

Abstract
Nonalcoholic fatty liver disease (NAFLD) represents a disease spectrum ranging from benign steatosis to life‐threatening cirrhosis and hepatocellular carcinoma. Elevated levels of reactive oxygen species (ROS) and exacerbated inflammatory responses have been implicated in NAFLD progression. Nicotinamide adenine dinucleotide phosphate (reduced) oxidase 2 (NOX2; also known as gp91Phox), the main catalytic subunit of the nicotinamide adenine dinucleotide phosphate (reduced) oxidase complex, modulates ROS production, immune responsiveness, and pathogenesis of obesity‐associated metabolic derangements. However, the role of NOX2 in the regulation of immune cell function and inflammatory vigor in NAFLD remains underdefined. Here, we demonstrate that obesogenic diet feeding promoted ROS production by bone marrow, white adipose tissue, and liver immune cells. Genetic ablation of NOX2 impeded immune cell ROS synthesis and was sufficient to uncouple obesity from glucose dysmetabolism and NAFLD pathogenesis. Protection from hepatocellular damage in NOX2‐deficient mice correlated with reduced hepatic neutrophil, macrophage, and T‐cell infiltration, diminished production of key NAFLD‐driving proinflammatory cytokines, and an inherent reduction in T‐cell polarization toward Th17 phenotype. Conclusion: Current findings demonstrate a crucial role of the NOX2–ROS axis in immune cell effector function and polarization and consequent NAFLD progression in obesity. Pharmacologic targeting of NOX2 function in immune cells may represent a viable approach for reducing morbidity of obesity‐associated NAFLD pathogenesis. (Hepatology Communications 2018;2:546‐560)
Abbreviations
- ALT
alanine aminotransferase
- BAT
brown adipose tissue
- BMDC
bone marrow derived dendritic cell
- BMDM
bone marrow derived macrophage
- BMN
bone marrow neutrophil
- CD
clusters of differentiation
- DCFDA
2′,7′‐dichlorofluorescin diacetate
- eWAT
epididymal white adipose tissue
- FBS
fetal bovine serum
- HFD
high‐fat diet
- IFN‐γ
interferon‐γ
- IL
interleukin
- iWAT
inguinal white adipose tissue
- LPS
lipopolysaccharide
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NOX
reduced nicotinamide adenine dinucleotide phosphate oxidase
- ROS
reactive oxygen species
- RPMI
Roswell Park Memorial Institute
- TG
triglyceride
- Th
T helper
- TLR
toll‐like receptor
- TNF‐α
tumor necrosis factor α
- UCP1
uncoupling protein 1
- WT
wild type
Nonalcoholic fatty liver disease (NAFLD) has emerged as the most prominent chronic liver disease. NAFLD, a hepatic manifestation of metabolic disease, represents a wide spectrum of liver diseases ranging from simple steatosis to nonalcoholic steatohepatitis (NASH) and cirrhosis. NAFLD progression involves a complex interplay between various biological processes, including obesity, dysbiosis of the intestinal microbiome, and exacerbation of proinflammatory immune responses.1, 2 Although NAFLD is a multifactorial disease, it is well accepted that diet‐induced inflammation, reactive oxygen species (ROS) production, and lipid peroxidation play central roles in NAFLD pathogenesis.3, 4, 5
ROS, the general byproduct of cellular metabolism, impacts macromolecular integrity, cell signaling, and innate immunity. Cell intrinsic imbalance between ROS production and antioxidant capability results in oxidative stress, while oxidative stress contributes to the pathogenesis of various metabolic diseases.6 In obesity, excess systemic nutrients induce oxidative stress and promote physiologic changes in key peripheral tissues including adipose tissue, liver, and skeletal muscle. The nicotinamide adenine dinucleotide phosphate (reduced) oxidase (NOX) enzyme complex, a central source of intracellular ROS, is composed of NOX2 (also known as gp91Phox); the cytosolic p40Phox, p47Phox, p67Phox units; and Rac family small guanosine triphosphatase 1 complexes.7 NOX2, which is highly expressed in immune cells,8 modulates insulin resistance, adipose tissue inflammation, hepatic stellate cell activation, and hepatic fibrosis.9, 10, 11 However, the role of NOX2 in modulating immune cell responsiveness in NAFLD has not been examined. A better understanding of mechanisms underlying activation of NOX2‐dependent ROS production may uncover unappreciated molecular targets for novel NAFLD therapies.
