

Abstract
Transfection and transduction using lentivirus has gained attention in biomedical research. To date, how to reach the maximum transfection and viral transduction efficiency is still challenging. Here we compared the transfection and viral transduction efficiency using commercially available transfection reagents including FuGENE 6, Lipofectamine 2000 and Lipofectamine 3000 in different cell lines and primary cultured cells. Enhanced green fluorescent protein (EGFP) was clearly seen in Eppendorf tubes from harvested cells using Lipofectamine 3000 without using a microscope and UV activation. Strong expression of EGFP was observed in HEK293 cells, mouse primary cortical neurons and human umbilical vein endothelial cells (HUVECs) using confocal microscopy. Western blot showed the strongest EGFP expression using cell lysates from Lipofectamine 3000 transfected HEK293 cells and transduced HUVECs compared with Lipofectamine 2000 or FuGENE 6 reagents. Using Cx43 shRNA lentivirus combined with Lipofectamine 3000 transfection reagent, we can achieve about 90% Cx43 knockdown efficacy in HUVECs. Therefore, our results suggest that a much higher transfection and viral transduction efficiency can be attained by using Lipofectamine 3000 transfection reagent.
Keywords: Transfection, viral transduction, Lipofectamine 3000, EGFP, connexin43
Introduction
Manipulation of gene expression holds much promise in the field of biomedical research. Transfection of exogenous gene in specific plasmids to various cells is considered as a major technique to manipulate gene expression. Various transfection reagents or devices including chemical and physical methods have been used to deliver gene transfer such as calcium phosphates, electroporation, nucleofection, lipids, polyethylenimine, nanoparticles carrying the genes [1-18]. Virus mediated transfection, also known as transduction, is also widely used in various in vitro and in vivo conditions [3].
Traditionally, many chemical methods have been successfully developed and used to deliver genes into cells, including cationic polymer, calcium phosphate and cationic lipid etc. [1-4]. Calcium phosphates has been used widely to enable the gene delivery to in vitro cells for more than 40 years since its first development in 1973. However, there has been a challenge in using calcium phosphates to transfer exogenous gene to cells, because of its relatively low transfection efficiency [2]. Physical methods such as electroporation and nucleofection techniques appeared to overcome the lower transfection efficiency by calcium phosphate transfection, but it needs to use a device to fulfil its task. In addition the electroporation or the necleofection device needs to be adjusted to reduce the substantial cell death [5,7,13,19,21]. Various nanoparticle-based gene delivery techniques, such as inorganic nanoparticles, polymer based nanoparticles, lipid based nanoparticles, and hybrid nanoparticles, look promising, but there are still disadvantages related to their efficiency, stability and toxic effects [22].
Another chemical method is using lipids to deliver nucleic acids into cells, such as Lipofectamine 2000 and Lipofectamine 3000 reagents, which are easy to use in many different cell types and generally achieve relatively high transfection efficiency. Therefore, they are widely used in the scientific field. The initial step for viral transduction is to produce the virus, which involves transfection of various viral vectors (adenovirus, lentivirus or adeno-associated virus or AAV vectors) [20,23] and packaging vectors into virus packaging cells such as HEK293 cells. Therefore, achieving high transfection efficiency is a key step to generate high titer viruses.
In this study, we evaluated the transfection and viral transduction efficiency using commercially available transfection reagents including FuGENE 6 transfection reagent, Lipofectamine 2000 and Lipofectamine 3000 reagents in different cell lines and primary cultured cells.
Materials and methods
Antibodies used included a rabbit polyclonal anti-GFP antibody (NB600-308), a rabbit polyclonal anti-beta actin antibody (NB600-503), which were purchased from Novus Biologicals (Littleton, Co, USA) and a rabbit polyclonal anti-Cx43 (Cat. No. 71-0700), which was obtained from ThermoFisher (Rockford, Il, USA). Transfection reagents lepofectamine 3000 and 2000 reagents were obtained from ThermoFisher, while FuGENE 6 was acquired from Promega (Madison, WI, USA).
Cell culture
HEK293 and human umbilical vein endothelial cells (HUVECs) were obtained from ATCC (American Type Culture Collection, Manassas, Virginia, USA).
Mouse cortical neuron culture. The isolation and culture of mouse cortical neurons was based on a previously described method [24]. The use of mice for isolating and culturing neuronal cell was approved by the Institutional Animal Care and Use Committee of Shanghai Tongji University. Embryonic day 16 Swiss mice were anesthetized with halothane followed by cervical dislocation. Fetal brains were rapidly removed and placed in an ice-cold Ca2+ and Mg2+-free Hank’s solution. Cerebral cortices were dissected and enzyme digested using 0.05% trypsin-EDTA for 10 min at 37°C, followed by trituration with fire-polished glass pipettes, and plated on poly-l-ornithine-coated culture dishes (35 mm in diameter), at a density of 1×106 cells per dish. Neurons were cultured with Neurobasal medium (ThermoFisher) supplemented with B27 and maintained at 37°C in a humidified 5% CO2 atmosphere incubator. Cultures were fed every three days and used for viral transduction 10 days after plating.
