

Abstract
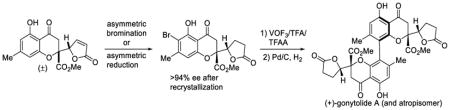
The first synthesis of the chromanone lactone dimer gonytolide A has been achieved employing vanadium(V)-mediated oxidative coupling of the monomer gonytolide C. An ortho-bromine blocking group strategy was employed to favor para-para coupling and to enable kinetic resolution of (±)-gonytolide C. Asymmetric conjugate reduction enabled practical kinetic resolution of a chiral, racemic precursor and the asymmetric synthesis of (+)-gonytolide A and its atropisomer.
Graphical Abstract
1. INTRODUCTION
Gonytolide A (1) is a dimeric chromanone natural product isolated by the Kikuchi group in 2011.1a In an effort to search for regulators of innate immunity from natural sources, an ex vivo culture system based on the Drosophila immune deficiency (IMD) signaling pathway2 identified gonytolide A as a potent innate immune promoter from the fungus Gonytrichum sp. Several related, non-active derivatives (gonytolides B-G, Figure 1) were co-isolated from the same fungal extract1b including the monomers gonytolide C (3) and gonytolide G (7). Gonytolide A (1) possesses four chiral centers and a chiral biaryl axis. The dimers gonytolide A (1), D (4) and F (6) share the same para-para linkage, whereas gonytolide B (2) and E (5) are connected by an ortho-para biaryl bond. The congeners gonytolides D (4) and E (5) are heterodimers in which one chromanone lactone is replaced by a tetrahydroxanthone moiety. It was shown that gonytolide A increases TNF-α-stimulated production of IL-8 in human umbilical vein endothelial cells (HUVECs).1a In later studies, the Kikuchi group prepared simplified bischromone and bisflavone analogues3 lacking the lactone moiety which also promoted the mammalian TNF-α signaling pathway. One key structural element observed for immune-promoting activity from their SAR studies is the requirement for bischromanone/bischromone moieties possessing a para-para linkage, which in the case of gonytolide A is a crowded chiral biaryl axis which presents a formidable synthetic challenge.
Figure 1.

Gonytolide A and Related Natural Products
We have previously developed methodology involving copper-mediated biaryl coupling of aryl stannanes to synthesize the related natural products secalonic acids A (8) and D (9) (Figure 1).4b However, in initial studies5 we were not successful in using this methodology to forge the crowded biaryl bond of gonytolide A, largely due to difficulty in accessing the requisite hindered aryl stannane reaction partner. Accordingly, we explored alternative oxidative coupling methods to construct this biologically active and synthetically challenging molecule in an atom- and step-economical fashion.6 The Kozlowski group has demonstrated the power of metal-catalyzed, oxidative coupling employing chiral copper/1,5-diaza-cis-decalin catalysts7 to synthesize a number of perylenequinone natural products including cercosporin and hypocrellin.8 The Pappo group has showcased use of iron-catalyzed, consecutive oxidative cross-couplings to prepare phlorotannin natural products.9 In oxidative coupling of phenols, control of chemo-, regio-, and stereoselectivity are significant challenges. Such issues have been recently studied in a number of different contexts.6 For example, the Waldvogel group has developed highly efficient, anodic cross-couplings to selectively access non-symmetric biphenols.10 The Pappo group has utilized chiral iron-phosphate catalysts for enantioselective homo- and cross-coupling of 2-naphthols.11 In a recent study, Pappo and coworkers reported oxidative coupling of halophenols with β-keto esters in which the halogenation site influenced C-O vs. C-C coupling.12 The Kozlowski group has also pioneered work on selective ortho-ortho, ortho-para, para-para oxidative homo-couplings of phenols using metal-salen complexes.13
Although para-para selective oxidative couplings have been documented using catalyst control by Kozlowski13 and others,14 substrates thus far have been limited to relatively simple phenols. In the current study, we wished to solve the regioselectivity issue for oxidative coupling of a complex phenolic substrate using a blocking group strategy. Our retrosynthetic analysis for gonytolide A is shown in Scheme 1. Dimeric chromanone 1 may be derived from para-para oxidative coupling of the monomer 3 in which the ortho position of the phenol is blocked with a halogen substituent such as bromine. It was anticipated that the installed halogen may also potentially stabilize developing metal phenoxyl radicals15 during the coupling process. We considered that the oxidative coupling substrate 10 may be obtained from (±)-gonytolide C (3) by an asymmetric bromination/kinetic resolution process. Recent reports have shown significant interest in stereoselective halogenation in natural product synthesis16 including examples involving dynamic kinetic resolution (DKR) via asymmetric bromination.17 Compound 3 may be accessed using vinylogous addition of siloxyfuran (12) to chromone (11)4a which we have previously used to access tetrahydroxanthone dimers including secalonic acids 8 and 9 and rugulotrosin A.4b–d
Scheme 1.

