

Abstract
Importance
Numerous studies have evaluated the prognostic value of minimal residual disease (MRD) in multiple myeloma (MM). Most studies were small and varied in terms of patient population, treatment, and MRD assessment methods.
Objective
To evaluate the utility of MRD detection in patients with newly diagnosed MM.
Data Sources
A Medline search was conducted for articles published in English between January 1990 and January 2016.
Study Selection
Eligible studies reported MRD status and progression-free survival (PFS) or overall survival (OS) in ≥ 20 patients following treatment. Among 405 articles identified, 21 met the initial eligibility criteria and were included in the analysis.
Data Extraction and Synthesis
Information on patient characteristics, treatment, MRD assessment, and outcomes were extracted using a standard form.
Main Outcome Measures
The impact of MRD status on PFS and OS was assessed by pooling data from relevant trials. Data were adjusted to allow for different proportions of patients with MRD in different studies, and analyzed using the Peto method. Forest plots were created based on Cox model analysis. Other pre-specified research questions were addressed qualitatively.
Results
Fourteen studies (n = 1,273) provided data on the impact of MRD on PFS, and 12 studies (n = 1,100) on OS. Results were reported specifically in patients who had achieved conventional complete response (CR) in 5 studies for PFS (n = 574) and 6 studies for OS (n = 616). MRD-negative status was associated with significantly better PFS overall (Hazard ratio [HR] 0.41; 95% confidence interval [CI] 0.36–0.48; P < .0001) and in studies specifically looking at CR patients (HR 0.44; 95% CI 0.34–0.56; P < .0001). OS was also favorable in MRD-negative patients overall (HR 0.57; 95% CI 0.46–0.71; P < .0001) and in CR patients (HR 0.47; 95% CI 0.33–0.67; P < .0001). Tests of heterogeneity found no significant differences among the studies for PFS and OS.
Conclusions and Relevance
MRD-negative status after treatment for newly diagnosed MM is associated with long-term survival. These findings provide quantitative evidence to support the integration of MRD assessment as an endpoint in clinical trials of MM.
INTRODUCTION
A substantial proportion of patients with multiple myeloma (MM) can now expect to achieve clinical complete response (CR), as a result of recent therapeutic advances.1,2 These advances include the combined use of immunomodulatory drugs (thalidomide, lenalidomide, or pomalidomide) and proteasome inhibitors (bortezomib or carfilzomib), along with high-dose therapy with autologous stem cell transplantation (ASCT) in eligible individuals. CR rates are likely to continue to increase with incorporation of novel combinations of therapies.3 Nevertheless, most patients who achieve CR eventually relapse,1 suggesting that a small but clinically relevant population of myeloma cells not detected by current techniques, persists. Assays with greater sensitivity have been developed to detect minimal residual disease (MRD), including multiparameter flow cytometry (MFC), allele-specific oligonucleotide quantitative polymerase chain reaction (ASO-qPCR), and next-generation sequencing (NGS) techniques.3,4
Potential applications of MRD assessment in MM management are numerous.1,3,5,6 It is already considered an important prognostic factor.7 MRD testing could be used to monitor response to therapy; the presence or absence of MRD may also inform subsequent treatment decisions, including consolidation and maintenance.7 Historically, due to the complexity of conventional MRD assays, evaluations were limited to a small number of patients. Recent development of MFC and NGS-based methods has allowed for MRD assessment in larger studies. To understand the real impact of MRD on outcomes from small-to-medium-sized studies, we performed a meta-analysis of all published data regarding the utility of MRD detection in patients with newly diagnosed MM (NDMM).
METHODS
Literature search and article selection
A Medline search was performed for articles published in English between January 1990 and January 2016, using the MeSH terms “multiple myeloma” AND “neoplasm, residual” and the non-MeSH terms “MRD”, “myeloma”, and “minimal residual disease”. Eligible articles included those that reported on controlled trials, randomized controlled trials, or patient cohort studies with MRD status and survival outcomes progression-free survival (PFS) or overall survival (OS) in 20 or more NDMM patients following therapy. Patients could have received any type of treatment except allogeneic stem cell transplantation (alloSCT), and MRD could be assessed by any method (MFC, ASO-qPCR, or NGS), but analysis was restricted to techniques with a limit of detection of 0.01% or lower. Trials were excluded if they: included only patients with relapsed/refractory MM (RRMM) or smoldering myeloma; assessed MRD in apheresis product; or reported on the same study population used in an already-included trial.
Data extraction
If primary data were not accessible, survival graphs from relevant trials were carefully measured and a computer program was written to reconstruct the individual survival and censoring times from these measurements. Articles were scrutinized to ensure that all P values, confidence intervals (CIs), hazard ratios (HRs), numbers of events/deaths, and median survival times and durations of patient follow-up matched those reported. There was a PFS curve but not an OS curve for one study.8 However, P values and percentages at particular times were provided for the OS data, which enabled censored values to be used from the PFS curves; it was therefore possible to use the additional information from the paper to derive the survival times. For the pooled analysis, data were adjusted to allow for the different proportions of patients with MRD in the different studies. P values are for adjusted log-rank χ2 tests.
