

Abstract
Pheochromocytomas and paragangliomas are highly vascular tumors of the autonomic nervous system. Germline mutations, including those in hypoxia-related genes, occur in one-third of the cases, but somatic mutations are infrequent in these tumors. Using exome sequencing of 6 paired constitutive and tumor DNA from sporadic pheochromocytomas and paragangliomas, we identified a somatic mutation in the HIF2A (EPAS1) gene. Screening of an additional 239 pheochromocytomas/paragangliomas uncovered three other HIF2A variants in sporadic (4/167, 2.3%), but not in hereditary tumors or controls. Three of the mutations involved proline 531, one of the two residues that controls HIF2α stability by hydroxylation. The fourth mutation, on Ser71, was adjacent to the DNA binding domain. No mutations were detected in the homologous regions of the HIF1A gene in 132 tumors. Mutant HIF2A tumors had increased expression of HIF2α target genes, suggesting an activating effect of the mutations. Ectopically expressed HIF2α mutants in HEK293, renal cell carcinoma 786-0 or rat pheochromocytoma PC12 cell lines showed increased stability, resistance to VHL-mediated degradation, target induction and reduced chromaffin cell differentiation. Furthermore, mice injected with cells expressing mutant HIF2A developed tumors, and those with Pro531Thr- and Pro531Ser mutations had shorter latency than tumors from mice with wild-type HIF2A. Our results support a direct oncogenic role for HIF2A in human neoplasia and strengthen the link between hypoxic pathways and pheochromocytomas and paragangliomas.
Keywords: pheochromocytoma, paraganglioma, hypoxia, cancer, mutations, HIF2A, EPAS1, somatic
Introduction
Pheochromocytomas and paragangliomas are sympathetic neural-derived tumors. Over one-third of these tumors carry germline mutations in one of 10 distinct genes (Jafri and Maher 2012). Remarkably, somatic events in pheochromocytomas have been limited to mutations of some of these susceptibility genes (Burnichon, et al. 2011).
A majority of hereditary pheochromocytomas and paragangliomas are related to the hypoxia pathway through mutations of the VHL gene and those encoding the succinate dehydrogenase (SDH) complex (Jafri and Maher 2012). Loss-of-function mutations in these genes lead to increased stability of main components of the response to low oxygen levels, the hypoxia inducible factor (HIF) proteins (Dahia, et al. 2005; Gimenez-Roqueplo, et al. 2001). The HIF transcription factors form heterodimers comprised of an inducible, oxygen-labile α-subunit (HIF1α, HIF2α or HIF3α) and a stable β-subunit (HIF1β/ARNT) that bind to the promoter of multiple genes associated with angiogenesis, glycolysis and cell growth. Stability of HIFα subunits is dependent on their hydroxylation at two specific prolines by oxygen-dependent prolyl hydroylases (PHDs)(Kaelin and Ratcliffe 2008). Hydroxylation of HIFα subunits at these two prolyl residues (Pro405 and Pro531 in HIF2α), located at the oxygen-dependent degradation domain (ODD), make it recognizable by VHL for binding and subsequent proteasome-mediated degradation. The activity of PHDs is regulated in part by the abundance of the SDH substrate, succinate, which accumulates when SDH is deficient(Selak, et al. 2005). As a result, mutations in VHL or SDH genes confer a distinctive pseudohypoxic profile to pheochromocytomas and paragangliomas(Burnichon et al. 2011; Dahia et al. 2005).
Deregulation of HIFα, specifically HIF2α, has long been implicated in tumorigenesis and in poor prognosis of various human cancers (Qing and Simon 2009). In particular, HIF2α activation was shown to be both necessary and sufficient for development of VHL-null renal cell carcinomas (Kondo, et al. 2003; Kondo, et al. 2002). In addition, other stimuli can lead to increased HIF2α activity in many malignancies (Qing and Simon 2009). However, HIF2α has also been proposed to function as a tumor suppressor in lung adenocarcinoma and gliomas (Acker, et al. 2005; Mazumdar, et al. 2010). Furthermore, germline HIF2A mutations confer susceptibility to familial erythrocytosis, a disorder not typically associated with tumors (Percy, et al. 2006). The absence of direct genetic evidence of HIF2A oncogenicity has contributed to fuel the controversy over its role in cancers. Here we report mutations of the HIF2A gene in pheochromocytomas identified by exome sequencing. Our findings extend recent candidate gene-based observations of HIF2A mutations in these tumors (Comino-Mendez, et al. 2013; Favier, et al. 2012; Lorenzo, et al. 2012; Pacak, et al. 2013; Zhuang, et al. 2012) by showing that these variants confer growth advantage in vivo, supporting their role as driver events in the pathogenesis of these tumors.
Materials and Methods
Patients and samples
Tumor and matching normal tissue were obtained from patients with pheochromocytomas and paragangliomas according to institutionally approved protocols. Details on this cohort of 239 tumors are provided in Supplementary Methods. Normal tissue was obtained from blood or adjacent normal tissue.
DNA and RNA isolation
Blood and tumor DNA and RNA were isolated using with Qiagen kits or Trizol, respectively, following manufacturers’ protocols.
