout a building zone. HVAC systems recirculate and disperse air throughout the multiple rooms of the zone served by the system, meaning that what happens in one room affects all the other rooms in the zone. For this reason, a room located near a smoking area with smoke penetration or a room occupied by a smoker can effectively expose the other occupants served by the same HVAC system to THS, even if they do not share space directly. Aerosols can uptake RdNS from concentrations of residual THS in a single room and then be dispersed through the air distribution system to other rooms in the shared zone, leading to unintentional exposures to other occupants of the building. This mechanism is the likely explanation for the observations of our study showing 29% of indoor aerosol mass in a nonsmoking classroom to be composed of THS species. For people who do not smoke and avoid areas where smoking occurs, this is an additional involuntary exposure route for ETS species. Although total exposure will be dependent on a variety of factors including the THS surface concentration, indoor aerosol composition and concentration, season, building ventilation, and other occupant smoking behavior, we can estimate the exposure for a student or an office worker based on the concentrations observed in this study. For a student taking an hour-long class with a breathing rate of 0.8 m3 hour−1 (26), and the average THS concentration being equal to 0.85 μg m−3, the hourly exposure would be 0.68 μg. For an office worker under similar conditions, an 8-hour (daily) exposure would be 5.44 μg. It is important to note that these exposures would only occur during the warm outdoor periods when the building HVAC is cooling and would not be observed during times of the year when the classroom or office was heated.
For individual exposure in other spaces, there are additional considerations that should also be noted. Residential buildings not only have more intermittent air recirculation by the HVAC system and typically no mechanical ventilation but also would likely show similar partitioning of THS species to indoor aerosols if there is a history of smoking indoors or simply the presence of a smoker in the residence. Other shared spaces, such as smoking rooms at hotels or rental cars that have been smoked in, present additional potential exposure routes for nonsmokers to THS by this mechanism. Finally, e-cigarettes, which volatilize liquids that are ~10% nicotine and are routinely used indoors where cigarettes are banned, present another opportunity for this exposure route because the key RdNS will make up a significant fraction of the e-cigarette effluent and deposited residue.
Although the observational results presented here apply to a single location, we expect the proposed chemical THS aerosol uptake mechanism to commonly occur in built environments for the following reasons: (i) With a controlled laboratory study, we were able to reproduce the observations from I/O measurements, and (ii) the various steps in the proposed mechanism are supported by other studies from the literature. Together, these support the universality of the individual steps of the THS partitioning to aerosol and suggest that these observations will be expected in other indoor spaces with a history of smoking or the presence of smokers and aqueous aerosol.
MATERIALS AND METHODS
Experimental design
Sampling for this project took place at Drexel University in Philadelphia in August of 2014. Gas and aerosol measurements were made from outdoor and indoor inlets, alternating every 4 min using a custom valve switching system (7). The outdoor inlet was positioned on the roof of the Alumni Engineering Laboratory building at Drexel University near the air intake for the HVAC system. The indoor inlet sampled from a classroom across the hall from the laboratory where the AMS and other instruments were sampling. When not sampling, an equal and continuous bypass flow was maintained for each inlet.
Indoor space description
The classroom was a ~150-m3 room with a tile floor and painted brick walls with approximately 25 student desks. In addition to the classroom, the HVAC system served student and faculty offices but no other classrooms. Summertime air exchange rates for the classroom air volume were 3.9 hour−o. Ventilation of the HVAC zone with outdoor air occurred more slowly because of the large fraction of recirculation of air within the zone. Air exchange rates with outdoor air were 0.4 hour−1.
Aerosol composition measurements
Aerosol particle chemical composition measurements were made using an Aerodyne Soot-Particle AMS (27) with the laser off. The AMS was operated in the V-ion optical mode (28). AMS data were processed using standard analysis procedures for unit mass resolution and high-resolution data. Individual ion signals were determined using the custom peak-fitting algorithm. The matrix of individual organic ion signals was used as an input to factor analysis by PMF (29, 30) and included the combined indoor and outdoor data sets. The individual organic ions were also separated into families (for example, CxHy, CxHyOz, CxHyNz, etc.), and relative signal intensity for each family in the indoor and outdoor environment was calculated. PMF analysis identified a four-factor solution, with three factors consistent with typical observations of outdoor aerosol types (31). The frequently observed factors included HOA (related to traffic and chemically similar to lubricating oil), OOA (associated with secondary chemistry in the atmosphere), and COA (peaking at meal times and chemically distinct from HOA) (32). The unique indoor factor containing a high fraction of reduced nitrogen species (THS) is the subject of the main body of the study. Mass spectra of the four identified PMF factors are shown in fig. S3. Indoor concentrations were lower for all species measured, with the exception of the THS factor, as shown in fig. S2. Mean indoor to mean outdoor concentration (mean I/O) ratios and median value for hourly I/O ratios for each species were as follows: SO4, 0.37 (0.37); NO3, 0.47 (0.55); NH4, 0.42 (0.44); Org, 0.36 (0.38) [HOA, 0.34 (0.37); COA, 0.29 (0.29); OOA, 0.19 (0.19)]. Organic PMF factors HOA, OOA, and COA, identified by PMF, showed temporal trends in the indoor environment that mirrored changes in the outdoor environment with a time lag due to ventilation time scales (7). Time series and I/O ratio data are presented in fig. S2, with average concentration of the submicrometer outdoor and indoor aerosol and histogram data for the I/O ratios for all chemical species given in Fig. 1.