Statistical analysis
For a pooled analysis of all studies reporting survival data, PFS and OS curves were generated.9 This method adjusts for the different proportions of MRD positivity and negativity in each study, thereby avoiding inappropriate bias potentially generated by studies with high or low proportions of MRD positivity. The method produces an adjusted log-rank χ2 statistic to evaluate the significance of any differences between MRD positivity and negativity. It also provides a non-proportional hazards-based equivalent to performing a Cox model analysis stratified by study or group. If the hazards are proportional, the results will be similar to such a Cox model analysis, which was the case in all such analyses in this report.
The overview methodology described in detail by Peto10 was applied. In brief, for PFS and OS, the expected number (E) of events was derived in the MRD-positive and MRD-negative groups for each study, assuming no difference between the MRD groups. This was compared with the observed number (O) of events and the differences (O-Es) were then tested for heterogeneity to see whether the scatter of results was unexpected. The sum of [O-E]2/variance should be distributed as χ2n-1 if the scatter is random, where n is the number of studies.
HR forest plots were then generated using the inverse variance weighting method, as described in detail by Whitehead and Whitehead.11 Cox proportional hazards model analysis was performed for each study, generating HR and CIs, and the required variance. An overall Cox model analysis was run on the whole dataset, stratified by study to generate similar statistics for the total of all the studies combined. The size of the solid squares (Figures 2A, 2B, 3A, and 3B) is proportional to the amount of information each trial contains (the inverse of the variance). 95% CIs are shown for the individual trials. For the overall result, 95% CIs are also given (open diamonds in the forest plot). The proportional hazards assumption was checked for the Cox model analyses using log–log plots and Schoenfeld residuals and any departures from proportionality were extremely minor.
Figure 2.
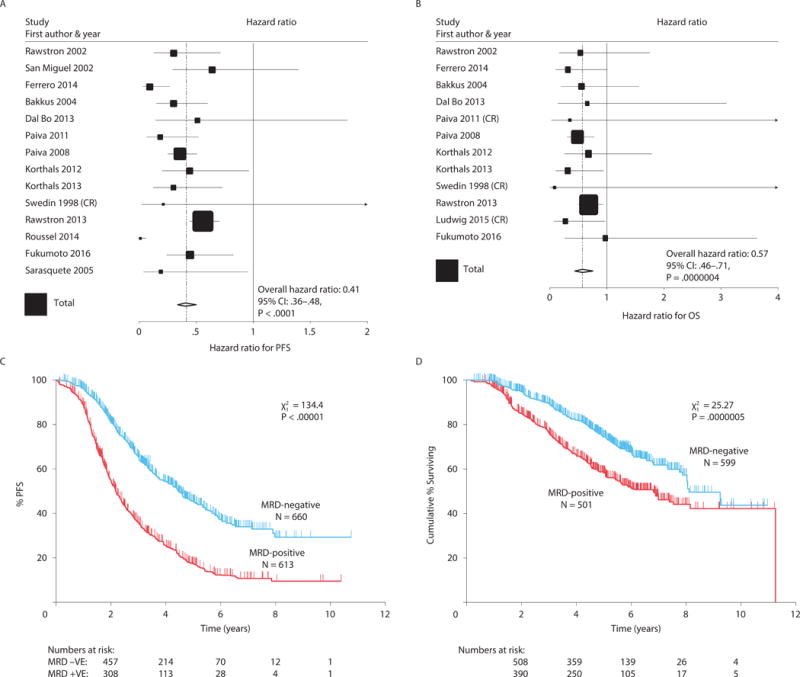
Overall effect of MRD status on PFS (A) and OS (B), indicating that MRD-negative patients had better outcomes. Tests for heterogeneity indicated no significant differences between the studies for both PFS and OS. Kaplan-Meier curves for PFS (C) and OS (D); data were adjusted to account for the different proportions of patients in each study being MRD-positive and MRD-negative. The sizes of the Forest plot squares represent the weighting of that trial in the meta-analysis, specifically the inverse variance of the Cox model estimate, and the horizontal lines represent the 95% CIs. CI, confidence interval; MRD, minimal residual disease; OS, overall survival; PFS, progression-free survival.
Figure 3.