Exome sequencing
We performed whole exome sequencing of matched germline and tumor DNA from six pheochromocytomas/paragangliomas without an identifiable PSG mutation. Libraries were prepared and exomes enriched using Agilent Sure Select 44Mbp at the Beijing Genomics Institute, as described (Gui, et al. 2011). Paired-end reads of enriched exomes were generated in Illumina HiSeq2000 (average depth of 50×) and aligned using the Mosaik algorithm (Hillier, et al. 2008). Variants detected at the somatic level were selected using a modified version of the GigaBayes/FreeBayes pipeline for somatic variants (Marth, et al. 1999). Synonymous sequence variants and those present in the dbSNP135 database were excluded.
Sanger sequencing
Primers flanking the 16 exons of the HIF2A gene and HIF1A exons 9, 12 and 16 (sites of three regulatory hydroxylation domains) were used for PCRs and Sanger sequencing as described in Suppl. Methods.
Microarray-based expression profiling
We used normalized transcription data from 126 pheochromocytomas that were generated using Affymetrix U133 microarrays (GEO accession number GSE28416 and GSE199877) (Dahia et al. 2005; Qin, et al. 2010). Two HIF2A mutant tumors (37 and 208) were available in this cohort and were used for comparisons with tumors from other well-established hereditary pheochromocytomas. Details are described in Suppl. Methods.
Quantitative Real-time PCR (qRT-PCR)
Quantitative Real-time PCR (qRT-PCR) was performed in cDNA isolated from tumors and cell lines (Qin et al. 2010). Additional details are provided in Supplementary Methods.
Clones and constructs
A HA-HIF2alpha-pBabe-puro construct containing the wild-type HIF2A coding sequence, originated from Dr. Willam Kaelin’s laboratory(Kondo et al. 2003), was obtained from Addgene. Site-direct mutagenesis was used to generate each of the HIF2A mutants as described, all maintaining the wild-type Pro405, to mimic the tumor-related mutations (Qin et al. 2010). A HA-tagged full-length VHL construct was cloned into the MSCV-GFP retroviral vector.
Cell Culture, Transductions and Transfections
Cell Culture, Transductions and Transfections HEK293, 786-0 and PC12 cell lines were cultured as described in Suppl. Methods. For hypoxia experiments cells were maintained in a hypoxia chamber at 1% O2 (InVivo200, Ruskinn) for 16hours. Retroviral transduction was performed as described(Qin et al. 2010). MSCV-HA-VHL constructs were sorted by flow cytometry based on GFP expression(Qin et al. 2010). Puromycin-resistant pools or clones were obtained.
Cycloheximide assay
HEK293 cells stably expressing wildtype or the various HIF2A mutants were treated with cycloheximide 100ug/ml for the indicated times.
NGF differentiation and withdrawal
PC12 cells were exposed to NGF 50ng/ml (Harlan) for 5-7days followed by its withdrawal for 24h, as previously described(Lee, et al. 2005).
Nude mouse xenografts
Nude mouse xenograft assays were performed as described(Rai, et al. 2010). Cells stably expressing empty vector, wild-type HIF2α, and 531 mutants were injected subcutaneously in nude mice (6 or 7 mice per group). Animals were sacrificed after 28 days and tumor weights were recorded. Two additional cohorts were performed and in these, animals that developed tumors (n=5-10 per group) were sacrificed when tumor reached maximum size, following UTHSCSA-IACUC approved protocols.
Statistics
The statistical significance of in vitro assays was determined with a 2-tailed Student’s t test; and one-way ANOVA or Kruskal-Wallis for the xenograft assay. In all instances, P<0.05 was considered significant. Data analyses were performed with the Prism software (GraphPad) and Excel (Microsoft).
Results
Sporadic pheochromocytomas and paragangliomas carry somatic mutations of the HIF2A but not HIF1A gene
To discover novel somatic mutations involved in pheochromocytomas and paragangliomas, we generated exome sequences of 6 paired constitutive and tumor DNA samples without mutation in the pheochromocytoma susceptibility genes. We identified 269 somatic variants and herein focus on a heterozygous HIF2A gene mutation affecting the hydroxyl-acceptor residue, proline 531 (Pro531Thr), identified in one of these patients (Table 1).
Table 1.
Clinical and genetic features of patients with pheochromocytomas and paragangliomas carrying somatic HIF2A mutations
| Sample ID |
Age | Gender | Tumor location | Malignancy status |
Other disease |
Familial | Sequence variant (nucleotide) |
Sequence variant (protein) |
Heterozygosity at the mutation site |
Polyphen2/ SIFT* prediction |
|---|---|---|---|---|---|---|---|---|---|---|
| 9 | 10 | M | periadrenal | lymph node metastases | no | N | c.1591C>AC** | p.Pro531Thr | Y | damaging |
| 208 | 47 | F | periaortic | unknown | no | N | c.1591C>TC | p.Pro531Ser | Y | damaging |
| 37 | 72 | F | adrenal | unknown | no | N | c.1592C>TC | p.Pro531Leu | Y | damaging |
| 100288# | ? | F | adrenal | unknown | ? | ? | c.212C>AC | p. Ser71Tyr | Y | damaging |
PolyPhen and Sift are softwares that predict the possible impact of an amino acid substitution on the structure and function of a protein.