Water content of the indoor aerosol at 90% (HVAC) and 50% (room) RH values was calculated using the average indoor aerosol composition and κ-Köhler theory (33), with an assumption of volume additivity for aerosol components according to ZSR (Zdanovskii, Stokes, and Robinson). Inorganic ion pairing was carried out following the study of Gysel et al. (34), and organic κ values were assumed to be 0.1.
Laboratory experiments of deposited ETS
Laboratory experiments were performed to investigate the partitioning behavior of deposited ETS and to identify the chemical signature of RdNS from THS partitioning into the aerosol phase. Tobacco smoke directly from a burning cigarette was drawn through a 2-liter Pyrex vessel, allowing smoke to deposit to the walls of the container. After the cigarette was extinguished, a high-efficiency particulate air (HEPA) filter was placed on the jar inlet, and air was pumped out of the jar until the jar was visually clear. Input air was filtered until low (<10 μg m−3) sampling mass was observed. Six hours of aerosol sampling with the AMS was performed with alternating measurements of ambient outdoor air, followed by ambient outdoor air drawn through the jar. In all experiments, sample air flow through the jar was ~100 ml/min, giving an air exchange rate of 20 min. We did not include these data because the distinction between SHS and THS was not clear. Following this sampling, a HEPA particle filter was placed on the inlet to the jar, providing particle-free air to the jar. The AMS sampled from this overnight, and the concentration of measured aerosol fell to 0 μg m−3. The next day and the following week (day 1 and day 8, respectively), the same sampling alternating between outdoor air and outdoor air passing through the Pyrex vessel was performed (see Fig. 2). Between sampling on days 1 and 8, the vessel was sealed and kept in the laboratory.
Aerosol mass spectral analysis
Mass spectral contributions from THS partitioning to the aerosol phase were determined using a sulfate-normalized mass spectral subtraction. Sulfate was assumed to be a nonvolatile tracer for particle loss while sampling through the Pyrex vessel. This also reasonably assumes that there is no source of sulfate in the Pyrex vessel. The mass spectrum measured after passing through the jar was multiplied by the ratio of the ambient sulfate to the sulfate concentration measured after passing through the jar. The ambient measured mass spectrum was then subtracted from this scaled mass spectrum to provide the approximate mass spectrum from partitioning of THS to the ambient outdoor aerosols on days 1 and 8. These mass spectra are shown in Fig. 2 (D and E).
Supplementary Material
Acknowledgments
Invaluable conversations with C. Weschler and others were facilitated by meetings organized by the Sloan Foundation’s Chemistry of Indoor Environments Program. Funding: Funding for this study was provided by the NSF award no. 1437916. Author contributions: P.F.D. and M.S.W. conceived the study. A.M.A. performed the I/O measurements and laboratory measurements. A.M.A. and P.F.D. analyzed and interpreted the data. All authors wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Processed QA/QC (Quality Assured/Quality Controlled)–level data from this study are accessible at Dryad (doi:10.5061/dryad.f25c87j).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaap8368/DC1
fig. S1. The fraction of CxHyNz ion signal for organic aerosol in the indoor and outdoor data as a function of season.
fig. S2. Time series of summer aerosol measurements.
fig. S3. Mass spectra of factors determined from PMF.
REFERENCES AND NOTES
- 1.U.S. Department of Health and Human Services, The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, 2014).
