

Abstract
NMDA receptors (NMDARs) mediate both long-term potentiation and long-term depression (LTD) and understanding how a single receptor can initiate both phenomena remains a major question in neuroscience. A prominent hypothesis implicates the NMDAR subunit composition, specifically GluN2A and GluN2B, in dictating the rules of synaptic plasticity. However, studies testing this hypothesis have yielded inconsistent and often contradictory results, especially for LTD. These inconsistent results may be due to challenges in the interpretation of subunit-selective pharmacology and in dissecting out the contributions of differential channel properties versus the interacting proteins unique to GluN2A or GluN2B. In this study, we address the pharmacological and biochemical challenges by using a single-neuron genetic approach to delete NMDAR subunits in conditional knock-out mice. In addition, the recently discovered non-ionotropic nature of NMDAR-dependent LTD allowed the rigorous assessment of unique subunit contributions to NMDAR-dependent LTD while eliminating the variable of differential charge transfer. Here we find that neither the GluN2A nor the GluN2B subunit is strictly necessary for either non-ionotropic or ionotropic LTD.
SIGNIFICANCE STATEMENT NMDA receptors are key regulators of bidirectional synaptic plasticity. Understanding the mechanisms regulating bidirectional plasticity will guide development of therapeutic strategies to treat the dysfunctional synaptic plasticity in multiple neuropsychiatric disorders. Because of the unique properties of the NMDA receptor GluN2 subunits, they have been postulated to differentially affect synaptic plasticity. However, there has been significant controversy regarding the roles of the GluN2 subunits in synaptic long term depression (LTD). Using single-neuron knock-out of the GluN2 subunits, we show that LTD requires neither GluN2A nor GluN2B.
Keywords: LTD, metabotropic, NR2A, NR2B, signaling, synaptic plasticity
Introduction
NMDA receptors (NMDARs) play prominent roles in bidirectional synaptic plasticity, mediating major forms of both long-term potentiation (LTP) and long-term depression (LTD; Collingridge et al., 1983; Dudek and Bear, 1992). Most NMDARs are heterotetramers containing two obligatory GluN1 subunits and two GluN2 subunits, with GluN2A and GluN2B being the predominant subunits in the mammalian forebrain, including the hippocampus (Gray et al., 2011). Because the functional and regulatory properties of NMDARs are largely determined by their GluN2 subunit composition (Cull-Candy and Leszkiewicz, 2004), many studies have explored the hypothesis that different NMDAR subunits dictate the rules of synaptic plasticity, though results have been inconsistent and often contradictory, especially for studies of LTD (for review, see Shipton and Paulsen, 2014).
There are a number of potential reasons for the inconsistencies in LTD studies. First, interpretation of GluN2 subunit-selective pharmacology is problematic. GluN2 subunit-selective antagonists are limited by poor subunit selectivity (e.g., the GluN2A “selective antagonist” NVP-AAM077 is only fivefold selective over GluN2B; Neyton and Paoletti, 2006), incomplete blockade (e.g., ifenprodil only reduces currents from pure GluN2B-containing receptors ∼80%; Fischer et al., 1997; Hatton and Paoletti, 2005; Gray et al., 2011), and complex effects on glutamate affinity (e.g., ifenprodil increases glutamate affinity and prolongs NMDAR synaptic currents; Kew et al., 1996; Gray et al., 2011; Tovar and Westbrook, 2012). Second, recent evidence has demonstrated that a high proportion of synaptic NMDARs are triheteromeric, containing GluN2A and GluN2B (Gray et al., 2011; Rauner and Köhr, 2011; Tovar et al., 2013). These triheteromeric receptors are only modestly responsive to GluN2-selective pharmacology (Hatton and Paoletti, 2005), further complicating the interpretation of these studies. Finally, conventional knock-out (KO) studies of GluN2 subunits have serious limitations as the GluN1 and GluN2B KO mice die perinatally (Forrest et al., 1994; Kutsuwada et al., 1996) and broad deletion of NMDARs results in altered network activity (Li et al., 1994; Iwasato et al., 2000).
Here we used a single-neuron genetic approach to isolate individual GluN2 subunits and assess their contributions to LTD. This approach avoids both the network-wide disruptions found in previous genetic manipulations as well as the difficult-to-interpret subunit-specific pharmacology. Importantly however, even the interpretation of the effects of pure GluN2A or GluN2B receptor populations on synaptic plasticity can be problematic. Specifically, effects of pure GluN2 subunit populations could be related to large differences in charge transfer (including Ca2+) or to critical associations with their divergent intracellular C-terminal tails. The inability to separate these variables further limits interpretations of NMDAR subunit-specific plasticity. Recently however, NMDAR-mediated LTD has been shown to occur in the absence of ion flux through the NMDAR (Nabavi et al., 2013; Stein et al., 2015; Carter and Jahr, 2016; but see Babiec et al., 2014), providing the opportunity to rigorously examine the GluN2 subunit-dependence of LTD while eliminating charge transfer as a variable. Surprisingly, we show no dependence of GluN2 subunit composition on either non-ionotropic or ionotropic NMDAR-dependent LTD.
Materials and Methods
Animals and postnatal viral injection.
Animals were housed according to IACUC guidelines at the University of California Davis. Grin2afl/fl (Gray et al., 2011), Grin2Bfl/fl (Mishina and Sakimura, 2007; Akashi et al., 2009), and Grin1fl/fl mice (Li et al., 1994; Adesnik et al., 2008) are all as previously described. Neonatal [postnatal day (P)0–P1] mice of both sexes were stereotaxically injected with high-titer rAAV1-Cre:GFP viral stock (∼1–5 × 1012 vg/ml) with coordinates targeting CA1 of hippocampus as previously described (Gray et al., 2011). Transduced neurons were identified by nuclear GFP expression. Cre expression was generally limited to the hippocampus within a sparse population of CA1 pyramidal neurons.
Electrophysiology.
