

Abstract
High von Willebrand factor (VWF) levels have been reported to be associated with an increased risk of cardiovascular events. However, the relationship between VWF levels and coronary atherosclerosis in patients with coronary artery disease (CAD) who have already received stain treatment is still unclear. We examined the association between VWF levels and coronary plaque as assessed by intravascular ultrasound (IVUS) in CAD patients treated with statins. Ninety-one CAD patients who underwent percutaneous coronary intervention under IVUS guidance, and who were already receiving statin treatment based on Japanese guidelines, were included. An IVUS examination was performed for the culprit lesion, and plasma VWF antigen levels were measured using enzyme-linked immuno sorbent assay. In all of the patients, the low-density lipoprotein cholesterol levels just before the IVUS examination were low (86 ± 26 mg/dL). The VWF levels were positively correlated with the plaque burden expressed as percent atheroma volume (PAV) (r = 0.39, P = .001), while there was no significant association between VWF and plaque composition. Multivariate stepwise regression analysis showed that higher VWF levels were independently associated with increased PAV (β=0.26, P = .01). In CAD patients who had already been treated with statins, higher VWF levels were associated with a higher coronary plaque burden, suggesting that a high VWF level may be a marker of the residual cardiovascular risk after statin treatment.
Keywords: coronary atherosclerosis, coronary plaque, intravascular ultrasound, von Willebrand factor
1. Introduction
von Willebrand factor (VWF) is a glycoprotein that is produced mainly by vascular endothelial cells in humans. VWF mediates platelet adhesion and aggregation, and plays a crucial role in primary hemostasis and the initial steps in thrombus formation.[1] VWF levels are considered to increase as a result of vascular endothelial damage, and VWF may be a marker of vascular injury.[2] In addition, high VWF levels have been reported to be associated with an increased risk of cardiovascular events.[3–7]
Statin is a standard treatment for the prevention of cardiovascular diseases and has favorable pleiotropic effects on atherosclerosis.[8] However, cardiovascular events continue to occur at an unacceptable frequency in patients who are receiving statins.[9] We hypothesized that a high VWF level may be a marker of the residual cardiovascular risk after statin treatment.
Intravascular ultrasound (IVUS) is an imaging modality that enables the precise quantification of coronary atherosclerosis.[10] The IVUS-derived coronary atheroma burden is considered to be a useful marker of the risk of cardiovascular events,[11] even in patients who are receiving statin therapy.[12] At present, little is known about the association between the level of VWF and coronary plaque as assessed by IVUS. Therefore, to elucidate whether VWF is a marker of the residual cardiovascular risk after statin treatment, we examined the relationship between VWF levels and coronary plaque using IVUS in coronary artery disease (CAD) patients who had been treated with statins.
2. Materials and methods
2.1. Patients and study design
A total of 1271 patients with CAD who underwent percutaneous coronary intervention (PCI) at Fukuoka University Hospital from February 2012 to January 2016 were screened according to the following exclusion criteria: patients who did not undergo IVUS-guided PCI; blood disease (e.g., von Willebrand disease); familial hypercholesterolemia; liver cirrhosis; severe infection; recent surgery or trauma; and contraindication to antiplatelet agents or statins. Of these, 153 patients in whom we could obtain consent for measurement of the VWF level were included. We also excluded 44 patients who had not received statins before PCI, 5 patients with acute coronary syndrome (ACS) and 13 patients with unanalyzable IVUS images. Ultimately, 91 stable CAD patients who had already received statin therapy before IVUS-guided PCI were included in this study.
According to the Japanese guidelines for the secondary prevention of myocardial infarction,[13] all of the patients received a standard antiplatelet therapy (aspirin and thienopyridines) before PCI. Statin treatment was based on the Japan Atherosclerosis Society guidelines for the diagnosis and prevention of atherosclerotic cardiovascular diseases, and the target level of low-density lipoprotein cholesterol (LDL-C) was <100 mg/dL.[14] Fasting blood samples for the measurement of clinical laboratory data, including plasma levels of VWF and high-sensitivity C-reactive protein (hs-CRP), were obtained just before PCI.
This study was approved by the ethics committee of Fukuoka University Hospital (EC/IRB: 15-3-12) and conducted according to the Declaration of Helsinki regarding investigations in humans.