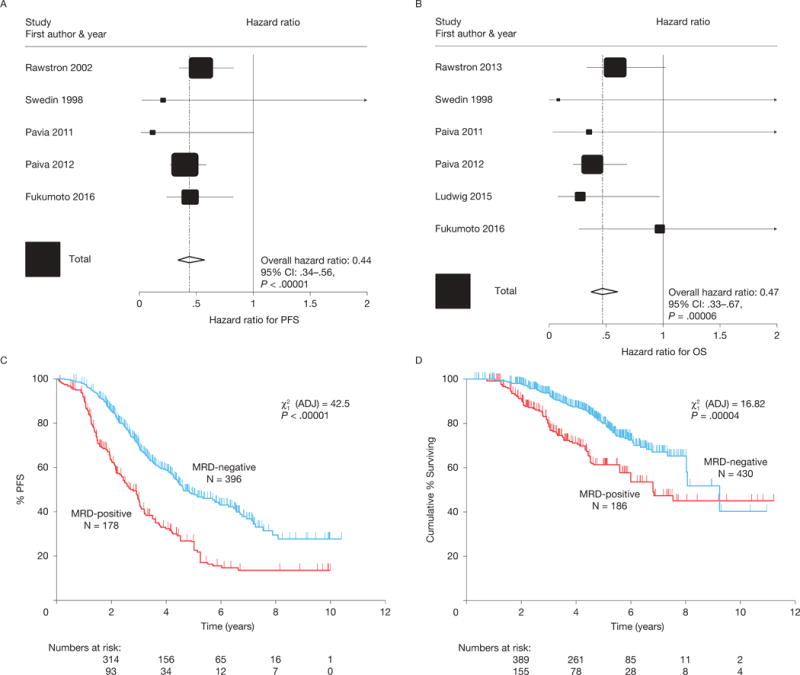
In CR patients, effect of MRD status on PFS (A) and OS (B), indicating that MRD-negative patients had better outcomes. Tests for heterogeneity indicated no significant differences between the studies for both PFS and OS. Kaplan-Meier curves for PFS (C) and OS (D); data were adjusted to account for the different proportions of patients in each study being MRD-positive and MRD-negative. The sizes of the Forest plot squares represent the weighting of that trial in the meta-analysis, specifically the inverse variance of the Cox model estimate, and the horizontal lines represent the 95% confidence intervals. CI, confidence interval; CR, complete response; MRD, minimal residual disease; OS, overall survival; PFS, progression-free survival.
There were no PFS events in the MRD-negative group in one study12 and no OS events in the MRD-negative group in another study,8 making it impossible to derive CIs and variance for Cox model HRs. In these two cases, an odds ratio approach was used to derive CIs and variance, incorporating the correlations between odds ratios and HRs which were all strong (r > .988).
Statistical analyses were performed with Stata v13.0, or purpose-written Digital Visual Fortran Version 6.0A software. Hypothesis tests were 2-sided.
RESULTS
Literature search
The initial search yielded 405 articles, and 25 additional articles were identified from the reference sections of recently published articles on the topic. After applying eligibility criteria 21 studies were included in the qualitative assessments (Figure 1).8,12–30 Of the 21 articles identified, 13 involved patients with NDMM and in nine articles it was not reported whether the population was limited to NDMM patients. Sixteen articles involved ASCT-eligible patients and one involved ASCT-ineligible patients; the remaining four studies included both ASCT-eligible and ASCT-ineligible patients. The primary MRD assay that was evaluated was MFC (n = 9); PCR (n = 11), or NGS (n = 1).
Figure 1.
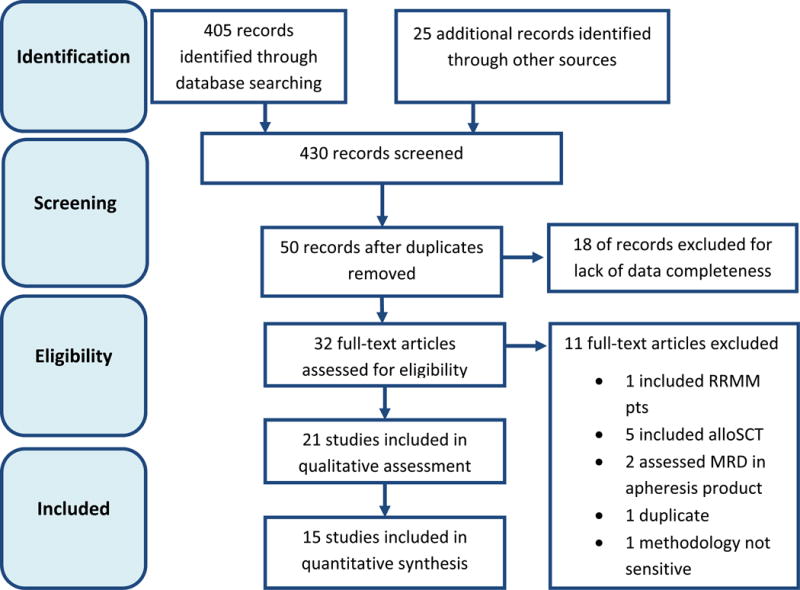
Article identification and selection. AlloSCT, allogeneic stem cell transplantation; MRD, minimal residual disease; RRMM, relapsed/refractory multiple myeloma.
