

Abstract
Recent epidemiological evidence suggests that exposure to antibiotics in early‐to‐middle adulthood is associated with an increased risk of colorectal adenoma. However, mechanistic studies in established preclinical cancer to examine these claims are extremely limited. Therefore, we investigated the effect of long‐term exposure of an antibiotic cocktail composed of Vancomycin, Neomycin, and Streptomycin, on tumor development and progression in the Apc Min/+ mouse, an established genetic model for familial adenomatous polyposis. Clinical pathologies related to tumor development as well as intestinal and colon tissue histopathology were studied at ages 8, 12, and 16 weeks of age, which correspond to the approximate ages of development of neoplasia, gut inflammation with polyposis, and cancer progression, respectively, in this animal model. We show that the antibiotics significantly increase the severity of clinical symptoms, including effects on intestinal histology and goblet cell numbers. In addition, they promote small intestinal polyposis. Finally, metagenomic analysis of fecal samples demonstrated that antibiotic exposure is associated with a significant but nonuniform depletion of the animal's natural gut flora. Overall, these findings support the premise that long‐term antibiotic exposure mediates the selected depletion of gut microbial communities and the concomitant thinning of the protective mucus layer, resulting in an increase in tumor development.
Keywords: Antibiotics, Colorectal cancer, Goblet cells, Microbiome
Introduction
The gut microbiome is integral to gastrointestinal tract function and is connected to a variety of health issues 1, 2, 3. Its role has been demonstrated in a number of conditions, such as diabetes 4, metabolic disorders 5, Alzheimer's disease 6, systemic lupus erythematosus 7, hypertension 8, mental disorders 9, obesity 10, pancreatic disorders 11, cardiovascular disorders 12, aging 13, inflammatory disorders 1, and cancer 14, 15 including colorectal cancer 16.
Colorectal cancer is the second leading cause of cancer‐related death in the United States and ranks fourth in estimated new cases 17. The functional link between gut microbial dysbiosis and colorectal cancer is supported by preclinical studies with animal models 18, 19, 20, 21 and by clinical investigations with patients predisposed to colorectal cancer 22, 23, 24, 25, 26. Hence, the significant increase in the use of antibiotics among adults and children in the United States 27, 28, 29, 30, 31 is a public health concern. Accumulating evidence supports the notion that long‐term antibiotic exposure alters the functional capacity of the gut microbiota 32, 33 resulting in an increased risk of chronic gut diseases such as inflammatory bowel disease 34 and celiac disease 35 as well as activation of the biological mechanisms that initiate or promote colorectal carcinogenesis 36.
Despite these lines of evidence, there are significant gaps in our understanding of how antibiotics increase the risk of colorectal cancer. Current studies show that tetracycline mediates upregulation of cyclooxygenase‐2 and prostaglandin production 37, which promote chronic inflammation‐induced colorectal cancer 38. Addressing this knowledge gap is critical prior to clinical recommendations and development of microbial therapies to counter the effects of long‐term antibiotic use.
In the current study, we address this knowledge gap by examining the effects of long‐term administration of an antibiotic cocktail of Vancomycin, Neomycin, and Streptomycin on gut polyposis in the Apc Min/+ mouse, an established genetic model for familial adenomatous polyposis 39. This model develops approximately 30–50 tumors in the gut at an age of 16–20 weeks with tumors mostly located toward the iliac part of the small intestine and the descending part of the colon 40. Reported here are our findings from the comparisons of clinical pathologies and the intestinal and colon tissue histopathologies related to colorectal cancer in antibiotic administered and control mice of at ages 8, 12, and 16 weeks. The ages, respectively, correspond to the approximate ages of neoplasia, gut inflammation with polyposis, and cancer progression in this animal model.
Materials and Methods
Experimental animal groups and diet
Apc Min/+ mice were obtained from Jackson Laboratories and bred in‐house at the Animal Resource Facility at the, University of South Carolina. Food (Purina chow) and drinking water were available to the mice ad libidum under a 12:12‐hour light–dark cycle and a low‐stress environment (22°C, 50% humidity, and low noise). At 4 weeks of age, littermates were randomly assigned to the following six groups (Fig. 1): (1) Apc Min/+ untreated controls sacrificed after 8 weeks (2) Apc Min/+ untreated controls sacrificed after 12 weeks, (3) Apc Min/+ untreated controls sacrificed after 16 weeks, (4) Apc Min/+ administered with antibiotics and sacrificed after 8 weeks, (5) Apc Min/+ administered with antibiotics and sacrificed after 12 weeks and (6) Apc Min/+ administered with antibiotics and sacrificed after 16 weeks. All procedures and animal care followed institutional guidelines and were approved by the Institutional Animal Care and Use Committee at the University of South Carolina. Four mice were assigned to each of the six treatment groups, and this sample size was based on statistical power analyses conducted in previous microbiome studies 41, 42, 43, which indicated that 3–5 mice is the required sample size for studying changes in the mice gut microbial communities following antibiotic treatment.
Figure 1.
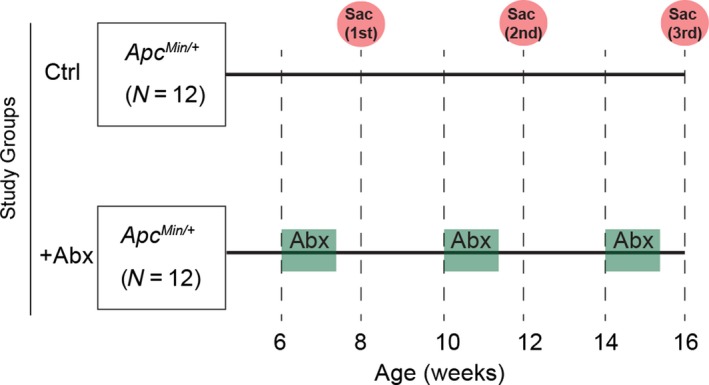
Study design. Two groups of Apc Min/+ mice (N = 12 per group) were used for this study. Antibiotics were added in the drinking water of ‘+Abx mice’ at age 6, 10, and 14 weeks for a period of 10 days (green highlight). Clinical pathology, tissue histopathology, and polyposis in +Abx mice were compared with the control (Ctrl) mice, at 8, 12, and 16 week time‐points upon sacrificing four mice per time‐point (red circles).