Inital Retrosynthetic Analysis for 1
2. RESULTS AND DISCUSSION
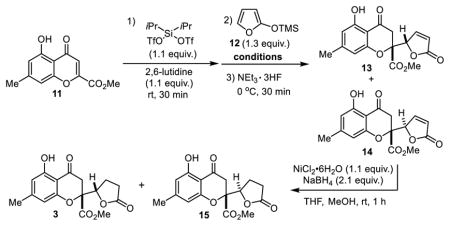
We first optimized the vinylogous addition by varying both solvent and temperature (Table 1). Chromone 11 was activated to a siloxybenzopyrylium species by treatment with diisopropylsilyl triflate and 2,6-lutidine which was followed by addition of siloxyfuran 12 under various reaction conditions. The corresponding chromanone-butenolides 13 and 14 (found to be unstable to silica gel chromatography) were produced as a mixture of diastereomers and were directly subjected to conjugate reduction without purification to afford gonytolide C (3)18 and its diastereomer 15, compounds known from our previous study4a to be the thermodynamic and kinetic products, respectively. Vinylogous addition in CH2Cl2 (0 °C) gave the best results to afford 3/15 in 66 % yield and in a 3:1 diasteromeric ratio (dr) (entry 1). Further raising the reaction temperature or use of other solvents resulted in diminished yield or negatively impacted the dr (entries 2-8).
Table 1.
Evaluation of the Vinylogous Addition
| ||||
|---|---|---|---|---|
| entry | temperature | solvent | overall yield (3+15) | dr(3 : 15) |
| 1 | 0°C | CH2Cl2 | 66% | 3 : 1 |
| 2 | rt | CH2Cl2 | 57% | 3 : 1 |
| 3 | reflux | CH2Cl2 | 54% | 3 : 1 |
| 4 | 0°C | THF | 60% | 1 : 5 |
| 5 | 0°C | Et2O/CH2Cl2(2 : 1) | 39% | 2 : 1 |
| 6 | 0°C | toluene | 47% | 1 : 4 |
| 7 | rt | benzene | 52% | 1 : 1 |
| 8 | rt | MeCN | 24% | 3 : 1 |
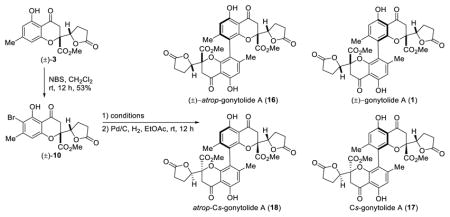
With (±)-gonytolide C (3) in hand, we next prepared (±)-bromo-gonytolide C (10) by bromination with N-bromosuccinimide in CH2Cl2 at room temperature (Table 2). We subsequently carried out the key oxidative coupling using (±)-10. After evaluation of an array of commonly used metal catalysts and oxidants (Table 2), including FeCl3 (entry 1),19 Cr-Salen (entry 3),13,20 Cu(OH)Cl-TMEDA (entry 4),21 Mn(OAc)3 (entry 5),22 PhI(OH)(OTs) (entry 6),23 MoCl5 (entry 7)24 and a number of vanadium oxidants,25 and electrochemical methods (entry 12),26 we were pleased to discover that the silica gel-supported ferric chloride (FeCl3/SiO2) (entry 2)19a and oxyvanadium trifluoride (VOF3) (entry 11) in TFA/TFAA27 both led to clean oxidative coupling to afford brominated dimers as indicated by UPLC-MS analysis.5 The crude dimers were subsequently dehalogenated with Pd/C under a hydrogen atmosphere to afford (±)-gonytolide A and its isomers. Theoretically, two homodimers (1 and 16) with C2-symmetry and two heterodimers (17 and 18) with Cs-symmetry derived from oxidative coupling of different enantiomers may be produced. The FeCl3/SiO2 conditions (entry 2) afforded only trace amounts of the desired dimer gonytolide A, and the reaction required considerable time to reach completion (6 days). Other conditions either gave no reaction or led to undesired byproducts.5 It should be noted that the VOF3-oxidative coupling of (±)-gonytolide C (3) largely afforded decomposition5 thereby demonstrating the importance of the halide blocking group. Attempts to install alternative blocking groups (e.g. benzyl, triisopropylsilyl, and tert-butyldiphenylsilyl ethers) on the phenol of 3 failed to give the desired dimers using VOF3/TFA/TFAA conditions. Instead, the blocking groups were cleaved during the reaction and afforded complex mixtures of dimers, trimers, and tetramers derived from gonytolide C.5
Table 2.
Oxidative Coupling of (±)-gonytolide C (3)
| ||
|---|---|---|
| entry | conditionsa | results |
| 1 | 0.1 equiv. FeCl3, 2.5 equiv. t-BuOOt-Bu, HFIP, rt | No reaction |
| 2 | FeCl3/SiO2, 70 °C, no solvent, 6 days | <5% 1, 19% 17, 28% 16 |
| 3 | 1.0 equiv. Cr-salen, DCE, O2, 70 °C | No reaction |
| 4 | 1.0 equiv. Cu(OH)Cl-TMEDA, DCE, O2, 70 °C | Decomposition |
| 5 | 1.0 equiv. Mn(OAc)3, AcOH, rt | No reaction |
| 6 | 0.5 equiv. PhI(OH)(OTs), HFIP, rt | Complex mixture |
| 7 | 4.0 equiv. MoCl5, CH2Cl2, rt | Para-chlorination of 10 |
| 8 | 2.0 equiv. VOCl3, THF, rt | Para-chlorination of 10 |
| 9 | 2.0 equiv. VOSO4, CH2Cl2, O2, rt | No reaction |
| 10 | 2.0 equiv. VOF3, HFIP, rt | No reaction |
| 11 | 2.5 equiv. VOF3, TFA/TFAA, rt | 11% 1, 23% 17, 23% 16 |
| 12 | Divided cell, graphite anode, TFE/H2O 4:1, 0.2 M LiClO4, 1.0 mA, 3.0~4.0 V | Decomposition |
Reaction conducted for 12 h unless specified otherwise.