Here, we demonstrated that high‐fat diet (HFD) feeding promoted immune cell ROS generation and that NOX2 modulated ROS synthesis, immune cell inflammatory capacity, and obesity pathogenesis. Specifically, we show that despite similar obesity, adiposity, and liver triglyceride (TG) accumulation, NOX2‐deficient mice were protected from obesity‐induced glucose dysmetabolism and hepatocellular damage. Reduced severity of obesity‐driven hepatocellular damage in NOX2‐deficient mice was associated with decreased hepatic infiltration of immune cells. Mechanistically, NOX2‐deficient hepatic neutrophils, macrophages, and T cells had diminished production of key NAFLD‐driving, proinflammatory cytokines, including interleukin (IL)‐17A, interferon‐γ (IFN‐γ), and tumor necrosis factor α (TNF‐α). Further, T‐cell intrinsic NOX2 expression was sufficient to modulate polarization and activation‐driven proinflammatory cytokine production in T cells. In sum, our data suggest that NOX2‐driven modulation of immune cell inflammatory capacity may play important roles in obesity‐driven NAFLD progression.
Materials and Methods
MICE
All studies were approved by the Cincinnati Children's Hospital Medical Center's Institutional Animal Care and Use Committee. All mice used in this study were on a C57BL/6 background, housed in the same room within a specific pathogen‐free facility but not as littermates, fed food and water ad libitum, and provided care in accordance with the Guide for the Care and Use of Laboratory Animals. Wild‐type (WT) mice (strain name C57BL/6J) and NOX2−/− mice (strain name B6.129S‐CybbtmDin/J) were obtained from Jackson Laboratories and were subsequently bred at the Cincinnati Children's Hospital Medical Center.
OBESITY AND NAFLD MODEL
Starting at 8 weeks of age, mice were fed ad libitum either chow diet (Lab diet #5010) or HFD (Research diets #D12492) for 8 or 22 weeks, with fresh food provided on a weekly basis. Body weight and food consumption were monitored weekly.
IMMUNE CELL ISOLATION AND CYTOKINE PRODUCTION
Peritoneal neutrophils (PN) were collected by peritoneal gavage after intraperitoneal injection of zymosan (10, 100, or 1,000 μg/mouse; 12 hours) as described.12 Bone marrow‐derived macrophages (BMDMs), bone marrow neutrophils (BMNs), and bone marrow‐derived dendritic cells (BMDCs) were isolated as described.12, 13, 14, 15 Briefly, bone marrow cells were differentiated into BMDMs or BMDCs using Roswell Park Memorial Institute (RPMI) medium (Gibco) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 1% glutamine, and 20 ng/mL macrophage colony stimulating factor or granulocyte‐macrophage colony stimulating factor (Biolegend) for 6 days at 37oC and 5% CO2. BMNs were purified using the anti‐Ly‐6G Microbead Kit (Miltenyi Biotec) and cultured in RPMI medium supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% glutamine. BMDMs (6 × 105 cells/mL), BMDCs (6 × 105 cells/mL), and BMNs (5 × 105 cells/mL) were stimulated with lipopolysaccharide (LPS; 10, 100, or 1,000 ng/mL) for 4 hours, followed by ROS quantification and collection of cell supernatant for cytokine analysis by enzyme‐linked immunosorbent assay (BD Opt EIA; BD Bioscience). For tissue immune cell isolation, single‐cell suspensions from epididymal white adipose tissue (eWAT) and liver were obtained using common methods.14, 16, 17 Minced eWAT tissue was digested for 45 minutes in collagenase cocktail (0.03 mg/mL liberase and 50 U/mL deoxyribonuclease I [Sigma‐Aldrich]), filtered using a 100‐μm nylon cell strainer, centrifuged at 1,000g for 10 minutes, treated with erythrocyte lysis buffer (Lonza), and washed in RPMI medium prior to ROS and cytokine‐level quantification. Liver tissue samples were minced using gentleMACS tubes (Miltenyi Biotec) according to the manufacturer's recommendation. Immune cells were purified using 33% Percoll (Sigma‐Aldrich) in RPMI medium followed by centrifugation at 250g for 20 minutes at room temperature and then treated with erythrocyte lysis buffer and washed in RPMI medium prior to ROS and cytokine‐level quantification.