Plasmids extraction and purification
Various plasmids were transformed to DH5α competent cells and amplified in LB medium containing specific antibiotic overnight using a 37°C shaker with speed at 225 rpm. Then the bacteria was harvested and plasmids were extracted and purified using a Qiagen plasmid extraction and purification kit according to the procedure provided by the manufacture.
Transient transfection
HEK293 cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum with 1% antibiotics and maintained in a tissue culture incubator at 37°C with 5% CO2. After 24 hours passaging in a 75 ml flask and reaching about 70% confluence, cells were transfected with FuGene 6 transfection reagent, Lipofectamine 2000 and Lipofectamine 3000, respectively according to manufacturer’s instructions (Invitrogen or Promega). Expression plasmids used for transfections included plenti CMV GFP DEST, lentivirus Cx43 shRNA, and packaging vectors including pLP1, pLP2 and pLP/VSVG plasmids. Briefly, 5.8 ug pLP1, 2.8 ug pLP2, 3.8 ug pLP/VSVG, 6.0 ug plenty CMV GFP DEST vector or 4.0 ug Cx43 shRNA plasmid were mixed with 30 ul Lipofectamine 3000 and 30 ul P3000 in 1.0 ml Opti MEM reduced serum medium (ThermoFisher). For using Lipofectamine 2000 reagent, the above plasmids were mixed with 30 ul Lipofectamine 2000 in 1.0 ml Opti MEM reduced serum medium (ThermoFisher). For using FuGENE 6 reagent, the above plasmids were mixed with 30 ul FuGENE 6 transfection reagent. After 15 mins incubation at room temperature, the mixture was added to the HEK293 cells in a 75 ml flask containing 10 ml DMEM medium with 10% fetal bovine serum and 1% antibiotics. After 48 hours of transfection, the cell culture medium was harvested, centrifuged for 5 mins with 1000g centrifugal force and filtered using a 0.45 um filter, and the supernatant were then equally distributed to HVEC cells (plenti CMV GFP DEST vector and lentivirus Cx43 shRNA plasmid, respectively) and mouse cortical neurons (plenti CMV GFP DEST vector). After 3 days of viral transduction, cells were subjected to GFP monitoring, western blotting or immunofluorescence labeling.
Cx43 shRNA was obtained from Santa Cruz Biotechnology (Dallas, TX, USA), the shRNA sequences targeting human Cx43 were consisted of pools of four different sequences, including: A: 5’-GATCCCTGCGAACCTACATCATCATTCAAGAGATGATGATGTAGGTTCGCAGTTTTT-3’; B: 5’-GATCCGAACCTACATCATCAGTATTTCAAGAGAATACTGATGATGTAGGTTCTTTTT-3’; C: 5’-GATCCGTTGGGATGTCACTTAACATTCAAGAGATGTTAAGTGACATCCCAACTTTTT-3’; D: 5’-GATCCCCTACTTAATACACAGTAATTCAAGAGATTACTGTGTATTAAGTAGGTTTTT-3’. Scrambled shRNA plasmids were used as control.
Immunofluorescence labeling and GFP detection
The procedure for immunofluorescence labeling was the same as we previously described (25-27). Briefly, After viral transduction, HUVECs was immunolabelled with polyclonal anti-Cx43 at a concentration of 1 μg/ml for 24 h at 4°C, with antibodies diluted in 50 mM Tris-HCl, pH 7.4, containing 1.5% sodium chloride and 0.3% Triton X-100 (TBSTr). The cells were then washed for 1 h in TBSTr, and incubated for 1 h at room temperature simultaneously using Alexa Flour 488-conjugated goat anti-rabbit IgG secondary antibody diluted at 1:1000 (Molecular Probes, Eugene, Oregon. After secondary antibody incubations, cells were washed in TBSTr for 20 min, then in 50 mM Tris-HCl buffer, pH 7.4 for 30 min, covered with prolong antifade medium and coverslipped. Control procedures included omission of one of the primary antibodies with inclusion of each of the secondary antibodies. Confocal immunofluorescence microscopy was conducted on an Olympus confocal microscope (Fluoview FV1000; Olympus, Waltham, MA, USA). For checking GFP expression, after two days of transfection using plenti CMV GFP DEST plasmid or three days after viral transduction, GFP fluorescence from various cells in the flasks or six-wells were monitored with the same lower power setting using 4× or 10× objective lens.
Western blot: For preparation of cell lysates, after 2 days of transient transfection or 3 days of viral transduction, cultured cells were rinsed with PBS, lysed in ice-cold immunoprecipitation (IP) buffer containing 20 mM Tris-HCl, pH 8.0, 140 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EGTA, 1.5 mM MgCl2, 1 mM dithiothreitol, 1 mM phenylmethylsulphonyl fluoride, and 5 μg/mL each of the protease inhibitors leupeptin, pepstatin A and aprotinin, and then briefly sonicated, centrifuged and resulting supernatants used for western blot analysis [25-27]. The protein concentration in all samples was determined using a microvolume Nanodrop 2000 spectrophotometer (Thermfisher, Rockford, IL, USA). For immunoblotting, 10 ug or 1 ug total protein per lane in loading buffer were separated by 12.5% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), and proteins were then transferred to polyvinylidene difluoride (PVDF) membranes. Following transfer, membranes were blocked for 60 min in TBS-Tw (20 mM Tris pH 7.4, 150 mM NaCl, and 0.2% Tween-20) containing 5% skim milk powder and then incubated overnight at 4°C with primary antibody (for GFP antibody, using 1:10000 dilution; for Cx43 antibody and beta actin antibody, using 1:1000 dilution) in TBS-Tw containing 1% skim milk. After washing in TBS-Tw, blots were incubated with horse radish peroxidase-conjugated secondary antibodies and reactive bands revealed by chemiluminescence (Fisher Scientific, Rockford, IL, USA). Alternatively, PVDF membranes were incubated with the appropriate species secondary antibody conjugated to either IRDye800 or IRDye700 (Rockland Immunochemicals, Gilbert, PA, USA), then monitored using the Odyssey IR imaging system (Licor, Lincoln, NE, USA).