2.2. IVUS procedure and analysis
Before IVUS-guided PCI, IVUS examination was performed for the culprit lesion of a coronary artery using an imaging catheter and a console (View IT and VISIWAVE, Terumo, Tokyo, Japan) (Fig. 1). An optimal dose of nitroglycerin was injected into the coronary artery just before IVUS examination to prevent coronary spasm. The IVUS catheter was advanced to the distal side of the culprit lesion and pulled back automatically at a speed of 0.5 mm/sec.
Figure 1.
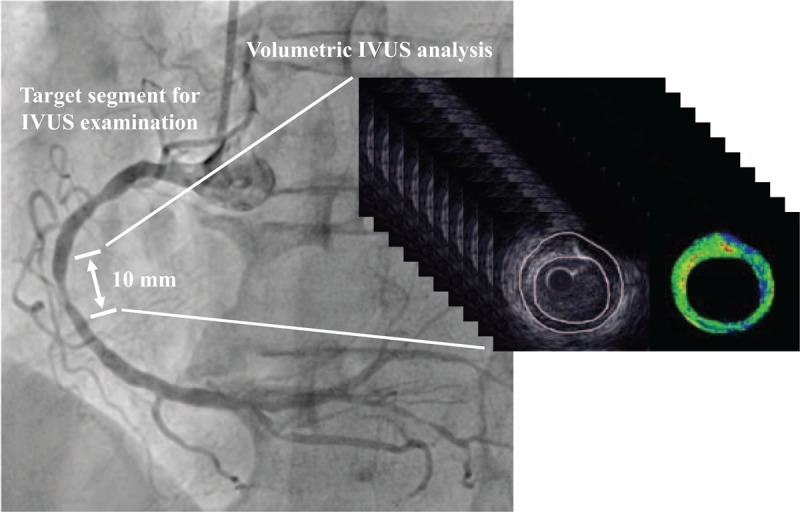
Representative coronary angiogram of the evaluated vessel for IVUS analysis. The target segment for IVUS analysis included the most-diseased cross-section before PCI, and its length was 10 mm. IVUS = intravascular ultrasound, PCI = percutaneous coronary intervention.
For IVUS analysis, first, the cross-section with the smallest minimum lumen area of the culprit lesion was identified. Next, the cross-sections that were located 5 mm proximal and distal to the most diseased cross-section were identified. This process identified the 10 mm segment that was considered for IVUS analysis (Fig. 1). IVUS analysis of the culprit lesion was conducted using an integrated backscatter (IB) IVUS analysis system (VISIATLAS, Terumo, Tokyo, Japan), which measures both plaque volume and plaque composition. This system has been reported to be comparable to a commonly used IVUS analysis system (echoPlaque, INDEC Systems, Santa Clara, CA) and to have excellent reliability and validity.[15] External elastic membrane (EEM) cross-sectional area (CSA) and lumen CSA were manually traced at 1-mm axis intervals for a length of 10 mm (Fig. 1), and atheroma CSA (EEM CSA minus lumen CSA) was calculated. The remodeling index was calculated as the EEM CSA of the lesion site divided by the average EEM CSA of the proximal and distal reference sites. The lesion length of the culprit lesion was calculated as the distance between the proximal and distal reference sites using the above-mentioned IVUS analysis system. Vessel volume and lumen volume were calculated automatically as Σ EEM CSA and Σ lumen CSA, respectively. Total atheroma volume and percent atheroma volume (PAV) were calculated as follows:
Total atheroma volume = Σ (EEM CSA – lumen CSA)
PAV = 100 × total atheroma volume/Σ EEM CSA
The tissue characteristics of coronary plaque were analyzed by the software for IB IVUS that came with the above-mentioned IVUS analysis system. The plaque components were divided into 4 classifications: lipid, fibrosis, dense fibrosis, and calcification, as reported previously.[16] The area and volume of each plaque classification were calculated automatically and presented as percentages.