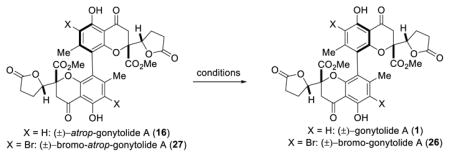
Using the VOF3/TFA/TFAA oxidative coupling conditions, (±)-gonytolide A 1 was produced in an 11% yield along with its atropisomer (16) in 23% yield after reductive debromination. In addition, the heterodimer Cs-gonytolide A (17) was produced in 23% yield; the corresponding Cs-atropisomer (18) was not observed. The structure of Cs-gonytolide A (17) was confirmed by X-ray crystallographic analysis (Figure 2). Interestingly, the crystal structure of 17 shows out-of-plane distortion (9.4°) of the biaryl axis. This distortion observed in the solid state appears to occur based on minimization of steric interactions (e.g. CH3 – CH3 and CH3 – CO2Me) between monomer units. Similar distortions have been observed in other strained biaryl systems.28
Figure 2.
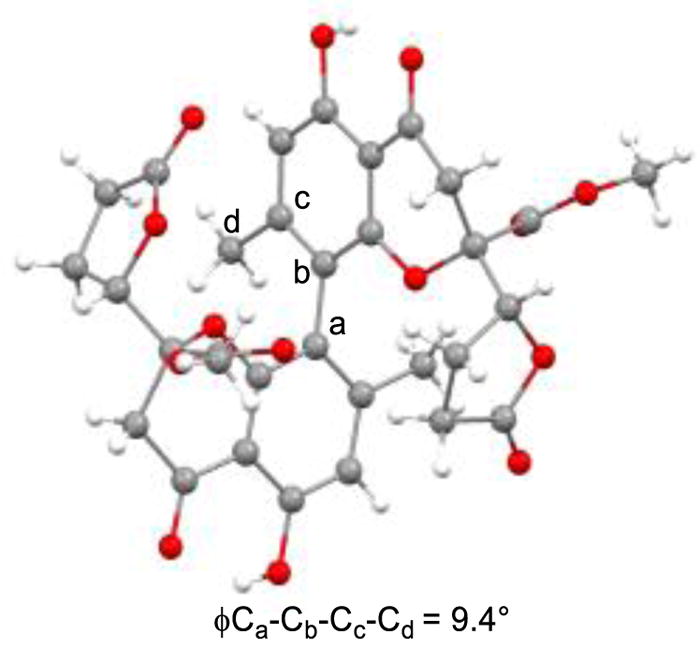
X-ray Crystal Structure of Cs-gonytolide A (17)
In order to eliminate the formation of a Cs-dimer during the oxidative coupling and achieve asymmetric synthesis of gonytolide A, access to enantiopure monomer 3 was required.4c Based on reported examples of asymmetric halogenation of alkenes and phenols,16,17 we evaluated cinchona alkaloid catalysts29 for asymmetric bromination of (±)-3. After considerable experimentation, we found that the dimeric catalyst (DHQD)2Pyr could achieve bromination of (±)-gonytolide C to effect kinetic resolution (Scheme 2). Under optimized conditions,5 the desired ortho-bromo gonytolide C 10 was obtained in 88:12 e.r. (16% yield) which after crystallization from iso-propanol slowly formed a single crystal which was characterized by X-ray crystallography to determine absolute stereochemistry. Given that the bromination site is far from point chirality, the asymmetric bromination represents an interesting example of remote asymmetric induction. Monomer (−)-10 bearing the incorrect absolute stereochemistry to access natural gonytolide was produced in an overall low yield due to formation of the para- and di-bromination byproducts 19 and 20.5
Scheme 2.

Attempted Asymmetric Bromination
As shown in reported examples of asymmetric fluorination with cinchona alkaloid catalysts, N-fluorinated-cinchona derivatives have been characterized and identified as catalytic species.30 We reasoned that NBS should first brominate the tertiary nitrogen29b of the cinchona catalyst and that the resulting N-Br species may have a preference to recognize substrate enantiomers and deliver bromine to the ortho-position of 3. Accordingly, we conducted DFT calculations of the complex of [(DHQD)2PyrBr]+ and both enantiomers of 3 (Figure 3). Geometry optimizations of the pre-transition state (pre-TS) at the B3LYP/LAV3P level showed that both enantiomers can be anchored to the N-brominated-cinchona catalyst through hydrogen-bonding interactions. The unnatural (−)-enantiomer of gonytolide C appears to be preferred in the catalyst pocket with an energy 3.2 kcal/mol lower in comparison to the natural (+)-enantiomer which is consistent with the observed reaction outcome. The difference in energy may be attributed in part to a OH-π interaction31 between the pyrimidine spacer and the phenol of (−)-3. For (+)-3, a weaker hydrogen bonding interaction is observed between the phenol and the methoxy group on the cinchona sidearm.
Figure 3.