Statistics analysis: Graphpad Prism software and student’s t-test was used to determine the differences in protein expression level under different transfection conditions.
Results
High level of EGFP is detected in HEK293 cells transfected with Lipofectamine 3000 reagent
HEK293 cells were transiently transfected with lentivirus expression vector plenti-CMV-GFP-DEST separately using Lipofectamine 3000, 2000 or FuGENE 6 reagents. After two days of transfection, the EGFP florescence in living cells were monitored using an Olympus microscope. The results showed that robust EGFP is detected in almost every cell transfected using Lipofectamine 3000 reagent (Figure 1A), while using the same microscopic condition with lower power at 4× objective lens, HEK293 cells transfected with Lipofectamine 2000 (Figure 1B) or FuGENE 6 (Figure 1C) reagents showed less EGFP expressing cells.
Figure 1.
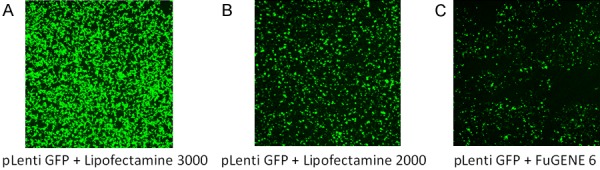
Comparison of transfection efficiency after transiently transfected lentivirus expression vector plenti CMV GFP DEST GFP into HEK293 cells using Lipofectamine 3000, 2000 or FuGENE 6 reagents. Two days after transfection, the GFP in living cells was monitored using the same lower power setting with 4× objective lens in an Olympus confocal microscope. It showed the highest GFP expression using Lipofectamine 3000 transfection reagent (A) as compared with using Lipofectamine 2000 (B) or FuGENE 6 (C) transfection reagents.
High level of EGFP is detected in HUVECs after viral transduction with lentivirus produced from medium of HEK293 cells utilizing Lipofectamine 3000 transfection reagent
HUVECs were transduced with lentivirus produced from HEK293 cells, which were transiently transfected with expression vector plenti-CMV-GFP-DEST for two days using Lipofectamine 3000, 2000, or FuGENE 6 transfection reagents, respectively. After three days of virus transduction, the EGFP florescence in HUVEC living cells were monitored using an Olympus microscope. The results showed that robust EGFP is detected in almost every cell with virus generated using Lipofectamine 3000 reagent (Figure 2A), while using the same microscopic condition with lower power setting utilizing 10× objective lens, HUVECs transduced using lentivirus produced with Lipofectamine 2000 (Figure 2B) or FuGENE 6 (Figure 2C) reagents showed less EGFP expressing cells.
Figure 2.
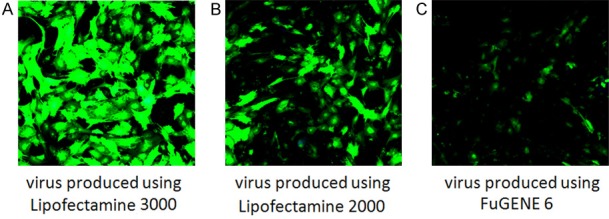
Comparison of viral transduction efficiency in HUVECs using lentivirus generated from HEK293 cells utilizing Lipofectamine 3000, 2000 or FuGENE 6 reagents. Three days after viral transduction, the GFP in living cells was monitored using the same lower power setting with 10× objective lens in an Olympus confocal microscope. It showed the highest GFP expression using lentivirus generated with Lipofectamine 3000 transfection reagent (A) as compared with using Lipofectamine 2000 (B) or FuGENE 6 (C) transfection reagents.
High level of EGFP is detected in cortical neurons after viral transduction with lentivirus generated from medium of HEK293 cells using Lipofectamine 3000 transfection reagent
Mouse cortical neurons at p10 stage were transduced with lentivirus produced from HEK293 cells, which were transiently transfected with expression vector plenti-CMV-GFP-DEST for two days using Lipofectamine 3000, 2000, or FuGENE 6 transfection reagents, respectively. After three days of virus transfduction, the EGFP florescence in cortical neurons was monitored using an Olympus microscope. The results showed that robust EGFP is detected in almost every cell with virus generated using using Lipofectamine 3000 reagent (Figure 3A), while using the same microscopic condition with lower power utilizing 10× objective lens, HUVECs transduced using lentivirus produced with Lipofectamine 2000 (Figure 3B) or FuGENE 6 (Figure 3C) reagents showed less EGFP expressing cells and most substantially, it appears that EGFP is only detectable at the soma of some cortical neurons.