IVUS analysis was performed by an experienced physician who was unaware of the patient characteristics according to the criteria described in the American College of Cardiology Clinical Expert Consensus document of IVUS.[10]
2.3. Clinical laboratory examinations
Serum levels of LDL-C, high-density lipoprotein cholesterol (HDL-C) and triglyceride, serum creatinine level, fasting blood sugar, hemoglobin A1C level, and uric acid level were measured at the Fukuoka University Hospital Laboratory Unit. Estimated glomerular filtration rate (eGFR) was calculated as follows: 194 × serum creatinine−1.094×age−0.287 (male), 194×serum creatinine−1.094×age−0.287×0.739 (female). The blood samples for the measurement of VWF and hs-CRP levels were prepared by centrifugation (20 minutes at 3000 rpm at 4°C) and stored at −80°C until use. Plasma levels of VWF (VWF antigen levels) and hs-CRP were measured at SRL Co Ltd. (Tokyo, Japan). For the measurement of VWF levels, enzyme-linked immuno sorbent assay (ELISA) microplates (STA-LIATEST VWF:Ag, DIAGNOSTICA STAGO S.A.S, Paris, France) were used.[17]
2.4. Statistical data analysis
The SAS software package (version 9.4, SAS Institute) at Fukuoka University (Fukuoka, Japan) was used for statistical data analyses. Categorical variables are presented as numbers (%), and continuous variables are presented as mean ± SD or median [interquartile range (IQR, 25th–75th percentile)]. Univariate regression analysis was performed to evaluate the correlation between continuous variables including the VWF level and IVUS parameters. To identify the factors that were associated with the coronary plaque burden presented as PAV, a multivariate stepwise regression analysis was conducted among conventional risk factors (age, gender, body mass index, current smoking, hypertension and diabetes mellitus), LDL-C and VWF level. A P value of <0.05 was considered to indicate statistical significance unless indicated otherwise.
3. Results
3.1. Characteristics of the patients and clinical laboratory data
Table 1 shows the patient characteristics in this study. The mean age was 70 ± 8 years, and 66 of the patients were male (73%). The frequencies of hypertension, diabetes, and dyslipidemia were 85%, 75% and 90%, respectively. The frequency of paroxysmal, persistent, or permanent atrial fibrillation (AF) was 10%, and all of the patients with AF were already being treated with oral anticoagulants (OACs). All of the patients in this study were already receiving statin and dual antiplatelet therapy (aspirin and thienopyridines) before PCI and IVUS examination.
Table 1.
Patient characteristics in CAD patients treated with statins.

Table 2 shows the clinical laboratory data for the subjects in this study. Lipid profiles were well controlled according to the Japanese guidelines; serum LDL-C levels were low (86 ± 26 mg/dL), and HDL-C and triglyceride levels were 47 ± 12 mg/dL and 138 ± 73 mg/dL, respectively. Systolic and diastole blood pressure were 128 ± 18 mm Hg and 70 ± 12 mm Hg, respectively.
Table 2.
Clinical laboratory data in CAD patients treated with statins.

3.2. Gray-scale and IB IVUS parameters at the culprit lesion
Gray-scale and IB IVUS parameters at the culprit lesion are summarized in Table 3. In the most diseased cross-section, the lumen area and the percentage of plaque area were 1.9 ± 0.5 mm2 and 81 ± 7%, respectively. The lesion length was 15.7 ± 7.7 mm, and the remodeling index was 0.96 ± 0.19. The percent atheroma volume was 68 ± 12%, and the percentages of lipid volume and fibrous volume were 56 ± 16% and 36 ± 11%, respectively.
Table 3.
Gray-scale IVUS and IB IVUS parameters.

3.3. Associations between clinical data and coronary plaque
Figure 2 shows the associations between VWF levels and IVUS parameters as assessed by univariate regression analysis. While VWF levels were positively correlated with PAV (r = 0.34, P = .003) (Fig. 2A), there was no significant association between VWF and plaque composition (Fig. 2 B–E).
Figure 2.
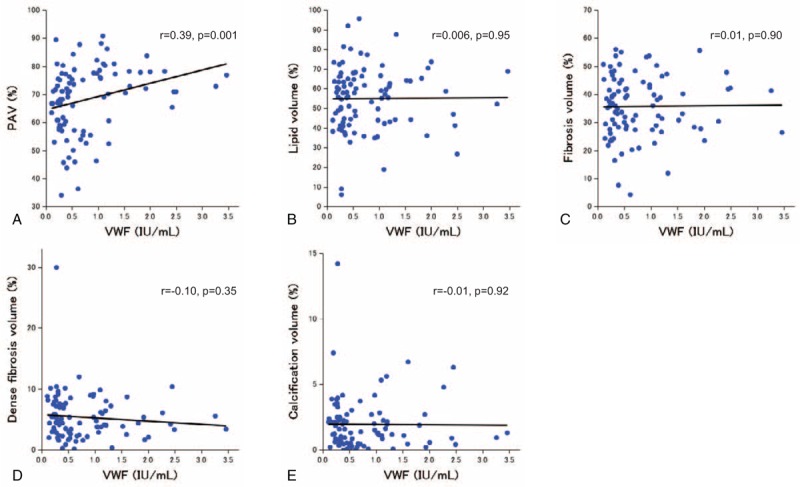
Associations between VWF levels and IVUS parameters. PAV (A), lipid volume (B), fibrosis volume (C), dense fibrosis volume (D), and calcification volume (E). IVUS = intravascular ultrasound, PAV = percent atheroma volume, VWF = von Willebrand factor.