Variant found by exome sequencing, confirmed by Sanger sequence;
sample obtained from tumor bank, limited data available
This mutation was confirmed by Sanger sequencing in the tumor and was absent from the patient’s germline DNA (Fig. 1A). We next extended the screening of the entire HIF2A coding sequence to 239 pheochromocytomas/paragangliomas, including 166 sporadic and 72 with a germline PSG mutation. We identified 32 variants (Suppl. Table 1), of which four, including the change originally detected in the exome, fulfilled our criteria for pathogenic significance: involved a conserved residue, were absent in controls, and were predicted to change protein function in silico (Table 1). Similar to the exome finding, two other variants also targeted the 531 codon (Pro531Ser and Pro531Leu). The remaining one was detected immediately distal to the DNA binding domain of HIF2α (Ser71Tyr). All four variants were found in sporadic, but not hereditary, pheochromocytomas and paragangliomas (4 of 167 tumors, 2.3%) and were absent in an ethnically-matched control group (n=214 alleles) or publicly available sequences from the SNP database (http://www.ncbi.nlm.nih.gov/snp/) and 1000 Genomes Project (http://www.1000genomes.org/). The mutations were exclusively detected in tumor DNA of two cases in which germline material was available (Fig. 1A). In two other samples, the somatic status of the mutations cannot be established due to unavailability of constitutive DNA. The homologous region of the HIF1A gene was also sequenced in 132 of these tumors, including the tumors with HIF2A mutations, but no pathogenic variants were identified (Suppl. Table 1).
Fig. 1. Somatic mutations of HIF2A in pheochromocytomas and paragangliomas.
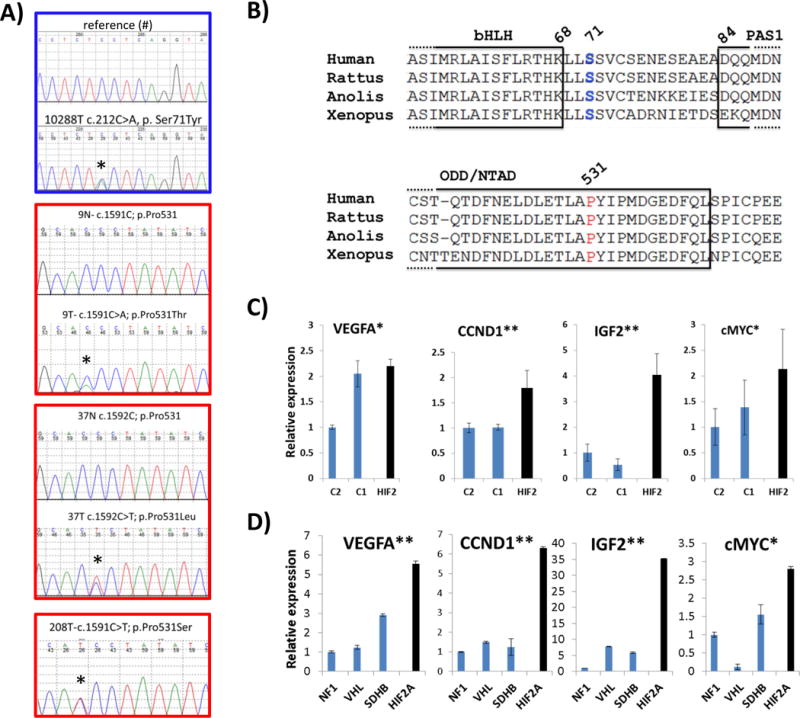
A) Sequence traces of tumor DNA showing the four mutations identified (Ser71Tyr, Pro531Thr, Pro531Leu, Pro531Ser) and either their respective normal sequences from germline DNA or a reference control (#).Mutations are indicated with an asterisk. Border coloring matches the mutated amino acid on HIF2α sequence shown in B. B) Amino acid sequence alignment of HIF2α showing high conservation at the DNA binding domain (basic helix-loop-helix=bHLH), PAS1 (PER-ARNT-SIM) domain and the N-terminal transactivation domain (N-TAD) oxygen-dependent degradation domain (ODD) across various species. The residues mutated in pheochromocytomas and paragangliomas are shown in color, matching the respective DNA sequences displayed on (A). C) Expression levels of VEGFA, CCND1, IGF2 and c-MYC transcripts obtained from microarray-based profiling of combined HIF2A-mutant tumors (37 and 208= HIF2) in comparison with pheochromocytomas mutated for NF1, TMEM17 genes (C2=cluster 2) and VHL, SDHB, SDHD genes (C1=cluster 1), as detailed in Methods. Values of the C2 cluster samples have been set to 1 (* indicates statistically significant difference between HIF2A-mutant and C2 tumors; ** indicates statistically significant difference between HIF2A-mutant samples and both C1- and C2- tumors, at p<0.05). D) Expression levels of VEGFA, CCND1, IGF2 and c-MYC transcripts in one HIF2A-mutated primary tumor (tumor from sample 9) along with other pheochromocytomas carrying mutations of VHL, SDHB or NF1 genes assayed by quantitative real time PCR (qRT-PCR). Samples were run in triplicate and the experiment was repeated at least twice. Results are shown as the average and bars represent standard error of the means. Values of NF1 samples were set to 1. (* indicates statistically significant difference between HIF2A-mutant samples and NF1 tumors, ** indicates statistically significant difference between HIF2A-mutant samples and NF1-, VHL- or SDHB-related tumors, at p<0.05).