ROS STAINING
ROS production was quantified ex vivo by 2′,7′‐dichlorofluorescin diacetate (DCFDA) staining. Isolated immune cells were incubated with 25 μM DCFDA at 37oC in complete RPMI medium for 30 minutes, washed with phosphate‐buffered saline, and analyzed by flow cytometry. For each experiment, at least 1 × 105 cells/mouse were analyzed to calculate mean fluorescence intensity of DCFDA staining.
SERUM PARAMETERS
TG quantification was performed as reported.14, 17, 18, 19 Briefly, 200 μL of triglyceride reagent (Pointe Scientific) was added to 10 μL of serum in a 96‐well, clear, flat‐bottom plate (Costar). Standards (Pointe Scientific) were prepared according to the manufacturer's instructions. Serum leptin (Millipore) and NO2 (Sigma‐Aldrich) levels were quantified using commercially available enzyme‐linked immunosorbent assay kits.
HISTOLOGY
Liver and eWAT samples were fixed in 10% formalin. Hematoxylin and eosin staining was performed on 5‐μm sections from the paraffin‐embedded tissue blocks for conventional light microscopy analysis. Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling staining of eWAT samples were deparaffinized and stained using the standard protocol. For immunohistochemical staining, liver sections were deparaffinized and immunostained with rabbit anti‐mouse clusters of differentiation (CD)68 antibody (Abcam) or rabbit monoclonal anti‐mouse CD3 antibody (Ventana Medical Systems, Inc.), using the automated Ventana immunostainer according to the manufacturer's recommendation.14, 17, 18
GLUCOSE AND INSULIN SENSITIVITY TESTS
All studies were performed on mice fasted overnight as reported.14, 17 Briefly, for the glucose tolerance test, mice were challenged with a bolus of dextrose (100 mg/kg body weight) by intraperitoneal injection and blood glucose levels were measured at 20, 40, 60, 90, and 120 minutes. For the insulin tolerance test, mice were challenged with insulin (0.3 U/mL of insulin/10 g of body weight) by intraperitoneal injection and blood glucose levels were measured at the time points specified above. All measurements were performed using the Accu‐chek kit (Roche).
LIVER TGS
Hepatic TG levels were quantified as described.14, 17, 18 Briefly, standards and samples were added to a 96‐well, clear, flat‐bottom plate (Costar) containing 200 μL of triglyceride reagent (Pointe Scientific). Hepatic TGs were quantified at 500‐520 nm using the vmax Microplate Reader (Molecular Devices) and SoftMax Pro version 5 software.
HEPATOCELLULAR DAMAGE
Alanine aminotransferase (ALT) levels were quantified as described.14, 18 Briefly, 10 μL of mouse serum was added to a 96‐well flat‐bottom plate (Costar) containing 200 μL ALT buffer that was prepared by mixing ALT Activator Reagent and ALT sample Diluent Reagent (Catachem Inc.). Catatrol I and II (Catachem Inc.) were used as controls and were prepared according to the manufacturer's instructions. ALT levels were quantified using the BioTek Synergy 2 Multi‐Mode Microplate Reader with Gen5 version 2 software.