Antibiotic administration
A mixture containing Vancomycin (1 mg/mL, active against Gram‐positive bacteria), Neomycin (1 mg/mL, active against Gram‐negative bacteria), and Ampicillin (1 mg/mL, active against both Gram‐positive and Gram‐negative bacteria) was administered to antibiotic treatment group (+Abx). The antibiotic mixture was added to the drinking water at age 6, 10, and 14 weeks (Fig. 1). Normal drinking water replaced the antibiotic containing water after 10 days of administration.
Clinical score, histopathological assessments and polyp counts
The use of a ‘clinical score’ has been previously used to quantitatively express disease symptoms has been described previously 44, 45. The cumulative clinical score for each mouse, with a maximum score of 12, was based on weight loss measurement, diarrhea, and fecal hemoccult. There was a maximum score of four within each of the three quantitative parameters. Score for the weight loss was based on the following published scale where 0 = 0–5% weight loss; 1 = 6–10% weight loss; 2 = 11–15% weight loss; 3 = 16–20% weight loss; and 4 = >20% weight loss. Scoring of diarrhea was as follows: 0 = well‐formed pellets, 2 = pasty and semi‐formed stools that do not adhere to the anus, 4 = liquid stools that adhere to the anus. Detection of blood in the stools was determined using hemoccult kit (Beckman coulter, Brea, CA), which is a hydrogen peroxide‐based kit that forms a visible blue colored complex with blood. The followings were the score rates for the fecal hemoccult: 0 = no blood, 2 = positive hemoccult, 4 = gross bleeding. The total clinical score was the summation of the individual score of weight loss, diarrhea, and fecal hemoccult. Tumor quantification was conducted manually upon observing the 1% methyl blue stained tissue sections under the light microscope as described previously 44. Histopathological analyses for colonic tissue inflammation were conducted using a scoring system as described previously 44. Quantitative comparison of intestinal inflammation was conducted by comparisons of crypt depth‐to‐villus height ratio (CVR). Goblet‐to‐epithelial ratio per crypt was quantified upon analyses of intestinal tissue samples stained with alcian blue (for staining mucus‐containing goblet cells) and counterstained with Nuclear Fast Red solution (for staining the epithelial cells of the mucosa) as described previously [44].
Fecal bacteriome analysis
Genomic DNA from fresh feces were isolated using the MoBio PowerSoil DNA Isolation Kit 46 and subjected to 16S rRNA gene analysis 47. The 16S metadata were demultiplexed with QIIME 48, 49. OTUs were shortlisted using OTUPipe analysis pipeline for error correction, chimera checking, UCLAST clustering and picking the optimal representative sequence centroid. Reference‐based chimera checking was conducted against a set of trusted sequences from the ‘Gold’ database 50. Taxonomy were assigned using the RDP classifier version 2.237 as described previously 51. The rendered OTU tables were checked for mislabeling and contamination as described previously 52. Finally, alpha‐diversity was estimated for each sample/sample pair using Chao1 (estimator of richness) and Shannon Diversity Index (richness and evenness). Also weighted UniFrac (dissimilarity based on phylogenetic differences and taxonomic abundance) and unweighted UniFrac (dissimilarity based on phylogenetic differences but not abundance) were used to express beta‐diversity 53 within and between the antibiotic‐administered and control mice.
Statistical analysis
Two‐way analysis of variance (ANOVA), two‐way repeated‐measure ANOVA, and one‐way ANOVA were used to analyze the data. A Tukey post hoc analysis was used to determine differences in physiological responses upon antibiotic‐administered mice and the controls. All statistical analyses were performed with SigmaStat 3.5 (SPSS, Chicago, IL). For fecal bacteriome analyses, Kruskal–Wallis and Mann–Whitney statistical analyses were performed to calculate significance in diversity and relative abundance, respectively. A P value of <0.05 was considered significant.
Results
Effect of antibiotic administration on total polyp counts and clinical pathology
To mimic long‐term antibiotic exposure, Apc Min/+ mice were exposed to the antibiotic cocktail beginning at 6, 10, and 14 weeks of age, and polyp numbers and sizes were assessed at 8, 12, and 16 weeks (Fig. 1). As shown in Figure 2A, total polyp counts were significantly higher for antibiotic‐administered mice at 12 and 16 weeks, as compared to control mice. This was predominantly due to larger polyps (i.e., ≥1 mm2), as there were no significant differences for polyps <1 mm2. Thus, antibiotic exposure promoted development of intestinal polyps.
Figure 2.
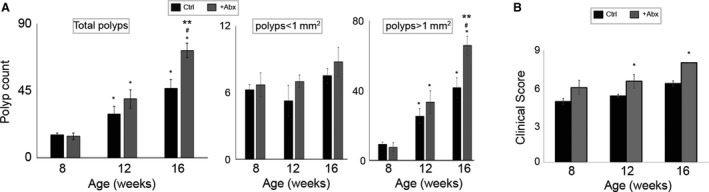
Effect of antibiotic exposure on polyposis and clinical scores: (A) Polyp counts. Graph representing the small intestine polyp in Control (Ctrl) and + Abx mice; left panel, total count, middle panel, count of polyps of size < 1 mm2, right panel, count of polyps of size >1 mm2. Two‐way repeated‐measure analysis of variance (ANOVA) was applied to calculate the significant difference between the polyp counts between +Abx and Ctrl groups at the different ages (**P < 0.01 Ctrl vs. +Abx within each time‐point, *P < 0.05 vs. 8 weeks in each group, #P < 0.05 16 weeks vs. 12 weeks in each group), (B) Clinical score. Comparison of clinical scores for +Abx and Ctrl groups at different time‐points during the study. Two‐way repeated‐measure analysis of variance (ANOVA) was applied to calculate the significant difference between the clinical scores between +Abx and Ctrl groups (*P < 0.05 +Abx vs. Ctrl).
As shown in Figure 2B, clinical scores correlated with the polyp counts in that antibiotic‐administered mice demonstrated significantly higher scores compared to the controls at 12 and 16 weeks of age.