Fourteen studies (n = 1,273) reported information on the impact of MRD on PFS and twelve assessed the impact of MRD on OS (n = 1,100); these studies were therefore included in the overall quantitative meta-analysis (Supplementary Table). Twelve publications reported conventional CR7 at the time of MRD measurement.6,8,19,21–27,31 However, further investigation identified potential duplication of data across some studies and led to the exclusion of five additional articles from the quantitative analysis in CR patients.21–24,27
The impact of MRD status on survival outcomes
The overall prognostic value of MRD status in terms of PFS was assessed in 14 studies involving 1,273 patients (660 MRD-negative, 613 MRD-positive).8,12–14,16–18,24,25,28–31 The impact of MRD status on OS was assessed in 12 studies involving 1,100 patients (599 MRD-negative, 501 MRD-positive).6,8,13,14,16–19,24,25,28,31 Compared with MRD positivity, MRD negativity was associated with better PFS (HR 0.41; 95% CI 0.36–0.48; P < .0001) (Figure 2A) and OS (HR 0.57; 95% CI 0.46–0.71; P < .0001) (Figure 2B). Median PFS was 54 months for MRD-negative patients and 26 months for MRD-positive patients (Figure 2C); median OS was 98 and 82 months, respectively (Figure 2D). Tests of heterogeneity found no significant differences among the studies for OS (χ2 = 8.81, 11 df; P = 0.64) but significant differences among the studies for PFS (χ2 = 42.1, 13df; P < 0.001). This was a result of 2 very small studies,12,16 which showed unusually large differences; the Roussel et al. study also had no events occurring in MRD negative patients. When these 2 studies were excluded the test for heterogeneity was no longer significant (χ2 = 10.1, 11df; P = 0.53).
MRD is a better predictor of PFS and OS than conventional complete response
To evaluate the impact of MRD status on PFS in patients who had achieved conventional CR, data were pooled from five studies involving 574 patients (396 MRD-negative, 178 MRD-positive.8,25,26,28,31 For OS, data were pooled from six studies involving 616 patients (430 MRD-negative, 186 MRD-positive).8,19,25,26,28,31 In patients achieving CR, the presence of MRD predicted shorter PFS (HR 0.44; 95% CI 0.34–0.56; P < 0.00001) (Figure 3A) and OS (HR 0.47; 95% CI 0.33–0.67; P = 0.00006) (Figure 3B). Median PFS was 56 months for MRD-negative patients and 34 months for MRD-positive patients (Figure 3C) and median OS was 112 and 82 months, respectively (Figure 3D); PFS rates were 70% and 46% at 3 years, 48% and 27% at 5 years, and 37% and 14% at 7 years, respectively. Similarly, the OS rate was higher for MRD-negative patients compared with MRD-positive patients at 3 years (94% vs 80%), 5 years (80% vs 61%), and 7 years (67% vs 47%). Tests of heterogeneity found no significant differences among the studies for PFS (χ2 = 2.68, 4 df; P = .61) and OS (χ2=4.22, 5 df; P = .62).
Among the published analyses that were not restricted to CR patients, the impact of MRD on outcomes was less clear.12–14,16–18,29,30 One study found no significant difference in outcomes between patients with or without detectable MRD.14 Others noted that MRD status did not correlate with standard response criteria.17,18 In the study conducted by Rawstron et al.,6 it was noted that 34 of 246 (26%) MRD-negative patients did not achieve conventional CR, including 29 (12%) who had less than very good partial response (VGPR). Patients who were MRD-negative but failed to achieve CR had similar PFS and OS as those who were MRD-positive. Further analyses by this group suggested that log reduction in MRD (assessed as a continuous variable, rather than using a threshold for MRD positivity vs negativity), negated the significance of response in multivariate analyses for both PFS and OS.32
None of the trials directly compared the ability of two different treatment approaches to induce MRD-negative status. However, five studies evaluated MRD status before and after ASCT.6,12,17,18,20 All five indicated that ASCT increased the proportion of patients with MRD-negative status.
The prognostic value of MRD status in relation to other prognostic factors, e.g., high-risk cytogenetics
Eleven articles reported results from univariate and/or multivariate analyses regarding the ability of MRD status to predict outcomes.13,16,18,21–26,28,30 In all 11 trials, MRD was shown to be a significant predictor of outcomes. Notably, only six articles mentioned cytogenetics: high-risk cytogenetics, defined as any t(4;14), t(14;16), or del(17p),33 was a negative predictor of PFS (or time to progression [TTP] or event free survival [EFS]) in 3 reports,18,24,26 and OS in 2 reports.18,26 In the study by Paiva et al.,26 the combination of MRD status and cytogenetics was highly predictive of TTP, and the combination of MRD status, cytogenetics, and age was predictive of OS. Only one study reported that MRD status predicted PFS and OS in patients with unfavorable cytogenetics (defined as gain[1q], del[1p32], t[4;14], t[14;20], t[14;16], and del[17p]).6 Our meta-analysis of these latter studies6,26 indicated that the best OS is seen in patients with favorable cytogenetics who achieve MRD negativity compared with patients who are either high-risk or MRD-positive; worst results are seen in patients with high-risk cytogenetics who remain MRD-positive (P < .001) (Supplementary Figure). In a more recent analysis, cytogenetics (favorable vs unfavorable vs unknown/not evaluable) and log reduction in MRD were the only significant predictors of both PFS and OS in multivariate analysis.32
The impact of maintenance therapy on MRD
Ten studies mentioned maintenance therapy,6,8,12,15,17,18,25–27,29 but only two specifically evaluated MRD status after maintenance therapy. In one article, lenalidomide maintenance therapy was reported to increase response status in 4 patients and MRD status in 5 patients12 In the MM-IX study, more MRD-positive patients became MRD-negative during thalidomide maintenance compared with patients on no maintenance (8/29 [28%] vs 1/29 [3%]).6 Furthermore, more MRD-negative patients remained MRD-negative with thalidomide maintenance than with no maintenance (24/25 [96%] vs 11/16 [69%]; P = .026).