Figure 3.
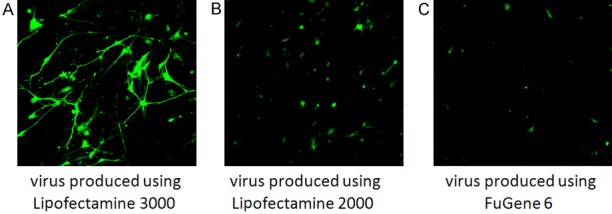
Comparison of viral transduction efficiency in mouse cortical neurons using lentivirus generated from HEK293 cells utilizing Lipofectamine 3000, 2000 or FuGENE 6 reagents. Three days after viral transduction, the GFP in living cells was monitored using the same lower power setting with 10× objective lens in an Olympus confocal microscope. It showed the highest GFP expression using lentivirus generated with Lipofectamine 3000 transfection reagent (A) as compared with using Lipofectamine 2000 (B) or FuGENE 6 (C) transfection reagents.
EGFP is clearly seen without using a microscope or without an UV stimulation condition
After two days of transient transfection using different transfection reagents, or three days of viral transduction, some of HEK293 cells and HUVECs were harvested in PBS buffer. Then, unexpectedly, even without using a microscope or without an UV stimulation, we can clearly see strong EGFP in tubes harvested from HEK293 cells transfected using Lipofectamine 3000 reagent (Figure 4A, tube 1) or from HUVECs with lentivirus generated using Lipofectamine 3000 reagent (Figure 4B, tube 1). However, we can not see any EGFP clearly in tubes harvested from HEK293 cells transiently transfected with lipofectaime 2000 (Figure 4A, tube 2), or with FuGENE 6 reagents (Figure 4A, tube 3) or non-transfected HEK293 cells (Figure 4A, tube 4). Similarly, we can not see any EGFP in tubes harvested from HEK293 cells transduced using virus produced with lipofectaime 2000 (Figure 4B, tube 2), or with FuGENE 6 reagents (Figure 4B, tube 3) or in tube harvested with non-transduced HUVECs (Figure 4B, tube 4).
Figure 4.

Without using a fluorescence microscope and without UV activation, GFP was clearly seen in harvested cells after 2 days of transfection in HEK293 cells using Lipofectamine 3000 or three days of viral transduction in HUVECs using lentivirus generated from HEK293 cells utilizing Lipofectamine 3000 reagent. Two days after transfection in HEK293 cells using Lipofectamine 3000 or three days after viral transduction in HUVECs with lentivirus generated from HEK293 cells utilizing Lipofectamine 3000 reagent, the cells were harvested, and GFP was clearly in these harvested HEK293 cells (A, tube 1) and HUVECs (B, tube 1) even without using a confocal microscope or without using UV activation. On the contrary, we can not see any obvious GFP fluorescence under the same condition but using Lipofectamine 2000 (A and B, tube 2), or FuGENE 6 (A and B, tube 3) or non-transfection or non-transduction controls (A and B, tube 4).
Western blot showing high level of GFP expression in cortical neuron lysates after viral transduction using Lipofectamine 3000 reagent
We first characterized anti-GFP antibody using cell lysates from HUVECs after three days of viral transduction with virus generated using Lipofectamine 3000 reagent. As shown in Figure 5D, lane 2, after SDS-PAGE, robust GFP was detected using 10 ug of total protein loading, however, there is absent of GFP detection using cell lysates without viral transduction (Figure 5D, lane 1). Therefore, we adjusted our total protein loading to 1 ug. As shown in Figure 5A, after SDS-PAGE, a single protein band at around 25 kDa is clearly seen using cell lysates from virus transduced mouse cortical neurons (Figure 5A, upper panel, lane 1), but the band is absent in cell lysates from mouse cortical neurons without undertaking viral transduction or using supernatant of lentivirus only (Figure 5A, upper panel, lane 2 and 3). The same membrane was striped and reprobed with anti-beta actin antibody which showed equal beta-actin detection (Figure 5A, lower panel, lane 1 and 2). As expected, there was no beta-actin band detection using supernatant containing lentivirus only (Figure 5A, lower panel, lane 3).
Figure 5.