Effect of antibiotic administration on intestinal histopathological scores and crypt‐to‐villus ratios
Clinical scores derived from an integral assessment of weight loss, diarrhea, and fecal hemoccult were observed to increase in antibiotic administered as compared to control mice at all three ages (Fig. 3A; a representation of the H&E stained tissues is shown in Fig. S1). Crypt‐to‐villus ratios were higher in antibiotic treated as compared to control mice at 12 and 16, but not 8, weeks (Fig. 3B). These results suggest an increase in intestinal and colonic inflammation upon long‐term antibiotic administration.
Figure 3.
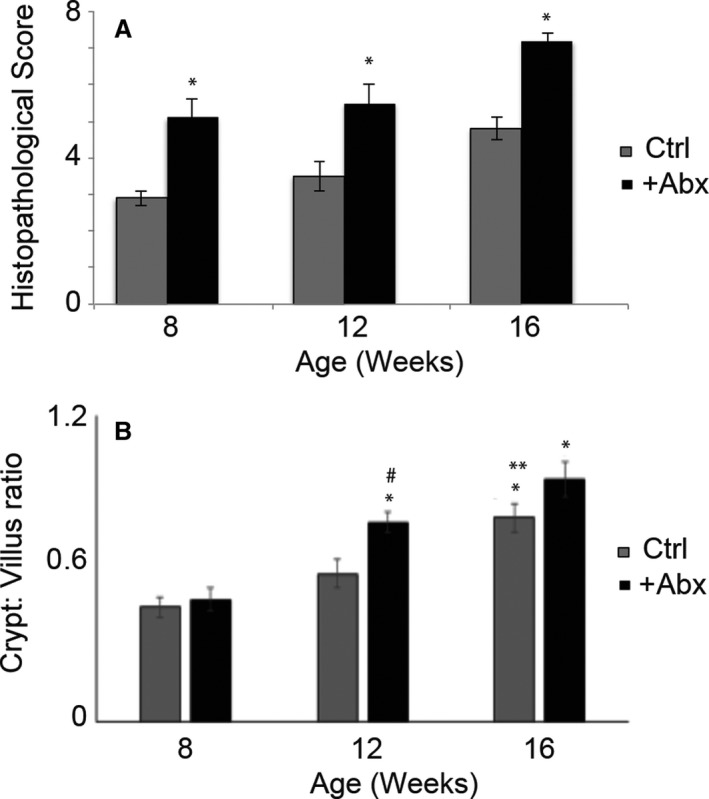
Analysis of tissue inflammation. (A) Histopathology scoring. The scoring was based on the analysis of 10 different sections determining the degree of inflammation and immune cell infiltration was plotted for Ctrl and +Abx groups at different ages. Two‐way analysis of variance (ANOVA) was used to calculate the significant difference between groups at different ages (*P < 0.05‐ +Abx vs. Ctrl). (B) Intestinal CVR. CVR was calculated as a measure of intestinal inflammation for all animals. Two‐way analysis of variance (ANOVA) was used to determine the significance of difference between Ctrl and +Abx mice at different ages. *P value<0.04 versus 8 weeks, # P < 0.01 +Abx versus Ctrl and **P < 0.05 16 versus 12 weeks within the same group.
Effects on goblet cell counts
As antibiotics impact the thickness of the intestinal mucus layer 54, we investigated the effects of the antibiotic treatment on the numbers of mucus‐producing goblet cells. Results (Fig. 4) indicated a significant drop in the ratio of goblet to epithelial cells in mice exposed to antibiotics at all three ages. These effects may be due to either an increase in goblet cell apoptosis or a decrease in production.
Figure 4.
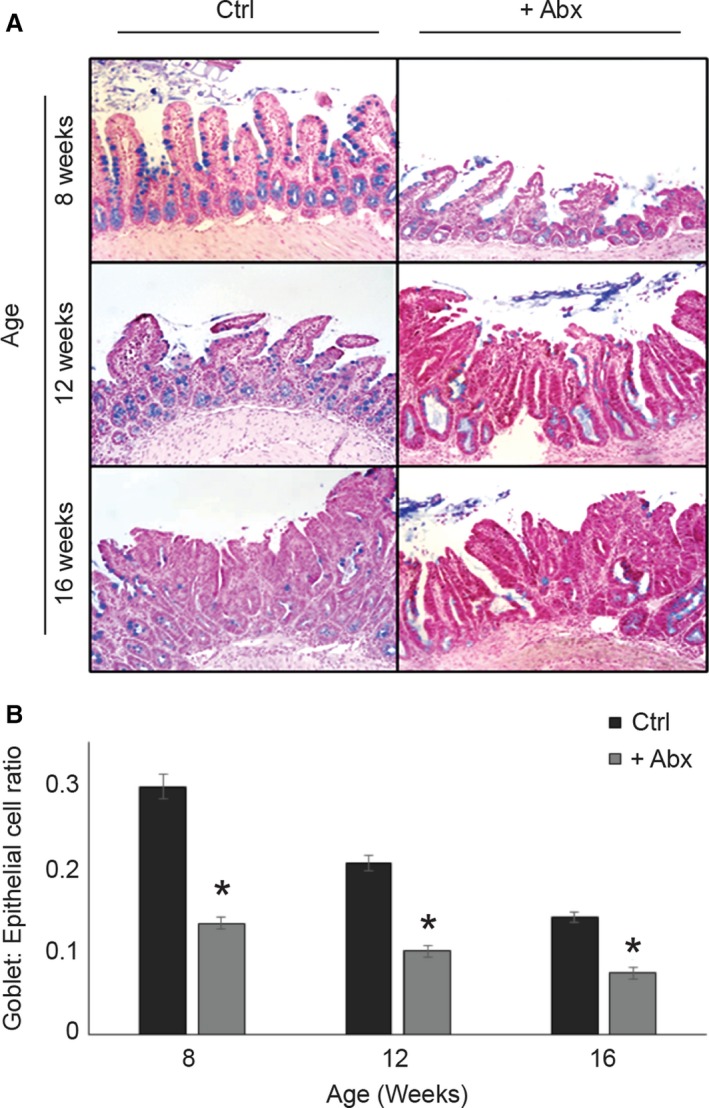
Effect of antibiotic exposure on goblet cell development. (A) Representative Alcian blue stained tissue sections from Ctrl and +Abx mice. Blue and pink staining indicates goblet and epithelial cells, respectively. (B) Graph showing goblet‐to‐epithelial cell ratio per view per group. Two‐way ANOVA was used to determine the significant difference in goblet to epithelial cell ratio between groups. *P < 0.01 Ctrl versus +Abx group.