P15–P21 mice were anesthetized in isoflurane and decapitated. Brains were rapidly removed and placed in ice-cold sucrose cutting buffer, containing the following (in mm): 210 sucrose, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 7 glucose, 7 MgCl2, and 0.5 CaCl2. Modified transverse 300 μm slices of dorsal hippocampus were prepared by performing a ∼10° angle blocking cut of the dorsal portion of each cerebral hemisphere (Bischofberger et al., 2006) then mounting the cut side down on a Leica VT1200 vibratome in ice-cold cutting buffer. Slices were recovered in 32°C artificial CSF (ACSF) solution containing the following (in mm): 119 NaCl, 26.2 NaHCO3, 11 glucose, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2, and 1.3 MgSO4, for 1 h before recording. Slices were transferred to a submersion chamber on an upright Olympus microscope, perfused in room temperature normal ACSF containing picrotoxin (0.1 mm) and saturated with 95%O2/5%CO2. CA1 neurons were visualized by infrared differential interference contrast microscopy and GFP+ neurons were identified by epifluorescence microscopy. Cells were patched with 3–5 MΩ borosilicate pipettes filled with intracellular solution, containing the following (in mm): 135 cesium methanesulfonate, 8 NaCl, 10 HEPES, 0.3 Na-GTP, 4 Mg-ATP, 0.3 EGTA, and 5 QX-314 (Sigma-Aldrich). EPSCs were evoked by electrical stimulation of Schaffer collaterals with a bipolar electrode (MicroProbes). AMPAR-EPSCs were measured at a holding potential of −70 mV, and NMDAR-EPSCs were measured at +40 mV in the presence of 10 μm NBQX. LTD was induced using a standard low-frequency stimulation protocol of 900 stimuli at 1 Hz (15 min) and holding the neuron at −40 mV. Series resistance was monitored and not compensated, and cells were discarded if series resistance varied >25%. All recordings were obtained with a MultiClamp 700B amplifier (Molecular Devices), filtered at 2 kHz, digitized at 10 Hz. Analysis was performed with the Clampex software suite (Molecular Devices).
Experimental design and statistical analysis.
All data represent the mean ± SEM of n = number of neurons or pairs of neurons. With the exception of the drug titrations, a minimum of three mice were used per group. All experimental groups include both males and females. Data were analyzed using Clampfit 10.4 (Axon Instruments) and Prism 7 software (GraphPad). LTD experiments were analyzed by averaging the final 10 min of the recording and normalizing as a percentage of the baseline AMPAR-EPSC amplitude. Paired amplitude and decay data were analyzed with a paired two-tailed t test and comparisons of LTD experiments were analyzed by unpaired two-tailed t test both with p < 0.05 considered significant.
Results
NMDAR glycine-site antagonists, which prevent channel opening, provide a key means to study non-ionotropic LTD. 7-Chlorokynuernic acid (7CK) is a competitive NMDAR glycine-site antagonist that we and others have previously used to examine non-ionotropic LTD (Nabavi et al., 2013; Dore et al., 2015; Stein et al., 2015; Carter and Jahr, 2016). However, at concentrations needed for complete NMDAR block in acute brain slices (100 μm), 7CK also significantly inhibits AMPAR-EPSCs (Fig. 1B,C, purple; 74.9 ± 6.0%, n = 4) making whole-cell LTD recordings challenging. Thus, we have characterized the use of L689,560 (L689), a competitive glycine-site antagonist with higher potency and selectivity than 7CK (Leeson et al., 1992; Grimwood et al., 1995). A dose–response of L689 on acute hippocampal slices found rapid, complete block of NMDAR-EPSCs by 10 μm L689 (Fig. 1A), a concentration that blocks only ∼10% of AMPAR-EPSCs (Fig. 1B,C; 10 μm L689: 10.7 ± 4.3%, n = 4).
Figure 1.

The NMDAR glycine-site antagonist L689 blocks NMDAR currents but not NMDAR-mediated LTD. A, Dose–response of NMDAR-EPSC block by L689 in acute mouse hippocampal slices. NMDAR-EPSCs were fully inhibited by 10 and 100 μm L689 within 5 min (n = 3 per dose). B, C, Inhibition of AMPAR-EPSCs by NMDAR glycine-site antagonists. B, Time course of AMPAR-ESPC inhibition by 7CK and L689 normalized to baseline amplitude. C, Percentage block of AMPAR-EPSCs by 7CK and L689 averaging from 20 to 30 min after drug application. One hundred micromolar 7CK and L689 inhibited AMPAR-EPSCs by 74.9 ± 6.0% and 55.2 ± 5.7%, respectively, whereas 10 μm L689 inhibited only 10.7 ± 4.3% (n = 4 for each condition). D–F, Non-ionotropic NMDAR-mediated LTD occurs in the presence of 10 μm L689 and is blocked by 50 μm D-AP5. D, Averaged whole-cell LTD experiments and representative traces (10 ms, 50 pA). E, Cumulative distribution of experiments in D. F, Ten micromolar L689 alone resulted in LTD (68.9 ± 3.6% of baseline, n = 8). In contrast, addition of AP5 significantly inhibits this LTD (100.7 ± 5.5% of baseline, n = 8; t(14) = 4.854, *p = 0.0003, t test). All data represents mean ± SEM.
Non-ionotropic LTD is NMDAR-dependent
Consistent with 100 μm 7CK (Nabavi et al., 2013; Stein et al., 2015), non-ionotropic LTD occurs in the presence of 10 μm L689 and remains NMDAR-dependent as it was blocked by concurrent incubation with the competitive glutamate-site antagonist AP5 (Fig. 1D–F; L689: 68.9 ± 3.6%, n = 8; +AP5: 100.7 ± 5.5%, n = 8; t(14) = 4.854, p = 0.0003, t test). To further test the NMDAR-dependence of non-ionotropic LTD, we removed the obligatory GluN1 subunit in a sparse subset of CA1 pyramidal neurons by P0 stereotaxic injection of adeno-associated virus, serotype 1 expressing a Cre recombinase GFP fusion protein (AAV1-Cre:GFP) into floxed GluN1 (Grin1fl/fl) mice for conditional deletion of GluN1 (Fig. 2A). This mosaic deletion allows for simultaneous whole-cell recordings from Cre-expressing (Cre:GFP+) and untransfected neighboring cells, providing a rigorous comparison while controlling for presynaptic input. Consistent with our previous work (Gray et al., 2011), GluN1 deletion (ΔGluN1) results in a complete loss of NMDAR-EPSCs by P15 (Fig. 2B,C; control, 82.1 ± 15.7 pA; ΔGluN1, 1.75 ± 0.53 pA; n = 5, t(4) = 5.021, p = 0.007, paired t test). As expected, deletion of GluN1 prevented LTD in the presence of L689 (Fig. 2D-F; control, 73.7 ± 3.5%, n = 8; ΔGluN1, 99.8 ± 5.2%, n = 8; t(14) = 4.194, p = 0.0009, t test). Together, these results demonstrate that non-ionotropic LTD is dependent on NMDARs.
