


Abstract
The dimer of the amyloid-β peptide Aβ of 42 residues is the smallest toxic species in Alzheimer’s disease, but its equilibrium structures are unknown. Here we determined the equilibrium ensembles generated by the four atomistic OPLS-AA, CHARMM22*, AMBER99sb-ildn, and AMBERsb14 force fields with the TIP3P water model. On the basis of 144 µs replica exchange molecular dynamics simulations (with 750 ns per replica), we find that the four force fields lead to random coil ensembles with calculated cross-collision sections, hydrodynamics properties, and small-angle X-ray scattering profiles independent of the force field. There are, however, marked differences in secondary structure, with the AMBERsb14 and CHARMM22* ensembles overestimating the CD-derived helix content, and the OPLS-AA and AMBER99sb-ildn secondary structure contents in agreement with CD data. Also the intramolecular beta-hairpin content spanning residues 17–21 and 30–36 varies between 1.5% and 13%. Overall, there are significant differences in tertiary and quaternary conformations among all force fields, and the key finding, irrespective of the force field, is that the dimer is stabilized by nonspecific interactions, explaining therefore its possible transient binding to multiple cellular partners and, in part, its toxicity.
Graphical abstract
1. INTRODUCTION
The wild-type (WT) Aβ42 peptide, DAEFRHDSGYEVHHQKLVFFAEDVGSN KGAIIGLMVGGVVIA, with two hydrophilic (residues D1-K16 and E22-G29) and hydrophobic (residues L17-A21 and A30-A42) regions, aggregates faster in vitro and is more toxic than the most abundant Aβ40 peptide.1 Toxicity arises from all Aβ42 aggregate sizes, though the Aβ42 monomer with the Artic mutation (E22G) has been reported to be toxic in contrast to the WT monomer.2 The WT Aβ dimers, which are the smallest toxic species leading to neuritic degeneration,3 are structurally heterogeneous in aqueous solution.4 High-resolution structure determination of dimers is, however, of paramount importance to understand the primary nucleation process and how they interact with all partners in the extracellular milieu.5
Atomistic or coarse-grained (GC) molecular dynamics (MD) and replica exchange molecular dynamics (REMD) simulations are often used to generate the conformational ensemble of intrinsically disordered proteins (IDPs), but the results vary with the protein force field and the analysis tools used.6–17 On one hand, all-atom simulations of Aβ40 and Aβ42 monomers11,12,18 show transient antiparallel interactions between the residues L17-A21 and A30-L34 in Aβ40 and Aβ42 and additional β-hairpin spanning residues 39–41 in Aβ42, but their probabilities vary between OPLS, AMBER99sb-ildn, and CHARMM22*. On the other hand, marked differences between simulations of the three force fields are observed for the N-terminus, where AMBER99sb-ildn predicts some β-sheet character, while OPLS and CHARMM22* predict more turn.11 Although there is difference in both compact maps and secondary structures between the force fields, all compare well to nuclear magnetic resonance (NMR) Aβ42 observables such as chemical shifts and J couplings.12 Interestingly, on the basis of a complete set of chemical shifts and J couplings, the 34 N-terminal residues behave identically in the two monomeric species at pH 7, and the Aβ40 and Aβ42 ensembles resemble random coil, with the possibility of transient heterogeneous structured conformations of low probability.19
Our understanding on how the equilibrium ensemble of the Aβ42 dimer is impacted by the protein force field is even more limited and contradictory, despite numerous CG simulations,7,8 all-atom MD simulations of 1 to 4 µs in explicit solvent using OPLS20,21 and AMBER99sb-ildn22 with TIP3P water model, and OPLS/TIP3P REMD simulations, with replicas varying between 200 ns23 and 400 ns.24 We recently reported the REMD results of Aβ42 dimer using AMBER99sb-ildn/TIP3P up to 530 ns per replica, with 48 replicas starting from different chain dimensions and secondary structure contents.25 We characterized hundreds of free energy minima, in contrast to previous simulations that identified a few dominant minima, with all predicted states differing from the U-shape or S-shape conformations of the peptides observed in the fibrils. So a natural question is whether these free energy minima are preserved with other protein force fields.
To this end, we determine the Aβ42 dimer ensembles obtained using four force fields coupled to extensive REMD simulations (48 replicas with 750 ns per replica and force field), and compare them with available experimental circular dichroism (CD) and ion-mobility mass-spectrometry (IM-MS) data. The force fields include OPLS-AA,26 CHARMM22*,27 AMBER99sb-ildn,28 and AMBERsb1429 coupled to TIP3P30 model. Note the three former protein force fields have already tested on the Aβ42 monomer, we use the modified TIP3P model for CHARMM22* simulation,31 and it is beyond the scope of this paper to explain the main differences between the four force fields. Then, we compare the generated equilibrium ensembles with experimental observables that could be measured, such as hydrodynamic properties from NMR diffusion experiments, small-angle X-ray scattering (SAXS), and six sets of NMR J couplings.
2. MATERIAL AND METHODS
The simulation protocol and analysis methods have been described elsewhere.25 In brief, the same ensemble of initial dimer structures for the 48 replicas was used and has a secondary structure content of 11.7% β-strand, 7% α-helix, and 82% coil-turn. Simulations were carried out at pH 7 in a cubic TIP3P box at peptide concentration of 14.8 mM, in which the peptides have NH3+ and CO2− termini, protonated Arg and Lys, deprotonated Glu and Asp, and neutral His with a protonated Nε atom, and are neutralized by Na+ ions resulting in 21858 atoms.
GROMACS with SHAKE allows a time step of 2 fs.32 Electrostatic interactions used particle mesh Ewald method with a cutoff of 1.1 nm,33 van der Waals interactions used a cutoff of 1.2 nm, and the velocity-rescaling thermostat was employed.34 REMD was performed with 48 replicas from 300 to 400 K, exchanges between two replicas attempted every 2 ps, leading to a mean acceptance ratio of 25–30% and each replica ran for 750 ns.
The conformations were analyzed by their secondary structure contents using STRIDE program35 (with helix content including 3–10 helix, Pi helix and α-helix), their solvent accessible surface areas (SASA) and radii of gyration using GROMACS tools, their side-chain–side-chain (SC–SC) contact maps (with a contact formed if the minimum distance between any carbon, oxygen and nitrogen side chain atoms is less than 4.5 Å), and their total numbers of SC–SC contacts calculated by Q values.36 A β-hairpin is considered formed if there are at least two backbone H-bonds between consecutive β-strands, with each strand consisting of three consecutive residues belonging to the Ramachandran β-region.36 A H-bond is formed if the acceptor–donor distance is <3.5 Å and the acceptor–donor–H angle is less than 30°.37
Rather than projecting the free energy surfaces on selected order parameters, we used our general method to identify accurately all the states of the dimer.38
First, the single-molecule states, i.e., states of the intramolecular conformation of one chain in the presence of another chain, are identified as the minima on the free energy landscape F(v1,v2) = −kBT[ln P(v1,v2) − ln Pmax]. Here, (v1,v2) are the first two principal components corresponding to the largest eigenvalues obtained from the dihedral principal component analysis (dPCA)37 of the backbone dihedral angles of two chains, and P(v1,v2) is the probability distribution obtained from a histogram of the four combined REMD trajectories (each per system) at 315 K. Pmax denotes the maximum of the distribution, which is subtracted to ensure that F(v1,v2) = 0 for the lowest free energy minimum. The single-molecule states were also analyzed using the standard Cα root-mean square deviations (RMSD) using GROMACS tools.