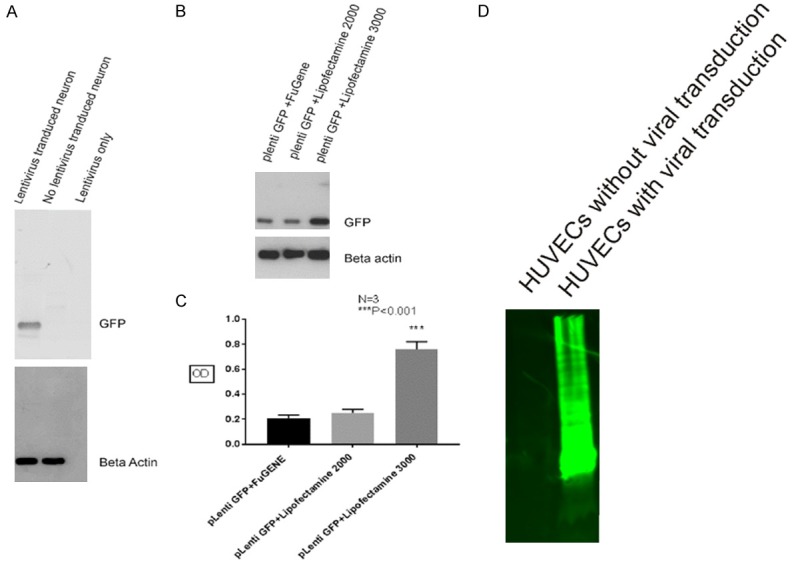
Western blot showing high level of GFP expression in cortical neuron cell lysates after viral transduction using Lipofectamine 3000 reagent. Using cell lysates from HUVECs after three days of viral transduction with virus generated utilizing Lipofectamine 3000 reagent, western blot showed a strong GFP band migrating from the top of the gel to the bottom after using 10 ug of total protein loading, however, there is absent of GFP detection using cell lysates without viral transduction (D). After SDS-PAGE using 1 ug protein loading, a single protein band migrating around 25 kDa is clearly seen using cell lysates from virus transduced mouse cortical neurons (A, upper panel, lane 1), but the band is absent using cell lysates from mouse cortical neurons without undertaking viral transduction or using supernatant of lentivirus only (A, upper panel, lane 2 and 3), the same membrane was striped and reprobed with anti-beta actin antibody showed equal beta-actin detection (A, lower panel, lane 1 and 2), but there was no beta-actin band detection using supernatant containing lentivirus only (A, lower panel, lane 3). Western blot also showed highest GFP protein expression in mouse cortical neurons using virus generated from HEK293 cells transfected with Lipofectamine 3000 reagent (B, upper panel, lane 3), while less GFP is detected using virus produced with FuGENE 6 or Lipofectamine 2000 transfection reagents (B, upper panel, lane 1 and 2), the same membrane was striped and reprobed with anti-beta actin antibody, which showed equal beta-actin detection (B, lower panel). Similarly, statistics analysis also showed that GFP protein level using lipofectaime 3000 reagent is at least three times higher than using Lipofectamine 2000 or FuGENE 6 transfection reagents (C).
After successfully characterized anti-GFP antibody, we compared the GFP protein expression level in cell lysates harvested from mouse cortical neurons transduced using lentivirus generated form HEK293 cells with different transfection reagents. As shown in Figure 5B, highest GFP protein expression in mouse cortical neurons is detected using virus generated from HEK293 cells transfected with Lipofectamine 3000 reagent (Figure 5B, upper panel, lane 3), while less GFP is detected using virus produced with Fugen 6 or Lipofectamine 2000 transfection reagents (Figure 5B, upper panel, lane 1 and 2). Three separate experiments showed the same results and the statistics analysis was summarized in Figure 5C, which showed that GFP protein level using lipofectaime 3000 reagent is at least three times higher than that using Lipofectamine 2000 or Fugen 6 transfection reagents.
High efficiency of Cx43 knockdown in HUVECs using lentivirus generated from HEK293 cells utilizing Lipofectamine 3000 transfection reagent
Cx43 has been shown to be expressed in HUVECs. We further determined Cx43 knockdown efficiency in HUVECs using lentivirus carrying Cx43 shRNA, which contains four different plasmids and is packaged in HEK293 cells using Lipofectamine 3000 transfection reagent. As shown in Figure 6A, relatively strong Cx43 punctate labeling in HUVECs was seen after using lentivirus produced from Cx43 scrambled shRNA control plasmids. However, after using lentivirus generated utilizing Cx43 shRNA combined with Lipofectamine 2000 transfection reagent, Cx43 labeling in HUVECs was significantly reduced (Figure 6B). Furthermore, the substantial Cx43 knockdown was seen in HUVECs using the lentivirus generated utilizing the same Cx43 shRNA plasmids combined with Lipofectamine 3000 reagent (Figure 6C).
Figure 6.
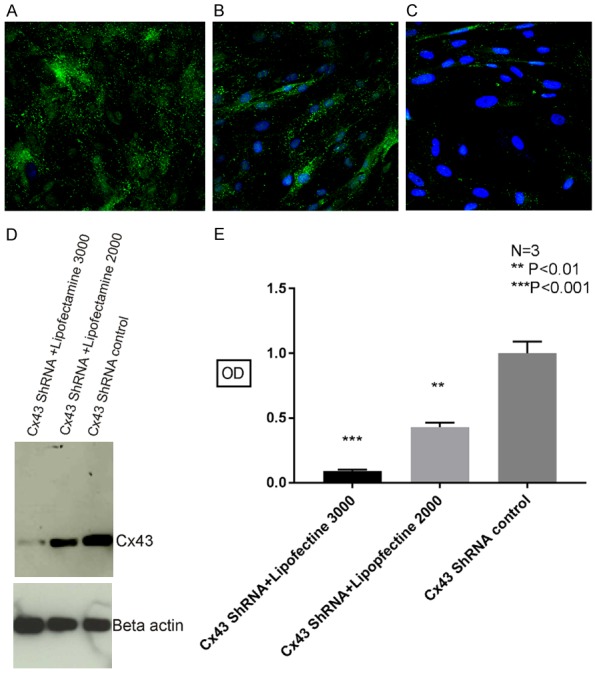
High efficiency of Cx43 knockdown in HUVECs using lentivirus generated from HEK293 cells utilizing Lipofectamine 3000 transfection reagent. Immunofluorescence labeling of Cx43 in HUVECs showed significantly reduced Cx43 punctate labeling using the lentivirus generated utilizing Cx43 shRNA plasmids combined with Lipofectamine 3000 reagent (C) compared with Cx43 labeling in HUVECs using Cx43 shRNA combined with Lipofectamine 2000 transfection reagent (B) or using scrambled Cx43 shRNA plasmids with Lipofectamine 3000 reagent (A). The same result was also verified using western blot (D), there was statistically significant reduced Cx43 detection in HUVECs cells using lentivirus produced with Cx43 ShRNA and Lipofectamine 3000 transfection reagents, which showed about 90% Cx43 knocked down, compared with about 60% Cx43 knockdown using lentivirus generated utilizing Cx43 ShRNA plasmids and Lipofectamine 2000 transfection reagent (E).