Models for Asymmetric Bromination of 3
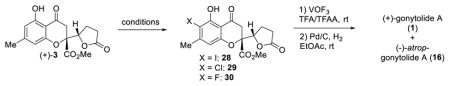
Although the asymmetric bromination of gonytolide C (3) provided access to an enantiopure, monomeric coupling substrate and enabled determination of absolute stereochemistry, the presence of para- and di-bromination byproducts made product purification somewhat tedious and lowered the isolated yield of ortho-bromide 10. Extensive screening of additives, solvents, bromine sources, temperature, and catalysts did not improve results.5 Accordingly, we considered asymmetric reduction of butenolide 13 for kinetic resolution, as asymmetric conjugate reductions of butenolides have been well documented by both the Lipshutz32 and Buchwald33 laboratories using copper hydride.34 In exploring these reductions, (Scheme 3) we found that butenolide 13 was very sensitive to silica gel chromatography and decomposed to afford an elimination product.5 Chiral HPLC analysis of (±)-butenolide 13 also showed partial decomposition. Further studies revealed that 13, 14, and the elimination product are interconvertible under basic conditions. Subsequent recrystallization was used to obtain pure samples of butenolides 13 and 14 and the elimination product from crude mixtures.5 After examination of various chiral ligands, we found that the Josiphos ligand was effective at promoting CuH reduction with useful levels of e.r. Under optimized conditions using Josiphos SL-J001-2, (+)-gonytolide C (3) was produced in 49% yield and in 89:11 e.r. from (±)-butenolide 13. This material was further enriched to 97:3 e.r. by recrystallization and was carried forward to the bromination/oxidative coupling/hydrogenation sequence. As expected, Cs-gonytolide A was not observed in the reaction, and gonytolide A was produced in 12% yield along with atrop-gonytolide A (35% yield).
Scheme 3.

Asymmetric Conjugate Reduction and Synthesis of (+)-Gonytolide A
In order to gain mechanistic insight into the VOF3-induced oxidative coupling and to understand the role of the halide blocking group, we carried out several mechanistic experiments. O-Methylation of monomer 10 afforded methyl ether substrate 21 (Scheme 3) which was subjected to standard oxidative coupling conditions to afford dimeric products, albeit in a much lower conversion than substrate 10.5 In line with literature precedents35 and based on the need for a blocking group and also the reactivity observed with the O-methylated substrate 21, we propose that the oxidative dimerization may proceed through nucleophilic addition of 10 to an electrophilic radical cation19b (Figure 4). We propose that the ortho-bromide 10 may be oxidized by VOF3/TFA/TFAA to form a phenoxyl radical cation 22, likely through a charge transfer complex.36 The ortho-bromine may potentially stabilize the radical cation through electronic effects. Moreover, calculations5,37 support such stabilization effects by showing that the spin density can reside on the bromine atom of 22. Unoxidized 10 may then add to the stabilized radical cation 22 to form radical cation intermediate 23 to initiate the coupling reaction. Intermediate 23 may undergo further oxidation to pre-biaryl structures 24 and 25.
Figure 4.

Proposed Mechanism for Oxidative Coupling
We further considered that atropdiastereoselection for production of either 26 or 27 may be thermodynamically controlled by the relative energies of cyclohexadienone intermediates 24 and 25. We reasoned that, during the rearomatization process, chiral information should be maintained. For example, conformers 24A and 25A should lead to 26 and 24B and 25B should afford 27, similar to the traceless chirality exchange involving rearomatization of dimeric 1,4-diketones reported by the Thomson group.38 In order to probe this hypothesis, we computed DFT energies5 for both conformers of 24/25 at the B3LYP/LACV3P level which showed that the precursors for bromo-atrop-gonytolide A (27) have lower relative energy, regardless of the C-8 configuration (Figure 4).
In addition, no interconversion of 1 and 16 was observed after thermolysis of either compound in refluxing toluene for 24 h. The rotational barrier for interconversion of atropisomers was calculated to be 47.5 kcal/mol using the B3LYP/6-31G(d) level of theory.5 Although the rotational barrier is high, the Pappo group11a and others39 have demonstrated that BINOL can undergo racemization under oxidative conditions. This prompted us to examine single electron transfer (SET) conditions with either atrop-gonytolide A (16) or its brominated precursor bromo-atrop-gonytolide A (27) (Table 3). Conditions reported by Pappo and coworkers11a (entries 1 and 2) did not reveal any reactivity. Use of an acridinium photocatalyst40 under visible light conditions was found to decompose substrate 16 (entry 3) and led to no reaction with substrate 27 (entry 4). Use of (NH4)2Ce(NO3)6 as single electron oxidant did not lead to conversion of 27 to 26 (entry 5). Finally, resubjection of dimer 27 to VOF3/TFA/TFAA conditions also led to decomposition (entry 6). These results are highly interesting given that the unnatural atropisomers 16/27 were favored during the oxidative coupling, but could not be isomerized to the natural atropisomers 1/26.
Table 3.
Attempted Atropisomerism of Gonytolide Dimers
| |||
|---|---|---|---|
| entry | X | conditionsa | results |
| 1 | H | 0.1 equiv. FeCl3, DCE/HFIP 1:1, rt | No reaction |
| 2 | Br | 0.1 equiv. FeCl3, 1.5 equiv. t-BuOOt-Bu, DCE/HFIP 1:1, rt | No reaction |
| 3 | H | 0.1 equiv. 9-mesityl-10-methylacridinium tetra-fluoroborate, CH3CN, 427 nm, rt | Complete decomposition |
| 4 | Br | 0.1 equiv. 9-mesityl-10-methylacridinium tetra-fluoroborate, CH3CN, 427 nm, rt | No reaction |
| 5 | Br | 0.5 equiv. (NH4)2Ce(NO3)6, CH3CN/H2O 1:1, rt | Decomposition |
| 6 | Br | 0.5 equiv. VOF3, TFA/TFAA 20:1, rt | Complete decomposition |
Reaction conducted for 12 h unless specified otherwise.