Table 4 shows the results of a univariate regression analysis to identify the factors associated with PAV at the culprit lesion. While the VWF level was significantly associated with PAV (r = 0.39, P = .001), age, male gender, body mass index, hypertension, diabetes mellitus, current smoking, LDL-C, HDL-C, triglyceride, eGFR, uric acid and hs-CRP were not. A multivariate stepwise regression analysis indicated that VWF levels (β=0.26, P = .01) was independently associated with PAV (Table 5).
Table 4.
Univariate factors associated with PAV.

Table 5.
Multivariate factors associated with PAV.

4. Discussion
The main finding of this study was that higher VWF levels were significantly associated with a higher coronary plaque burden in CAD patients who had already received statin treatment (Fig. 2A, Tables 4 and 5).
VWF is produced almost exclusively by vascular endothelial cells,[2,18] and mediates platelet adhesion and aggregation.[1] VWF and platelets play important roles in the initiation of atherosclerotic plaque formation. Platelet adhesion to plaque-prone sites of arteries, which requires vascular endothelial cell activation but not actual endothelial denudation, is the initial step in plaque formation. This process is inhibited by the inactivation of VWF in a rabbit model.[19] In addition, the formation of fatty streaks in VWF-deficient mice is delayed compared with that in control mice.[20] VWF and/or the state of endothelial activation reflected by increased VWF levels may contribute to the pathogenesis of early atherosclerotic plaques.
Endothelial dysfunction, which is characterized by impaired bioavailability of endothelial-derived nitric oxide (NO), is involved in the development of atherosclerosis. Endothelial NO plays a crucial role in vasodilation, platelet aggregation, vascular smooth muscle proliferation, and endothelial-leukocyte interactions.[21] Several studies have demonstrated that atherosclerotic arteries decrease NO production.[22–24] It has been reported that blockade of NO production increased VWF levels in humans.[25,26] NO may be an inhibitor of endothelial VWF secretion.[18] Several human studies have reported a positive association between VWF levels and the risk of CAD.[3–7] Furthermore, a strong association between the extent of atherosclerosis in both the aortic arch and the carotid arteries and VWF levels in patients with transient ischemic attack or ischemic stroke has been reported.[27] A recent study using IVUS also showed that higher VWF levels were associated with a higher coronary plaque burden in CAD patients.[28] Similarly, a positive association between the VWF level and coronary plaque burden was found in our study. Elevated VWF levels may reflect impaired endothelial NO generation in vascular endothelial dysfunction and advanced atherosclerosis in patients with atherosclerotic cardiovascular diseases.
Statin is an established treatment for the prevention of cardiovascular events and has several pleiotropic effects. Statin increases endothelial NO production and improves vascular endothelial injury.[8,29] In addition, statin has a beneficial effect on coagulation and the fibrinolysis cascade. It has been reported that statin therapy reduced VWF levels in ACS patients.[30] Bruni et al[31] reported that statin decreased VWF levels in patients with hypercholesterolemia, and the change in plasminogen activator inhibitor 1 level, which is a marker of endothelial function, was directly related to the reduction in the VWF level. NO may suppress VWF secretion[18] and statins increase endothelial NO generation.[29] Therefore, the effect of statins on the reduction in VWF may be partially attributed to the improvement of endothelial dysfunction after statin therapy. In our study, all of the patients had already received statins, and LDL-C levels were appropriately lowered (86 ± 26 mg/dL) according to the Japanese guidelines.[13,14] Although VWF levels before the initiation of statin treatment were unclear, the mean VWF level in our study (0.78 ± 0.71 IU/mL) (Table 2) is lower than that of other CAD patients reported in a large-scale study (1.15 ± 2.00 to 1.39 ± 3.00 IU/mL).[6] Since statins produce coronary plaque regression, and may decrease VWF levels, our results may have been influenced by the statin treatment regimen. However, in our study, the LDL-C levels were not associated with either the coronary plaque burden (Table 4) or the VWF levels (data not shown). In addition, the positive association between the VWF levels and the plaque burden was independent of both LDL-C levels and conventional risk factors. These results suggest that high VWF levels may be a marker of the residual cardiovascular risk after statin treatment in CAD patients.