Clinically, two of the mutations occurred in paragangliomas, one metastatic and the other recurrent, while the other two were in pheochromocytomas (Table 1). All four tumor DNA sequence traces had a heterozygous appearance, indicating retention of both alleles (Fig. 1A). In one case, sequence of tumor cDNA showed that both alleles were transcribed (Suppl. Fig. 1).
HIF2A mutations induce target genes in primary tumors and cell lines and increase HIF2α stability
The clustering of the mutations (3/4) at the primary site of HIF2α hydroxylation suggested that these variants are functionally relevant (Fig. 1B). To explore the consequences of these HIF2A mutations, we measured expression of HIF target genes in primary tumor samples. Two of the tumors, carrying a mutation on Pro531Ser and Pro531Leu, had been previously included in our global transcriptional study of pheochromocytomas (Dahia et al. 2005). Reassuringly, both HIF2A-mutant tumors aligned with the pseudohypoxic group, comprised of samples carrying germline VHL and SDHB gene mutations (known as cluster 1), by supervised clustering analysis (Dahia et al. 2005). Normalized data from probes encoding HIF target genes were extracted from this dataset and plotted against expression data from other pheochromocytomas carrying either cluster 1 or cluster 2 (RET, NF1 or TMEM127) mutations (Fig. 1C). RNA was also obtained from another HIF2A mutant tumor and compared to genetically defined pheochromocytomas or paragangliomas. Quantitative real-time polymerase chain reaction (qRT-PCR) of HIF-responsive genes encoding VEGFA, cyclin D1 (CCND1), IGF2 and c-MYC, was performed in these tumors (Fig 1D). The HIF2A-mutant tumors, both by microarray and qRT-PCR analyses, displayed increased target gene expression (Fig 1C, 1D). Moreover, cyclin D1 (also known as CCND1) and c-MYC, considered HIF2α-specific growth-promoting determinants in renal cell carcinomas (Gordan, et al. 2007; Raval, et al. 2005), and IGF2, associated with hypoxic neuroblastomas (Pietras, et al. 2009), were also induced, suggesting that these effectors may contribute to the protumorigenic effects of HIF2A mutations in pheochromocytomas/paragangliomas.
The site of the mutations on the critical 531 prolyl residue suggested that these variants affected HIF2A stability. We examined these effects by generating HEK293 cell lines stably expressing each mutation, created by site-directed mutagenesis in a retroviral backbone. Constructs containing wild-type 531 sequence or a HIF2A mutation (Pro531Ala), previously shown to be both resistant to PHD action and defective in VHL binding (Kondo et al. 2002), were also used. All constructs, analogous to the primary tumors, were wild-type for proline 405, the secondary HIF2α hydroxylation site. Stable expression of the HIF2A 531 mutations led to increased stability and prolonged half-life of HIF2A in vitro under normoxia to levels similar to those seen with the Pro531Ala mutant, as shown by a time course assay with cycloheximide (Fig. 2A and Suppl Fig 2). These findings are consistent with structural studies of HIF1α indicating that replacement of the equivalent proline (Pro564) with other residues prevents hydroxylation at this site (Min, et al. 2002). The expression levels of the juxta-DNA binding domain mutation, Ser71Tyr, in contrast, did not differ from the levels of the wild-type construct, suggesting that this mutation does not affect HIF2α stability (Fig. 2A).
Fig. 2. HIF2A mutations lead to prolonged stability of the protein, activation of downstream targets and resistance to VHL-mediated degradation in vitro.
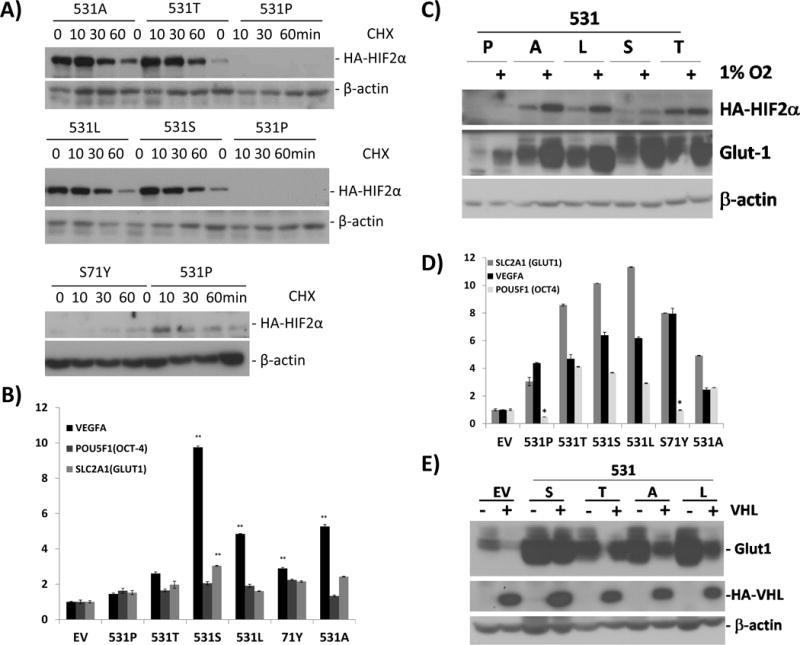
A) HEK293 cell lines were retrovirally infected to stably produce HA-tagged wild-type or the various HIF2A mutants (Pro531Thr=531T, Pro531Leu=531L, Pro531Ser=531S, Ser71Tyr=S71Y) or wild-type (531P) as indicated. The Pro531Ala (P531A) mutant construct is included as a positive control (Kondo et al. 2002). Cell extracts were prepared after cycloheximide exposure for the indicated times and immunoblotted with anti-HA antibody or beta-actin as a loading control. B) Transcript levels of VEGFA, POU5F1(OCT-4) and SLC2A1(GLUT1) of HEK293 cells stably expressing the indicated HIF2A mutants and controls, measured by quantitative real time PCR (qRT-PCR). Results are shown as the average and bars represent standard error of the means. Values were normalized to those of EV control (*indicates statistical significance at p <0.05; **indicates statistical significance at p<0.005 between cells expressing each HIF2A construct vs. EV). C) HEK293 cells expressing HIF2A mutants or controls as above (empty