DISCUSSION
This large-cohort meta-analysis confirms that MRD status has prognostic value and is a valid surrogate marker for both PFS and OS in patients with MM, including those who had achieved a CR. All studies, irrespective of the therapies used, uniformly confirmed the impact of MRD status on outcome, indicating that the predictive value of MRD status was independent of the type of treatment used. This is consistent with the results of a recent study demonstrating that the depth of MRD is the determining factor for subsequent outcome.34 Findings from this meta-analysis provide quantitative evidence to support the conceptual basis for integrating MRD assessment into the management of MM.35
One of the main strengths of this analysis of pooled data from different clinical trials is the method used to generate the PFS and OS curves. These curves were adjusted for each study or group to allow for different proportions of patients with MRD positivity and negativity in the different studies, using methods described in detail elsewhere.9 This approach avoids the creation of curves that were biased inappropriately by studies with very high or very low proportions of patients with MRD positivity.
This analysis did not account for the type of MRD test used in each study. Approaches to testing vary widely36; the sensitivity of different protocols also varies.4,27,36,37 However, this may represent a strength of the analysis as the results are method-agnostic, i.e., it suggests that if MRD is undetectable with a certain level of sensitivity, the results have similar significance irrespective of the method used. MFC is the most widely used method for MRD testing in MM thus far due to its broad availability, short turnaround time, and relatively low cost.3 The main limitations of this technique are its lower sensitivity (up to 1 × 10−4 or −5) and lack of standardization among laboratories. ASO-qPCR, although sensitive, is cumbersome and is being replaced by NGS-based MRD assessment which is more sensitive than MFC38,39 or ASO-qPCR,40 and feasible in up to 90% of MM patients.41 To assess whether differences in the method of MRD assessment across the studies would impact our findings, we performed additional analyses comparing HRs for OS and PFS according to the two major methods of MRD assessment, flow cytometry and PCR. The HR for OS in the MFC studies (n = 923) was 0.60 (95% CI 0.47–0.76); in the PCR studies (n = 177) it was 0.44 (95% CI 0.26–0.77). The HR for PFS (n = 1072) in the MFC studies was 0.44 (95% CI 0.37–0.52); and in the PCR studies (n = 201) it was 0.27 (95% CI 0.18–0.40). As expected, the HR is slightly greater in the PCR studies as it provides a more sensitive measurement.
The studies in this analysis included primarily NDMM patients, most of whom were undergoing ASCT. The applicability of the results of this analysis in other populations, such as those with transplant-ineligible NDMM, RRMM, or high-risk cytogenetic features, is unclear. In addition, the timing of MRD assessment varied among the studies. For example, among the 14 trials included in the overall PFS meta-analysis, 5 assessed MRD before ASCT and 12 assessed MRD after ASCT. Among the trials assessing MRD after ASCT, most assessed patients after 3 months (or day 100), but some continued to assess patients every 3 to 6 months thereafter. Despite these differences, all studies showed large and consistent effects of MRD, confirmed by the non-significant χ2 statistic for heterogeneity, suggesting that any methodological variations between studies have a relatively minor influence on the overall MRD effect. In addition, there is always a risk in meta-analyses that negative results are less likely to have been reported, e.g. lack of effect of MRD status on OS and/or PFS. Lastly, this analysis did not isolate the prognostic effect of MRD from those of post-transplant treatments patients may have received. Future trials will need to focus on some of these questions to determine the clinical utility of MRD assessment as well as its ability to inform treatment decisions.
Assessment of MRD has several important potential applications in MM.1,42 In clinical trials, MRD assessment after initial treatment could be a useful surrogate endpoint for PFS and/or OS. It is in fact becoming an important component of the recommendations for uniform reporting of clinical trials.7 In clinical practice MRD testing may aid in prognostication; help make decisions regarding subsequent treatment, especially consolidation treatment; and, in the near future, guide the type and duration of maintenance therapy. Importantly, as the frequency of CR has increased, MRD negativity is emerging as a key endpoint for clinical studies. Integration of MRD testing into standard practice requires optimization and standardization of MRD assessment and standardization of its timing.4,7,42 Test standardization includes establishing optimal assay methods, timing of sample collection, sensitivity requirements, thresholds for MRD-positive status, and other factors.36 For example, recent evidence suggests that MRD quantitation may be more informative than MRD status: MRC Myeloma IX trial32 demonstrated a 1-year survival benefit for each 1-log depletion in tumor burden by MFC. The questions to be addressed in future include determining the impact of different treatment approaches on MRD status (e.g., consolidation or maintenance therapy); and the prognostic importance of MRD status in relation to other known prognostic factors.