Finally, we evaluated various halogen blocking groups on the monomer gonytolide C (+)-3 for examination in the VOF3-oxidative coupling in an effort to tune atropselectivity (Table 4). Various halogen blocking groups were introduced using conventional methods, resulting in nearly equal amounts of para/ortho halogenation with the exception of BnMe3NICl2 which preferentially afforded the ortho-iodide. Interestingly, a trend for atropselectivity was observed. Use of an ortho-iodo blocking group afforded exclusively atrop-gonytolide A (entry 1), while a 1:3 ratio of atropisomers in favor of atrop-gonytolide A was observed using a bromine blocking group (entry 2). The ratio of atropisomers increased to nearly 1:1 employing an ortho-chlorine blocking group (entry 3). In addition, we found that use of an ortho-fluorine blocking group completely inhibited reactivity (entry 4). These results showed the critical role of halogen blocking group in the oxidative coupling, and the possibility to tune atropselectivity by choice of halogen. Moreover, DFT computations5 provided insight on how the halogen blocking group may influence atropselectivity to produce either 1 or 16. Figure 5 shows the disfavored model of 24A showing close proximity of the aryl bromide and the appended lactone moiety as well as the favored model of 24B where the bromine is proximal to a methyl ester. This steric analysis is also consistent with the atropselectivity trend observed from I → Br → Cl blocking groups in which greater amounts of gonytolide A are observed. Unfortunately, attempts to use chiral ligands41 or a chiral oxovanadium complexes42 in oxidative coupling of 10 to further tune the atropselectivity failed to yield dimers.5 These results are not surprising as the mechanism suggests an outer-sphere oxidation process.43
Table 4.
Oxidative Coupling Using Various Halogen Blocking Groups
| |||
|---|---|---|---|
| Entry | Conditionsa | Halogenation results | Coupling results |
| 1 | BnMe3NICl2, CaCO3, CH2Cl2/CH3OH, rt | 70%, X= I | 0% 1, 25% 16 |
| 2 | NBS, CH2Cl2, rt | 53%, X= Br | 11% 1, 35% 16 |
| 3 | NCS, DMF, 60 °C 4 h | 44%, X= Cl | 23% 1, 27% 16 |
| 4 | Selectfluor®, CH3CN, rt | 31%, X= F | No reaction |
Reaction conducted for 12 h unless specified otherwise.
Figure 5.

Space Filling Models for Biaryl Precursors
3. CONCLUSION
In conclusion, we have synthesized the natural product (+)-gonytolide A and its unnatural atropisomer (-)-atrop-gonytolide A in five steps from a chromone ester monomer. The synthesis features vinylogous addition to rapidly construct a chromanone butenolide substrate, asymmetric conjugate reduction/asymmetric bromination to resolve a chiral, racemic butenolide monomer, and oxidative coupling using a halide blocking group to construct the crowded biaryl linkage of the dimer gonytolide A and its atropisomer. The halogen blocking group appears to have several different roles in the oxidative coupling: 1) stabilization of radical cation intermediates, 2) control of para-para coupling selectivity, and 3) steric tuning of atropdiastereoselection providing access to both natural and unnatural atropisomers in varying ratios depending on the halogen blocking group. Further studies on the syntheses and biology of dimeric chromanones and derivatives are currently ongoing in our lab and will be reported in due course.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (R35 GM-118173) for research support. Work at the BU-CMD is supported by the NIH R24 grant GM-111625. We thank Prof. Scott Miller and Dr. Anthony Metrano (Yale University) for helpful discussions and preliminary experiments. We thank the Uehara Memorial Foundation for a postdoctoral fellowship to T.I., the American Cancer Society for a postdoctoral fellowship to K.D.R. (PF-16-235-01-CDD), and Dr. Jeffrey Bacon (Boston University) for X-ray crystal structure analyses. We thank Prof. Haruhisa Kikuchi (Tohoku University) for providing a natural sample of gonytolide A. NMR (CHE-0619339) and MS (CHE-0443618) facilities at Boston University are supported by the NSF.
Footnotes
Notes
The authors declare no competing financial interests.
Experimental and computational details, and CIF files for 6 and 13. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kikuchi H, Isobe M, Sekiya M, Abe Y, Hoshikawa T, Ueda K, Kurata S, Katou Y, Oshima Y. Org Lett. 2011;13:4624. doi: 10.1021/ol2018449. [DOI] [PubMed] [Google Scholar]; (b) Kikuchi H, Isobe M, Kurata S, Katou Y, Oshima Y. Tetrahedron. 2012;68:6218. [Google Scholar]; (c) Maha A, Rukachaisirkul V, Phongpaichit S, Poonsuwan W, Sakayaroj J. Tetrahedron. 2016;72:2874. doi: 10.1016/j.phytochem.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 2.(a) Yajima M, Takada M, Takahashi N, Kikuchi H, Natori S, Oshima Y, Kurata S. Biochem J. 2003;371:205. doi: 10.1042/BJ20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sekiya M, Ueda K, Fujita T, Kitayama M, Kikuchi H, Oshima Y, Kurata S. Life Sci. 2006;80:113. doi: 10.1016/j.lfs.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 3.Kikuchi H, Hoshikawa T, Kurata S, Katou Y, Oshima Y. J Nat Prod. 2016;79:1259. doi: 10.1021/acs.jnatprod.5b00829. [DOI] [PubMed] [Google Scholar]
- 4.(a) Qin T, Johnson RP, Porco JA., Jr J Am Chem Soc. 2011;133:1714. doi: 10.1021/ja110698n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qin T, Porco JA., Jr Angew Chem Int Ed. 2014;53:3107. doi: 10.1002/anie.201311260. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Qin T, Skraba-Joiner SL, Khalih ZG, Johnson RP, Capon RJ, Porco JA., Jr Nat Chem. 2015;7:234. doi: 10.1038/nchem.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Qin T, Iwata T, Ransom TT, Beutler JA, Porco JA., Jr J Am Chem Soc. 2015;137:15225. doi: 10.1021/jacs.5b09825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.See Supporting Information for complete experimental details.