Second, the double-molecule states, i.e., describing the intermolecular positions and orientations of two chains, are determined in the same way, except that the PCA was performed on the inverse distances between interchain Cα atoms.
Third, the overall states of the Aβ42 dimer are described as the product of the single- and double-molecule states.38 The population and the conformational center of each minimum on all free energy landscapes were determined by k-means clustering method as reported elsewhere.25,36,37
The collision cross sections (CCS) of Aβ42 dimer were calculated using MOBCAL39 and the trajectory method as reported in other simulations.36,37 Small-angle X-ray scattering (SAXS) curves of Aβ42 dimer were calculated using WAXSiS and these predicted SAXS profiles can be directly compared to experimental profiles.40 The translational diffusion and rotational diffusion constants of Aβ42 dimer were calculated using HYDROPRO.41 The six sets of J-coupling constants of Aβ42 dimer were calculated using the Karplus equations and the parameters of refs 42 and 43.
In what follows, the force fields will be referred to as FOP (OPLS-AA), F14 (AMBERsb14), F22 (CHARMM22*) and F99 (AMBER99sb-ildn). The Nter spans residues 1–15, the loop spans residues 23–30, and the Cter spans residues 31–42. Our central hydrophobic core, CHC, includes residues 16–22 and not only the five hydrophobic residues L17–A21 as in literature. This choice is motivated by our definition of Q36 to avoid too small values and the fact that K16 and E22 also contain a hydrophobic side chain component.
3. RESULTS
REMD convergence for each force field was controlled by the metrics used in ref25., namely the radius of gyration, Cα end-to-end distance, total Q number of intermolecular SC–SC contacts, SASA and turn propensity along the sequence. The results using the first-half (100–415 ns) and second-half (415– 750 ns) trajectories overlap (Figures S1 and S2 in the Supporting Information), confirming the convergence of each simulation. In what follows, and unless mentioned explicitly, all observables are ensemble-averaged data at 315 K and include all predicted conformations of each force field within the time 100–750 ns. This temperature was selected because it is near to the physiological temperature and allows comparison with Aβ40 WT dimer simulation.37
Figure 1 compares the distributions of the radii of gyration, Rg, of both individual chains and the dimer, the end-to-end distance between the Cα atoms of the first and last residues (dee), and the total Q number of intermolecular SC–SC contacts obtained using the four force fields. The Rg distributions, averaged over both chains, with FOP and F14 are identical with a peak at 1.1 nm vs a peak at 1.2 nm with F99, and a plateau between 1.1 and 1.2 nm with F22 (Figure 1A). The Rg distributions of dimers are superposable with a peak at 1.3 nm (Figure 1B) and there is little difference between the four RMSD distributions using residues 17–42 with respect to the U-shape (PDB entry 2BEG44) and S-shape (PDB entry 2NA045) Aβ42 fibril models (Figure 1, parts E and F). Differences are noted for (a) end-to-end distributions where FOP and F14 display a peak at 0.5 and 1.0 nm with probabilities (P) of 6% and 7.5%, while F99 and F22 show maxima at 2.1 and 2.5 nm with P of 5.5% (Figure 1C) and (b) Q intermolecular contact distributions where the values span 2–9 with F14 vs 2–12 with the other three force fields (Figure 1D).
Figure 1.

Distribution of various quantities including the radius of gyration of single chains (A) and dimer (B), the Cα end-to-end distance (C), the total Q number of intermolecular side-chain contacts (D), the Cα root-mean-square deviation (RMSD) with respect to two fibril structures (E, PDB: 2BEG) and (F, PDB: 2NAO) using residues 17–42. Shown are results at 315 K obtained from the four force fields using 100–750 ns.
The mean and error bar of the secondary structures averaged over all residues are reported in Table 1 for each force field. In all cases, turn dominates varying between 38 ± 1.7% (F14) and 47 ± 1.4% (FOP), and coil remains at 27–29 ± 2%. In F14 and F22, a helix is preferred (20 ± 1.6%, 19 ± 2.2%) over β-strand (14% and 11%), while in F99 and FOP, the β-strand is preferred (24 ± 2.8%, 20 ± 1.5%) over helix (6% and 4%).
Table 1.
Mean and Standard Deviation of Secondary Structures of Aβ42 Dimer at 315 Ka
| SS [%] | ||||
|---|---|---|---|---|
|
|
||||
| F14 | F22 | F99 | FOP | |
| β | 13.8 ± 1.5 | 11.3 ± 1.4 | 24.3 ± 2.8 | 20.2 ± 1.5 |
| α | 20.0 ± 1.6 | 18.8 ± 2.2 | 5.5 ± 1.2 | 3.5 ± 0.7 |
| turn | 38.3 ± 1.7 | 43.3 ± 2.0 | 41.1 ± 2.6 | 47.5 ± 1.4 |
| coil | 27.9 ± 1.6 | 26.6 ± 2.1 | 29.1 ± 2.7 | 28.8 ± 1.6 |
The standard deviation is calculated using two independent time intervals (100–425 and 425–750 ns).
Figure 2 shows the secondary structure profile along the sequence. Error bars are not shown, but they are similar in magnitude for the four force fields to those reported in ref 25. The four coil profiles show differences of 10–20% in the region 9–40. All turn profiles show four peaks at residues: 6–9 with very similar probabilities (P = 75% for residue 7), 14–16 and 23–29 (with differences up to 17% and 40% for residues 15 and 39), and 34–38 in F99, FOP, and F14 with P = 40% vs 36–39 in F22 with P = 65% for residue 38. Looking at the helix propensity per residue, FOP displays a signal of 14% in the region 13–18, F99 shows a signal of 8% in the region 13–37, and F22 and F14 display a signal of 22%–55% within residues 14–36. The maximum is, however, observed at CHC by F14 vs residues 32–33 by F22.
Figure 2.

Secondary structure propensities of each amino acid of Aβ42 dimer at 315 K obtained from the four force fields. F14 (black), F22 (red), F99 (green), and FOP (blue).
All β-strand profiles show five regions spanning residues 2–5, 11–13, 17–21, 30–36 (for F14, FOP and F99, but only 34–36 for F22) and 39–41. While the mean β-strand contents in F99 and FOP are rather similar (24% and 20%), there are differences in the β-character of the residues 2–5, 17–21, 36 and 39–40 with maxima in (F99, FOP) of (43%, 22%), (38%, 22%), (67%, 45%) and (47%, 30%), respectively. The main feature of the F22 strand profile with respect to the other force fields is the very low β-strand content of the residues 30–34. The F14 strand profile follows that of FOP, except at positions 31–32 and 33–36, with a difference of −25%.