In summary, the results of this large analysis showed that MRD negativity, as determined by various high-sensitivity methods, predicted better PFS and OS in patients with MM, including those who had achieved CR. MRD status is a marker of long-term outcomes in patients with MM. It should therefore be considered a new endpoint in clinical trials and clearly has a role as a surrogate marker of OS.
Supplementary Material
Supplementary Figure. Overall survival in patients achieving CR according to cytogenetic risk category (FISH) and MRD status. CR, complete response; FISH, fluorescent in situ hybridization; MRD, minimal residual disease; OS, overall survival.
Acknowledgments
The authors received editorial assistance in the preparation of this manuscript from Excerpta Medica (Daniel Gilmartin, PhD). The authors are fully responsible for all content and editorial decisions. The work was partly supported by NIH grants PO1-155258, and P50-100707 (NCM, HAL and KCA). Celgene provided financial support for statistical analysis and editorial assistance for this study. The authors thank all the investigators and patients who participated in the clinical trials referred to in the paper.
The data analysis were performed by Prof. Walter Gregory, University of Leeds, Leeds, UK, and Dr. Mehmet Kemal Samur, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA. Prof. Walter Gregory had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding/Support
This study was partly supported by NIH grants PO1-155258 and P50-100707 (NCM and KCA), and an MRC UK grant G0100132 (JAC) and partly funded by Celgene.
Role of the Sponsors
Celgene had no role in the design and conduct of the study; or the collection, management, analysis, and interpretation of the data; Celgene conducted a review of the draft manuscript for medical accuracy but was not involved in the final decision to the content or to submit the manuscript for publication. Celgene has minor investment in Adaptive biotechnologies Inc., a company that performs MRD assessment.
Footnotes
Conflicts of Interest Disclosure
N.M.: Celgene, Takeda, Janssen, and Merck – consultancy, advisory committee membership.
K.A.: Celgene, Millennium, Gilead – consultancy
H.A.-L.: Celgene, Takeda – consultancy, advisory committee membership.
A.C.R.: Janssen – research funding; Gilead – research funding, consultancy, honoraria; Celgene, Roche, Abbvie, Genzyme, GSK – honoraria; BD biosciences – other remuneration.
R.O.: Celgene – consultancy, honoraria, research funding; Janssen – consultancy; Takeda-honoraria
A.T., P.S.: Celgene – employment and equity ownership.
J.A.C.: Celgene – research funding.
A.G.: Excerpta Medica – employment.
W.G.: Celgene – consultancy; Janssen – honoraria.
M.K.S.: no conflicts of interest.
Author Contributions
Design and conduct of the study: Munshi, Avet-Loiseau, Anderson, Gregory, Georgieva.
Collection, management, analysis, and interpretation of the data: Gregory, Samur, Owen, Rawstron, Child, Avet-loiseau, Munshi, Anderson.
Preparation, review, or approval of the manuscript. Munshi, Avet-Loiseau, Rawstron, Owen, Child, Thakurta, Georgieva, Sherrington, Samur, Anderson, Gregory.
References
- 1.Munshi N, Anderson K. Minimal residual disease in multiple myeloma. J Clin Oncol. 2013;31(20):2523–2526. doi: 10.1200/JCO.2013.49.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson KC. Oncogenomics to target myeloma in the bone marrow microenvironment. Clin Cancer Res. 2011;17(6):1225–1233. doi: 10.1158/1078-0432.CCR-10-3366. [DOI] [PubMed] [Google Scholar]
- 3.Mailankody S, Korde N, Lesokhin AM, et al. Minimal residual disease in multiple myeloma: brining the bench to the bedside. Nat Rev Clin Oncol. 2015;12(5):286–295. doi: 10.1038/nrclinonc.2014.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biran N, Ely S, Chari A. Controversies in the assessment of minimal residual disease in multiple myeloma: clinical significance of minimal residual disease negativity using highly sensitive techniques. Curr Hematol Malig Rep. 2014;9(4):368–378. doi: 10.1007/s11899-014-0237-y. [DOI] [PubMed] [Google Scholar]
- 5.Rawstron AC, de Tute RM, Haughton J, Owen RG. Measuring disease levels in myeloma using flow cytometry in combination with other laboratory techniques: Lessons from the past 20 years at the Leeds Haematological Malignancy Diagnostic Service. Cytometry B Clin Cytom. 2016;90(1):54–60. doi: 10.1002/cyto.b.21271. [DOI] [PubMed] [Google Scholar]
- 6.Rawstron AC, Child JA, de Tute RM, et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. J Clin Oncol. 2013;31(20):2540–2547. doi: 10.1200/JCO.2012.46.2119. [DOI] [PubMed] [Google Scholar]
- 7.Rajkumar SV, Harousseau JL, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel. Blood. 2011;117(18):4691–4695. doi: 10.1182/blood-2010-10-299487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swedin A, Lenhoff S, Olofsson T, et al. Clinical utility of immunoglobulin heavy chain gene rearrangement identification for tumour cell detection in multiple myeloma. Br J Haematol. 1998;103(4):1145–1151. doi: 10.1046/j.1365-2141.1998.01075.x. [DOI] [PubMed] [Google Scholar]