Effect of antibiotics on the fecal bacteriome
The composition of the fecal bacteriome was compared between control and antibiotic‐administered mice. As shown in Figure 5A, alpha‐diversity did not change significantly with age in either the control or the antibiotic exposed mice, suggesting that Apc Min/+ mice retained a stable microbiome during the time window of 8–16 weeks. However, for all age groups, alpha‐diversity was significantly decreased by antibiotic treatment, as measured by total operational taxonomic units (OTUs), the Chao1 index, or the Shannon index (Fig. 5A). Similar decreases were observed in 8, 12, and 16 weeks.
Figure 5.
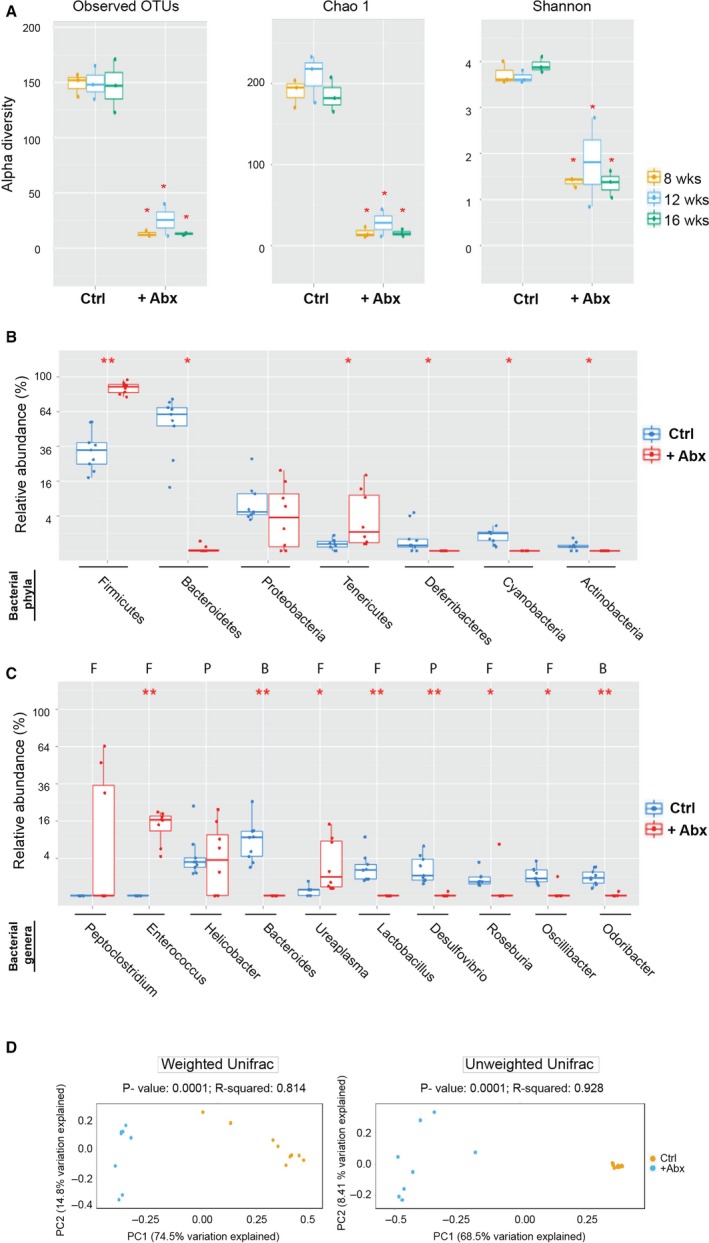
Effect of antibiotic administration on fecal bacteriome. (A) Alpha‐diversity measures in fecal samples. Depletion of fecal bacteriome in +Abx mice was demonstrated using observed OTUs and Chao1 diversity measures and depletion of bacteriome richness was demonstrated using Shannon diversity measures. Comparisons to determine age‐dependent variance were performed using Kruskal–Wallis analyses of variance. P‐values determined from these analyses for OTU, Chao, and Shannon indices were 0.96, 0.29, and 0.29, respectively, for Ctrl mice and 0.5, 0.3, and 0.97, respectively, for +Abx mice. P‐values to assess significance in change of alpha‐diversity indices were determined using Mann–Whitney U‐test, (*) P < 0.05, +Abx versus Ctrl for all age groups. (B) Relative abundance of top seven predominant bacterial phyla in Ctrl and +Abx mice. Statistical significance was assessed by Mann–Whitney U‐test (*) represents P < 0.05, (**) represents P ≤ 0.0001. (C) Relative abundance of top seven bacterial genera in Ctrl and +Abx mice. F, genus belongs to phylum Firmicutes, P, genus belongs to phylum Proteobacter, B, genus belongs to phylum Bacteroidetes. Statistical significance was assessed by Mann–Whitney U‐test (*) represents P < 0.05, (**) represents P < 0.001. As panel A demonstrated that alpha‐diversity was age independent, the 16S rRNA reads from mice of all time‐points were pooled for analysis while comparing Ctrl and +Abx mice in panels B and C. (D) Weighted and unweighted beta‐diversity for Ctrl and +Abx fecal samples. Plots of PCoA based on weighted and unweighted UniFrac distance matrices of microbial communities in fecal samples of Ctrl and +Abx mice. For weighted and unweighted beta‐diversity, PC1 (x‐axis) explained 74.5% and 68.5% of variability, respectively, and PC2 (y‐axis) explained 14.8% and 8.4% of variability, respectively.
As age had no significant effect on alpha‐diversity of either control or treated groups, we pooled the 16S rRNA reads from all the time‐points to determine the predominant phyla within each group. For both control and treated mice, the 16S rRNA reads were assigned to seven phyla, of which Bacteroidetes, Firmicutes, and Proteobacteria constituted were most predominant. As shown in Figure 5B, Bacteroidetes and Firmicutes showed significant shifts in relative abundance upon antibiotic exposure. Abundance for Bacteriodetes decreased from 60% to <5%, while that for Firmicutes increased by nearly threefold.