Figure 2.

Single-neuron deletion of GluN1 prevents non-ionotropic LTD. A, Schematic of experimental preparation. GluN1fl/fl mice were injected with AAV1-Cre:GFP at P0 for conditional deletion of GluN1. After 15–21 d, dual whole-cell recordings were made from neighboring transduced and control neurons. Representative image of the sparse transduction of CA1 pyramidal cells with AAV1-Cre:GFP counterstained with DAPI. Scale bar, 100 μm. B, C, NMDAR-EPSCs are eliminated by 15–21 d. B, Scatterplot of individual neuron pairs (open circles) and averaged pair ± SEM (solid circle). Sample trace scale bars indicate 100 ms, 40 pA. C, Average NMDAR-EPSC amplitudes for control (82.1 ± 15.7 pA, n = 5) and Cre:GFP+ neurons (1.75 ± 0.53 pA, n = 5; t(4) = 5.021, p = 0.007, paired t test). D–F, Deletion of GluN1 prevents LTD. D, Averaged whole-cell LTD experiments and representative traces (10 ms, 50 pA). E, Cumulative distribution of experiments in D. F, Average percentage depression relative to baseline; control neurons (73.7 ± 3.5%, n = 8), Cre:GFP+ neurons (ΔGluN1: 99.8 ± 5.2%, n = 8; t(14) = 4.194, *p = 0.0009, t test).
Non-ionotropic LTD is independent of GluN2 subtype
We next assessed the contribution of individual GluN2 subtypes to non-ionotropic LTD using single-neuron deletion of GluN2A and GluN2B. As with GluN1, we performed simultaneous whole-cell recordings of CA1 pyramidal neurons in Grin2Afl/fl and Grin2Bfl/fl mice transduced with AAV1-Cre:GFP at P0. Deletion of GluN2A (ΔGluN2A) resulted in no change in the NMDAR-EPSC amplitude (Fig. 3A,B; control: 102.8 ± 15.2 pA; ΔGluN2A: 96.4 ± 11.9 pA; n = 6, t(5) = 0.9913, p = 0.367, paired t test) but a greatly prolonged EPSC decay (Fig. 3A,C; control: 230.4 ± 8.5 ms; ΔGluN2A: 414.4 ± 13.3 ms; n = 6, t(5) = 13.35, p < 0.0001, paired t test). This is consistent with our previous results (Gray et al., 2011) and represents a pure population of GluN2B-containing NMDARs. Deletion of GluN2A did not affect the expression of non-ionotropic LTD (Fig. 3D–F; control: 77.1 ± 5.8%, n = 6; ΔGluN2A: 65.1 ± 6.2%, n = 6; t(10) = 1.431, p = 0.183, t test). Importantly, in interleaved experiments, AP5 continued to block LTD (Fig. 3G–I; control: 97.0 ± 9.0%, n = 6; ΔGluN2A: 96.7 ± 6.0%, n = 6; t(10) = 0.0274, p = 0.979, t test) demonstrating that NMDAR dependence is maintained.
Figure 3.

Single-neuron deletion of GluN2A does not prevent non-ionotropic LTD. A–C, Single-neuron deletion of GluN2A. A, Scatterplot of individual neuron pairs (open circles) and averaged pair ± SEM (solid circle). Sample trace scale bars indicate 100 ms, 40 pA. B, Average NMDAR-EPSC amplitudes for control (102.8 ± 15.2 pA, n = 6) and Cre:GFP+ neurons (96.4 ± 11.9 pA, n = 6); p = 0.48. C, GluN2A deletion results in significantly longer decay kinetics (control: 230.4 ± 8.5 ms, Cre:GFP+: 414.4 ± 13.3 ms; p < 0.0001). D–F, GluN2A deletion does not block LTD. D, Averaged whole-cell LTD experiments and representative traces (10 ms, 50 pA). E, Cumulative distribution of experiments in D. F, Average percentage depression relative to baseline; control neurons (77.1 ± 5.8%, n = 6), Cre:GFP+ neurons (ΔGluN2A: 65.1 ± 6.2%, n = 6; t(10) = 1.431, p = 0.183, t test) G–I, LTD after GluN2A deletion is still blocked by AP5. G, Averaged whole-cell LTD experiments and representative traces (10 ms, 50 pA). H, Cumulative distribution of experiments in G. I, Summary graph of average percentage depression relative to baseline; control neurons (97.0 ± 9.0%, n = 6), Cre:GFP+ neurons (ΔGluN2A: 96.7 ± 6.0%, n = 6; t(10) = 0.0274, p = 0.979, t test).
Single-neuron deletion of GluN2B (ΔGluN2B) resulted in a significant speeding of the NMDAR-EPSC decay time (Fig. 4A,C; control: 233.7 ± 8.2 ms; ΔGluN2B: 79.0 ± 2.9 ms; n = 6, t(5) = 20.10, p < 0.0001, paired t test) consistent with a pure population of GluN2A-containing NMDARs (Gray et al., 2011). Additionally, there was also a 30–40% reduction in the NMDAR-EPSC amplitude (Fig. 4A,B; control: 90.1 ± 12.8 pA; ΔGluN2B: 58.1 ± 7.2 pA; n = 6, t(5) = 3.078, p = 0.028, paired t test), as described previously (Gray et al., 2011). The simultaneous changes in NMDAR-EPSC amplitude and decay leads to a large decrease in charge transfer that could affect the interpretation of subunit dependence in LTD. However, deletion of GluN2B did not affect the expression of non-ionotropic LTD (Fig. 4D–F; control: 76.1 ± 6.8%, n = 8; ΔGluN2B: 74.3 ± 8.1%, n = 9; t(15) = 0.1662, p = 0.870, t test) and this LTD remained NMDAR-dependent (Fig. 4G–I; control: 98.8 ± 7.3%, n = 4; ΔGluN2B: 96.9 ± 8.6%, n = 4; t(6) = 0.1717, p = 0.869, t test). Together, these results show that the expression of NMDAR-dependent non-ionotropic LTD requires neither GluN2A nor GluN2B.