- 6.Kozlowski MC. Acc Chem Res. 2017;50:638. doi: 10.1021/acs.accounts.6b00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Li X, Yang J, Kozlowski MC. Org Lett. 2001;3:1137. doi: 10.1021/ol015595x. [DOI] [PubMed] [Google Scholar]; (b) Hewgley JB, Stahl SS, Kozlowski MC. J Am Chem Soc. 2008;130:12232. doi: 10.1021/ja804570b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Mulrooney CA, Li X, Divirgilio ES, Kozlowski MC. J Am Chem Soc. 2003;125:6856. doi: 10.1021/ja027745k. [DOI] [PubMed] [Google Scholar]; (b) Divirgilio ES, Dugan EC, Mulrooney CA, Kozlowski MC. Org Lett. 2007;9:385. doi: 10.1021/ol062468y. [DOI] [PubMed] [Google Scholar]; (c) O’Brien EM, Morgan BJ, Kozlowski MC. Angew Chem Int Ed. 2008;47:6877. doi: 10.1002/anie.200800734. [DOI] [PubMed] [Google Scholar]; (d) Morgan BJ, Dey S, Johnson SW, Kozlowski MC. J Am Chem Soc. 2009;131:9413. doi: 10.1021/ja902324j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Morgan B, Mulrooney CA, O’Brien EM, Kozlowski MC. J Org Chem. 2010;75:30. doi: 10.1021/jo901384h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vershinin V, Dyadyuk A, Pappo D. Tetrahedron. 2017;73:3660. [Google Scholar]
- 10.(a) Riehl B, Dyballa K, Franke R, Waldvogel SR. Synthesis. 2016;49:252. [Google Scholar]; (b) Wiebe A, Schollmeyer D, Dyballa KM, Franke R, Waldvogel SR. Angew Chem Int Ed. 2016;55:11801. doi: 10.1002/anie.201604321. [DOI] [PubMed] [Google Scholar]; (c) Lips S, Wiebe A, Elsler B, Schollmeyer D, Dyballa KM, Franke R, Waldvogel SR. Angew Chem Int Ed. 2016;55:10872. doi: 10.1002/anie.201605865. [DOI] [PubMed] [Google Scholar]; (d) Schulz L, Enders M, Elsler B, Schollmeyer D, Dyballa KM, Franke R, Waldvogel SR. Angew Chem Int Ed. 2017;56:4877. doi: 10.1002/anie.201612613. [DOI] [PubMed] [Google Scholar]
- 11.(a) Narute S, Parnes R, Toste FD, Pappo D. J Am Chem Soc. 2016;138:16553. doi: 10.1021/jacs.6b11198. [DOI] [PubMed] [Google Scholar]; (b) Forkosh H, Vershinin V, Reiss H, Pappo D. Org Lett ASAP. doi: 10.1021/acs.orglett.8b00800. [DOI] [PubMed] [Google Scholar]
- 12.Regev A, Shalit H, Pappo D. Synthesis. 2015;47:1716. [Google Scholar]
- 13.Lee YE, Cao T, Torruellas C, Kozlowski MC. J Am Chem Soc. 2014;136:6782. doi: 10.1021/ja500183z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Carrick WL, Karapinka GL, Kwiatkowski GT. J Org Chem. 1968;33:2388. [Google Scholar]; (b) Sperry J, Sejberg JJP, Stiemke FM, Brimble MA. Org Biomol Chem. 2009;7:2599. doi: 10.1039/b905077a. [DOI] [PubMed] [Google Scholar]; (c) Shalit H, Libman A, Pappo D. J Am Chem Soc. 2017;139:13404. doi: 10.1021/jacs.7b05898. [DOI] [PubMed] [Google Scholar]
- 15.Shimazaki Y, Yamauchi O. Indian J Chem. 2011;50A:383. [Google Scholar]
- 16.(a) Hu DX, Shibuya GM, Burns NZ. J Am Chem Soc. 2013;135:12960. doi: 10.1021/ja4083182. [DOI] [PubMed] [Google Scholar]; (b) Hu DX, Seidl FJ, Bucher C, Burns NZ. J Am Chem Soc. 2015;137:3795. doi: 10.1021/jacs.5b01384. [DOI] [PubMed] [Google Scholar]; (c) Bucher C, Deans RM, Burns NZ. J Am Chem Soc. 2015;137:12784. doi: 10.1021/jacs.5b08398. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Landry ML, Hu DX, McKenna GM, Burns NZ. J Am Chem Soc. 2016;138:5150. doi: 10.1021/jacs.6b01643. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Burckle AJ, Vasilev VH, Burns NZ. Angew Chem Int Ed. 2016;55:11476. doi: 10.1002/anie.201605722. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Samanta R, Yamamoto H. J Am Chem Soc. 2017;139:1460. doi: 10.1021/jacs.6b13193. [DOI] [PubMed] [Google Scholar]