Alterations at the genus level were also assessed (Fig. 5C). While a significant elevation in abundance of three Firmicutes genera (Enterococcus, Ureaplasma, and Peptoclostridium) was measured in response to antibiotic treatment, several probiotic genera (Bacteroides, Lactobacillus, Desulfovibrio, among others) were nearly eliminated. The shifts in bacterial abundance upon antibiotic administration were also reflected by the beta‐diversity patterns (Fig. 5D), in that significant differences were observed at all ages between the antibiotic‐administered and control groups in both unweighted and weighted UniFrac distance metrics (P = 0.0001).
Overall, the results demonstrate that administration of antibiotics eliminated the majority of bacterial flora, with the most drastic depletion being observed for the phylum Bacteroides and several beneficial genera within Firmicutes.
Discussion
In the current study, we provide direct experimental evidence in support of the notion that long‐term exposure to antibiotics can promote polyp development in the gut of a genetically susceptible mouse strain, as suggested by previous epidemiological reports that linked colorectal carcinogenesis to antibiotic use in humans 36, 55, 56, 57, 58. We emphasize that the significance of our findings expands beyond colorectal cancer, as a number of recent studies have shown that disruption of the innate microbiota by low‐dose antibiotic exposures, even if limited to transient perturbations early in life, can have long‐term metabolic alterations and affect ileal expression of genes involved in immunity 59. Recent reports from Boursi et al. 60. also indicate that antibiotic exposures increase the risk of diabetes, which in turn increases the risk of developing colorectal cancer 61, 62, as well as lung, prostrate, gastric, and breast cancers 63.
An intriguing and novel aspect of the current study is the observation that depletion of the natural bacterial flora upon antibiotic exposure correlates with reduction in mucus‐producing goblet cell numbers, along with an increase in both colon histopathological scores and intestinal crypt‐to‐villus ratios. The functional link between antibiotic‐mediated microbial dysbiosis and the inhibitory effects of antibiotics on development of goblet cells is of particular interest, as these cells are integral to protection against inflammation and polyposis. In all, this is consistent with previous findings suggesting that gut microbiota impact the thickness of the mucus layer 54, which provides nutrition and energy to the intestinal microflora by enabling them to break down and utilize the glycans present in the mucus 64.
Our results imply that the intestinal microbiome has a profound impact on the global physical properties of the gut, which in turn determines the severity of inflammation and cancer. This is illustrated by our ‘cyclical microbiome protection model’ depicted in Figure 6. According to this model, factors such as long‐term antibiotic exposures lead to gut microbial dysbiosis and decrease goblet cell counts. This in turn increases the severity of microbial dysbiosis, eventually leading to inflammation and cancer.
Figure 6.
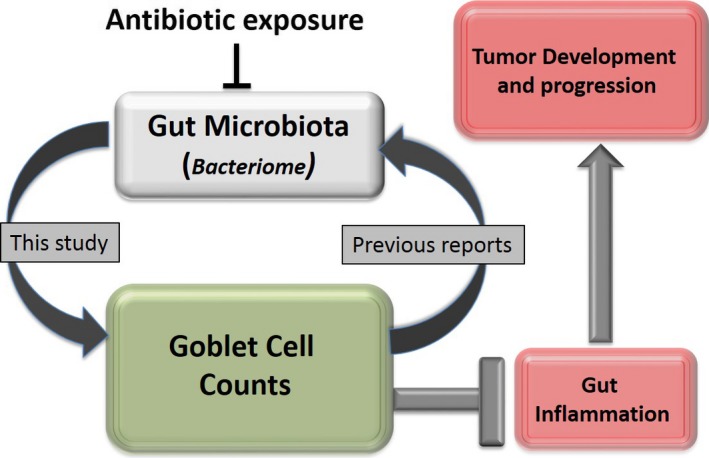
Microbiome‐goblet cell protection model. According to this model, the composition of the natural gut bacterial flora is associated with gut mucosal goblet cell counts. Depletion of bacterial communities reduced goblet counts that could be attributed to either regulation of goblet cell apoptosis or goblet cell development or both. Goblet cells are integral to the maintenance of the mucus layer, which in turn, regulates the composition of the gut microflora (findings from previous studies 64). As mucus layer offer protection against inflammation and tumor progression 44, factors that disrupts this protection cycle will lead to gut inflammation and carcinogenesis.
The choice of the antibiotics for this study was based on their known mechanisms of action in bacterial cells and their minimal nontoxic effects on the host. Neomycin is used against both Gram‐positive and Gram‐negative bacteria. To minimize nephrotoxic effects of the antibiotic, it is prescribed as an oral antibiotic. Its inhibits bacterial growth by binding RNA duplexes 65 and is water soluble with a low toxicity in animals 66. Vancomycin, which inhibits Gram‐positive bacteria 67, 68, is not absorbed by the intestinal mucosa 69 and acts by inhibiting cell wall formation through binding to the terminal D‐alanyl‐D‐alanine moieties of the N‐acetyl glucosamine/N‐acetyl muramic acid peptide 70. Ampicillin is active against both Gram‐positive and Gram‐negative bacteria and is typically nontoxic 71. It penetrates bacterial cell walls and irreversibly inhibits the enzyme transpeptidase that is needed for cell wall synthesis, thereby leading to cell lysis 71. Based on a plethora of evidence on direct immunomodulatory effects of antibiotics 72, we cannot rule out the notion that the antibiotics used in this study, in addition to having antibacterial effects, act directly on the physiology of the animal host. However such alternate possibilities do not undermine the primary conclusions of the current study.
Interestingly, the antibiotic cocktail used currently selectively depleted the genus Bacteroides and some ‘beneficial’ genera such as Lactobacillus and Desulfovibrio. This implicates the possible role of these genera in protection against cancer progression. The increase in larger polyps during cancer progression may be due to selective depletion of these bacterial genera. Alternatively, the dramatic increase in relative abundance of Firmicutes upon antibiotic administration may underlie increased polyposis. Distinguishing between these and other possibilities will require follow‐up studies with gnotobiotic rodent models. Our findings are different from observations described by Hamoya et al. 73., in which low‐dose erythromycin exposure (0.5 mg/mL) reduced polyposis in Apc Min/+ mice. It may be that different antibiotic exposures may result in distinct microbial profiles, which in turn, may lead to variation in disease outcomes.