FLOW CYTOMETRY
Immune cells were stained directly with conjugated monoclonal antibodies or isotype controls (all eBioscience). For quantification of immune cell cytokine production, isolated immune cells were stimulated for 4 hours with 50 ng/mL phorbol 12‐myristate 13‐acetate (Sigma‐Aldrich) and 1 μg/mL ionomycin (Calbiochem), in the presence of brefeldin A (10 μg/mL; Sigma‐Aldrich) and analyzed by intracellular flow cytometry. Briefly, cells were treated with fluorescence‐activated cell sorting buffer (BD Biosciences) for 15 minutes, washed, and stained with live/dead stain (Zombie UV Dye: Biolegend) and with directly conjugated monoclonal antibodies to CD45‐PEDazzle (clone 104), CD4‐allophycocyanin (APC)ef780 (clone RM4‐5), CD3‐Alexa Fluor 700 (clone 17A2), CD11b‐ef450 (clone M1/70), CD11c‐APC (clone N418), Gr1‐ fluorescein isothiocyanate (FITC) (clone RB6‐8C5), and B220‐BV605 (clone RA3‐6B2) in phosphate‐buffered saline supplemented with 2% FBS for 30 minutes. Cells were subsequently fixed, permeabilized, and stained with IFN‐γ–phycoerythrin (PE)–Cy7 (clone XMG1.2), TNF‐α–BV650 (clone MP6‐XT22), and IL‐17A–Percp Cy5.5 (clone eBio17B7) (all antibodies from e‐Bioscience) for 30 minutes. LSR Fortessa (BD Biosciences) and FlowJo software (version X0.7) were used for flow cytometry data collection and analysis.14, 17, 18
T‐HELPER 17 POLARIZATION AND T‐CELL ACTIVATION
T‐cell isolation kits (Miltenyi Biotec) were used to isolate naive splenic T cells according to the manufacturer's protocol. Isolated CD4+CD62L+ T cells were cultured in a goat anti‐hamster immunoglobulin G‐coated plate and differentiated for 6 days with anti‐CD3 (1 μg/mL) and anti‐CD28 (0.5 μg/mL) and with or without recombinant human IL‐1β (1 ng/mL), recombinant mouse IL‐6 (10 ng/mL), recombinant human IL‐23 (20 ng/mL), or recombinant human TGF‐β1 (1 ng/mL). T cells were stimulated using phorbol 12‐myristate 13‐acetate (50 ng/mL), ionomycin (1 μg/mL), and brefeldin A (10 μg/mL) for 4 hours, and IL‐17A, IFN‐γ, and TNF‐α production were quantified by flow cytometry. For each experiment, 1 × 105 to 2 × 105 cells per mouse were analyzed.
PERIPHERAL BLOOD CELLULAR COMPOSITION
Blood samples were collected in heparin tubes (BD Biosciences) and analyzed in Hemavet 950 (Drew Scientific Inc.) using the manufacturer's program for mouse blood analysis.
STATISTICAL ANALYSIS
Data were analyzed using the unpaired Student t test. All values are represented as means + SE and graphed with Prism 7 software (GraphPad Software Inc.).
Results
OBESITY AND INFLAMMATION, IN PART BY NOX2 ACTIVATION, PROMOTE IMMUNE CELL ROS PRODUCTION
Obesity‐associated inflammation and oxidative stress, in part by ROS production, modulate the pathogenesis of obesity‐associated sequelae, including type 2 diabetes and NAFLD.1, 2, 4, 5, 20 Here, we examined the contribution of obesogenic diet feeding on immune cell ROS production in various tissues. HFD feeding compared to the chow diet significantly enhanced ROS production by primary BMNs (Fig. 1A). However, HFD feeding failed to promote ROS production in primary BMDMs and BMDCs, both being immune cells derived by prolonged in vitro culture and polarization (Fig. 1A; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1162/full). Further, HFD feeding significantly enhanced ROS production by neutrophils (CD11b+Gr1high) and macrophages (CD11b+F4/80+) infiltrating eWAT and liver (Fig. 1B,C), which are tissues relevant to NAFLD pathogenesis. Genetic ablation of NOX2 was sufficient to impede obesity‐driven ROS induction in BMNs and macrophages in the context of inflammatory challenge (Fig. 1D). Obesity is associated with metabolic endotoxemia17, 21 and various inflammatory triggers, including endotoxin (LPS), that drive ROS and proinflammatory cytokine production.2, 4 NOX2 is a central player in immune cell ROS synthesis. Hence, the impact of NOX2 on immune cell ROS production in response to endotoxin sensing was examined next. NOX2‐deficient neutrophils, BMDMs, and BMDCs compared to WT controls exhibited reduction in LPS‐induced ROS production in a dose‐dependent manner (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1162/full). Further, LPS‐induced ROS levels were higher in BMNs isolated from HFD‐fed mice compared to chow‐fed controls (data not shown). These effects were not limited to LPS as zymosan treatment of NOX2‐deficient mice resulted in similarly decreased neutrophil ROS production (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1162/full). Further, the NOX2 impact on zymosan‐induced ROS production was independent of differential neutrophil recruitment into the peritoneal cavity (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1162/full). Congruently, stimulation of NOX2‐deficient macrophages with LPS resulted in reduced IL‐6 production (data not shown), a proinflammatory mediator known to play a role in pathogenesis of obesity‐associated sequelae.22 Collectively, these data suggest that obesity augments immune cell ROS production in tissues that contribute to the pathogenesis of obesity‐associated sequelae. Further, our data suggest that NOX2, in the context of HFD‐induced obesity or an obesity‐associated inflammatory environment, likely promotes immune cell‐driven ROS synthesis.