Figure 4.

Single-neuron deletion of GluN2B does not prevent non-ionotropic LTD. A–C, Single-neuron deletion of GluN2B. A, Scatterplot of individual neuron pairs (open circles) and averaged pair ± SEM (solid circle). Sample trace scale bars indicate (100 ms, 40 pA). B, Average NMDAR-EPSC amplitudes for control (90.1 ± 12.8 pA, n = 6) and Cre:GFP+ neurons (58.1 ± 7.2 pA, n = 6); p = 0.016. C, GluN2A deletion results in significantly faster decay kinetics (control: 233.7 ± 8.2 ms, Cre:GFP+: 79.0 ± 2.9 ms; p < 0.0001). D–F, GluN2B deletion does not block LTD. D, Averaged whole-cell LTD experiments and representative traces (10 ms, 50 pA). E, Cumulative distribution of experiments in D. F, Average percentage depression relative to baseline; control neurons (76.1 ± 6.8%, n = 8), Cre:GFP+ neurons (ΔGluN2B: 74.3 ± 8.1%, n = 9; t(15) = 0.1662, p = 0.870, t test). G–I, LTD after GluN2B deletion is still blocked by AP5. G, Averaged whole-cell LTD experiments and representative traces (10 ms, 50 pA). H, Cumulative distribution of experiments in G. I, Summary graph of average percentage depression relative to baseline; control neurons (98.8 ± 7.3%, n = 4), Cre:GFP+ neurons (ΔGluN2B: 96.9 ± 8.6%, n = 4; t(6) = 0.1717, p = 0.869, t test).
Ionotropic LTD is independent of GluN2 subtype
As some groups have failed to detect non-ionotropic LTD (Babiec et al., 2014; Gray et al., 2016), we examined the role of GluN2A and GluN2B in classical “ionotropic” LTD experiments in the absence of L689. Again, we found that both GluN2A-lacking and GluN2B-lacking neurons expressed LTD that was indistinguishable from control neurons (Fig. 5; control: 75.5 ± 5.7%, n = 8; ΔGluN2A: 68.9 ± 6.5%, n = 9; ΔGluN2B: 81.6 ± 8.9%, n = 9; control:ΔGluN2A: t(15) = 0.7582, p = 0.460, t test; control:ΔGluN2B: t(15) = 0.5557, p = 0.556, t test). Together, these findings provide rigorous evidence that NMDAR-mediated LTD is independent of GluN2 subunit composition.
Figure 5.
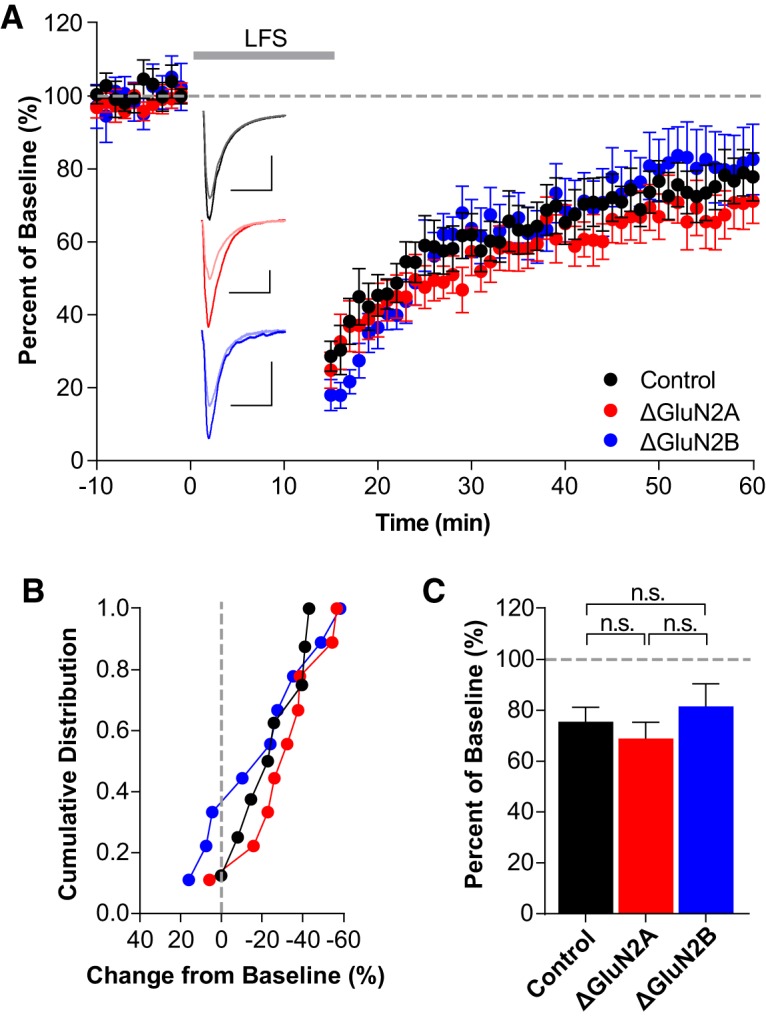
Single-neuron deletion of either GluN2A or GluN2B does not prevent ionotropic LTD. A, Averaged whole-cell LTD experiments and representative traces (10 ms, 50 pA). B, Cumulative distribution of experiments in A. C, Average percentage depression relative to baseline; control neurons (75.5 ± 5.7%, n = 8), ΔGluN2A neurons (68.9 ± 6.5%, n = 9), ΔGluN2B neurons (81.6 ± 8.9%, n = 9). There were no significant differences between control experiments and either GluN2A deletion (t(15) = 0.7582, p = 0.460, t test) or GluN2B deletion (t(15) = 0.5557, p = 0.556, t test), or between ΔGluN2A and ΔGluN2B (t(16) = 1.151, p = 0.267, t test).