- 17.(a) Barrett KT, Miller SJ. J Am Chem Soc. 2013;135:2963. doi: 10.1021/ja400082x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mori K, Ichikawa Y, Kobayashi M, Shibata Y, Yamanaka M, Akiyama T. J Am Chem Soc. 2013;135:3964. doi: 10.1021/ja311902f. [DOI] [PubMed] [Google Scholar]; (c) Barrett KT, Metrano AJ, Rablen PR, Miller SJ. Nature. 2014;509:71. doi: 10.1038/nature13189. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Miyaji R, Asano K, Matsubara S. J Am Chem Soc. 2015;137:6766. doi: 10.1021/jacs.5b04151. [DOI] [PubMed] [Google Scholar]; (e) Diener ME, Metrano AJ, Miller SJ. J Am Chem Soc. 2015;137:12369. doi: 10.1021/jacs.5b07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Syntheses of gonytolide C: Tietze LF, Jackenkroll S, Hierold J, Ma L, Waldecker B. Chem Eur J. 2014;20:8628. doi: 10.1002/chem.201402495.Sudhakar G, Bayya S, Kadam VD, Nanubolu JB. Org Biomol Chem. 2014;12:5601. doi: 10.1039/c4ob00950a.Liu J, Li Z, Tong P, Xie Z, Zhang Y, Li Y. J Org Chem. 2015;80:1632. doi: 10.1021/jo502571r.Li FF, Atkinson DJ, Furkert DP, Brimble MA. Eur J Org Chem. 2016;1145
- 19.(a) Li H-Y, Nehira T, Hagiwara M, Harada N. J Org Chem. 1997;62:7222. doi: 10.1021/jo970670w. [DOI] [PubMed] [Google Scholar]; (b) Libman A, Shalit H, Vainer Y, Narute S, Kozuch S, Pappo D. J Am Chem Soc. 2015;137:11453. doi: 10.1021/jacs.5b06494. [DOI] [PubMed] [Google Scholar]
- 20.(a) Katsuki T. Chem Soc Rev. 2004;33:437. doi: 10.1039/b304133f. [DOI] [PubMed] [Google Scholar]; (b) Cozzi PG. Chem Soc Rev. 2004;33:410. doi: 10.1039/b307853c. [DOI] [PubMed] [Google Scholar]
- 21.(a) Noji M, Nakajima M, Koga K. Tetrahedron Lett. 1994;35:7983. [Google Scholar]; (b) Nakajima M, Miyoshi I, Kanayama K, Hashimoto S, Noji M, Koga K. J Org Chem. 1999;64:2264. [Google Scholar]; (c) O’Brien EM, Morgan BJ, Kozlowski MC. Angew Chem Int Ed. 2008;47:6877. doi: 10.1002/anie.200800734. [DOI] [PubMed] [Google Scholar]; (d) Yan P, Sugiyama Y, Takahashi Y, Kinemuchi H, Temma T, Habaue S. Tetrahedron. 2008;64:4325. [Google Scholar]
- 22.Wang K, Hu Y, Li Z, Wu M, Liu Z, Su B, Yu A, Liu Y, Wang Q. Synthesis. 2010;7:1083. [Google Scholar]
- 23.(a) Dohi T, Ito M, Morimoto K, Iwata M, Kita Y. Angew Chem Int Ed. 2008;47:1301. doi: 10.1002/anie.200704495. [DOI] [PubMed] [Google Scholar]; (b) Morimoto K, Ohnishi Y, Koseki D, Nakamura A, Dohi T, Kita Y. Org Biomol Chem. 2016;14:8947. doi: 10.1039/c6ob01764a. [DOI] [PubMed] [Google Scholar]
- 24.Schubert M, Leppin J, Wehming K, Schollmeyer D, Heinze K, Waldvogel SR. Angew Chem Int Ed. 2014;53:2494. doi: 10.1002/anie.201309287. [DOI] [PubMed] [Google Scholar]
- 25.Hirao T. Chem Rev. 1997;97:2707. doi: 10.1021/cr960014g. [DOI] [PubMed] [Google Scholar]
- 26.Kirste A, Elsler B, Schnakenburg G, Waldvogel SR. J Am Chem Soc. 2012;134:3571. doi: 10.1021/ja211005g. [DOI] [PubMed] [Google Scholar]
- 27.(a) Kupchan SM, Dhingra OP, Kim CK. J Org Chem. 1978;43:4076. doi: 10.1021/jo00395a025. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Wood MR, Trotter BW, Richardson TI, Barrow JC, Katz JL. Angew Chem Int Ed. 1998;37:2700. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2700::AID-ANIE2700>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 28.(a) Colquhoun HM, Dudman CC, Thomas M, O’Mahoney CA, Williams DJ. J Chem Soc, Chem Commun. 1990. p. 336. [Google Scholar]; (b) Burns N, Krylova IN, Hannoush RN, Baran PS. J Am Chem Soc. 2009;131:9172. doi: 10.1021/ja903745s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Konkol LC, Guo F, Sarjeant AA, Thomson RJ. Angew Chem Int Ed. 2011;50:9931. doi: 10.1002/anie.201104726. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liau BB, Milgram BC, Shair MD. J Am Chem Soc. 2012;134:16765. doi: 10.1021/ja307207q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mitra NK, Meudom R, Corzo HH, Gorden JD, Merner BL. J Am Chem Soc. 2016;138:3235. doi: 10.1021/jacs.6b00538. [DOI] [PubMed] [Google Scholar]