Our follow‐up studies will investigate the effect of these potential ‘therapeutic microbes’ on cancer development and progression. Other aspects to be investigated in our follow‐up studies are use of different antibiotics, and the timing of antibiotic exposures relative to polyp development. Such studies will provide additional mechanistic information on the association of antibiotic exposure, effects on the gut microbiome, and development of colorectal carcinogenesis.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Representative Hemeatoxylin and eosin (H&E) stained colon tissues belonging to control and +Abx mice at different ages.
Acknowledgments
A pilot project award to A.C. from the Center for Colon Cancer Research (CCCR) at University of South Carolina, Columbia, funded this study. This pilot project was funded by the parent grant # 5P30GM103336‐02 from the National Institutes of Health (NIH) and the National Cancer Institute (NCI) to F.G.B
Cancer Medicine 2018; 7(5):2003–2012
References
- 1. Sundin, J. , Ohman L., and Simren M.. 2017. Understanding the Gut microbiota in inflammatory and functional gastrointestinal diseases. Psychosom. Med. 79:857–867. [DOI] [PubMed] [Google Scholar]
- 2. Rojo, D. , Mendez‐Garcia C., Raczkowska B. A., Bargiela R., Moya A., Ferrer M., et al. 2017. Exploring the human microbiome from multiple perspectives: factors altering its composition and function. FEMS Microbiol. Rev. 41:453–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tropini, C. , Earle K. A., Huang K. C., and Sonnenburg J. L.. 2017. The Gut Microbiome: Connecting Spatial Organization to Function. Cell Host Microbe 21:433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bordalo Tonucci, L. , Dos Santos K. M., De Luces Fortes Ferreira C. L., Ribeiro S. M., De Oliveira L. L., and Martino H. S.. 2017. Gut microbiota and probiotics: focus on diabetes mellitus. Crit. Rev. Food Sci. Nutr. 57:2296–2309. [DOI] [PubMed] [Google Scholar]
- 5. Altuntas, Y. , and Batman A.. 2017. Microbiota and metabolic syndrome. Turk. Kardiyol. Dern. Ars. 45:286–296. [DOI] [PubMed] [Google Scholar]
- 6. Jiang, C. , Li G., Huang P., Liu Z., and Zhao B.. 2017. The Gut Microbiota and Alzheimer's Disease. J. Alzheimers Dis. 58:1–15. [DOI] [PubMed] [Google Scholar]
- 7. Katz‐Agranov, N. , and Zandman‐Goddard G.. 2017. The microbiome and systemic lupus erythematosus. Immunol. Res. 65:432–437. [DOI] [PubMed] [Google Scholar]
- 8. Pevsner‐Fischer, M. , Blacher E., Tatirovsky E., Ben‐Dov I. Z., and Elinav E.. 2017. The gut microbiome and hypertension. Curr. Opin. Nephrol. Hypertens. 26:1–8. [DOI] [PubMed] [Google Scholar]
- 9. Rieder, R. , Wisniewski P. J., Alderman B. L., and Campbell S. C.. 2017. Microbes and mental health: a review. Brain Behav. Immun. 66:9–17. [DOI] [PubMed] [Google Scholar]
- 10. Serino, M. , Nicolas S., Trabelsi M. S., Burcelin R., and Blasco‐Baque V.. 2017. Young microbes for adult obesity. Pediatr. Obes. 12:e28–e32. [DOI] [PubMed] [Google Scholar]
- 11. Signoretti, M. , Roggiolani R., Stornello C., Delle Fave G., and Capurso G.. 2017. Gut microbiota and pancreatic diseases. Minerva Gastroenterol. Dietol. 63:399–410. [DOI] [PubMed] [Google Scholar]
- 12. Tang, W. H. , Kitai T., and Hazen S. L.. 2017. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 120:1183–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vaiserman, A. M. , Koliada A. K., and Marotta F.. 2017. Gut microbiota: a player in aging and a target for anti‐aging intervention. Ageing Res. Rev. 35:36–45. [DOI] [PubMed] [Google Scholar]
- 14. Dzutsev, A. , Badger J. H., Perez‐Chanona E., Roy S., Salcedo R., Smith C. K., et al. 2017. Microbes and Cancer. Annu. Rev. Immunol. 35:199–228. [DOI] [PubMed] [Google Scholar]
- 15. Rajagopala, S. V. , Vashee S., Oldfield L. M., Suzuki Y., Venter J. C., Telenti A., et al. 2017. The human microbiome and cancer. Cancer Prev. Res. (Phila.) 10:226–234. [DOI] [PubMed] [Google Scholar]
- 16. Gao, R. , Gao Z., Huang L., and Qin H.. 2017. Gut microbiota and colorectal cancer. Eur. J. Clin. Microbiol. Infect. Dis. 36:757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Siegel, R. L. , Miller K. D., Fedewa S. A., Ahnen D. J., Meester R. G., Barzi A., et al. 2017. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 67:177–193. [DOI] [PubMed] [Google Scholar]
- 18. Weisburger, J. , Reddy B. S., Narisawa T., and Wynder E.. 1975. Germ‐free status and colon tumor induction by N‐methyl‐N'‐nitro‐N‐nitrosoguanidine. Proc. Soc. Exp. Biol. Med. 148:1119–1121. [DOI] [PubMed] [Google Scholar]
- 19. Li, Y. , Kundu P., Seow S. W., de Matos C. T., Aronsson L., Chin K. C., et al. 2012. Gut microbiota accelerate tumor growth via c‐jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis 33:1231–1238. [DOI] [PubMed] [Google Scholar]
- 20. Hu, B. , Elinav E., Huber S., Strowig T., Hao L., Hafemann A., et al. 2013. Microbiota‐induced activation of epithelial IL‐6 signaling links inflammasome‐driven inflammation with transmissible cancer. Proc. Natl. Acad. Sci. U S A. 110:9862–9867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zackular, J. P. , Baxter N. T., Iverson K. D., Sadler W. D., Petrosino J. F., Chen G. Y., et al. 2013. The gut microbiome modulates colon tumorigenesis. MBio 4:e00692–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sanapareddy, N. , Legge R. M., Jovov B., McCoy A., Burcal L., Araujo‐Perez F., et al. 2012. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J. 6:1858–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sobhani, I. , Tap J., Roudot‐Thoraval F., Roperch J. P., Letulle S., Langella P., et al. 2011. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 6:e16393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang, T. , Cai G., Qiu Y., Fei N., Zhang M., Pang X., et al. 2012. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 6:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu, N. , Yang X., Zhang R., Li J., Xiao X., Hu Y., et al. 2013. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 66:462–470. [DOI] [PubMed] [Google Scholar]