Discussion
Because major forms of both LTP and LTD are mediated by the NMDAR, it has long been hypothesized that the GluN2 subunit composition dictates the directionality of synaptic plasticity. This was an attractive hypothesis for a number of reasons. First, GluN2A and GluN2B confer distinct kinetic properties to synaptic NMDARs (Cull-Candy and Leszkiewicz, 2004) that could lead to the different levels of postsynaptic Ca2+ influx thought to underlie LTP and LTD (Dudek and Bear, 1992; Cummings et al., 1996; Yang et al., 1999; Rubin et al., 2005). Second, there is an activity-dependent developmental switch in synaptic NMDAR subunit composition in which predominantly GluN2B-containing NMDARs are replaced or supplemented by GluN2A (Sheng et al., 1994; Roberts and Ramoa, 1999). This subunit switch is thought to be a form of metaplasticity that alters the threshold and possibly the directionality of NMDAR-mediated synaptic plasticity (Quinlan et al., 1999; Dumas, 2005; Yashiro and Philpot, 2008; Gray et al., 2011). Third, GluN2A and GluN2B have long, highly divergent intracellular C-terminal domains that mediate an array of distinct protein-protein interactions that could be coupled to different downstream signaling pathways (Sanz-Clemente et al., 2013).
Numerous studies have set out to test the hypothesis that bidirectional plasticity is dictated by the GluN2 subunit composition, but their results have been inconsistent and conflicting, especially for LTD (for review, see Shipton and Paulsen, 2014). These inconsistent results are likely due to issues with GluN2 subunit-selective pharmacology (Neyton and Paoletti, 2006). Thus we have used a mosaic genetic approach to delete NMDAR subunits in individual hippocampal neurons. Importantly however, genetically dissecting the relative roles of GluN2 subunits in synaptic plasticity is further complicated by altering two variables simultaneously: (1) differential postsynaptic Ca2+ dynamics between GluN2A and GluN2B, and (2) unique protein–protein interactions with their highly divergent C-terminal domains. The recent discovery of non-ionotropic NMDAR-mediated LTD (Nabavi et al., 2013) in which conformational changes in response to repetitive glutamate binding, but not channel opening or Ca2+ influx, is posited to trigger LTD has provided a unique opportunity to reexamine the relative roles of GluN2A and GluN2B in synaptic plasticity. By removing Ca2+ influx as a variable, non-ionotropic LTD allows for a rigorous analysis of the subunit dependence of LTD. Our results here demonstrate conclusively that neither GluN2A nor GluN2B is strictly necessary for NMDAR-dependent LTD at the Schaffer collateral-to-CA1 synapse in the hippocampus during the third postnatal week. At earlier ages, before significant GluN2A expression, deletion of GluN2B would be expected to eliminate all NMDAR-dependent plasticity, including the LTD-like maintenance of silent synapses (Gray et al., 2011). In addition, because synaptic NMDARs in CA1 contain only GluN2A and GluN2B (Gray et al., 2011), GluN2C and GluN2D were not examined, and thus, a role for GluN2C or GluN2D at other excitatory synapses has not been ruled out (Mirante et al., 2014; Qi and Yang, 2015).
Role of GluN2B in LTD
GluN2 subunit selective inhibition is confounded by poor selectivity, incomplete blockade, and complex effects on glutamate affinity (Kew et al., 1996). For GluN1/GluN2B receptors, ifenprodil and Ro 25-6881 are selective negative allosteric modulators that bind to the extracellular N-terminal domains (Hatton and Paoletti, 2005). Some studies have reported block of LTD by ifenprodil or Ro 25-6981 (Liu et al., 2004; Massey et al., 2004; Fox et al., 2006; Izumi et al., 2006; Gerkin et al., 2007; Ge et al., 2010; Dong et al., 2013; Izumi and Zorumski, 2015; Mizui et al., 2015; Yasuda and Mukai, 2015), though others report no effect (Hendricson et al., 2002; Bartlett et al., 2007; Li et al., 2007; Morishita et al., 2007; Kollen et al., 2008; Hanson et al., 2015; Yasuda and Mukai, 2015). However, these inhibitors display partial activity dependence and only block a fraction (∼80%) of synaptic GluN1/GluN2B diheteromers (Fischer et al., 1997; Hatton and Paoletti, 2005; Gray et al., 2011), which could result in variable effects based on drug concentration, slice activity, and preincubation time. Furthermore, N-terminal domain inhibitors only block approximately one-quarter of the current in triheteromeric NMDARs (Hatton and Paoletti, 2005; Hansen et al., 2014) that make up a large proportion of synaptic NMDARs (Gray et al., 2011; Rauner and Köhr, 2011; Tovar et al., 2013). Another interesting consideration is that N-terminal domain inhibitors like ifenprodil decrease the glutamate dissociation rate (Kew et al., 1996; Gray et al., 2011; Tovar and Westbrook, 2012) that may have unknown effects on non-ionotropic LTD. For example, increasing glutamate affinity while preventing channel opening may promote non-ionotropic LTD, and one study reported that ifenprodil actually enhanced the magnitude of LTD (Hendricson et al., 2002). Together, the complexity of GluN2B-selective pharmacology makes firm conclusions on the role of GluN2B in LTD difficult.
Role of GluN2A in LTD
For GluN2A-containing NMDARs, subunit-selective pharmacology is even more problematic. The most widely used antagonist, NVP-AAM007 (NVP), is a competitive glutamate-site antagonist that has only fivefold selectivity for GluN2A over GluN2B (Neyton and Paoletti, 2006). As such, many LTD studies have used concentrations of NVP that antagonize a significant proportion of GluN2B (Liu et al., 2004; Massey et al., 2004; Izumi et al., 2006; Li et al., 2007). By titrating NVP to concentrations that block LTP, some groups found no inhibition of LTD (Liu et al., 2004; Gerkin et al., 2007; Ge et al., 2010), suggesting a key role for GluN2A in LTD, though other studies contradict this finding (Bartlett et al., 2007; Li et al., 2007). Given that NVP is a competitive glutamate site antagonist, NVP should consistently block LTD if only GluN2A is required; however, it remains unknown how NVP affects the triheteromeric receptors that predominate at earlier developmental points when LTD is most reliable. At “selective” concentrations, NVP should bind to the GluN2A glutamate site in triheteromers and block channel opening and LTP. However, it is unknown whether non-ionotropic LTD requires both glutamate sites to be occupied. Thus, continued glutamate binding to the GluN2B subunit in triheteromers could be sufficient to induce non-ionotropic LTD. Indeed, higher NVP concentrations consistently block LTD (Fox et al., 2006; Bartlett et al., 2007). Recently, more selective GluN2A inhibitors have been developed (e.g., TCN201; Bettini et al., 2010; McKay et al., 2012) that block LTD (Izumi and Zorumski, 2015). Interestingly, these inhibitors have been shown to bind allosterically to the dimer interface between GluN1 and GluN2 (Hansen et al., 2012), which may impair conformational-based signaling. Overall, there remains no clear consensus on the role of GluN2A in LTD.