vector=EV) were incubated overnight under normoxia or 1% O2, as shown. Cell extracts were prepared and probed with anti-HA, anti-GLUT1 or a loading control, beta-actin. D) Transcript levels of VEGFA, POU5F1(OCT-4) and SLC2A1(GLUT1) in VHL-reconstituted 786-0 cells stably expressing the indicated HIF2A mutants and controls, measured by qRT-PCR. Results were normalized to those of EV control (*indicates non-significance; all remaining values showed statistical significance at p<0.005 between cells expressing each HIF2A construct vs. EV). E) 786-0 renal carcinoma cells stably expressing the designated 531 HIF2A mutants were reconstituted with an MSCV-HA-VHL retroviral construct or control. Cell extracts were prepared and immunoblotted with GLUT1, HA and beta actin as a loading control.
Mutant HIF2α is stabilized by hypoxia and is resistant to VHL-mediated degradation
Despite already high normoxic levels of mutant HIF2A in HEK293 cells, these constructs could be further stabilized and activated by hypoxia, as measured by an increase in HA-HIF2A and GLUT1 protein abundance, respectively (Fig 2B). This response likely results from the effect of hypoxia on the intact proline 405, which leads to HA-HIF2A product escape from VHL-mediated proteasome degradation.
Similar to the primary tumors, HEK293 cell lines expressing HIF2A mutations showed increased expression of VEGFA, OCT-4/POU5F1 and GLUT1/SLC2A1 transcripts (Fig. 2C). These results suggest that the 531 mutations are associated with constitutive HIF2α activation.
We next stably expressed the HIF2A mutants in the VHL-null 786-0 renal carcinoma cells reconstituted with VHL and measured transcription of target genes. Again, mutants showed increased expression of VEGFA, OCT-4/POU5F1 and GLUT1/SLC2A1 (Fig. 2D). We also compared GLUT1 protein levels in 786-0 cells expressing the various HIF2A mutations without VHL or after its reconstitution. VHL expression normalized GLUT1 in cells expressing endogenous HIF2α and an empty vector control, as expected, but it was unable to overcome the effect of the new mutations or the positive control HIF2A Pro531Ala on this target (Fig. 2E). These results suggest that mutations of this site abrogate VHL binding to the HIF2α ODD (Kondo et al. 2002). Unlike the 531 mutants, HIF2A-Ser71Tyr mutant expression was undetectable in the presence of VHL similar to wild-type HIF2A (Suppl. Fig. 3), suggesting that binding to VHL was not altered by this variant.
HIF2A mutations changes the differentiation pattern of chromaffin cells
To explore the effect of the HIF2A mutations in a more tissue-specific context, we used rat pheochromocytoma PC12 cells. Expression of the four HIF2A mutants also led to increased target transcription in comparison with empty vector-expressing cells (Fig. 3A). PC12 cells are capable of differentiating into neurons after exposure to nerve growth factor (NGF) (Lee et al. 2005). Under these conditions, cells extended neurites and increased expression of chromaffin cell markers chromogranin A (CHGA) and neuropeptide Y (NPY), while transcription of HIF2A, its target, VEGFA and a marker of cell dedifferentiation, Nanog, were decreased (Suppl Fig. 4A). In contrast, exposure of PC12 cells expressing the various HIF2A mutations to NGF for 7 days led to reduced transcription of CHGA and NPY in comparison with cells expressing empty vector (Fig. 3B). Likewise, chromaffin markers NPY and dHAND were downregulated in a HIF2A mutant tumor (Fig. 3C), suggesting that the PC12 model recapitulates in vivo effects of these mutations. Furthermore, markers of cell dedifferentiation or stemness, Nanog, OCT4, JAGGED2, KRT19, SOX9 and CHD2 were increased in HIF2A-mutant pheochromocytomas/paragangliomas (Fig. 3C, 3D). This differential transcription pattern is reminiscent of the effects of hypoxia, and specifically, HIF2α, in promoting an immature, stem-cell like phenotype of the sympathetic-derived tumors neuroblastomas (Pietras et al. 2009). Thus, increased HIF2α activation caused by these mutations may lead to a shift in the NGF-induced transcriptional program. Parenthetically, we did not detect differences in apoptosis of these cells following NGF removal (Suppl. Fig. 4B), in agreement with earlier findings of a HIF-independent, inhibitory effect of pheochromocytoma mutations in sympathetic precursor cell death (Lee et al. 2005). The downregulation of chromaffin markers in both mutant HIF2A cell lines and tumors accompanied by the increased expression of dedifferentiation markers are consistent with the notion that mutant HIF2α may contribute to a less differentiated phenotype of pheochromocytomas and paragangliomas.