- 26. Wu, Y. J. , Wu C. T., Zhang X. B., Ou W. T., and Huang P.. 2012. Clinical study of different bowel preparations on changes of gut flora in patients undergoing colorectal resection. Zhonghua. Wei. Chang. Wai. Ke. Za. Zhi. 15:574–577. [PubMed] [Google Scholar]
- 27. Hicks, L. A. , Taylor T. H. Jr, and Hunkler R. J. U. S.. 2013. outpatient antibiotic prescribing, 2010. N. Engl. J. Med. 368:1461–1462. [DOI] [PubMed] [Google Scholar]
- 28. Lee, G. C. , Reveles K. R., Attridge R. T., Lawson K. A., Mansi I. A., Lewis J. S., et al. 2014. Outpatient antibiotic prescribing in the United States: 2000 to 2010. BMC Med. 12:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marra, F. , George D., Chong M., Sutherland S., and Patrick D. M.. 2016. Antibiotic prescribing by dentists has increased: why? J. Am. Dent. Assoc. 147:320–327. [DOI] [PubMed] [Google Scholar]
- 30. Tamma, P. D. , and Cosgrove S. E.. 2016. Addressing the appropriateness of outpatient antibiotic prescribing in the United States: an important first step. JAMA 315:1839–1841. [DOI] [PubMed] [Google Scholar]
- 31. Vaz, L. E. , Kleinman K. P., Raebel M. A., Nordin J. D., Lakoma M. D., Dutta‐Linn M. M., et al. 2014. Recent trends in outpatient antibiotic use in children. Pediatrics 133:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ponziani, F. R. , Scaldaferri F., Petito V., Sterbini F. P., Pecere S., Lopetuso L. R., et al. 2016. The role of antibiotics in gut microbiota modulation: the eubiotic effects of rifaximin. Dig. Dis. 34:269–278. [DOI] [PubMed] [Google Scholar]
- 33. Puiman, P. , Stoll B., Molbak L., de Bruijn A., Schierbeek H., Boye M., et al. 2013. Modulation of the gut microbiota with antibiotic treatment suppresses whole body urea production in neonatal pigs. Am. J. Physiol. Gastrointest. Liver Physiol. 304:G300–G310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kerman, D. H. , and Deshpande A. R.. 2014. Gut microbiota and inflammatory bowel disease: the role of antibiotics in disease management. Postgrad. Med. 126:7–19. [DOI] [PubMed] [Google Scholar]
- 35. Rahmoune, H. , Boutrid N., Amrane M., and Bioud B.. 2017. Antibiotics, intestinal dysbiosis and risk of celiac disease. Dig. Liver Dis. 49:106. [DOI] [PubMed] [Google Scholar]
- 36. Cao, Y. , Wu K., Mehta R., Drew D. A., Song M., Lochhead P., et al. 2018. Long‐term use of antibiotics and risk of colorectal adenoma. Gut 67:672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Attur, M. G. , Patel R. N., Patel P. D., Abramson S. B., and Amin A. R.. 1999. Tetracycline up‐regulates COX‐2 expression and prostaglandin E2 production independent of its effect on nitric oxide. J. Immunol. 162:3160–3167. [PubMed] [Google Scholar]
- 38. Dixon, D. A. , Blanco F. F., Bruno A., and Patrignani P.. 2013. Mechanistic aspects of COX‐2 expression in colorectal neoplasia. Recent Results Cancer Res. 191:7–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moser, A. R. , Pitot H. C., and Dove W. F.. 1990. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247:322–324. [DOI] [PubMed] [Google Scholar]
- 40. Tong, Y. , Yang W., and Koeffler H. P.. 2011. Mouse models of colorectal cancer. Chin J. Cancer 30:450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oh, J. E. , Kim B. C., Chang D. H., Kwon M., Lee S. Y., Kang D., et al. 2016. Dysbiosis‐induced IL‐33 contributes to impaired antiviral immunity in the genital mucosa. Proc. Natl. Acad. Sci. U S A. 113:E762–E771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Russell, S. L. , Gold M. J., Hartmann M., Willing B. P., Thorson L., Wlodarska M., et al. 2012. Early life antibiotic‐driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 13:440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Strzepa, A. , Majewska‐Szczepanik M., Kowalczyk P., Wozniak D., Motyl S., and Szczepanik M.. 2016. Oral treatment with enrofloxacin early in life promotes Th2‐mediated immune response in mice. Pharmacol. Rep. 68:44–50. [DOI] [PubMed] [Google Scholar]
- 44. Saxena, A. , Fayad R., Kaur K., Truman S., Greer J., Carson J. A., et al. 2017. Dietary selenium protects adiponectin knockout mice against chronic inflammation induced colon cancer. Cancer Biol. Ther. 18:257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saxena, A. , Chumanevich A., Fletcher E., Larsen B., Lattwein K., Kaur K., et al. 2012. Adiponectin deficiency: role in chronic inflammation induced colon cancer. Biochim. Biophys. Acta 1822:527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aagaard, K. , Petrosino J., Keitel W., Watson M., Katancik J., Garcia N., et al. 2013. The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J. 27:1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Berry, D. , Schwab C., Milinovich G., Reichert J., Mahfoudh K. B., Decker T., et al. 2012. Phylotype‐level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 6:2091–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Seedorf, H. , Kittelmann S., Henderson G., and Janssen P. H.. 2014. RIM‐DB: a taxonomic framework for community structure analysis of methanogenic archaea from the rumen and other intestinal environments. PeerJ 2:e494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Caporaso, J. G. , Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. 2010. QIIME allows analysis of high‐throughput community sequencing data. Nat. Methods 7:335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Haas, B. J. , Gevers D., Earl A. M., Feldgarden M., Ward D. V., Giannoukos G., et al. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454‐pyrosequenced PCR amplicons. Genome Res. 21:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McDonald, D. , Price M. N., Goodrich J., Nawrocki E. P., DeSantis T. Z., Probst A., et al. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6:610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Knights, D. , Kuczynski J., Koren O., Ley R. E., Field D., Knight R., et al. 2011. Supervised classification of microbiota mitigates mislabeling errors. ISME J. 5:570–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. He, Q. , Li X., Liu C., Su L., Xia Z., Li X., et al. 2016. Dysbiosis of the fecal microbiota in the TNBS‐induced Crohn's disease mouse model. Appl. Microbiol. Biotechnol. 100:4485–4494. [DOI] [PubMed] [Google Scholar]
- 54. Jakobsson, H. E. , Rodriguez‐Pineiro A. M., Schutte A., Ermund A., Boysen P., Bemark M., et al. 2015. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 16:164–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Boursi, B. , Haynes K., Mamtani R., and Yang Y. X.. 2015. Impact of antibiotic exposure on the risk of colorectal cancer. Pharmacoepidemiol. Drug Saf. 24:534–542. [DOI] [PubMed] [Google Scholar]
- 56. Dik, V. K. , van Oijen M. G., Smeets H. M., and Siersema P. D.. 2016. Frequent use of antibiotics is associated with colorectal cancer risk: results of a nested case‐control study. Dig. Dis. Sci. 61:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kilkkinen, A. , Rissanen H., Klaukka T., Pukkala E., Heliövaara M., Huovinen P., et al. 2008. Antibiotic use predicts an increased risk of cancer. Int. J. Cancer 123:2152–2155. [DOI] [PubMed] [Google Scholar]
- 58. Wang, J. L. , Chang C. H., Lin J. W., Wu L. C., Chuang L. M., and Lai M. S.. 2014. Infection, antibiotic therapy and risk of colorectal cancer: a nationwide nested case‐control study in patients with Type 2 diabetes mellitus. Int. J. Cancer 135:956–967. [DOI] [PubMed] [Google Scholar]
- 59. Cox, L. M. , Yamanishi S., Sohn J., Alekseyenko A. V., Leung J. M., Cho I., et al. 2014. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158:705–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Boursi, B. , Mamtani R., Haynes K., and Yang Y. X.. 2015. The effect of past antibiotic exposure on diabetes risk. Eur. J. Endocrinol. 172:639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de Kort, S. , Masclee A. A. M., Sanduleanu S., Weijenberg M. P., van Herk‐Sukel M. P., Oldenhof N. J., et al. 2017. Higher risk of colorectal cancer in patients with newly diagnosed diabetes mellitus before the age of colorectal cancer screening initiation. Sci. Rep. 7:46527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhu, B. , Wu X., Wu B., Pei D., Zhang L., and Wei L.. 2017. The relationship between diabetes and colorectal cancer prognosis: a meta‐analysis based on the cohort studies. PLoS ONE 12:e0176068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Boursi, B. , Mamtani R., Haynes K., and Yang Y. X.. 2015. Recurrent antibiotic exposure may promote cancer formation–Another step in understanding the role of the human microbiota? Eur. J. Cancer 51:2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kaur, K. , Saxena A., Larsen B., Truman S., Biyani N., Fletcher E., et al. 2015. Mucus mediated protection against acute colitis in adiponectin deficient mice. J. Inflamm. (Lond.) 12:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kaul, M. , and Pilch D. S.. 2002. Thermodynamics of aminoglycoside‐rRNA recognition: the binding of neomycin‐class aminoglycosides to the A site of 16S rRNA. Biochemistry 41:7695–7706. [DOI] [PubMed] [Google Scholar]
- 66. Waksman, S. A. , and Lechevalier H. A.. 1949. Neomycin, a New Antibiotic Active against Streptomycin‐Resistant Bacteria, including Tuberculosis Organisms. Science 109:305–307. [DOI] [PubMed] [Google Scholar]
- 67. Small, P. M. , and Chambers H. F.. 1990. Vancomycin for Staphylococcus aureus endocarditis in intravenous drug users. Antimicrob. Agents Chemother. 34:1227–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gonzalez, C. , Rubio M., Romero‐Vivas J., Gonzalez M., and Picazo J. J.. 1999. Bacteremic pneumonia due to Staphylococcus aureus: A comparison of disease caused by methicillin‐resistant and methicillin‐susceptible organisms. Clin. Infect. Dis. 29:1171–1177. [DOI] [PubMed] [Google Scholar]
- 69. Van Bambeke, F. 2006. Glycopeptides and glycodepsipeptides in clinical development: a comparative review of their antibacterial spectrum, pharmacokinetics and clinical efficacy. Curr. Opin. Investig. Drugs 7:740–749. [PubMed] [Google Scholar]
- 70. Chakraborty, S. P. , Sahu S. K., Mahapatra S. K., Santra S., Bal M., Roy S., et al. 2010. Nanoconjugated vancomycin: new opportunities for the development of anti‐VRSA agents. Nanotechnology 21:105103. [DOI] [PubMed] [Google Scholar]
- 71. Stemp, G. , Pascoe S., and Gatehouse D.. 1989. In vitro and in vivo cytogenetic studies of three beta‐lactam antibiotics (penicillin VK, ampicillin and carbenicillin). Mutagenesis 4:439–445. [DOI] [PubMed] [Google Scholar]
- 72. Buret, A. G. 2010. Immuno‐modulation and anti‐inflammatory benefits of antibiotics: the example of tilmicosin. Can. J. Vet. Res. 74:1–10. [PMC free article] [PubMed] [Google Scholar]
- 73. Hamoya, T. , Miyamoto S., Tomono S., Fujii G., Nakanishi R., Komiya M., et al. 2017. Chemopreventive effects of a low‐side‐effect antibiotic drug, erythromycin, on mouse intestinal tumors. J. Clin. Biochem. Nutr. 60:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative Hemeatoxylin and eosin (H&E) stained colon tissues belonging to control and +Abx mice at different ages.