- 2.Northrup T. F., Jacob P. III, Benowitz N. L., Hoh E., Quintana P. J., Hovell M. F., Matt G. E., Stotts A. L., Thirdhand Smoke: State of the science and a call for policy expansion. Public Health Rep. 131, 233–238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hang B., Snijders A. M., Huang Y., Schick S. F., Wang P., Xia Y., Havel C., Jacob P. III, Benowitz N., Destaillats H., Gundel L. A., Mao J.-H., Early exposure to thirdhand cigarette smoke affects body mass and the development of immunity in mice. Sci. Rep. 7, 41915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sleiman M., Gundel L. A., Pankow J. F., Jacob P. III, Singer B. C., Destaillats H., Formation of carcinogens indoors by surface-mediated reactions of nicotine with nitrous acid, leading to potential thirdhand smoke hazards. Proc. Natl. Acad. Sci. U.S.A. 107, 6576–6581 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekö G., Morrison G., Weschler C. J., Koch H. M., Pälmke C., Salthammer T., Schripp T., Eftekhari A., Toftum J., Clausen G., Dermal uptake of nicotine from air and clothing: Experimental verification. Indoor Air 28, 247–257 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Becquemin M. H., Bertholon J. F., Bentayeb M., Attoui M., Ledur D., Roy F., Roy M., Annesi-Maesano I., Dautzenberg B., Third-hand smoking: Indoor measurements of concentration and sizes of cigarette smoke particles after resuspension. Tob. Control 19, 347–348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson A. M., Waring M. S., DeCarlo P. F., Real-time transformation of outdoor aerosol components upon transport indoors measured with aerosol mass spectrometry. Indoor Air 27, 230–240 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Singer B. C., Hodgson A. T., Nazaroff W. W., Gas-phase organics in environmental tobacco smoke: 2. Exposure-relevant emission factors and indirect exposures from habitual smoking. Atmos. Environ. 37, 5551–5561 (2003). [Google Scholar]
- 9.Sleiman M., Logue J. M., Luo W., Pankow J. F., Gundel L. A., Destaillats H., Inhalable constituents of thirdhand tobacco smoke: Chemical characterization and health impact considerations. Environ. Sci. Technol. 48, 13093–13101 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Singer B. C., Revzan K. L., Hotchi T., Hodgson A. T., Brown N. J., Sorption of organic gases in a furnished room. Atmos. Environ. 38, 2483–2494 (2004). [Google Scholar]
- 11.Lunden M. M., Kirchstetter T. W., Thatcher T. L., Hering S. V., Brown N. J., Factors affecting the indoor concentrations of carbonaceous aerosols of outdoor origin. Atmos. Environ. 42, 5660–5671 (2008). [Google Scholar]
- 12.Weschler C. J., Nazaroff W. W., Semivolatile organic compounds in indoor environments. Atmos. Environ. 42, 9018–9040 (2008). [Google Scholar]
- 13.Pankow J., A consideration of the role of gas/particle partitioning in the deposition of nicotine and other tobacco smoke compounds in the respiratory tract. Chem. Res. Toxicol. 14, 1465–1481 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Pankow J., Luo W., Tavakoli A., Chen C., Isabelle L., Delivery levels and behavior of 1,3-butadiene, acrylonitrile, benzene, and other toxic volatile organic compounds in mainstream tobacco smoke from two brands of commercial cigarettes. Chem. Res. Toxicol. 17, 805–813 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Weber R. J., Guo H., Russell A. G., Nenes A., High aerosol acidity despite declining atmospheric sulfate concentrations over the past 15 years. Nat. Geosci. 9, 282–285 (2016). [Google Scholar]
- 16.Struckmeier C., Drewnick F., Fachinger F., Gobbi G., Borrmann S., Atmospheric aerosols in Rome, Italy: Sources, dynamics and spatial variations during two seasons. Atmos. Chem. Phys. 16, 15277–15299 (2016). [Google Scholar]
- 17.Borduas N., Murphy J. G., Wang C., da Silva G., Abbatt J. P. D., Gas phase oxidation of nicotine by OH radicals: Kinetics, mechanisms, and formation of HNCO. Environ. Sci. Technol. Lett. 3, 327–331 (2016). [Google Scholar]
- 18.Sleiman M., Destaillats H., Smith J. D., Liu C.-L., Ahmed M., Wilson K. R., Gundel L. A., Secondary organic aerosol formation from ozone-initiated reactions with nicotine and secondhand tobacco smoke. Atmos. Environ. 44, 4191–4189 (2010). [Google Scholar]
- 19.Petrick L., Destaillats H., Zouev I., Sabach S., Dubowski Y., Sorption, desorption, and surface oxidative fate of nicotine. Phys. Chem. Chem. Phys. 12, 10356–10364 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Ongwandee M., Morrison G. C., Influence of ammonia and carbon dioxide on the sorption of a basic organic pollutant to carpet and latex-painted gypsum board. Environ. Sci. Technol. 42, 5415–5420 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Ongwandee M., Sawanyapanich P., Influence of relative humidity and gaseous ammonia on the nicotine sorption to indoor materials. Indoor Air 22, 54–63 (2012). [DOI] [PubMed] [Google Scholar]
- 22.A. M. Webb, B. C. Singer, W. W. Nazaroff, Effect of gaseous ammonia on nicotine sorption, in Proceedings of the 9th International Conference on Indoor Air Quality and Climate, H. Levin, Ed. (Indoor Air, 2002), pp. 512–517. [Google Scholar]
- 23.Furukawa S., Sekine Y., Kimura K., Umezawa K., Asai S., Miyachi H., Simultaneous and multi-point measurement of ammonia emanating from human skin surface for the estimation of whole body dermal emission rate. J. Chromatogr. B 1053, 60–64 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Liang C., Pankow J. F., Gas/particle partitioning of organic compounds to environmental tobacco smoke: Partition coefficient measurements by desorption and comparison to urban particulate material. Environ. Sci. Technol. 30, 2800–2805 (1996). [Google Scholar]
- 25.Donahue N. M., Robinson A. L., Stanier C. O., Pandis S. N., Coupled partitioning, dilution, and chemical aging of semivolatile organics. Environ. Sci. Technol. 40, 2635–2643 (2006). [DOI] [PubMed] [Google Scholar]
- 26.U.S. Environmental Protection Agency (EPA), “Exposure Factors Handbook 2011 Edition (Final Report),” (EPA/600/R-09/052F, EPA, 2011).