The SASA profiles of backbone and side-chain atoms do not show any significant differences between the force fields (data not shown). Similarly, the propensity of all intramolecular (separated by at least by four residues) and intermolecular salt bridges does not show population differences exceeding ±7% (Figure S3). The D23–K28 salt bridge has a population of 4 ± 2% with all force fields.
Figure 3 compares the intrapeptide SC–SC contact maps. Noteworthy is the observation of high probability (P = 18%) off-diagonal contacts between residues 17–21 and residues 30– 32 (in F99 and FOP) or 30–35 in F14, almost absent in F22 (P = 6%). The F22 force field therefore weakens substantially the CHC–Cter contacts. Contacts within the 12 N-terminal residues are observed with P = 70% by F99, FOP and F14 and P = 45% by F22.
Figure 3.

Intrapeptide side-chain-side-chain contact probabilities of Aβ42 dimer at 315 K obtained from four force fields. For clarity, contacts with P < 10% are not shown.
Figure 4 reports the intermolecular SC–SC contact maps. Differences between the four maps are highlighted in Figure S4. With the exception of interactions between the 2 N-terminus residues in F14 with P = 16%, all contact maps show probabilities between 9 and 12%. There are, however, four main differences between the maps: (i) CHC–CHC interactions are observed in F99, F14 and F22, but not in FOP, (ii) the density of interactions between CHC and Cter is much higher in F14 and F22 than in F99 and FOP, (iii) the density of interactions between Cter and Cter is much higher in F22 and FOP than in F14 and F99, and (iv) FOP and F14 show interactions between Nter and Cter/CHC that are not generated by F22. Overall the four force fields display distinct maps and change substantially the involvement of the Cter region in dimer formation.
Figure 4.

Interpeptide side-chain-side-chain contact probabilities of Aβ42 dimer at 315 K using the four force fields. For clarity, contacts with P < 3% are not shown.
Table S1 characterizes the ten free energy minima of the single-molecule states, denoted as s1–s10, using the four force fields, and Figure S5 shows the conformation closest to the center of each minimum. There is not a single dominant state, the s1 population varying between 17.6% (FOP) and 12.5% (F22). The error bars of the populations are on the order of ±2%. F14 and F22 single-molecule ensembles are dominated by disordered states with one to three α or 3–10 helices: 9 states in F14 and 7 states (e.g., s1 and s4) in F22. Also, F22 and F14 show one and three α–β conformations, respectively: s10 with a β-hairpin at the Cter in F22, s10 and s6 with β-hairpins at different locations in F14. FOP and F99 consist of five and six states, respectively with short β-hairpins spanning various CHC–Cter, Cter, or loop–Cter mixed with very short helical fragments for F99 only.
Differences in the single-molecule states can be further revealed by the percentages of common structures shared by the four force fields. Clustering of the conformations generated by the combined four trajectories and based on a standard Cα RMSD cutoff of 0.3 nm46 shows there is no similarity using the full sequences (Figure 5A). Using residues 9–42, only the first four clusters are recognized by at most three force fields (Figure 5B). The first cluster is recognized by FOP, F14, and F99 with probabilities of 8%, 5%, and 2.5%, so differences statistically significant. This cluster is associated with a β-hairpin spanning CHC–Cter. Using residues 17–42, we see that the second cluster, characterized by a 3-stranded β-sheet spanning CHC– Cter–Cter, is shared by the four force fields (Figure 5C) with probabilities varying, however, from 7.5 to 0.4%. To have a more global picture, Figure 6 reports the populations of intramolecular β-sheets at different positions with error bars varying at most by ±1%. It is observed that the population of β-hairpin varies from 11% (F99) to 6% (F22), 3% (F14) and 1% (FOP) in Nter, the population of β-hairpin increases from 1% (FOP and F99) to 3% (F14) and 8% (F22) in Cter–Cter, and the population of β-hairpin decreases from 13% (F99) to 8% (FOP), 4.4% (F22), and 1.5% (F14) in CHC–Cter. The force fields have marginal probabilities for β-hairpins spanning the Nter–CHC (except F22, 3.8%) and small probabilities for intramolecular 3-stranded β-sheets spanning either Nter– CHC–Cter (<1%) or CHC–Cter–Cter (1.5–2.5% with F99, F14 and FOP and negligible with F22). Overall, the differences in the intramolecular beta-hairpin populations at Nter, spanning Cter–Cter and CHC–Cter are statistically different between the force fields.
Figure 5.

Population of the first 20 most populated clusters of the single-molecule states obtained from RMSD clustering of the combined four force fields trajectories. For each cluster, the contribution of each force field is shown: F14 (black), F22 (red), F99 (green), and FOP (blue). The RMSD is calculated using the residues 1–42 (A), 9–42 (B), and 17–42 (C). Structures of the first two clusters are shown with the ball indicating the first residue.
Figure 6.

Probability of Aβ42 dimer to form β-hairpins only at Nter (HPNter–Nter), between Nter and CHC (HPNter–CHC), at Cter (HPCter–CHC), and between CHC and Cter (HPCHC–Cter). The populations of intramolecular three-stranded β-sheets formed at Nter–CHC–Cter (βSNter–CH–Cter) and CHC–Cter–Cter (βSCHC–Cter–Cter) are also shown. Error bars are calculated using two independent time windows (100–425 ns and 425–750 ns).
The first ten clusters of Aβ42 dimer, denoted as to S1–S10, are displayed in Figure 7 for each force field. They are analyzed in Table S2 by their populations, two single-molecule states, secondary structure compositions, Q values of interpeptide SC–SC contacts between various regions, and their CCS values. The error bar of each population is on the order of ±1.5%. Because of the heterogeneous structural character of each ensemble, e.g. the total population of the first 200 clusters representing 98% (F14), 87% (F22), 92% (F99) and 97% (FOP) of all conformations, we limit the detailed comparison to the first three states or clusters shown on Figure 8 and described by their contact maps. We also define that the two chains have parallel, antiparallel or perpendicular orientations if the two turn regions (residues 23–30) point in the same, opposite or perpendicular directions
Figure 7.

Centers of the first 10 overall states (Si) of the Aβ dimer obtained from 4 different force fields. The ball indicates the first residue. The population of each state is shown.
Figure 8.

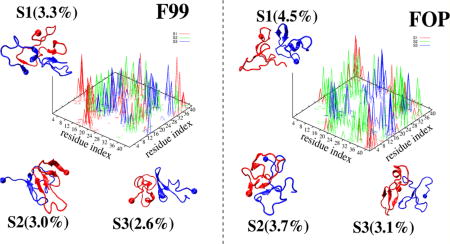
Representative structures of the first three most populated dimer states (Si) of Aβ42 dimer, with the ball indicating the first residue. The interpeptide side-chain–side-chain contact probability map of each state, S1 (red), S2 (green), and S3 (blue), is also shown.