After Cx43 knockdown in HUVECs, the cells were lysed using IP buffer and subjected to Western blot analysis. As shown in Figure 6D, there was statistically significant reduction of Cx43 level in HUVECs cells using lentivirus produced with Cx43 ShRNA and Lipofectamine 3000 transfection reagents, which showed about 90% Cx43 knockdown, compared with about 60% Cx43 knockdown using lentivirus generated utilizing Cx43 ShRNA plasmids and Lipofectamine 2000 transfection reagent.
Discussion
Our present results demonstrate that we have established an easy method to achieve the highest transfection and viral transduction efficiency using Lipofectamine 3000 transfection reagent combined with viral transduction, which is shown by the highest EGFP expression using Western blot even with 1:10,000 anti-GFP antibody dilution and with only 1 ug total protein loading. More convincingly, GFP is easily seen from harvest cells without utilizing UV light activation or confocal microscopy. It appears that the lentiviral transduction efficiency is not only higher in HUVECs, but also in primary cultured cortical neurons, which is generally considered difficult to transfect and transduce with virus. Therefore, there is possibility that our protocol can also be used for other cell types as well. In addition, we can knockdown gap junction protein Cx43 in HUVECs. It is also possible we can use this method combined with other gene target shRNA lentivirus plasmids to knockdown other genes in cultured cells. CRISPR Cas9 technique is also widely used for knockdown gene expression using the NHEJ (non homologus end joining) in both dividing and non dividing cells [28], we speculate that there is possibility that our protocol also can be used to manipulate gene expression using CRISPR-Cas9 system, further, it is also possible we can use the same protocol to generate lentivirus for in vivo studies [29].
After transient transfection or viral transduction, we used the same very low power setting to take all living GFP fluorescence with 4× or 10× objective lens. There is possibility that some fixation or immunological procedure may have an effect on EGFP labeling [30]. For instance, GFP labeled living cells treated with cold methanol, 4% paraformaldehyde or ethanol may weak the GFP labeling, therefore the fixation condition, duration time may worth special attention.
From our knowledge, this is the first time we can see EGFP even without using a microscopy and an UV activation in our laboratories, the strong EGFP expression is also demonstrated by western blot, which showed robust GFP migration pattern using 10 ug total protein loading with reduced concentration of primary anti-GFP antibody dilution at 1:10,000, this promoted us to adjust total protein loading at 1 ug.
The high efficiency of EGFP transfection and lentiviral transduction may be contributed by the following factors. First, the cultured cells appear very healthy and no apparent toxic effect after utilizing all three transfection reagents 48 hours after plasmids transient transfection; Second, we used freshly made lentivirus without any concentration or purification after 48 hours transfection; Third, we checked the results after 72 hours of viral transduction, not 48 hours. Our unpublished data showed that 72 hours viral transduction induces higher EGFP expression in transduced cells than using 48 hours viral transduction. Therefore, we conclude that the high quality of cell culture combined with fresh virus and appropriate viral transduction time is critical for achieving the highest transfection efficiency. In addition, we did not concentrate or purify the lentiviruses after transient transfection. There is possibility that if we concentrate or purify the lentivirus, we can achieve even higher transduction efficiency.
In our experiments, we used lentivirus expression vectors that carry an EGFP or Cx43 shRNA plasmids. We speculate there is possibility that this method may also be useful when combined with other expression vectors that carry an EGFP or without EGFP marker, such as overexpression gene expression vectors, siRNA, CRISPR Cas9 or Cpf1 systems for gene knockdown or specific amino acid replacement.
Another very interesting point is due to the high efficiency of gene transfection and viral transduction using EGFP as an indicator, there is also possible that our procedure may be used to generate stably overexpressed gene expression system even without further selection using specific resistant gene markers. It is well known that generation of a gene specific stably expressing cell lines is a time consuming process, like we did for mouse Cx36 gene stable HeLa cell lines, which needed for at least four weeks to complete [25]. Further studies are needed to evaluate the feasibility of producing stable over expression cell lines without antibiotic selection.