Fig. 3. HIF2A mutations activate downstream targets and reduce expression of nerve growth factor (NGF)-induced chromaffin differentiation markers in pheochromocytoma cells.
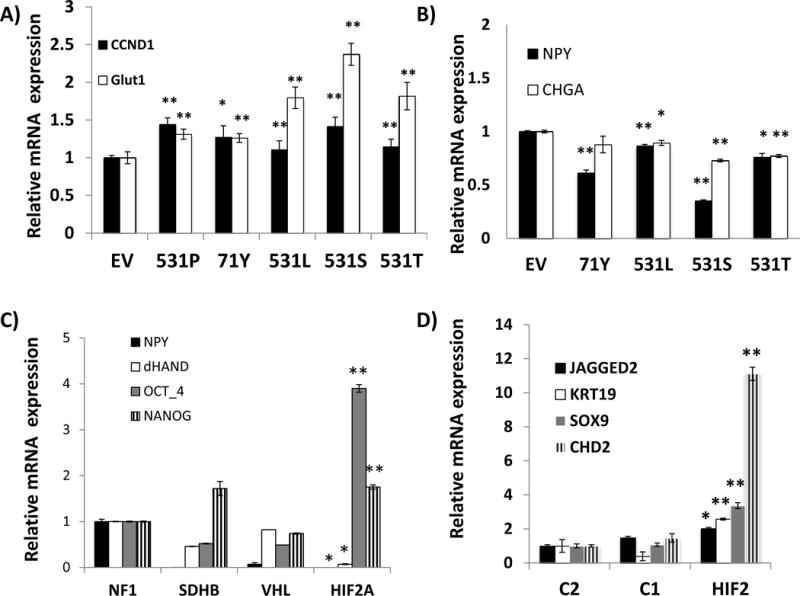
A) Expression levels of CCND1 and GLUT1 (SLC2A1) transcripts of undifferentiated PC12 cells stably expressing an empty vector (EV), the indicated HIF2A mutants (Ser71Tyr=71Y, Pro531Thr=T, Pro531Leu=L, Pro531Ser=S) or a wild-type (P) HIF2A construct by quantitative real time PCR (qRT-PCR). Samples were run in triplicate and experiment was repeated twice. Results are shown as the average and bars represent standard error of the means. Values of EV-expressing cells were set to 1(*indicates statistical significance between each HIF2A-mutant and empty vector at p <0.05; ** indicates statistical significance between each HIF2A-mutant and an empty vector at p <0.001). B) Expression levels of NPY and CHGA transcripts by qRT-PCR of PC12 cells stably expressing the designated HIF2A constructs or an empty vector (EV) after 7 days exposure to 50ng/ml NGF (*p<0.001 EV vs. all mutants, **p<0.0001 EV vs. all mutants), performed as in (A). C) Expression of NPY, dHAND, OCT4 and NANOG transcripts in HIF2A-mutated paraganglioma from sample 9 along with other pheochromocytomas carrying mutations of VHL, SDHB or NF1 genes. Samples were run in triplicate. Results are shown as the average and bars represent standard error of the means. Values of NF1 samples were set to 1. All results were statistically significant between the HIF2A-mutant tumor and NF1 at p<0.01* or p<0.001**) D) Expression of stemness-related genes JAGGED2, KRT19, SOX9 and CHD2 in transcripts obtained from microarray-based profiling of combined HIF2A-mutant tumors (37 and 208= HIF2) in comparison with pheochromocytomas mutated for NF1, TMEM17 genes (C2=cluster 2) and VHL, SDHB, SDHD genes (C1= cluster 1), as detailed in Methods. Values of the C2 cluster samples have been set to 1 (* indicates statistically significant difference between HIF2A-mutant and C2 tumors; ** indicates statistically significant difference between HIF2A-mutant samples and both C1- and C2- tumors, at p<0.05).
HIF2A mutations promote tumor growth in mice
No changes were seen in HIF2A-mutant cell proliferation in vitro, similar to reports of HIF2A overexpression in renal carcinomas (Suppl. Fig 5)(Kondo et al. 2002). Thus we explored the transforming properties of the 531 mutations with a murine xenograft model. Six to 7 nude mice per group were injected subcutaneously with HEK293 cells expressing empty vector, wild-type or mutant HIF2A constructs. While mice carrying vector-expressing cells did not form tumors after 28 days, most HIF2A-injected mice developed macroscopic tumors (Fig. 4A). In particular, animals injected with Pro531Thr-expressing cells produced significantly larger tumors than any other cell group (Fig. 4A, 4B, p<0.05). These results were validated in two other cohorts, in which 5-10 mice per group developed tumors. In these groups, the tumor latency in mice carrying HIF2A Pro531Thr and Pro531Ser-expressing cells was shorter than those injected with HIF2A without these mutations (p<0.05, Fig 4C). In our model, the remaining mutants, Pro531Leu and the positive control Pro531Ala, did not yield significantly more aggressive tumors (Fig. 4A, 4B). The mild phenotype of the Pro531Ala-injected cells might suggest a cell-type dependent effect. It is also possible that the transcriptional transactivation properties of the amino acid sequence surrounding the Pro531 region may contribute to mutation-specific differences (Kondo et al. 2003; Yan, et al. 2007). Additional in vitro and in vivo models may elucidate the reasons for this genotype-phenotype variability.