- 9.Gregory WM. Adjusting survival curves for imbalances in prognostic factors. Br J Cancer. 1988;58(2):202–204. doi: 10.1038/bjc.1988.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peto R. Why do we need systematic overviews of randomised trials? Stat Med. 1987;6:233–240. doi: 10.1002/sim.4780060306. [DOI] [PubMed] [Google Scholar]
- 11.Whitehead A, Whitehead J. A general parametric approach to the meta-analysis of randomized clinical trials. Stat Med. 1991;10(11):1665–1677. doi: 10.1002/sim.4780101105. [DOI] [PubMed] [Google Scholar]
- 12.Roussel M, Lauwers-Cances V, Robillard N, et al. Front-line transplantation program with lenalidomide, bortezomib, and dexamethasone combination as induction and consolidation followed by lenalidomide maintenance in patients with multiple myeloma: a phase II study by the Intergroupe Francophone du Myélome. J Clin Oncol. 2014;32:2712–2717. doi: 10.1200/JCO.2013.54.8164. [DOI] [PubMed] [Google Scholar]
- 13.Bakkus MH, Bouko Y, Samson D, et al. Post-transplantation tumour load in bone marrow, as assessed by quantitative ASO-PCR, is a prognostic parameter in multiple myeloma. Br J Haematol. 2004;126(5):665–674. doi: 10.1111/j.1365-2141.2004.05120.x. [DOI] [PubMed] [Google Scholar]
- 14.Dal Bó S, Pezzi A, Amorin B, et al. Detection of minimal residual disease by flow cytometry for patients with multiple myeloma submitted to autologous hematopoietic stem cell transplantation. ISRN Hematol. 2013;2013:847672. doi: 10.1155/2013/847672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies FE, Forsyth PD, Rawstron AC, et al. The impact of attaining a minimal disease state after high-dose melphalan and autologous transplantation for multiple myeloma. Br J Haematol. 2001;112(3):814–819. doi: 10.1046/j.1365-2141.2001.02530.x. [DOI] [PubMed] [Google Scholar]
- 16.Ferrero S, Ladetto M, Drandi D, et al. Long-term results of the GIMEMA VEL-03-096 trial in MM patients receiving VTD consolidation after ASCT: MRD kinetics’ impact on survival. Leukemia. 2015;29(3):689–695. doi: 10.1038/leu.2014.219. [DOI] [PubMed] [Google Scholar]
- 17.Korthals M, Sehnke N, Kronenwett R, et al. Molecular monitoring of minimal residual disease in the peripheral blood of patients with multiple myeloma. Biol Blood Marrow Transplant. 2013;19(7):1109–1115. doi: 10.1016/j.bbmt.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 18.Korthals M, Sehnke N, Kronenwett R, et al. The level of minimal residual disease in the bone marrow of patients with multiple myeloma before high-dose therapy and autologous blood stem cell transplantation is an independent predictive parameter. Biol Blood Marrow Transplant. 2012;18(3):423–431. doi: 10.1016/j.bbmt.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 19.Ludwig H, Greil R, Masszi T, et al. Bortezomib, thalidomide and dexamethasone, with or without cyclophosphamide, for patients with previously untreated multiple myeloma: 5-year follow-up. Br J Haematol. 2015;171(3):344–354. doi: 10.1111/bjh.13582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludwig H, Viterbo L, Greil R, et al. Randomized phase II study of bortezomib, thalidomide, and dexamethasone with or without cyclophosphamide as induction therapy in previously untreated multiple myeloma. J Clin Oncol. 2013;31(2):247–255. doi: 10.1200/JCO.2011.39.5137. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Lopez J, Fernández-Redondo E, García-Sánz R, et al. Clinical applicability and prognostic significance of molecular response assessed by fluorescent-PCR of immunoglobulin genes in multiple myeloma. Results from a GEM/PETHEMA study. Br J Haematol. 2013;163(5):581–589. doi: 10.1111/bjh.12576. [DOI] [PubMed] [Google Scholar]
- 22.Martinez-Lopez J, Lahuerta JJ, Pepin F, et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood. 2014;123(20):3073–3079. doi: 10.1182/blood-2014-01-550020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martínez-Sánchez P, Montejano L, Sarasquete ME, et al. Evaluation of minimal residual disease in multiple myeloma patients by fluorescent-polymerase chain reaction: the prognostic impact of achieving molecular response. Br J Haematol. 2008;142(5):766–774. doi: 10.1111/j.1365-2141.2008.07263.x. [DOI] [PubMed] [Google Scholar]
- 24.Paiva B, Vidriales MB, Cerveró J, et al. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood. 2008;112(10):4017–4023. doi: 10.1182/blood-2008-05-159624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paiva B, Martinez-Lopez J, Vidriales MB, et al. Comparison of immunofixation, serum free light chain, and immunophenotyping for response evaluation and prognostication in multiple myeloma. J Clin Oncol. 2011;29(12):1627–1633. doi: 10.1200/JCO.2010.33.1967. [DOI] [PubMed] [Google Scholar]