The first three F14 dimer states, S1–S3, with populations of 8.3, 4.8 and 4.1% and characterized by (21, 12%), (22, 5%) and (7, 31%) of (β-strand, helix), are stabilized by different interfaces involving Nter–Nter and Nter–Cter, and Cter– Cter interactions in all cases (but with a much higher density of Cter–Cter contacts in S3), and CHC–CHC contacts only in S1 and S2. While the two chains are random coil in S1, one chain has either strong beta-character in S2 or high helical content in S3 from residues 17–42. The two chains are side-by side in S1, and face-to-face parallel in S2 and antiparallel in S3. The three states have CCS values of 1246, 1210, and 1158 Å2.
The first three F22 dimer states, with the lowest populations among all force fields (6.9% vs 17%. with F14), are characterized by (6, 24%), (18, 32%) and (8, 28%) of (β-strand, helix). The contact maps of S1–S3 are more delocalized than for those in F14, with S1 and S3 free of Cter–Nter interactions, and S2 free of CHC–CHC contacts. The three states with F22 have CCS values of 1228, 1176, and 1301 Å2. The S1–S3 states in F99 and FOP with a total population of 9% and 11%, respectively have no well-defined structures and interfaces, and display various topologies ranging from a 5-stranded β-sheet with an intermolecular parallel β-sheet in the Cter spanning residues 37–40 and 31–33 in S2 of F99 to two mainly disordered chains in S2 of FOP. The two chains in S1 and S3 are side-by side (parallel) with F99 while they are side-by-side (antiparallel) with FOP. The S1, S2, and S3 states have CCS values of (1272, 1212, and 1248 Å2) for F99 and (1266, 1173, and 1274 Å2) for FOP.
4. DISCUSSION
We compare the secondary structure content generated by each force field to CD experiments and previous simulations. On the basis of one stringent sample preparation using many rounds of filtration, CD on Aβ42 WT peptide at pH 7.5 at time zero, and thus consisting of monomers and small oligomers, led to α-helix of 3% and β-strand of 13%,47 consistent with an earlier study.48 A more recent study using PICUP and SDS-PAGE shows an increase to 10% of α-helix and 39% of β-strand for pure Aβ40 WT dimer and no data were provided for Aβ42 WT dimer.49 It should be stressed that SDS-PAGE has been recently questioned to characterize small Aβ oligomers,50 and cross-linking can change the secondary structure content in a way that is still not well understood. Indeed, it has been shown that cross-linking can in fact stabilize oligomers by inhibiting their conversion into fibrils.51 So in this context, and even if differences of ±3% in absolute levels of helix are observed with different convolution protocols,47 the F14 and F22 force fields with 20 ± 1.6% and 18.8 ± 2.2% of helix content clearly overestimate the experimental CD helix content, and only the F99 and FOP calculated values with (6% and 24%) and (4% and 20%) of (helix and β-strand) are in agreement with CD (3% and 13%).
At the simulation levels, earlier atomistic OPLS/TIP3P REMD18 based on replicas of 400 ns and implicit solvent CG OPEP-REMD8 which lead to (<3% of helix, 15–19% of β-strand) and (5% of helix, 20% of β-strand) are in agreement with our FOP contents (4% and 20%). These values run in contrast with (i) previous OPLS MD simulation starting from multiple conformations with 0.9% of helix and 6.6% of β-strand,20 (ii) recent standard and accelerated MD trajectories using AMBER99sb-ildn/TIP3P24 with 4% of helix and 5% of β-strand,22 vs 6% and 24% with our F99 contents, and (iii) to a lesser extent to extensive DMD simulations of Aβ42 peptides coupled to an implicit solvent four-bead CG model with 0% of helix and 15.7% of β-strand.52
Experimentally, the cross collision section (CCS) of Aβ42 dimer was estimated by an IM-MS study to be of 1252 (±20) Å2.53 The CCS values obtained using the solvated conformations of the first 200 clusters are 1255 Å2 for F14, 1286 Å2 for F22, 1282 Å2 for F99, and 1257 Å2 for FOP. All these calculated CCS values match experiment. Note we do not attempt to reproduce the ESI (electrospray ionization)-IM-MS CCS values, i.e., in gas phase with a charge state +5 (the most populated charge state).50
Second, we calculate other experimental properties, not determined yet, to assess whether they are dependent on the equilibrium ensembles. Using the conformational centers of the first 200 clusters and with each conformation subject to a MD trajectory of 160 ps by the WAXSiS server, the four SAXS profiles are nearly superposable to each other (Figure 9). Note that SAXS analysis has been reported for a sample of Aβ42 monomers and various oligomeric species at time 0.1 and 3 h,54 and Cu2+-induced Aβ42 oligomers with measured Rg values varying between 20 and 80 Å, respectively, making comparison with our predicted profiles impossible.55
Figure 9.

SAXS predicted curves of Aβ42 dimer using the four force fields with error bars.
Using the conformational centers of the first 200 clusters, the calculated rotational diffusion coefficient is 2.86, 2.80, 2.78, and 2.82 × 107s−1 for F14, F22, F99, and FOP, respectively, and the calculated translational diffusion coefficient is 12.6, 12.5, 12.5, and 12.6 × 10−7 cm2 s−1 for F14, F22, F99, and FOP. All these diffusion coefficients are identical within the experimental uncertainties of ±0.1 units.56,57 Taken together, these predictions on SAXS and hydrodynamics, which are sensitive to global structural features, clearly indicate that these measurements will not contribute to our understanding of Aβ dimers.
We also calculate six sets of NMR scalar couplings, which report on ensemble averaged backbone dihedral angles, as reported for the Aβ40/42 monomers.19 These scalar couplings include 3JHN–Ha, 3JHa–C′, 3JHN–C′, and 3JC′–C′, which report on the phi dihedral angle, and 1JN–Cα and 2JN–Cα, which report on the ψ dihedral angle. As can be seen on Figure 10, and assuming an uncertainty of 5% in the experimental values,17 the F99 and FOP 3JHN-Ha constants along the sequence are very similar and clearly different from the F22 and F14 corresponding J-profiles, with the largest deviation involving the amino acid region 16–39. This similarity between these scalar couplings obtained by F99 and FOP is reminiscent of their secondary structure profiles (Figure 2) and it is beyond the scope of this paper to discuss which aspects of the two force fields are similar.
Figure 10.

Backbone J-coupling constants along residues of Aβ42 dimer using the four force fields. The calculation is based on the Karplus relations of the type J(η) = A cos2(η + δ) + B cos2(η + δ) + C, where η denotes either the ϕ (for 3J) or the ψ (for 1J and 2J) backbone dihedral angle. The A, B, and C parameters and δ, the phase shift, are taken from refs 42 and 43.