Figure 1.

HFD modulates ROS production by immune cells. (A) ROS production by bone marrow neutrophils and macrophages from either chow diet or HFD‐fed WT mice. (B) ROS production by hepatic neutrophils and macrophages from either chow‐ or HFD‐fed WT mice. (C) ROS production by epididymal white adipose tissue neutrophils and macrophages from either chow‐ or HFD‐fed WT mice. (D) ROS production by bone marrow neutrophils and macrophages from HFD‐fed WT and NOX2−/− mice. (A‐D) Data presented as relative mean fluorescence intensity. Gray bars denote WT chow‐fed mice; white bars denote WT HFD‐fed mice; black bars denote NOX2−/− HFD‐fed mice. (A,D) A single experiment, n = 3‐4 mice/condition. (B,C) Data combined from three independent experiments, n = 6‐8 mice/condition. Data represent means + SE. Student t test, *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: CD, chow diet; MFI, mean fluorescence intensity.
NOX2 UNCOUPLES OBESITY AND ADIPOSITY FROM GLUCOSE DYSMETABOLISM
As NOX2 modulated immune cell ROS and proinflammatory cytokine production with HFD feeding, the impact of NOX2 in obesity development and pathogenesis of obesity‐associated sequelae was examined. In agreement with a published report,11 both WT and NOX2−/− mice exhibited similar HFD‐driven weight gains (Fig. 2A). Comparable weight gain correlated with similar food intake, systemic TG, leptin and NO2 levels, brown adipose tissue (BAT) mass, and BAT uncoupling protein 1 (Ucp1) expression (Fig. 2B‐F). Notably, analogous BAT characteristics are suggestive of similar overall energy expenditure between WT and NOX2−/− mice and are in agreement with published findings.23 Altered adipose tissue mass distribution (e.g., decreased eWAT and increased inguinal white adipose tissue [iWAT]), increased adipose tissue inflammation, and adipocyte death have been correlated with advancement of obesity‐related sequelae, including NAFLD.24 Notably, obese NOX2−/− mice compared to WT controls exhibited increased eWAT weight and decreased iWAT weight (Fig. 2G). The difference in eWAT mass was independent of adipocyte size (Fig. 2H) but correlated with decreased eWAT adipocyte cell death as determined by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling staining (Fig. 2I).
Figure 2.

NOX2 modulates HFD‐driven white adipose tissue distribution. Eight‐week‐old WT and NOX2−/− mice were fed an HFD for 22 weeks. (A) Body weight; (B) cumulative weekly food intake; (C) serum leptin level; (D) serum triglyceride level; (E) serum nitrite level; (F) BAT weight and Ucp1 expression; (G) WAT weight; (H) hematoxylin and eosin staining of eWAT; (I) TUNEL staining of eWAT and TUNEL‐positive cell number per field of view. White bars/squares denote WT mice; black bars/squares denote NOX2−/− mice. (A‐I) A representative of two individual experiments, n = 4‐6 mice/condition. Data represent means + SE. Student t test, *P < 0.05, **P < 0.01. Abbreviations: AU, arbitrary unit (relative); TUNEL, terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling.
The role of NOX2‐dependent ROS in modulating the severity of obesity‐associated glucose dysmetabolism is controversial.9, 10, 25, 26 Hence, the development of glucose dysmetabolism and insulin resistance was examined. Although obese NOX2−/− mice compared to WT controls had reduced fasting glucose levels and improved glucose tolerance (Fig. 3A,B), lack of NOX2 did not alter systemic insulin levels or insulin sensitivity (Fig. 3C,D). Further, genetic ablation of NOX2 did not modulate the expression of mediators central for insulin‐induced glucose homeostasis in liver and eWAT (e.g., glucose transporter 1 [Glut1], Glut4, carbohydrate‐responsive element‐binding protein β, and sterol regulatory element binding protein 1c; data not shown).27, 28 Overall, these data suggest that NOX2 regulates glucose but not insulin metabolism in HFD‐induced obesity. Of note, these data are in agreement with reported NOX2‐independent pathways of glucotoxicity in pancreatic islets in mice.29
Figure 3.