Genetic studies of GluN2 subunits in LTD
In addition to pharmacological studies, a few genetic studies have addressed the GluN2 subunits in LTD. GluN2B KO mice die perinatally due to loss of suckling (Kutsuwada et al., 1996), but can survive by handfeeding. A loss of LTD was observed in hippocampal slices from 3-d-old GluN2B KO mice (Kutsuwada et al., 1996), though at this age, a loss of GluN2B would result in a near complete loss of synaptic NMDARs (Gray et al., 2011). Selective deletion of GluN2B impaired LTD (Brigman et al., 2010) in 14- to 22-week-old mice, though LTD required block of glutamate transporters to induce spillover, presumably to activate extrasynaptic receptors. Importantly, these studies were at the developmental time points that widely deviate from the standard LTD literature making generalization difficult. Interestingly, acute disruption of the interaction of GluN2B with PSD95 using a cell-permeable peptide reduced synaptic GluN2B levels and impaired LTP but had no effect on LTD (Gardoni et al., 2009), consistent with our findings that GluN2B is not necessary. Fewer studies have examined GluN2A, though germline GluN2A KO mice have normal NMDAR-dependent LTD in CA1 (Longordo et al., 2009; Kannangara et al., 2015).
Mechanism of non-ionotropic LTD
The widely accepted model for bidirectional synaptic plasticity mediated by NMDAR activation posits that large, rapid increases in synaptic Ca2+ leads to LTP and prolonged, modest increases in Ca2+ leads to LTD (Lisman, 1989; Malenka, 1994; Neveu and Zucker, 1996). This model has recently been challenged with the finding that repetitive glutamate binding to the NMDAR is sufficient to induce LTD and spine shrinkage, independent of Ca2+ influx (Nabavi et al., 2013; Stein et al., 2015; Carter and Jahr, 2016; Gray et al., 2016), though this remains controversial (Babiec et al., 2014). Importantly, a role for Ca2+ in LTD remains, as intracellular Ca2+ chelators inhibit non-ionotropic LTD (Nabavi et al., 2013). However, clamping intracellular Ca2+ at baseline concentrations while preventing Ca2+ elevations rescued the expression of non-ionotropic LTD (Nabavi et al., 2013). These findings suggest that non-ionotropic LTD involves glutamate-mediated conformational changes in the NMDAR (Dore et al., 2015).
Conformation-based signaling by the NMDAR suggests modulation of receptor interacting partner(s), and the long intracellular C-terminal tails of the GluN2 subunits were the most likely candidates. For example, the death-associated protein kinase 1 (DAPK1) competes with the binding of CaMKII to GluN2B promoting LTD over LTP (Goodell et al., 2017). However, our current results suggest that these interactions are not strictly necessary for LTD and that the minimum sufficient LTD signal is not based on the divergence of the GluN2 subunits. So, without Ca2+ influx or unique GluN2 interacting proteins, what could be the crucial receptor-proximal factor for LTD? Possibilities include shared interactions between GluN2A and GluN2B, interactions with GluN1, or transmembrane or extracellular interactions. For example, protein phosphatase 1 is a key intermediary protein, which is displaced from GluN1 following NMDA binding suggesting a GluN1-proximal mechanism (Aow et al., 2015). Further studies are needed to identify the minimum NMDAR determinates necessary for LTD and to examine whether ionotropic and non-ionotropic LTD are identical or parallel processes.
Footnotes
This work was supported by NIH Grant K08MH100562 and a pilot Grant through the UC Davis Alzheimer's Disease Center P30AG010129. We thank Zaiyang “Sunny” Zhang and Haley Martin for their assistance with mouse breeding and genotyping; and Jamie Krueger, Jalina Graham, and the Wiltgen laboratory for their imaging assistance.
The authors declare no competing financial interests.
References
- Adesnik H, Li G, During MJ, Pleasure SJ, Nicoll RA (2008) NMDA receptors inhibit synapse unsilencing during brain development. Proc Natl Acad Sci U S A 105:5597–5602. 10.1073/pnas.0800946105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi K, Kakizaki T, Kamiya H, Fukaya M, Yamasaki M, Abe M, Natsume R, Watanabe M, Sakimura K (2009) NMDA receptor GluN2B (GluR epsilon 2/NR2B) subunit is crucial for channel function, postsynaptic macromolecular organization, and actin cytoskeleton at hippocampal CA3 synapses. J Neurosci 29:10869–10882. 10.1523/JNEUROSCI.5531-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aow J, Dore K, Malinow R (2015) Conformational signaling required for synaptic plasticity by the NMDA receptor complex. Proc Natl Acad Sci U S A 112:14711–14716. 10.1073/pnas.1520029112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiec WE, Guglietta R, Jami SA, Morishita W, Malenka RC, O'Dell TJ (2014) Ionotropic NMDA receptor signaling is required for the induction of long-term depression in the mouse hippocampal CA1 region. J Neurosci 34:5285–5290. 10.1523/JNEUROSCI.5419-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir ZI, Collingridge GL, Lodge D (2007) Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology 52:60–70. 10.1016/j.neuropharm.2006.07.013 [DOI] [PubMed] [Google Scholar]
- Bettini E, Sava A, Griffante C, Carignani C, Buson A, Capelli AM, Negri M, Andreetta F, Senar-Sancho SA, Guiral L, Cardullo F (2010) Identification and characterization of novel NMDA receptor antagonists selective for NR2A- over NR2B-containing receptors. J Pharmacol Exp Ther 335:636–644. 10.1124/jpet.110.172544 [DOI] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Li L, Geiger JR, Jonas P (2006) Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat Protoc 1:2075–2081. 10.1038/nprot.2006.312 [DOI] [PubMed] [Google Scholar]
- Brigman JL, Wright T, Talani G, Prasad-Mulcare S, Jinde S, Seabold GK, Mathur P, Davis MI, Bock R, Gustin RM, Colbran RJ, Alvarez VA, Nakazawa K, Delpire E, Lovinger DM, Holmes A (2010) Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J Neurosci 30:4590–4600. 10.1523/JNEUROSCI.0640-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BC, Jahr CE (2016) Postsynaptic, not presynaptic NMDA receptors are required for spike-timing-dependent LTD induction. Nat Neurosci 19:1218–1224. 10.1038/nn.4343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H (1983) Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol 334:33–46. 10.1113/jphysiol.1983.sp014478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN (2004) Role of distinct NMDA receptor subtypes at central synapses. Sci STKE 2004:re16. 10.1126/stke.2552004re16 [DOI] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC (1996) Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron 16:825–833. 10.1016/S0896-6273(00)80102-6 [DOI] [PubMed] [Google Scholar]