Despite the four ensembles closely resemble random coil, and are highly heterogeneous, they have a few common structural features. First, at the intramolecular level, all force fields show a small β-strand signal at residues 2–5, 11–13, and CHC, and a high β-strand signal at 34–36 and 39–41. They show a signal of 1.5–13% and 1–8% for a β-hairpin spanning CHC–Cter and within Cter, respectively and a signal of 0–2.5% for a double β-hairpin spanning residues 17–23, 29–36, and 39–41. This overall β-strand profile and the observation of β-hairpins spanning the Cter region and the CHC–Cter is similar to Aβ42 monomer simulations.11 The β-hairpin spanning CHC–Cter has been already discussed in many observed structures along the aggregation pathways58 or interacting with inhibitors.59,60 Upon association to the homodimeric ZAβ3 protein of 58 residues, the Aβ1–40 monomer forms a β-hairpin spanning CHC and the residues 31–36.59 Upon association to a beta-hairpin mimics containing a piperidine–pyrrolidine semi rigid β-turn inducer located between the Aβ residues CHC and 33–37 (GLMVG), Aβ42 peptide remains monomeric as can be seen by gel electrophoresis.60 This intramolecular antiparallel β-sheet has also been discussed by CG OPEP61,62 REMD simulations of dimers63 and trimers64 of Aβ variants, and there is a significant energy barrier for the formation of intermolecular parallel β-sheet.63,65 The formation of a rather stable turn at the Cter, discussed by other studies,25,52 and the formation of a double β-hairpin in the dimers, albeit with a small probability, is consistent with the fact that the Aβ39–42 peptide can modulate Aβ oligomerization and toxicity.66,67
Second, at the quaternary level, none dimer state resembles the U- and S-shapes of the fibrils, and the four force fields share a heterogeneous ensemble of interfaces. They display with low probabilities either intermolecular parallel β-sheets (e.g., S2 of F99 with residues 39–40 and 31–33 in Figure 8), intermolecular orthogonal (e.g., S9 of F99 and S4 of FOP in Figure 7) or antiparallel β-sheets (e.g., S2 of FOP with residues 30–31 and 35–36, S3 of FOP with residues 17–18 and 39–40 in Figure 8, S5 of F99 with residues 11–13 and 30–33 and S9 of F22 with residues 34–40 and 17–20 in Figure 7). This mixture of intermolecular β-sheet organization is not specific to Aβ42 WT dimer in explicit solvent. It has been already discussed by atomistic simulations of the dimers of Aβ40 WT, A2 V, WT/A2 V, and WT/A2T in explicit solvent,36,37,68 and previous CG simulations with implicit solvent of Ab42 peptides,8,52 but with significant different probabilities. Similarly, while the two previous CG simulations point to the strong involvement of the C-terminal region in dimer formation, our simulations reveal that this implication varies substantially between all-atom force fields, and overall our simulations provide a different ensemble of equilibrium structures.
In addition to variations in interfaces and propensities to form helical segments, the four ensembles are particularly different in the N-terminus region, as can be seen by the J-coupling constants (Figure 10) and the different propensities to form β-hairpins within the Nter (varying from 1 to 11%) and in the Nter–CHC region (varying from 0.2 to 2.5%). This difference in the conformations of the N-terminus is problematic since familial mutations in the hydrophilic Nter makes Alzheimer’s disease either more aggressive (e.g., H6R, D7N, and D7H) or protective (A2 V in heterozygous form and A2T in both homo- and heterozygous forms).4,69–71
5. CONCLUSION
It is the first time that the high-resolution structures of Aβ1–42 dimers, which play an important role in Alzheimer’s disease, have been characterized and compared by using four atomistic force fields, extensive REMD simulations (48 replicas, each of 0.75 µs), and the same analysis tools. Knowledge of this structural ensemble is interesting not only to design inhibitors,72,73 but also to understand the interactions with many cellular partners in early stage Alzheimer’s disease.4,74–77
Overall, the four simulations lead to ensembles resembling random coil and ensemble-average shows that the cross-collision sections, hydrodynamics properties and small-angle X-ray scattering profiles are independent of the force field. There are, however, marked differences in secondary (as can be seen by the calculated NMR J couplings) and tertiary structural properties among the four force fields.
The AMBERsb14 and CHARMM22* ensembles significantly overestimate the CD-derived helix content, while the OPLS-AA and AMBER99sb-ildn secondary structure contents are in agreement with CD data. The beta-hairpin content in the CHC–Cter region (residues 17–21 and 30–36) varies between 8% (OPLS-AA) and 13% (AMBER99sb-ildn), while it is between 1.5 (AMBERsb14) and 5% (CHARMM22*). These significant differences in the beta-hairpin population spanning 17–36 among the force fields are particularly important, because this region can contain: (i) aggressive FAD mutations (A21G, E22Q, E22G, D23N),4 (ii) chemical modifications either preventing (phosphorylation of S26)4 or accelerating (lactam bridge between D23 and K28)4 amyloid formation, or modulating toxicity (replacement of G25–S26– N27 by an azobenzene photoswitch),78 and (iii) mutations producing less toxic fibrils (L34T)79 or oligomers (L34T,80 and G33I81).
Overall, there are also statistical conformational differences in the N-terminus and C-terminal and in the quaternary structures among the four force fields, and an important key finding of all simulations is that the dimer is stabilized by nonspecific interactions, explaining therefore its possible transient binding to multiple cellular partners and in part its toxicity.
Supplementary Material
Acknowledgments
This study was supported by grants from GRAL ANR SIMI 12-BS07-0017-01, the “Initiative d’Excellence” program from the French State (Grant “DYNAMO”, ANR-11-LABX-0011-01), National Institute of Health (NIH-R01GM118508), National Science Foundation (NSF, SI2-SEE-1534941), and computer facilities provided by the NC state HPC, and the IDRIS, CINES and TGCC centers (grant x2016077604).
Footnotes
ASSOCIATED CONTENT
- Tables S1 and S2 characterize the single-molecule and overall states of the dimer. Figures S1 and S2 are related to the convergence of the REMD simulations. Figures S3 and S4 show differences in the salt-bridge and interpeptide side-chain side-chain populations. Figure S5 shows representative structures of the 10 single-molecule states of Aβ42 dimer obtained using the four force fields. (PDF)
The authors declare no competing financial interest.