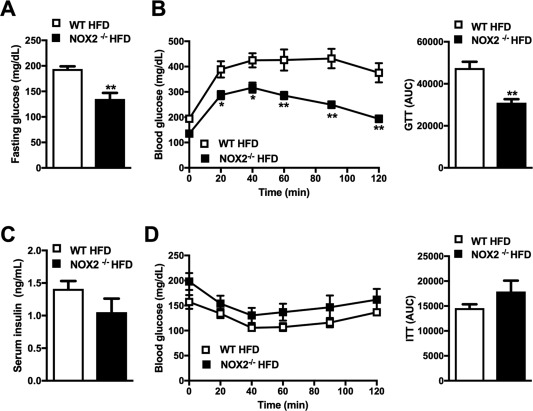
NOX2 regulates HFD‐induced glucose dysmetabolism. Eight‐week‐old WT and NOX2−/− mice were fed an HFD for 22 weeks. (A) Fasting glucose; (B) glucose tolerance test after 13 weeks of HFD feeding; (C) fasting insulin; (D) insulin tolerance test performed after 15 weeks of HFD feeding. White bars/squares denote WT mice; black bars/squares denote NOX2−/− mice. (A‐D) A representative of two individual experiments, n = 4‐6 mice/condition. Data represent means + SE. Student t test, *P < 0.05, **P < 0.01. Abbreviations: AUC, area under the curve; GTT, glucose tolerance test; ITT, insulin tolerance test.
NOX2 REGULATES HFD‐INDUCED HEPATOCELLULAR DAMAGE AND NAFLD PROGRESSION
Obesity and glucose dysmetabolism contribute to NAFLD pathogenesis. Thus, we examined the impact of NOX2 on NAFLD development. HFD‐fed WT and NOX2−/− mice exhibited similar liver weight (Fig. 4A) and hepatic TG accumulation (Fig. 4B). However, in contrast to obese WT controls, which had macrovesicular steatosis (a characteristic that is highly prevalent in most patients with NAFLD), obese NOX2−/− mice primarily exhibited microvesicular steatosis (Fig. 4C,D).30, 31 Attenuated macrovesicular steatosis observed in obese NOX2−/− mice correlated with reduced serum ALT levels (Fig. 4E) and hepatic lipid peroxidation (Fig. 4F). Of note, elevated hepatic TG and ROS levels are key characteristics of macrosteatotic livers, something that renders hepatocytes susceptible to secondary insults and cellular death.32 Such findings are in agreement with the notion that, along with mitochondrial ROS, NOX2‐dependent chronic oxidative stress and ROS‐induced lipid peroxidation are central to NAFLD progression.33
Figure 4.
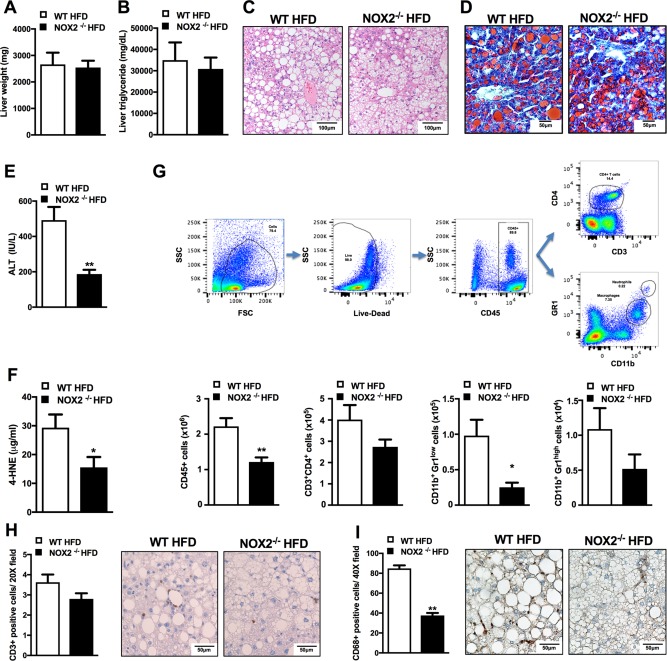
NOX2 regulates HFD‐induced NAFLD pathogenesis. Eight‐week‐old WT and NOX2−/− mice were fed an HFD for 22 weeks. (A) Liver weight; (B) liver triglyceride content; (C) hematoxylin and eosin staining of liver; (D) Oil‐Red‐O staining of liver; (E) ALT levels; (F) liver 4‐HNE levels. (G) Gating strategy for quantification of hepatic immune cell infiltration by flow cytometry and hepatic immune cell composition. CD11b+ Gr1low macrophage populations were further gated by forward side scatter to differentiate between CD11b+Gr1low macrophages and leukocytes. (H) Liver CD3+ staining; (I) liver CD68+ staining. White bars denote WT mice; black bars denote NOX2−/− mice. (A‐I) A representative of two individual experiments, n = 4‐6 mice/condition. Data represent means + SE. Student t test, *P < 0.05, **P < 0.01. Abbreviations: 4‐HNE, 4‐hydroxynonenal; FSC, Forward scatter; SSC, Side scatter.