It has been reported that VWF levels are elevated in patients with AF.[32] Also, anticoagulation therapy may influence the VWF level in AF patients.[32] In this study, 9 patients had AF and were receiving OACs (Table 1). However, there was no significant difference in the level of VWF between patients with and without (data not shown). In addition, antiplatelet agents have been reported to affect VWF levels.[33] In this study, all of the patients were already receiving dual antiplatelet therapy (aspirin and thienopyridines) before measurement of the VWF level and the IVUS examination (Table 1). VWF level may be affected by a thrombogenic status including AF and antithrombotic therapy. Further studies will be needed to clarify these issues.
Since VWF levels are considered to be strongly increased in the acute phase of vascular diseases,[2] ACS patients were not included in this study. It has been reported that there was no significant association between VWF levels and the coronary plaque burden in ACS patients.[28] Increased VWF levels as a result of an acute-phase reaction in ACS may not be useful as a marker of coronary atherosclerosis.
In most previous studies, VWF antigen levels were measured as representative of plasma VWF levels.[17] It is possible that the functional activity of VWF might be more important for investigating the association between VWF and coronary atherosclerosis. However, it is not currently feasible to evaluate VWF functionality.
Recently, different classes of drugs that directly interfere with the VWF pathway, such as monoclonal antibodies, nanobodies, and aptamers, have been developed. These novel drugs have been reported to safely exert powerful antithrombotic effects.[34] To assess the effectiveness of these new drugs on coronary atherosclerosis, large-scale, prospective and randomized clinical trials are required.
The present study has several limitations. First, since the sample size in this single-center study is small, our results should be interpreted carefully. A large-scale multicenter study using IVUS will be needed to confirm our results. Second, CAD patients who did not undergo IVUS-guided PCI, who had unanalyzable IVUS findings, who were not receiving statins before PCI or whose blood samples could not be corrected for the measurement of VWF levels were not included in this study. In addition, the duration of enrollment in this study was 4 years, which may have been too long for the enrollment of only 91 patients. Therefore, the results may be affected by selection bias and may not be applicable to all CAD patients. Third, we performed IVUS examination only at the PCI site. Ethical considerations precluded the assessment of multiple plaques using IVUS in 3-vessel coronary trees, due to concerns regarding complications of IVUS examination such as coronary spasm and transient ischemia. Fourth, this study did not examine the relationships between the changes in VWF levels and the progression/regression of coronary plaque. A prospective study with serial IVUS examinations will be needed to investigate this issue. Fifth, both the type and dose of statin and the duration of statin treatment before IVUS examination are unknown. At present, it is unclear what type of statin is the best choice or whether a higher dose of statin is useful for more strongly decreasing the VWF level.[30] The type, dose, and duration of other medications such as antihypertensive and antiglycemic agents before enrollment are also unclear. These factors may affect the association between the coronary plaque burden and VWF levels.
5. Conclusions
Higher VWF levels were associated with a higher coronary plaque burden in CAD patients who had been treated with statins, suggesting that a high VWF level may be a marker of the residual cardiovascular risk after statin treatment.
Author contributions
Conceptualization: Yuta Kato, Atsushi Iwata.
Data curation: Yuta Kato, Makito Futami, Motoki Yamashita.
Formal analysis: Yuta Kato, Makito Futami, Motoki Yamashita.
Investigation: Satoshi Imaizumi, Takashi Kuwano, Amane Ike, Makoto Sugihara, Hiroaki Nishikawa, Shin’ichiro Yasunaga.
Project administration: Atsushi Iwata.
Supervision: Keijiro Saku, Shin-ichiro Miura.
Validation: Atsushi Iwata, Bo Zhang.
Writing – original draft: Atsushi Iwata.
Footnotes
Abbreviations: ACS = acute coronary syndrome, AF = atrial fibrillation, CAD = coronary artery disease, CSA = cross-sectional area, EEM = external elastic membrane, eGFR = estimated glomerular filtration rate, ELISA = enzyme-linked immuno sorbent assay, HDL-C = high-density lipoprotein cholesterol, hs-CRP = high-sensitivity C-reactive protein, IB = integrated backscatter, IVUS = intravascular ultrasound, LDL-C = low-density lipoprotein cholesterol, NO = nitric oxide, OACs = oral anticoagulants, PAV = percent atheroma volume, PCI = percutaneous coronary intervention, VWF = von Willebrand factor.