References
- 1.Selkoe DG, Hardy J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, et al. Beta-amyloid Monomers are Neuroprotective. J. Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble Amyloid beta-protein Dimers Isolated from Alzheimer Cortex directly Induce Tau Hyperphosphorylation and Neuritic Degeneration. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nasica-Labouze J, Nguyen PH, Sterpone F, Berthoumieu O, Buchete NV, Coté S, De Simone A, Doig AJ, Faller P, Garcia A, et al. Amyloid beta Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chem. Rev. 2015;115:3518–3563. doi: 10.1021/cr500638n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morris M, Maeda S, Vossel K, Mucke L. The Many Faces of Tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sterpone F, Melchionna S, Tuffery P, Pasquali S, Mousseau N, Cragnolini T, Chebaro Y, St-Pierre J, Kalimeri M, Barducci A, et al. The OPEP Protein Model: From Single Molecules, Amyloid Formation, Crowding and Hydrodynamics to DNA/RNA Systems. Chem. Soc. Rev. 2014;43:4871–4893. doi: 10.1039/c4cs00048j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urbanc B, Cruz L, Yun S, Buldyrev SV, Bitan G, Teplow DB, Stanley HE. In Silico Study of Amyloid beta-Protein Folding and Oligomerization. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17345–17350. doi: 10.1073/pnas.0408153101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Côté S, Laghaei R, Derreumaux P, Mousseau N. Distinct Dimerization for Various Alloforms of the amyloid-beta Protein: Abeta(1–40), Abeta(1–42), and Abeta(1–40)(D23N) J. Phys. Chem. B. 2012;116:4043–4055. doi: 10.1021/jp2126366. [DOI] [PubMed] [Google Scholar]
- 9.Côté S, Derreumaux P, Mousseau N. Distinct Morphologies for Amyloid Beta Protein Monomer: Aβ1–40, Aβ1–42, and Aβ1–40(D23N) J. Chem. Theory Comput. 2011;7:2584–2592. doi: 10.1021/ct1006967. [DOI] [PubMed] [Google Scholar]
- 10.Melquiond A, Mousseau N, Derreumaux P. Structures of Soluble Amyloid Oligomers from Computer Simulations. Proteins: Struct., Funct., Genet. 2006;65:180–191. doi: 10.1002/prot.21100. [DOI] [PubMed] [Google Scholar]
- 11.Rosenman DJ, Wang C, García AE. Characterization of Aβ Monomers through the Convergence of Ensemble Properties among Simulations with Multiple Force Fields. J. Phys. Chem. B. 2016;120:259–277. doi: 10.1021/acs.jpcb.5b09379. [DOI] [PubMed] [Google Scholar]
- 12.Carballo-Pacheco M, Strodel B. Comparison of Force Fields for Alzheimer’s Aβ42: A Case Study for Intrinsically Disordered Proteins. Protein Sci. 2017;26:174–185. doi: 10.1002/pro.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen PH, Li MS, Derreumaux P. Effects of All-atom Force Fields on Amyloid Oligomerization: Replica Exchange Molecular Dynamics Simulations of the Aβ(16–22) Dimer and Trimer. Phys. Chem. Chem. Phys. 2011;13:9778–9788. doi: 10.1039/c1cp20323a. [DOI] [PubMed] [Google Scholar]
- 14.Gerben SR, Lemkul JA, Brown AM, Bevan DR. Comparing Atomistic Molecular Mechanics Force Fields for a Difficult Target: A Case Study on the Alzheimer’s Amyloid β-peptide. J. Biomol. Struct. Dyn. 2014;32:1817–1832. doi: 10.1080/07391102.2013.838518. [DOI] [PubMed] [Google Scholar]
- 15.Fluitt AM, de Pablo JJ. An Analysis of Biomolecular Force Fields for Simulations of Polyglutamine in Solution. Biophys. J. 2015;109:1009–1018. doi: 10.1016/j.bpj.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henriques J, Cragnell C, Skepö M. Molecular Dynamics Simulations of Intrinsically Disordered Proteins: Force Field Evaluation and Comparison with Experiment. J. Chem. Theory Comput. 2015;11:3420–3431. doi: 10.1021/ct501178z. [DOI] [PubMed] [Google Scholar]
- 17.Rauscher S, Gapsys V, Gajda MJ, Zweckstetter M, de Groot BL, Grubmüller H. Structural Ensembles of Intrinsically Disordered Proteins Depend Strongly on Force Field: A Comparison to Experiment. J. Chem. Theory Comput. 2015;11:5513–5524. doi: 10.1021/acs.jctc.5b00736. [DOI] [PubMed] [Google Scholar]
- 18.Das P, Murray B, Belfort G. Alzheimer’s Protective A2T Mutation Changes the Conformational Landscape of the Aβ1–42 Monomer Differently than Does the A2V Mutation. Biophys. J. 2015;108:738–747. doi: 10.1016/j.bpj.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roche J, Shen Y, Lee JH, Ying J, Bax A. Monomeric Aβ(1–40) and Aβ(1–42) Peptides in Solution Adopt Very Similar Ramachandran Map Distributions That Closely Resemble Random Coil. Biochemistry. 2016;55:762–775. doi: 10.1021/acs.biochem.5b01259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barz B, Urbanc B. Dimer Formation Enhances Structural Differences between Amyloid β-protein (1–40) and (1–42): an Explicit-solvent Molecular Dynamics Study. PLoS One. 2012;7:e34345. doi: 10.1371/journal.pone.0034345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viet MH, Nguyen PH, Ngo ST, Li MS, Derreumaux P. Effect of the Tottori Familial Disease Mutation (D7N) on the Monomers and Dimers of Aβ40 and Aβ42. ACS Chem. Neurosci. 2013;4:1446–1457. doi: 10.1021/cn400110d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Hashemi M, Lv Z, Lyubchenko YL. Self-assembly of the Full-length Amyloid Aβ42 Protein in Dimers. Nanoscale. 2016;8:18928–18937. doi: 10.1039/c6nr06850b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang T, Zhang J, Derreumaux P, Mu Y. Molecular Mechanism of the Inhibition of EGCG on the Alzheimer Aβ(1–42) Dimer. J. Phys. Chem. B. 2013;117:3993–4002. doi: 10.1021/jp312573y. [DOI] [PubMed] [Google Scholar]
- 24.Das P, Chacko AR, Belfort G. Alzheimer’s Protective Cross-interaction between Wild-type and A2T Variants Alters Abeta42 Dimer Structure. ACS Chem. Neurosci. 2017;8:606–618. doi: 10.1021/acschemneuro.6b00357. [DOI] [PubMed] [Google Scholar]