While biological processes responsible for progression of NAFL to NASH are not fully understood, augmented infiltration of immune cells and enhanced production of proinflammatory mediators by liver‐infiltrating immune cells are believed to play an important role.2, 4 Hence, hepatic immune cell composition in HFD‐fed WT and NOX2−/− mice was examined. A reduction in total hepatic immune cell infiltration (CD45+ cells) observed in obese NOX2−/− mice largely correlated with robustly lowered numbers of liver‐infiltrating macrophages (CD11b+Gr1Low) and CD4+ T cells (Fig. 4G), a finding further confirmed by a significant reduction in CD68 and a trend toward reduced CD3 immunostaining of liver sections (Fig. 4H,I). Of note, altered hepatic immune infiltration observed in obese NOX2−/− mice was independent of differential circulating immune cell numbers or composition in the context of HFD feeding or following systemic LPS challenge (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1162/full).
NOX2 MODULATES HEPATIC IMMUNE CELL INFLAMMATORY VIGOR
Hepatic accumulation of neutrophils, macrophages, and CD4+ T cells (e.g., T helper [Th]1 and Th17 subsets) correlates with NAFLD progression.34, 35 NOX2 regulates hepatic immune cell infiltration and immune cell cytokine production.36 Notably, proinflammatory cytokines (e.g., IFN‐γ, TNF‐α, IL‐17A) are well‐established modulators of NAFLD pathogenesis.1, 2, 4, 20 We therefore examined the role of NOX2 in the modulation of immune cell inflammatory vigor. Obese NOX2−/− mice compared to WT controls exhibited attenuated production of IFN‐γ and TNF‐α by liver‐infiltrating neutrophils (CD11b+Gr1high; Fig 5A), macrophages (CD11b+Gr1low; Fig 5B), and IL‐17A, IFN‐γ, and TNF‐α by CD4+ T cells (CD3+CD4+; Fig. 5C). Together, these data suggest that NOX2 modulates inflammatory vigor in liver‐infiltrating immune cells.
Figure 5.

NOX2 regulates hepatic immune cell inflammatory vigor. Eight‐week‐old WT and NOX2−/− mice were fed an HFD for 22 weeks, and cytokine production in liver infiltrating immune cells was quantified by flow cytometry. Cells were exclusively identified from neutrophil, macrophage, and CD3+CD4+ gates described in Fig. 4G. (A) Cytokine production by liver‐infiltrating neutrophils (CD11b+GR1high). (B) Cytokine production by liver‐infiltrating macrophages (CD11b+GR1low). (C) Cytokine production by liver‐infiltrating T cells (CD3+CD4+). White bars denote WT mice; black bars denote NOX2−/− mice. (A‐C) A representative of two individual experiments, n = 4‐6 mice/condition. Data represent means + SE. Student t test, *P < 0.05, **P < 0.01.
Although NOX2 is predominantly associated with myeloid immune cell function, the impact of NOX2 on T‐cell cytokine production and whether such effects are T‐cell intrinsic are unknown. Th17 cells are the primary producers of IL‐17A, a proinflammatory cytokine known to propagate NAFLD pathogenesis.17 Thus, we examined the role of NOX2 on Th17 cell polarization. Compared to WT cells, NOX2−/− primary naive splenic CD4+ T cells cultured under Th17 polarizing conditions exhibited reduced Th17 polarization and IL‐17A production (Fig. 6A). The effects on cytokine production were not unique to Th17 cells as primary naive splenic CD4+ T cells isolated from NOX2−/− mice were less poised to produce IFN‐γ and TNF‐α following polyclonal T‐cell receptor stimulation using antiCD3/antiCD28 (Fig. 6B). In sum, these data suggest that NOX2 plays an intrinsic role in T‐cell responsiveness and Th17 cell polarization. Importantly, such effects may confer modulation of the hepatic inflammatory milieu in NAFLD progression.