Fig. 4. Oncogenic effects of the HIF2A mutants in vivo.
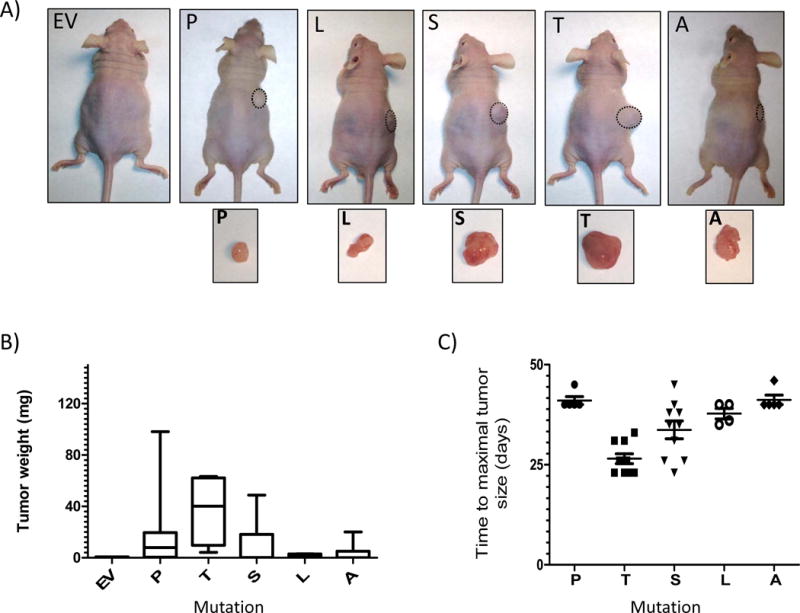
Forty-two nude mice (n=6 or 7 per group) were subcutaneously injected with 5×106 HEK293 cells stably expressing the indicated constructs (empty vector=EV, Pro531Thr=T, Pro531Leu=L, Pro531Ser=S, Pro531Ala=A) or a wild-type Pro531 (P) HIF2A construct. Animals were sacrificed after 28 days and tumors were removed. A) Representative mice and respective tumors are shown. None of the EV-injected mice developed tumors within the observed period. B) Weight (in mg) of tumors plotted by the type of mutation (*EV vs. T p<0.05 by Kruskal Wallis). C) Two additional cohorts (n= 5-10 mice per group) were used and animals were sacrificed when tumors reached maximum size in accordance with IACUC approved protocols. Mice that did not develop tumors were excluded from this analysis. Time from injection until maximum size (in days) was plotted (*P vs. T, and *P vs. S p<0.05).
Discussion
Our data support an oncogenic role for HIF2A in cancer and confirm recent reports of somatic mutations of this gene in pheochromocytomas and paragangliomas, which became available as we were completing this study (Burnichon, et al. 2012; Comino-Mendez et al. 2013; Favier et al. 2012; Lorenzo et al. 2012; Zhuang et al. 2012). Furthermore, our work expands on these observations by showing the tumorigenic effect of mutations of the 531 residue in vivo. Moreover, our finding of the inhibitory effect of these mutations on chromaffin marker expression suggest that activated HIF2α may contribute to a more aggressive phenotype of target cells, similar to poorly oxygenated areas of various cancers(Semenza 2012). Despite its effect on increased transcription of target genes, expression of the Ser71Tyr mutant did not enhance HIF2α protein stability or resistance to VHL degradation, so a potential ‘passenger’ effect of this mutation cannot be ruled out.
Despite notable recent advances, the genetic basis of most pheochromocytomas, including those with a pseudohypoxic profile, still remains unresolved. Future investigations may uncover other genetic variations that can strengthen the link between the hypoxia pathways and cancer.
Supplementary Material
Acknowledgments
This work is dedicated to the memory of Alberto Antônio Dahia. We thank the International Familial Consortium members for their continuing collaboration and the patients and their families for their participation.
Funding
This work was supported by a Voelcker Fund Award and a CTSA-IIMS pilot project as part of the NIH-8UL1TR000149 to P.L.M.D; a Voelcker Fund and NCI-5R01CA138747 to R.C.T.A. The Flow Cytometry and DNA Nucleic Acid Core are supported by UTHSCSA, NIH-NCI P30-CA54174 to the Cancer Therapy and Research Center (CTRC at UTHSCSA).
Footnotes
Declaration of Interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Author Contributions
R.A.T. designed experiments, processed and extracted nucleic acids from tissues and cell lines, validated mutations, performed quantitative gene expression and copy-number studies, statistical analyses and preliminary exome analysis. Y.Q. was responsible for generating most stable cell line pools and clones, and for performing cellular and biochemical studies. S.S. generated cell lines and performed biochemical analyses. N.P.M. assisted with sample processing and sequencing and generated mutagenized clones. Q.L. and S-W.K. performed the mouse xenograft studies, Y.D. assisted with tumor and cell lines expression analysis. M.A.P.P. provided clinical information. S.P.A.T. provided clinical samples and reviewed patients’ records. X.S. performed bioinformatic and somatic analysis of exomes. R.C.T.A. provided reagents, designed and performed experiments, managed xenograft studies and analyzed the data. P.L.M.D. conceived the study, designed and performed experiments, analyzed the data and wrote the manuscript, with input from R.A.T., R.C.T.A and the other authors.