Gap junction protein Cx43 has been reported to be expressed in HUVECs. Our immunofluorescence labeling in HUVECs and western blot result using anti-Cx43 antibody was in line with the previous result [31]. Besides high Cx43 shRNA viral transduction efficiency, our high level of Cx43 knockdown using Cx43 shRNA in HUVECs may also be contributed by using the mixture of four different shRNAs that target different Cx43 sequences, and healthy HUVECs with lentivirus incubation for 72 hours. Considering that Cx43 associates with many different protein binding partners and its functional role on cell growth, cell differentiation and maintenance of homeostasis [31,32], further study is needed to investigate the functional consequence of Cx43 knockdown in HUVECs.
In conclusion, we can achieve high level of transfection and transduction efficiency using Lipofectamine 3000 transfection reagent compared with Lipofectamine 2000 or FuGENE 6 reagents. This method may have potential to be used not only in other cells combined with other expression vectors systems, but also may be useful for quick generating stable cell expression clones combined with viral transduction system.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grants no: 81471174 and 81520108011), supported by National Institutes of Health/National Eye Institute grants EY010572 (P30 Casey Eye Institute Core facility grant), and an unrestricted grant to the Casey Eye Institute from Research to Prevent Blindness (New York, NY, USA). We would like to thank Dr. James I Nagy from Department of Physiology and Pathophysiology at the University of Manitoba for providing the invaluable anti-Cx43 antibody and his critical comments on this manuscript.
Disclosure of conflict of interest
None.
References
- 1.Sandbichler AM, Aschberger T, Pelster B. A method to evaluate the efficiency of transfection reagents in an adherent zebrafish cell line. Biores Open Access. 2013;2:20–27. doi: 10.1089/biores.2012.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mostaghaci B, Loretz B, Lehr CM. Calcium phosphate system for gene delivery: historical background and emerging opportunities. Curr Pharm Des. 2016;22:1529–1533. doi: 10.2174/1381612822666151210123859. [DOI] [PubMed] [Google Scholar]
- 3.Jin L, Zeng X, Liu M, Deng Y, He N. Current progress in gene delivery technology based on chemical methods and nano-carriers. Theranostics. 2014;4:240–255. doi: 10.7150/thno.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham FL, Van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 5.Potter H, Heller R. Transfection by electroporation. Curr Protoc Mol Biol. 2003 doi: 10.1002/0471142727.mb0903s92. Chapter: Unit-9.3. [DOI] [PubMed] [Google Scholar]
- 6.Horibe T, Torisawa A, Akiyoshi R, Hatta-Ohashi Y, Suzuki H, Kawakami K. Transfection efficiency of normal and cancer cell lines and monitoring of promoter activity by single-cell bioluminescence imaging. Luminescence. 2014;29:96–100. doi: 10.1002/bio.2508. [DOI] [PubMed] [Google Scholar]
- 7.Hornstein BD, Roman D, Arevalo-Soliz LM, Engevik MA, Zechiedrich L. Effects of circular DNA length on transfection efficiency by electroporation into HeLa cells. PLoS One. 2016;11:e0167537. doi: 10.1371/journal.pone.0167537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jain S, Kumar S, Agrawal AK, Thanki K, Banerjee UC. Enhanced transfection efficiency and reduced cytotoxicity of novel lipid-polymer hybrid nanoplexes. Mol Pharm. 2013;10:2416–2425. doi: 10.1021/mp400036w. [DOI] [PubMed] [Google Scholar]
- 9.Meng Z, Kang Z, Sun C, Yang S, Zhao B, Feng S, Meng Q, Liu K. Enhanced gene transfection efficiency use of peptide vectors containing laminin receptor-targeting sequence YIGSR. Nanoscale. 2018;10:1215–1227. doi: 10.1039/c7nr05843h. [DOI] [PubMed] [Google Scholar]
- 10.Vankayala R, Chiang CS, Chao JI, Yuan CJ, Lin SY, Hwang KC. A general strategy to achieve ultra-high gene transfection efficiency using lipid-nanoparticle composites. Biomaterials. 2014;35:8261–8172. doi: 10.1016/j.biomaterials.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 11.Romoren K, Thu BJ, Bols NC, Evensen O. Transfection efficiency and cytotoxicity of cationic liposomes in salmonid cell lines of hepatocyteand macrophage origin. Biochim Biophys Acta. 2004;1663:127–134. doi: 10.1016/j.bbamem.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Nouri F, Sadeghpour H, Heidari R, Dehshahri A. Preparation, characterization, and transfection efficiency of low molecular weightpolyethylenimine-based nanoparticles based nanoparticles for delivery of the plasmid encoding CD200 gene. Int J Nanomedicine. 2017;12:5557–5569. doi: 10.2147/IJN.S140734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiefer K, Clement J, Garidel P, Peschka-Suss R. Transfection efficiency and cytotoxicity of nonviral gene transfer reagents in human smooth muscle and endothelial cells. Pharm Res. 2004;21:1009–1017. doi: 10.1023/b:pham.0000029291.62615.ec. [DOI] [PubMed] [Google Scholar]