Conflict of interest: KS and SM are Directors of NPO Clinical and Applied Science, Fukuoka, Japan. KS and SM received a grant from the Public Interest Incorporated Foundation of “Clinical Research Promotion Foundation” in Fukuoka, Japan, and part of this work was transferred to NPO Clinical and Applied Science, Fukuoka, Japan. KS has an Endowed Department of Molecular Cardiovascular Therapeutics (SM), Fukuoka University, supported by MSD Co., Ltd, and an Endowed Department of Community and Emergency Medicine, Fukuoka University, supported by Izumi City, Kagoshima, Japan.
References
- [1].Ruggeri ZM. The role of von Willebrand factor in thrombus formation. Thromb Res 2007;120(suppl 1):S5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lip GY, Blann A. von Willebrand factor: a marker of endothelial dysfunction in vascular disorders? Cardiovasc Res 1997;34:255–65. [DOI] [PubMed] [Google Scholar]
- [3].Tzoulaki I, Murray GD, Lee AJ, et al. Relative value of inflammatory, hemostatic, and rheological factors for incident myocardial infarction and stroke: the Edinburgh Artery Study. Circulation 2007;115:2119–27. [DOI] [PubMed] [Google Scholar]
- [4].Willeit P, Thompson A, Aspelund T, et al. Hemostatic factors and risk of coronary heart disease in general populations: new prospective study and updated meta-analyses. PLoS One 2013;8:e55175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sonneveld MA, de Maat MP, Leebeek FW. Von Willebrand factor and ADAMTS13 in arterial thrombosis: a systematic review and meta-analysis. Blood Rev 2014;28:167–78. [DOI] [PubMed] [Google Scholar]
- [6].Folsom AR, Wu KK, Shahar E, et al. Association of hemostatic variables with prevalent cardiovascular disease and asymptomatic carotid artery atherosclerosis. The Atherosclerosis Risk in Communities (ARIC) Study Investigators. Arterioscler Thromb 1993;13:1829–36. [DOI] [PubMed] [Google Scholar]
- [7].Thompson SG, Kienast J, Pyke SD, et al. Hemostatic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group. N Engl J Med 1995;332:635–41. [DOI] [PubMed] [Google Scholar]
- [8].Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res 2017;120:229–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet (London, England) 2005;366:1267–78. [DOI] [PubMed] [Google Scholar]
- [10].Mintz GS, Nissen SE, Anderson WD, et al. American College of Cardiology Clinical Expert Consensus Document on Standards for Acquisition, Measurement and Reporting of Intravascular Ultrasound Studies (IVUS). A report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol 2001;37:1478–92. [DOI] [PubMed] [Google Scholar]
- [11].Nicholls SJ, Hsu A, Wolski K, et al. Intravascular ultrasound-derived measures of coronary atherosclerotic plaque burden and clinical outcome. J Am Coll Cardiol 2010;55:2399–407. [DOI] [PubMed] [Google Scholar]
- [12].Puri R, Nissen SE, Shao M, et al. Coronary atheroma volume and cardiovascular events during maximally intensive statin therapy. Eur Heart J 2013;34:3182–90. [DOI] [PubMed] [Google Scholar]
- [13].Guidelines for Secondary Prevention of Myocardial Infarction (JCS 2011). Circ J 2013;77:231–48. [DOI] [PubMed] [Google Scholar]
- [14].Teramoto T, Sasaki J, Ueshima H, et al. Executive summary of Japan Atherosclerosis Society (JAS) guideline for diagnosis and prevention of atherosclerotic cardiovascular diseases for Japanese. J Atheroscler Thromb 2007;14:45–50. [DOI] [PubMed] [Google Scholar]
- [15].Nakayama N, Hibi K, Endo M, et al. Validity and reliability of new intravascular ultrasound analysis software for morphological measurement of coronary artery disease. Circ J 2013;77:424–31. [DOI] [PubMed] [Google Scholar]
- [16].Kawasaki M, Takatsu H, Noda T, et al. In vivo quantitative tissue characterization of human coronary arterial plaques by use of integrated backscatter intravascular ultrasound and comparison with angioscopic findings. Circulation 2002;105:2487–92. [DOI] [PubMed] [Google Scholar]