- 27.Onasch T. B., Trimborn A., Fortner E. C., Jayne J. T., Kok G. L., Williams L. R., Davidovits P., Worsnop D. R., Soot particle aerosol mass spectrometer: Development, validation, and initial application. Aerosol Sci. Tech. 46, 804–817 (2012). [Google Scholar]
- 28.DeCarlo P. F., Kimmel J. R., Trimborn A., Northway M. J., Jayne J. T., Aiken A. C., Gonin M., Fuhrer K., Horvath T., Docherty K. S., Worsnop D. R., Jimenez J. L., Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 78, 8281–8289 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Lanz V. A., Alfarra M. R., Baltensperger U., Buchmann B., Hueglin C., Prévôt A. S. H., Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra. Atmos. Chem. Phys. 7, 1503–1522 (2007). [Google Scholar]
- 30.Ulbrich I. M., Canagaratna M. R., Zhang Q., Worsnop D. R., Jimenez J. L., Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data. Atmos. Chem. Phys. 9, 2891–2918 (2009). [Google Scholar]
- 31.Jimenez J. L., Canagaratna M. R., Donahue N. M., Prevot A. S., Zhang Q., Kroll J. H., DeCarlo P. F., Allan J. D., Coe H., Ng N. L., Aiken A. C., Docherty K. S., Ulbrich I. M., Grieshop A. P., Robinson A. L., Duplissy J., Smith J. D., Wilson K. R., Lanz V. A., Hueglin C., Sun Y. L., Tian J., Laaksonen A., Raatikainen T., Rautiainen J., Vaattovaara P., Ehn M., Kulmala M., Tomlinson J. M., Collins D. R., Cubison M. J., Dunlea E. J., Huffman J. A., Onasch T. B., Alfarra M. R., Williams P. I., Bower K., Kondo Y., Schneider J., Drewnick F., Borrmann S., Weimer S., Demerjian K., Salcedo D., Cottrell L., Griffin R., Takami A., Miyoshi T., Hatakeyama S., Shimono A., Sun J. Y., Zhang Y. M., Dzepina K., Kimmel J. R., Sueper D., Jayne J. T., Herndon S. C., Trimborn A. M., Williams L. R., Wood E. C., Middlebrook A. M., Kolb C. E., Baltensperger U., Worsnop D. R., Evolution of organic aerosols in the atmosphere. Science 326, 1525–1529 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Mohr C., Mohr C., DeCarlo P. F., Heringa M. F., Chirico R., Slowik J. G., Richter R., Reche C., Alastuey A., Querol X., Seco R., Peñuelas J., Jiménez J. L., Crippa M., Zimmermann R., Baltensperger U., Prévôt A. S. H., Identification and quantification of organic aerosol from cooking and other sources in Barcelona using aerosol mass spectrometer data. Atmos. Chem. Phys. 12, 1649–1665 (2012). [Google Scholar]
- 33.Petters M. D., Kreidenweis S. M., A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 7, 1961–1971 (2007). [Google Scholar]
- 34.Gysel M., Crosier J., Topping D. O., Whitehead J. D., Bower K. N., Cubison M. J., Williams P. I., Flynn M. J., McFiggans G. B., Coe H., Closure study between chemical composition and hygroscopic growth of aerosol particles during TORCH2. Atmos. Chem. Phys. 7, 6131–6144 (2006). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaap8368/DC1
fig. S1. The fraction of CxHyNz ion signal for organic aerosol in the indoor and outdoor data as a function of season.
fig. S2. Time series of summer aerosol measurements.
fig. S3. Mass spectra of factors determined from PMF.