References
- Acker T, Diez-Juan A, Aragones J, Tjwa M, Brusselmans K, Moons L, Fukumura D, Moreno-Murciano MP, Herbert JM, Burger A, et al. Genetic evidence for a tumor suppressor role of HIF-2alpha. Cancer Cell. 2005;8:131–141. doi: 10.1016/j.ccr.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Burnichon N, Cascon A, Schiavi F, Morales NP, Comino-Mendez I, Abermil N, Inglada-Perez L, de Cubas AA, Amar L, Barontini M, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res. 2012;18:2828–2837. doi: 10.1158/1078-0432.CCR-12-0160. [DOI] [PubMed] [Google Scholar]
- Burnichon N, Vescovo L, Amar L, Libe R, de Reynies A, Venisse A, Jouanno E, Laurendeau I, Parfait B, Bertherat J, et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet. 2011;20:3974–3985. doi: 10.1093/hmg/ddr324. [DOI] [PubMed] [Google Scholar]
- Comino-Mendez I, de Cubas AA, Bernal C, Alvarez-Escola C, Sanchez-Malo C, Ramirez-Tortosa CL, Pedrinaci S, Rapizzi E, Ercolino T, Bernini G, et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt069. Online February 14, 2013. [DOI] [PubMed] [Google Scholar]
- Dahia PL, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M, Kung AL, Sanso G, Powers JF, Tischler AS, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005;1:72–80. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier J, Buffet A, Gimenez-Roqueplo AP. HIF2A mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367:2161. doi: 10.1056/NEJMc1211953. author reply 2161-2162. [DOI] [PubMed] [Google Scholar]
- Gimenez-Roqueplo AP, Favier J, Rustin P, Mourad JJ, Plouin PF, Corvol P, Rotig A, Jeunemaitre X. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet. 2001;69:1186–1197. doi: 10.1086/324413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, Barnett D, Fox P, Glasscock JI, Hickenbotham M, Huang W, et al. Whole-genome sequencing and variant discovery in C. elegans. Nat Methods. 2008;5:183–188. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- Jafri M, Maher ER. The genetics of phaeochromocytoma: using clinical features to guide genetic testing. Eur J Endocrinol. 2012;166:151–158. doi: 10.1530/EJE-11-0497. [DOI] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–246. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG, Jr, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: Developmental culling and cancer. Cancer Cell. 2005;8:155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Lorenzo FR, Yang C, Ng Tang Fui M, Vankayalapati H, Zhuang Z, Huynh T, Grossmann M, Pacak K, Prchal JT. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl) 2012 doi: 10.1007/s00109-012-0967-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth GT, Korf I, Yandell MD, Yeh RT, Gu Z, Zakeri H, Stitziel NO, Hillier L, Kwok PY, Gish WR. A general approach to single-nucleotide polymorphism discovery. Nat Genet. 1999;23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- Mazumdar J, Hickey MM, Pant DK, Durham AC, Sweet-Cordero A, Vachani A, Jacks T, Chodosh LA, Kissil JL, Simon MC, et al. HIF-2alpha deletion promotes Kras-driven lung tumor development. Proc Natl Acad Sci U S A. 2010;107:14182–14187. doi: 10.1073/pnas.1001296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Jr, Pavletich NP. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- Pacak K, Jochmanova I, Prodanov T, Yang C, Merino M, Fojo T, Prchal JT, Tischler AS, Lechan RM, Zhuang Z. A new syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol. 2013 doi: 10.1200/JCO.2012.47.1912. 2013 Mar 18. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH, McMullin MF, Lee FS. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci U S A. 2006;103:654–659. doi: 10.1073/pnas.0508423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras A, Hansford LM, Johnsson AS, Bridges E, Sjolund J, Gisselsson D, Rehn M, Beckman S, Noguera R, Navarro S, et al. HIF-2alpha maintains an undifferentiated state in neural crest-like human neuroblastoma tumor-initiating cells. Proc Natl Acad Sci U S A. 2009;106:16805–16810. doi: 10.1073/pnas.0904606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–233. doi: 10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing G, Simon MC. Hypoxia inducible factor-2alpha: a critical mediator of aggressive tumor phenotypes. Curr Opin Genet Dev. 2009;19:60–66. doi: 10.1016/j.gde.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai D, Kim SW, McKeller MR, Dahia PL, Aguiar RC. Targeting of SMAD5 links microRNA-155 to the TGF-{beta} pathway and lymphomagenesis. Proc Natl Acad Sci U S A. 2010;107:3111–3116. doi: 10.1073/pnas.0910667107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak MA, Armour SM, Mackenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends in Pharmacological Sciences. 2012;33:207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Bartz S, Mao M, Li L, Kaelin WG., Jr The hypoxia-inducible factor 2alpha N-terminal and C-terminal transactivation domains cooperate to promote renal tumorigenesis in vivo. Mol Cell Biol. 2007;27:2092–2102. doi: 10.1128/MCB.01514-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, Popovic V, Stratakis CA, Prchal JT, Pacak K. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367:922–930. doi: 10.1056/NEJMoa1205119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.