- [17].Holmberg L, Berntorp E, Donner M, et al. von Willebrand's disease characterized by increased ristocetin sensitivity and the presence of all von Willebrand factor multimers in plasma. Blood 1986;68:668–72. [PubMed] [Google Scholar]
- [18].Vischer UM. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J Thromb Haemost 2006;4:1186–93. [DOI] [PubMed] [Google Scholar]
- [19].Theilmeier G, Michiels C, Spaepen E, et al. Endothelial von Willebrand factor recruits platelets to atherosclerosis-prone sites in response to hypercholesterolemia. Blood 2002;99:4486–93. [DOI] [PubMed] [Google Scholar]
- [20].Methia N, Andre P, Denis CV, et al. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood 2001;98:1424–8. [DOI] [PubMed] [Google Scholar]
- [21].Gauthier TW, Scalia R, Murohara T, et al. Nitric oxide protects against leukocyte-endothelium interactions in the early stages of hypercholesterolemia. Arterioscler Thromb Vasc Biol 1995;15:1652–9. [DOI] [PubMed] [Google Scholar]
- [22].Guerra R, Jr, Brotherton AF, Goodwin PJ, et al. Mechanisms of abnormal endothelium-dependent vascular relaxation in atherosclerosis: implications for altered autocrine and paracrine functions of EDRF. Blood Vessels 1989;26:300–14. [DOI] [PubMed] [Google Scholar]
- [23].Sreeharan N, Jayakody RL, Senaratne MP, et al. Endothelium-dependent relaxation and experimental atherosclerosis in the rabbit aorta. Can J Physiol Pharmacol 1986;64:1451–3. [DOI] [PubMed] [Google Scholar]
- [24].Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987;327:524–6. [DOI] [PubMed] [Google Scholar]
- [25].Pernerstorfer T, Stohlawetz P, Kapiotis S, et al. Partial inhibition of nitric oxide synthase primes the stimulated pathway of vWF-secretion in man. Atherosclerosis 2000;148:43–7. [DOI] [PubMed] [Google Scholar]
- [26].Jilma B, Pernerstorfer T, Dirnberger E, et al. Effects of histamine and nitric oxide synthase inhibition on plasma levels of von Willebrand factor antigen. J Lab Clin Med 1998;131:151–6. [DOI] [PubMed] [Google Scholar]
- [27].Sonneveld MA, van Dijk AC, van den Herik EG, et al. Relationship of Von Willebrand Factor with carotid artery and aortic arch calcification in ischemic stroke patients. Atherosclerosis 2013;230:210–5. [DOI] [PubMed] [Google Scholar]
- [28].Sonneveld MA, Cheng JM, Oemrawsingh RM, et al. Von Willebrand factor in relation to coronary plaque characteristics and cardiovascular outcome. Results of the ATHEROREMO-IVUS study. Thromb Haemost 2015;113:577–84. [DOI] [PubMed] [Google Scholar]
- [29].Laufs U, La Fata V, Plutzky J, et al. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 1998;97:1129–35. [DOI] [PubMed] [Google Scholar]
- [30].Ordulu E, Erdogan O. Early effects of low versus high dose atorvastatin treatment on coagulation and inflammation parameters in patients with acute coronary syndromes. Int J Cardiol 2008;128:282–4. [DOI] [PubMed] [Google Scholar]
- [31].Bruni F, Pasqui AL, Pastorelli M, et al. Effect of atorvastatin on different fibrinolyis mechanisms in hypercholesterolemic subjects. Int J Cardiol 2004;95:269–74. [DOI] [PubMed] [Google Scholar]
- [32].Wysokinski WE, Cohoon KP, Konik EA, et al. Effect of atrial fibrillation duration on plasma von Willebrand factor level. Eur J Haematol 2017;99:569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Matsuura K, Imamaki M, Ishida A, et al. The effect of preoperative aspirin administration on postoperative level of von Willebrand factor in off-pump coronary artery bypass surgery. Heart Vessels 2009;24:169–74. [DOI] [PubMed] [Google Scholar]
- [34].Gragnano F, Golia E, Natale F, et al. Von Willebrand Factor and cardiovascular disease: from a biochemical marker to an attractive therapeutic target. Curr Vasc Pharmacol 2017;15:404–15. [DOI] [PubMed] [Google Scholar]