- 14.Peng L, Xiong W, Cai Y, Chen Y, He Y, Yang J, Jin J, Li H. A simple, rapid method for evaluation of transfection efficiency based on fluorescent dye. Bioengineered. 2017;8:225–231. doi: 10.1080/21655979.2016.1222995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villa-Diaz LG, Garcia-Perez JL, Krebsbach PH. Enhanced transfection efficiency of human embryonic stem cells by the incorporation of DNA liposomes in extracellular matrix. Stem Cells Dev. 2010;19:1949–1957. doi: 10.1089/scd.2009.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamano S, Dai J, Moursi AM. Comparison of transfection efficiency of nonviral gene transfer reagents. Mol Biotechnol. 2010;46:287–300. doi: 10.1007/s12033-010-9302-5. [DOI] [PubMed] [Google Scholar]
- 17.Kami D, Kitani T, Kishida T, Mazda O, Toyoda M, Tomitaka A, Ota S, Ishii R, Takemura Y, Watanabe M, Umezawa A, Gojo S. Pleiotropic functions of magnetic nanoparticles for ex vivo gene transfer. Nanomedicine. 2014;10:1165–1174. doi: 10.1016/j.nano.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 18.Aravindan L, Bicknell KA, Brooks G, Khutoryanskiy VV, Williams AC. A comparison of thiolated and disulfide-crosslinked polyethylenimine for nonviral gene delivery. Macromol Biosci. 2013;13:1163–1173. doi: 10.1002/mabi.201300103. [DOI] [PubMed] [Google Scholar]
- 19.Haberl S, Kanduser M, Flisar K, Hodzic D, Bregar VB, Miklavcic D, Escoffre JM, Rols MP, Pavlin M. Effect of different parameters used for in vitro gene electrotransfer on gene expression efficiency, cell viability and visualization of plasmid DNA at the membrane level. J Gene Med. 2013;15:169–181. doi: 10.1002/jgm.2706. [DOI] [PubMed] [Google Scholar]
- 20.Hama S, Akita H, Ito R, Mizuguchi H, Hayakawa T, Harashima H. Quantitative comparison of intracellular trafficking and nuclear transcription between adenoviral and lipoplex systems. Mol Ther. 2006;13:786–794. doi: 10.1016/j.ymthe.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Iversen N, Birkenes B, Torsdalen K, Djurovic S. Electroporation by nucleofector is the best nonviral transfection technique in human endothelial and smooth muscle cells. Genet Vaccines Ther. 2005;3:2. doi: 10.1186/1479-0556-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dizaj SM, Jafari S, Khosroushahi AY. A sight on the current nanoparticle-based gene delivery vectors. Nanoscale Res Lett. 2014;9:252. doi: 10.1186/1556-276X-9-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Djurovic S, Iversen N, Jeansson S, Hoover F, Christensen G. Comparison of nonviral transfection and adeno-associated viral transduction on cardiomyocytes. Mol Biotechnol. 2004;28:21–32. doi: 10.1385/MB:28:1:21. [DOI] [PubMed] [Google Scholar]
- 24.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Olson C, Lu S, Kamasawa N, Yasumura T, Rash JE, Nagy JI. Neuronal connexin36 association with zonula occludens-1 protein (ZO-1) in mouse brain and interaction with the first PDZ domain of ZO-1. Eur J Neurosci. 2004;19:2132–2146. doi: 10.1111/j.l460-9568.2004.03283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Kamasawa N, Ciolofan C, Olson C, Lu S, Davidson KG, Yasumura T, Shigemoto R, Rash JE, Nagy JI. Connexin45-containing neuronal gap junctions in rodent retina also contain connexin36 in both apposing hemiplaques, forming bi-homotypic gap junctions, with scaffolding contributed by zonula occludens-1. J Neurosci. 2008;28:9769–9789. doi: 10.1523/JNEUROSCI.2137-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Lu S, Nagy JI. Direct association of connexin36 with zonula occludens-2 and zonula occludens-3. Neurochem Int. 2009;54:393–402. doi: 10.1016/j.neuint.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolli N, Lu M, Maiti P, Rossignol J, Dunbar GL. Application of the gene editing tool, CRISPR-Cas9, for treating neurodegenerative diseases. Neurochem Int. 2018;112:187–196. doi: 10.1016/j.neuint.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Fricano-Kugler CJ, Williams MR, Salinaro JR, Li M, Luikart B. Designing, packaging, and delivery of high titer CRISPR retro and lentiviruses via stereotaxic injection. J Vis Exp. 2016:111. doi: 10.3791/53783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Penes M, Odermatt B, Willecke K, Nagy JI. Ablation of Cx47 in transgenic mice leads to the loss of MUPP1, ZONAB and multiple connexins at oligodendrocyte-astrocyte gap junctions. Eur J Neurosci. 2008;28:1503–1517. doi: 10.1111/j.1460-9568.2008.06431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Radin JN, González-Rivera C, Frick-Cheng AE, Sheng J, Gaddy JA, Rubin DH, Algood HM, McClain MS, Cover TL. Role of connexin43 in Helicobacter pylori VacA-induced cell death. Infect Immun. 2014;82:423–432. doi: 10.1128/IAI.00827-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leithe E, Mesnil M, Aasen T. The connexin 43 C-terminus: a tail of many tales. Biochim Biophys Acta. 2018;1860:48–64. doi: 10.1016/j.bbamem.2017.05.008. [DOI] [PubMed] [Google Scholar]