- 25.Man VH, Nguyen PH, Derreumaux P. Conformational Ensembles of the Wild-type and S8C Abeta1–42 Dimers. J. Phys. Chem. B. 2017;121:2434–2442. doi: 10.1021/acs.jpcb.7b00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jorgensen WL, Tirado-Rives J. The OPLS [optimized potentials for liquid simulations] Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic peptides and Crambin. J. Am. Chem. Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 27.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How Fast-folding Proteins Fold. Science. 2011;334:517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 28.Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, et al. Atomic-level Characterization of the Structural Dynamics of Proteins. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 29.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 31.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 32.Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 33.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 34.Bussi G, Donadio D, Parrinello M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 35.Frishman D, Argos P. Knowledge-based Protein Secondary Structure Assignment. Proteins: Struct., Funct., Genet. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen PH, Sterpone F, Campanera JM, Nasica-Labouze J, Derreumaux P. Impact of the A2V Mutation on the Heterozygous and Homozygous Aβ1–40 Dimer Structures from Atomistic Simulations. ACS Chem. Neurosci. 2016;7:823–832. doi: 10.1021/acschemneuro.6b00053. [DOI] [PubMed] [Google Scholar]
- 37.Tarus B, Tran TT, Nasica-Labouze J, Sterpone F, Nguyen PH, Derreumaux P. Structures of the Alzheimer’s Wild-Type Aβ1–40 Dimer from Atomistic Simulations. J. Phys. Chem. B. 2015;119:10478–10487. doi: 10.1021/acs.jpcb.5b05593. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen PH, Li MS, Derreumaux P. Amyloid Oligomer Structure Characterization from Simulations: a General Method. J. Chem. Phys. 2014;140:094105. doi: 10.1063/1.4866902. [DOI] [PubMed] [Google Scholar]
- 39.Mesleh MF, Hunter JM, Shvartsburg AA, Schatz GC, Jarrold MF. Structural Information from Ion Mobility Measurements: Effects of the Long-Range Potential. J. Phys. Chem. 1996;100:16082–16086. [Google Scholar]
- 40.Knight CJ, Hub JS. WAXSiS: A Web Server for the Calculation of SAXS/WAXS Curves Based on Explicit-solvent Molecular Dynamics. Nucleic Acids Res. 2015;43(W1):W225–230. doi: 10.1093/nar/gkv309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ortega A, Amorós D, García de la Torre J. Prediction of Hydrodynamic and Other Solution Properties of Rigid Proteins from Atomic- and Residue-level Models. Biophys. J. 2011;101:892–898. doi: 10.1016/j.bpj.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu J-S, Bax A. Determination of φ and χ1 Angles in Proteins from 13C–13C Three-Bond J Couplings Measured by Three-Dimensional Heteronuclear NMR. How Planar Is the Peptide Bond? J. Am. Chem. Soc. 1997;119:6360–6368. [Google Scholar]
- 43.Wirmer J, Schwalbe J. Angular Dependence of 1J(Ni,Calphai) and 2J(Ni,Calpha(i-1)) Coupling Constants Measured in J-modulated HSQCs. J. Biomol. NMR. 2002;23:47–55. doi: 10.1023/a:1015384805098. [DOI] [PubMed] [Google Scholar]
- 44.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. 3D Structure of Alzheimer’s Amyloid-beta(1–42) Fibrils. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, Güntert P, Meier BH, Riek R. Atomic-resolution Structure of a Disease-relevant Aβ(1–42) Amyloid Fibril. Proc. Natl. Acad. Sci. U. S. A. 2016;113:E4976–E4984. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen Y, Maupetit J, Derreumaux P, Tufféry P. Improved PEP-FOLD Approach for Peptide and Miniprotein Structure Prediction. J. Chem. Theory Comput. 2014;10:4745–58. doi: 10.1021/ct500592m. [DOI] [PubMed] [Google Scholar]
- 47.Kirkitadze MD, Condron MM, Teplow DB. Identification and Characterization of Key Kinetic Intermediates in Amyloid beta-protein Fibrillogenesis. J. Mol. Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 48.Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid-b Protein (Ab) Assembly: Ab40 and Ab42 Oligomerize Through Different Pathways. Proc. Natl. Acad. Sci. U. S. A. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ono K, Condron MM, Teplow DB. Structure-neurotoxicity Relationships of Amyloid beta-protein Oligomers. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pujol-Pina R, Vilaprinyó-Pascual S, Mazzucato R, Arcella A, Vilaseca M, Orozco M, Carulla N. SDS-PAGE Analysis of Aβ Oligomers is Disserving Research into Alzheimeŕs Disease: Appealing for ESI-IM-MS. Sci. Rep. 2015;5:14809. doi: 10.1038/srep14809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams TL, Serpell LC, Urbanc B. Stabilization of Native Amyloid β-protein Oligomers by Copper and Hydrogen Peroxide Induced Cross-linking of Unmodified Proteins (CHICUP) Biochim. Biophys. Acta, Proteins Proteomics. 2016;1864:249–259. doi: 10.1016/j.bbapap.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 52.Urbanc B, Betnel M, Cruz L, Bitan G, Teplow DB. Elucidation of Amyloid beta-protein Oligomerization Mechanisms: Discrete Molecular Dynamics Study. J. Am. Chem. Soc. 2010;132:4266–80. doi: 10.1021/ja9096303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, et al. Amyloid-β Protein Oligomerization and the Importance of Tetramers and Dodecamers in the Aetiology of Alzheimer’s Disease. Nat. Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Messa M, Colombo L, del Favero E, Cantù L, Stoilova T, Cagnotto A, Rossi A, Morbin M, Di Fede G, Tagliavini F, et al. The Peculiar Role of the A2V Mutation in Amyloid-β (Aβ) 1–42 Molecular Assembly. J. Biol. Chem. 2014;289:24143–24152. doi: 10.1074/jbc.M114.576256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ryan TM, Kirby N, Mertens HD, Roberts B, Barnham KJ, Cappai R, Pham CLL, Masters CL, Curtain CC, Curtain CC. Small Angle X-ray Scattering Analysis of Cu(2+)-induced Oligomers of the Alzheimer’s Amyloid β Peptide. Metallomics. 2015;7:536–543. doi: 10.1039/c4mt00323c. [DOI] [PubMed] [Google Scholar]
- 56.Dubin SB, Clark NA, Benedek GB. Measurement of the Rotational Diffusion Coefficient of Lysozyme by Depolarized Light Scattering: Configuration of Lysozyme in Solution. J. Chem. Phys. 1971;54:5158. [Google Scholar]
- 57.Danielsson J, Andersson A, Jarvet J, Gräslund A. 15N Relaxation Study of the Amyloid beta-peptide: Structural Propensities and Persistence Length. Magn. Reson. Chem. 2006;44:S114–S121. doi: 10.1002/mrc.1814. [DOI] [PubMed] [Google Scholar]
- 58.Abelein A, Abrahams JP, Danielsson J, Gräslund A, Jarvet J, Luo J, Tiiman A, Wärmländer SK. The Hairpin Conformation of the Amyloid β Peptide is an Important Structural Motif along the Aggregation Pathway. JBIC J. Biol. Inorg. Chem. 2014;19:623–634. doi: 10.1007/s00775-014-1131-8. [DOI] [PubMed] [Google Scholar]