- 26.Paiva B, Gutiérrez NC, Rosiñol L, et al. High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood. 2012;119(3):687–691. doi: 10.1182/blood-2011-07-370460. [DOI] [PubMed] [Google Scholar]
- 27.Puig N, Sarasquete ME, Balanzategui A, et al. Critical evaluation of ASO RQ-PCR for minimal residual disease evaluation in multiple myeloma. A comparative analysis with flow cytometry. Leukemia. 2014;28(2):391–397. doi: 10.1038/leu.2013.217. [DOI] [PubMed] [Google Scholar]
- 28.Rawstron AC, Davies FE, DasGupta R, et al. Flow cytometric disease monitoring in multiple myeloma: the relationship between normal and neoplastic plasma cells predicts outcome after transplantation. Blood. 2002;100(9):3095–3100. doi: 10.1182/blood-2001-12-0297. [DOI] [PubMed] [Google Scholar]
- 29.San Miguel JF, Almeida J, Mateo G, et al. Immunophenotypic evaluation of the plasma cell compartment in multiple myeloma: a tool for comparing the efficacy of different treatment strategies and predicting outcome. Blood. 2002;99(5):1853–1856. doi: 10.1182/blood.v99.5.1853. [DOI] [PubMed] [Google Scholar]
- 30.Sarasquete ME, García-Sanz R, González D, et al. Minimal residual disease monitoring in multiple myeloma: a comparison between allelic-specific oligonucleotide real-time quantitative polymerase chain reaction and flow cytometry. Haematologica. 2005;90(10):1365–1372. [PubMed] [Google Scholar]
- 31.Fukumoto K, Fujisawa M, Suehara Y, et al. Prognostic impact of immunophenotypic complete response in patients with multiple myeloma achieving better than complete response. Leuk Lymphoma. 2016 Jan 13;:1–7. doi: 10.3109/10428194.2015.1121262. [DOI] [PubMed] [Google Scholar]
- 32.Rawstron AC, Gregory WM, de Tute RM, et al. Minimal residual disease in myeloma by flow cytometry: independent prediction of survival benefit per log reduction. Blood. 2015;125(12):1932–1935. doi: 10.1182/blood-2014-07-590166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munshi NC, Anderson KC, Bergsagel PL, Shaughnessy J, et al. Consensus recommendations for risk stratification in multiple myeloma: report of the International Myeloma Workshop Consensus Panel 2. Blood. 2015;117(18):4696–700. doi: 10.1182/blood-2010-10-300970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Tute RM, Rawstron AC, Gregory WM, Child JA. Minimal residual disease following autologous stem cell transpant in myeloma: impact on outcome is independent of induction regimen. Haematologica. 2016;101(2):e69–71. doi: 10.3324/haematol.2015.128215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paiva B, Van Dongen JJM, Orfao A. New criteria for response assessment: role of minimal residual disease in multiple myeloma. Blood. 2015;125(20):3059–3068. doi: 10.1182/blood-2014-11-568907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flanders A, Stetler-Stevenson M, Landgren O. Minimal residual disease testing in multiple myeloma by flow cytometry: major heterogeneity. Blood. 2013;122(6):1088–1089. doi: 10.1182/blood-2013-05-506170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.San-Miguel JF, Paiva B, Gutierrez C. New tools for diagnosis and monitoring of multiple myeloma. Am Soc Clin Oncol Educ Book. 2013 doi: 10.14694/EdBook_AM.2013.33.e313. Available at: http://meetinglibrary.asco.org/sites/meetinglibrary.asco.org/files/edbook_pdf/2013_EdBook.pdf. Accessed March 2016. [DOI] [PubMed]
- 38.Jasielec J, Dytfeld D, Griffith KA, et al. Minimal residual disease status predicts progression-free survival in newly diagnosed multiple myeloma (NDMM) patients treated with carfilzomib, lenalidomide, and low-dose dexamethasone (KRd) Blood. 2014:124. abstract 2127. [Google Scholar]
- 39.Korde N, Mailankody S, Roschewski M, et al. Minimal residual disease (MRD) testing in newly diagnosed multiple myeloma (MM) patients: a prospective head-to-head assessment of cell-based, molecular, and molecular-imaging modalities. Blood. 2014:124. abstract 2105. [Google Scholar]
- 40.Takamatsu, Murata R, Zheng J, et al. Prognostic value of sequencing-based minimal residual disease detection in multiple myeloma. Blood. 2014:124. abstract 2003. [Google Scholar]
- 41.Avet-Loiseau H, Corre J, Maheo S, et al. Identification rate of myeloma-specific clonotypes in multiple diagnostic sample types from patients with multiple myeloma using next-generation sequencing method. Blood. 2014:124. abstract 2036. [Google Scholar]
- 42.Fernández de Larrea, Delforge M, Davies F, et al. Response evaluation and monitoring of multiple myeloma. Expert Rev Hematol. 2014;7(1):33–42. doi: 10.1586/17474086.2014.876899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure. Overall survival in patients achieving CR according to cytogenetic risk category (FISH) and MRD status. CR, complete response; FISH, fluorescent in situ hybridization; MRD, minimal residual disease; OS, overall survival.