- Dong Z, Bai Y, Wu X, Li H, Gong B, Howland JG, Huang Y, He W, Li T, Wang YT (2013) Hippocampal long-term depression mediates spatial reversal learning in the Morris water maze. Neuropharmacology 64:65–73. 10.1016/j.neuropharm.2012.06.027 [DOI] [PubMed] [Google Scholar]
- Dore K, Aow J, Malinow R (2015) Agonist binding to the NMDA receptor drives movement of its cytoplasmic domain without ion flow. Proc Natl Acad Sci U S A 112:14705–14710. 10.1073/pnas.1520023112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF (1992) Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci U S A 89:4363–4367. 10.1073/pnas.89.10.4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas TC. (2005) Developmental regulation of cognitive abilities: modified composition of a molecular switch turns on associative learning. Prog Neurobiol 76:189–211. 10.1016/j.pneurobio.2005.08.002 [DOI] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA (1997) Ro 25–6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit characterization in vitro. J Pharmacol Exp Ther 283:1285–1292. [PubMed] [Google Scholar]
- Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T (1994) Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron 13:325–338. 10.1016/0896-6273(94)90350-6 [DOI] [PubMed] [Google Scholar]
- Fox CJ, Russell KI, Wang YT, Christie BR (2006) Contribution of NR2A and NR2B NMDA subunits to bidirectional synaptic plasticity in the hippocampus in vivo. Hippocampus 16:907–915. 10.1002/hipo.20230 [DOI] [PubMed] [Google Scholar]
- Gardoni F, Mauceri D, Malinverno M, Polli F, Costa C, Tozzi A, Siliquini S, Picconi B, Cattabeni F, Calabresi P, Di Luca M (2009) Decreased NR2B subunit synaptic levels cause impaired long-term potentiation but not long-term depression. J Neurosci 29:669–677. 10.1523/JNEUROSCI.3921-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Dong Z, Bagot RC, Howland JG, Phillips AG, Wong TP, Wang YT (2010) Hippocampal long-term depression is required for the consolidation of spatial memory. Proc Natl Acad Sci U S A 107:16697–16702. 10.1073/pnas.1008200107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerkin RC, Lau PM, Nauen DW, Wang YT, Bi GQ (2007) Modular competition driven by NMDA receptor subtypes in spike-timing-dependent plasticity. J Neurophysiol 97:2851–2862. 10.1152/jn.00860.2006 [DOI] [PubMed] [Google Scholar]
- Goodell DJ, Zaegel V, Coultrap SJ, Hell JW, Bayer KU (2017) DAPK1 mediates LTD by making CaMKII/GluN2B binding LTP specific. Cell Rep 19:2231–2243. 10.1016/j.celrep.2017.05.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA (2011) Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron 71:1085–1101. 10.1016/j.neuron.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Zito K, Hell JW (2016) Non-ionotropic signaling by the NMDA receptor: controversy and opportunity. F1000Res 5:F1000. 10.12688/f1000research.8366.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwood S, Kulagowski JJ, Mawer IM, Rowley M, Leeson PD, Foster AC (1995) Allosteric modulation of the glutamate site on the NMDA receptor by four novel glycine site antagonists. Eur J Pharmacol 290:221–226. 10.1016/0922-4106(95)00081-X [DOI] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Traynelis SF (2012) Subunit-selective allosteric inhibition of glycine binding to NMDA receptors. J Neurosci 32:6197–6208. 10.1523/JNEUROSCI.5757-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H, Traynelis SF (2014) Distinct functional and pharmacological properties of triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 81:1084–1096. 10.1016/j.neuron.2014.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Pare JF, Deng L, Smith Y, Zhou Q (2015) Altered GluN2B NMDA receptor function and synaptic plasticity during early pathology in the PS2APP mouse model of Alzheimer's disease. Neurobiol Dis 74:254–262. 10.1016/j.nbd.2014.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton CJ, Paoletti P (2005) Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron 46:261–274. 10.1016/j.neuron.2005.03.005 [DOI] [PubMed] [Google Scholar]
- Hendricson AW, Miao CL, Lippmann MJ, Morrisett RA (2002) Ifenprodil and ethanol enhance NMDA receptor-dependent long-term depression. J Pharmacol Exp Ther 301:938–944. 10.1124/jpet.301.3.938 [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knöpfel T, Erzurumlu RS, Itohara S (2000) Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406:726–731. 10.1038/35021059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF (2015) Sensitivity of N-methyl-d-aspartate receptor-mediated excitatory postsynaptic potentials and synaptic plasticity to TCN 201 and TCN 213 in rat hippocampal slices. J Pharmacol Exp Ther 352:267–273. 10.1124/jpet.114.220582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Auberson YP, Zorumski CF (2006) Zinc modulates bidirectional hippocampal plasticity by effects on NMDA receptors. J Neurosci 26:7181–7188. 10.1523/JNEUROSCI.1258-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannangara TS, Eadie BD, Bostrom CA, Morch K, Brocardo PS, Christie BR (2015) GluN2A−/− mice lack bidirectional synaptic plasticity in the dentate gyrus and perform poorly on spatial pattern separation tasks. Cereb Cortex 25:2102–2113. 10.1093/cercor/bhu017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew JN, Trube G, Kemp JA (1996) A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. J Physiol 497:761–772. 10.1113/jphysiol.1996.sp021807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollen M, Dutar P, Jouvenceau A (2008) The magnitude of hippocampal long term depression depends on the synaptic location of activated NR2-containing N-methyl-d-aspartate receptors. Neuroscience 154:1308–1317. 10.1016/j.neuroscience.2008.04.045 [DOI] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M (1996) Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron 16:333–344. 10.1016/S0896-6273(00)80051-3 [DOI] [PubMed] [Google Scholar]