- 59.Sandberg A, Luheshi LM, Söllvander S, Pereira de Barros T, Macao B, Knowles TP, Biverstål H, Lendel C, Ekholm-Petterson F, Dubnovitsky A, et al. Stabilization of Neurotoxic Alzheimer Amyloid-beta Oligomers by Protein Engineering. Proc. Natl. Acad. Sci. U. S. A. 2010;107:15595–15600. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pellegrino S, Tonali N, Erba E, Kaffy J, Taverna M, Contini A, Taylor M, Allsop D, Gelmi ML, Ongeri S. β-Hairpin Mimics Containing a Piperidine–pyrrolidine Scaffold Modulate the β-amyloid Aggregation Process Preserving the Monomer Species. Chem. Sci. 2017;8:1295–1302. doi: 10.1039/c6sc03176e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Derreumaux P. Folding a 20 Amino Acid alpha beta Peptide with the Diffusion Process-controlled Monte Carlo Method. J. Chem. Phys. 1997;107:1941–1947. [Google Scholar]
- 62.Chebaro Y, Pasquali S, Derreumaux P. The Coarse-grained OPEP Force Field for Non-amyloid and Amyloid Proteins. J. Phys. Chem. B. 2012;116:8741–8752. doi: 10.1021/jp301665f. [DOI] [PubMed] [Google Scholar]
- 63.Chebaro Y, Mousseau N, Derreumaux P. Structures and Thermodynamics of Alzheimer’s Amyloid-beta Abeta(16–35) Monomer and Dimer by Replica Exchange Molecular Dynamics Simulations: Implication for Full-length Abeta Fibrillation. J. Phys. Chem. B. 2009;113:7668–7675. doi: 10.1021/jp900425e. [DOI] [PubMed] [Google Scholar]
- 64.Chebaro Y, Jiang P, Zang T, Mu Y, Nguyen PH, Mousseau N, Derreumaux P. Structures of Aβ17–42 Trimers in Isolation and with Five Small-molecule Drugs using a Hierarchical Computational Procedure. J. Phys. Chem. B. 2012;116:8412–8422. doi: 10.1021/jp2118778. [DOI] [PubMed] [Google Scholar]
- 65.Nagel-Steger L, Owen MC, Strodel B. An Account of Amyloid Oligomers: Facts and Figures Obtained from Experiments and Simulations. ChemBioChem. 2016;17:657–76. doi: 10.1002/cbic.201500623. [DOI] [PubMed] [Google Scholar]
- 66.Zheng X, Wu C, Liu D, Li H, Bitan G, Shea JE, Bowers MT. Mechanism of C-Terminal Fragments of Amyloid β-Protein as Aβ Inhibitors: Do C-Terminal Interactions Play a Key Role in Their Inhibitory Activity? J. Phys. Chem. B. 2016;120:1615–1623. doi: 10.1021/acs.jpcb.5b08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Urbanc B, Betnel M, Cruz L, Li H, Fradinger EA, Monien BH, Bitan G. Structural Basis for Aβ1–42 Toxicity Inhibition by Aβ C-terminal Fragments: Discrete Molecular Dynamics Study. J. Mol. Biol. 2011;410:316–328. doi: 10.1016/j.jmb.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nguyen PH, Sterpone F, Pouplana R, Derreumaux P, Campanera JM. Dimerization Mechanism of Alzheimer Aβ40 Peptides: The High Content of Intrapeptide-Stabilized Conformations in A2V and A2T Heterozygous Dimers Retards Amyloid Fibril Formation. J. Phys. Chem. B. 2016;120:12111–12126. doi: 10.1021/acs.jpcb.6b10722. [DOI] [PubMed] [Google Scholar]
- 69.Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, et al. A Mutation in APP Protects against Alzheimer’s Disease and Age-related Cognitive Decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 70.Di Fede G, Catania M, Morbin M, Rossi G, Suardi S, Mazzoleni G, Merlin M, Giovagnoli AR, Prioni S, Erbetta A, et al. A Recessive Mutation in the APP Gene with Dominant-negative Effect on Amyloidogenesis. Science. 2009;323:1473–1477. doi: 10.1126/science.1168979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murray B, Sharma B, Belfort G. N-Terminal Hypothesis for Alzheimer’s Disease. ACS Chem. Neurosci. 2017;8:432–434. doi: 10.1021/acschemneuro.7b00037. [DOI] [PubMed] [Google Scholar]
- 72.Doig AJ, Derreumaux P. Inhibition of Protein Aggregation and Amyloid Formation by Small Molecules. Curr. Opin. Struct. Biol. 2015;30:50–56. doi: 10.1016/j.sbi.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 73.Nguyen PH, Derreumaux P. Understanding Amyloid Fibril Nucleation and Aβ Oligomer/Drug Interactions from Computer Simulations. Acc. Chem. Res. 2014;47:603–611. doi: 10.1021/ar4002075. [DOI] [PubMed] [Google Scholar]
- 74.Lei M, Xu H, Li Z, Wang Z, O’Malley TT, Zhang D, Walsh DM, Xu P, Selkoe DJ, Li S. Soluble Aβ Oligomers Impair Hippocampal LTP by Disrupting glutamatergic/GABAergic Balance. Neurobiol. Dis. 2016;85:111–121. doi: 10.1016/j.nbd.2015.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kanekiyo T, Xu H, Bu G. ApoE and Aβ in Alzheimer’s Disease: Accidental Encounters or Partners? Neuron. 2014;81:740–54. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu NW, Nicoll AJ, Zhang D, Mably AJ, O’Malley T, Purro SA, Terry C, Collinge J, Walsh DM, Rowan MJ. mGlu5 Receptors and Cellular Prion Protein Mediate Amyloid-β-Facilitated Synaptic Long-term Depression in vivo. Nat. Commun. 2014;5:3374. doi: 10.1038/ncomms4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Di Scala C, Yahi N, Boutemeur S, Flores A, Rodriguez L, Chahinian H, Fantini J. Common Molecular Mechanism of Amyloid Pore Formation by Alzheimer’s β-amyloid Peptide and α-Synuclein. Sci. Rep. 2016;6:28781. doi: 10.1038/srep28781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Doran TM, Anderson EA, Latchney SE, Opanashuk LA, Nilsson BL. An Azobenzene Photoswitch Sheds Light on Turn Nucleation in Amyloid-β Self-assembly. ACS Chem. Neurosci. 2012;3:211–220. doi: 10.1021/cn2001188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bonhommeau S, Talaga D, Hunel J, Cullin C, Lecomte S. Tip-Enhanced Raman Spectroscopy to Distinguish Toxic Oligomers from Aβ1–42 Fibrils at the Nanometer Scale. Angew. Chem., Int. Ed. 2017;56:1771–1774. doi: 10.1002/anie.201610399. [DOI] [PubMed] [Google Scholar]
- 80.Das AK, Rawat A, Bhowmik D, Pandit R, Huster D, Maiti S. An Early Folding Contact between Phe19 and Leu34 is Critical for Amyloid-β Oligomer Toxicity. ACS Chem. Neurosci. 2015;6:1290–195. doi: 10.1021/acschemneuro.5b00074. [DOI] [PubMed] [Google Scholar]
- 81.Harmeier A, Wozny C, Rost BR, Munter LM, Hua H, Georgiev O, Beyermann M, Hildebrand PW, Weise C, Schaffner W, et al. Role of Amyloid-beta Glycine 33 in Oligomerization, Toxicity, and Neuronal Plasticity. J. Neurosci. 2009;29:7582–7590. doi: 10.1523/JNEUROSCI.1336-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.