- 29.(a) Chen ZM, Zhang QW, Chen ZH, Li H, Tu YQ, Zhang FM, Tian JM. J Am Chem Soc. 2011;133:8818. doi: 10.1021/ja201794v. [DOI] [PubMed] [Google Scholar]; (b) Zhang X, Li J, Tian H, Shi Y. Chem Eur J. 2015;21:11658. doi: 10.1002/chem.201502133. [DOI] [PubMed] [Google Scholar]
- 30.(a) Shibata N, Suzuki E, Takeuchi Y. J Am Chem Soc. 2000;122:10728. [Google Scholar]; (b) Shibata N, Suzuki E, Asahi T, Shiro M. J Am Chem Soc. 2001;123:7001. doi: 10.1021/ja010789t. [DOI] [PubMed] [Google Scholar]; (c) Shibata N, Ishimaru T, Suzuki E, Kirk K. J Org Chem. 2003;68:2494. doi: 10.1021/jo026792s. [DOI] [PubMed] [Google Scholar]
- 31.(a) Mileni M, Garfunkle J, Ezzili C, Kimball FS, Cravatt BF, Stevens RC, Boger DL. J Med Chem. 2010;53:230. doi: 10.1021/jm9012196. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Howie R, Lima C, Kaiser C, de Souza M, Wardell J, Wardell S. Z Kristallogr. 2010;225:349. [Google Scholar]
- 32.(a) Lipshutz BH, Servesko JM, Taft BR. J Am Chem Soc. 2004;126:8352. doi: 10.1021/ja049135l. [DOI] [PubMed] [Google Scholar]; (b) Lipshutz BH, Frieman BA. Angew Chem Int Ed. 2005;44:6345. doi: 10.1002/anie.200500800. [DOI] [PubMed] [Google Scholar]
- 33.(a) Hughes G, Kimura M, Buchwald SL. J Am Chem Soc. 2003;125:11253. doi: 10.1021/ja0351692. [DOI] [PubMed] [Google Scholar]; (b) Rainka MP, Milne JE, Buchwald SL. Angew Chem Int Ed. 2005;44:6177. doi: 10.1002/anie.200501890. [DOI] [PubMed] [Google Scholar]
- 34.(a) Rendler S, Oestreich M. Angew Chem Int Ed. 2007;46:498. doi: 10.1002/anie.200602668. [DOI] [PubMed] [Google Scholar]; (b) Deutsch C, Krause N, Lipshutz B. Chem Rev. 2008;108:2916. doi: 10.1021/cr0684321. [DOI] [PubMed] [Google Scholar]
- 35.For radical cation mechanisms in biaryl coupling, see: Tanaka M, Nakashima H, Fujiwara M, Ando H, Souma Y. J Org Chem. 1996;61:788. doi: 10.1021/jo950814b.Schubert M, Franzmann P, Wunsche von Leupoldt A, Koszinowski K, Heinze K, Waldvogel SR. Angew Chem Int Ed. 2016;55:1156. doi: 10.1002/anie.201508035.
- 36.Hamamoto H. Chem Pharm Bull. 2012;60:799. doi: 10.1248/cpb.c112001. [DOI] [PubMed] [Google Scholar]
- 37.Morofuji T, Shimizu A, Yoshida J. Angew Chem Int Ed. 2012;51:7259. doi: 10.1002/anie.201202788. [DOI] [PubMed] [Google Scholar]
- 38.Guo F, Konkol LC, Thomson RJ. J Am Chem Soc. 2011;133:18. doi: 10.1021/ja108717r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.(a) Brussee J, Jansen AC. Tetrahedron. 1983;24:3261. [Google Scholar]; (b) Smrcina M, Polakova J, Vyskocil S, Kocovsky P. J Org Chem. 1993;58:4534. [Google Scholar]; (c) Qi C, Wang W, Reichl KD, McNeely J, Porco JA., Jr Angew Chem Int Ed. 2018;57:2101. doi: 10.1002/anie.201711535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukuzumi S, Kotani H, Ohkubo K, Ogo S, Tkachenko NV, Lemmetyinen H. J Am Chem Soc. 2004;126:1600. doi: 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]
- 41.Habaue S, Murakami S, Higashimura H. J Polym Sci A Polym Chem. 2005;43:5872. [Google Scholar]
- 42.(a) Takizawa S, Katayama T, Sasai H. Chem Commun. 2008;0:4113. doi: 10.1039/b806016a. [DOI] [PubMed] [Google Scholar]; (b) Kang H, Lee YE, Reddy PVG, Dey S, Allen SE, Niederer KA, Sung P, Kirsten H, Torruellas C, Herling MR, Kozlowski MC. Org Lett. 2017;19:5505. doi: 10.1021/acs.orglett.7b02552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.(a) Dick AR, Sanford MS. Tetrahedron. 2006;62:2439. [Google Scholar]; (b) Hashizume S, Oisaki K, Kanai M. Chem Rec. 2011;11:236. doi: 10.1002/tcr.201100024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.