- Leeson PD, Carling RW, Moore KW, Moseley AM, Smith JD, Stevenson G, Chan T, Baker R, Foster AC, Grimwood S (1992) 4-Amido-2-carboxytetrahydroquinolines: structure–activity relationships for antagonism at the glycine site of the NMDA receptor. J Med Chem 35:1954–1968. 10.1021/jm00089a004 [DOI] [PubMed] [Google Scholar]
- Li R, Huang FS, Abbas AK, Wigström H (2007) Role of NMDA receptor subtypes in different forms of NMDA-dependent synaptic plasticity. BMC Neurosci 8:55. 10.1186/1471-2202-8-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Erzurumlu RS, Chen C, Jhaveri S, Tonegawa S (1994) Whisker-related neuronal patterns fail to develop in the trigeminal brainstem nuclei of NMDAR1 knockout mice. Cell 76:427–437. 10.1016/0092-8674(94)90108-2 [DOI] [PubMed] [Google Scholar]
- Lisman J. (1989) A mechanism for the hebb and the anti-hebb processes underlying learning and memory. Proc Natl Acad Sci U S A 86:9574–9578. 10.1073/pnas.86.23.9574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT (2004) Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304:1021–1024. 10.1126/science.1096615 [DOI] [PubMed] [Google Scholar]
- Longordo F, Kopp C, Mishina M, Luján R, Lüthi A (2009) NR2A at CA1 synapses is obligatory for the susceptibility of hippocampal plasticity to sleep loss. J Neurosci 29:9026–9041. 10.1523/JNEUROSCI.1215-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC. (1994) Synaptic plasticity in the hippocampus: LTP and LTD. Cell 78:535–538. 10.1016/0092-8674(94)90517-7 [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI (2004) Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci 24:7821–7828. 10.1523/JNEUROSCI.1697-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay S, Griffiths NH, Butters PA, Thubron EB, Hardingham GE, Wyllie DJ (2012) Direct pharmacological monitoring of the developmental switch in NMDA receptor subunit composition using TCN 213, a GluN2A-selective, glycine-dependent antagonist. Br J Pharmacol 166:924–937. 10.1111/j.1476-5381.2011.01748.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirante O, Brandalise F, Bohacek J, Mansuy IM (2014) Distinct molecular components for thalamic- and cortical-dependent plasticity in the lateral amygdala. Front Mol Neurosci 7:62. 10.3389/fnmol.2014.00062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina M, Sakimura K (2007) Conditional gene targeting on the pure C57BL/6 genetic background. Neurosci Res 58:105–112. 10.1016/j.neures.2007.01.004 [DOI] [PubMed] [Google Scholar]
- Mizui T, Ishikawa Y, Kumanogoh H, Lume M, Matsumoto T, Hara T, Yamawaki S, Takahashi M, Shiosaka S, Itami C, Uegaki K, Saarma M, Kojima M (2015) BDNF pro-peptide actions facilitate hippocampal LTD and are altered by the common BDNF polymorphism Val66Met. Proc Natl Acad Sci U S A 112:E3067–E3074. 10.1073/pnas.1422336112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC (2007) Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology 52:71–76. 10.1016/j.neuropharm.2006.07.005 [DOI] [PubMed] [Google Scholar]
- Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, Malinow R (2013) Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc Natl Acad Sci U S A 110:4027–4032. 10.1073/pnas.1219454110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveu D, Zucker RS (1996) Postsynaptic levels of [Ca2+]i needed to trigger LTD and LTP. Neuron 16:619–629. 10.1016/S0896-6273(00)80081-1 [DOI] [PubMed] [Google Scholar]
- Neyton J, Paoletti P (2006) Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J Neurosci 26:1331–1333. 10.1523/JNEUROSCI.5242-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Yang Y (2015) Hunger states control the directions of synaptic plasticity via switching cell type-specific subunits of NMDA receptors. J Neurosci 35:13171–13182. 10.1523/JNEUROSCI.0855-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF (1999) Bidirectional, experience-dependent regulation of N-methyl-d-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci U S A 96:12876–12880. 10.1073/pnas.96.22.12876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauner C, Köhr G (2011) Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-d-aspartate receptor population in adult hippocampal synapses. J Biol Chem 286:7558–7566. 10.1074/jbc.M110.182600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EB, Ramoa AS (1999) Enhanced NR2A subunit expression and decreased NMDA receptor decay time at the onset of ocular dominance plasticity in the ferret. J Neurophysiol 81:2587–2591. 10.1152/jn.1999.81.5.2587 [DOI] [PubMed] [Google Scholar]
- Rubin JE, Gerkin RC, Bi GQ, Chow CC (2005) Calcium time course as a signal for spike-timing-dependent plasticity. J Neurophysiol 93:2600–2613. 10.1152/jn.00803.2004 [DOI] [PubMed] [Google Scholar]
- Sanz-Clemente A, Nicoll RA, Roche KW (2013) Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 19:62–75. 10.1177/1073858411435129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY (1994) Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368:144–147. 10.1038/368144a0 [DOI] [PubMed] [Google Scholar]
- Shipton OA, Paulsen O (2014) GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos Trans R Soc Lond B Biol Sci 369:20130163. 10.1098/rstb.2013.0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein IS, Gray JA, Zito K (2015) Non-ionotropic NMDA receptor signaling drives activity-induced dendritic spine shrinkage. J Neurosci 35:12303–12308. 10.1523/JNEUROSCI.4289-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (2012) Amino-terminal ligands prolong NMDA receptor-mediated EPSCs. J Neurosci 32:8065–8073. 10.1523/JNEUROSCI.0538-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, McGinley MJ, Westbrook GL (2013) Triheteromeric NMDA receptors at hippocampal synapses. J Neurosci 33:9150–9160. 10.1523/JNEUROSCI.0829-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS (1999) Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol 81:781–787. 10.1152/jn.1999.81.2.781 [DOI] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD (2008) Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55:1081–1094. 10.1016/j.neuropharm.2008.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Mukai H (2015) Turning off of GluN2B subunits and turning on of CICR in hippocampal LTD induction after developmental GluN2 subunit switch. Hippocampus 25:1274–1284. 10.1002/hipo.22435 [DOI] [PubMed] [Google Scholar]