Figure 6.
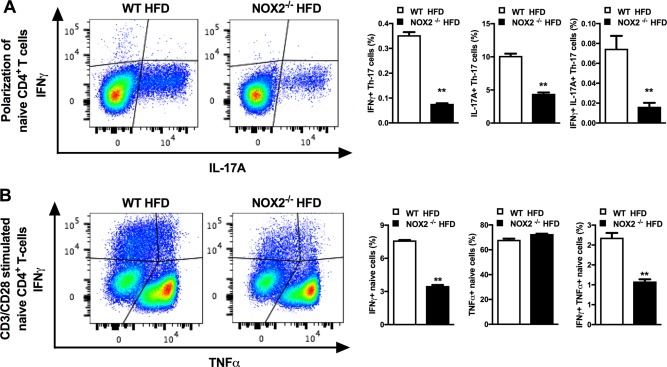
NOX2 modulates T‐cell polarization and effector function. Eight‐week‐old WT and NOX2−/− mice were fed an HFD for 8 weeks and used for analysis of T‐cell responses. (A) Representative flow plot of naive splenic CD4+ T cells toward Th17 differentiation and percentage of IL‐17A+ and IFN‐γ+ in polarized Th17 cells. (B) Representative flow plot of CD3/CD28‐stimulated naive CD4+ T cells and percentage of TNF‐α+ and IFN‐γ+ production by CD4+ T cells. White bars denote WT mice; black bars denote NOX2−/− mice. (A,B) A single experiment, n = 3 mice/condition. Data represent means + SE. Student t test, **P < 0.01.
Discussion
NAFLD, the most prominent liver disease worldwide, is directly linked with obesity, insulin resistance, and type 2 diabetes mellitus.20 Despite the clinical significance, the underlying mechanisms regulating NAFLD development and pathogenesis remain underdefined and represent a significant gap in knowledge. It is well appreciated that oxidative stress and subsequent alterations to inflammatory cascades contribute to the pathogenesis of obesity‐associated sequelae,37 but whether NOX2 regulates NAFLD pathogenesis and NAFLD‐relevant immune cell inflammatory vigor has not been determined. Our data provide novel insights about the role of NOX2 in obesity and obesity‐associated NAFLD pathogenesis. Specifically, here we show that in the context of HFD‐induced obesity, NOX2 modulates (a) immune cell ROS production, (b) immune cell inflammatory capacity pivotal for NAFLD progression, (c) glucose dysmetabolism, and (d) development of steatohepatitis and hepatocellular damage.
Under obesogenic stress, various immune cells produce elevated levels of ROS in bone marrow, WAT, and liver. Genetic ablation of NOX2 correlated with attenuated ROS generation and proinflammatory cytokine production in bone marrow immune cells following HFD feeding and endotoxin‐driven activation of toll‐like receptor (TLR)4 signaling. However, whether NOX2 modulates neutrophil and macrophage ROS expression in WAT and liver has not been determined. Future studies focused on the role of NOX2 in ROS synthesis, proinflammatory cytokine production, neutrophil recruitment, and macrophage polarization in obesity/NAFLD‐relevant tissues are clearly warranted. Notably, obesity is associated with metabolic endotoxemia,2, 21 and TLR4 signaling plays an important role in NAFLD pathogenesis.2, 14, 38 Hence, the interplay between TLR4 and NOX2 activation and ROS production in obesity warrants further investigation.
Our data also suggest that the NOX2 effects on immune cell ROS production are not endotoxin and/or TLR4‐signaling specific. Along with TLR4, TLR2 is a key contributor of development of NASH through inflammasome activation.39 Notably, neutrophil‐driven ROS production in the context of zymosan sensing, a component of fungal cell walls and an activator of TLR2 signaling, induced similar outcomes. Although zymosan levels are not commonly quantified in obesity, intestinal colonization with fungi has been implicated in NAFLD pathogenesis.40, 41 Whether other TLRs or innate immune receptors regulate activation of the NOX2–ROS axis and the contribution of such activation in NAFLD pathogenesis are unknown and should be examined.
Experimental and clinical evidence suggest a complex interplay between TLRs, metabolic endotoxemia, and intestinal microbiome in NAFLD progression.1, 2, 14, 21, 41 Interactions between the microbiome, alcohol‐producing bacteria, ROS, and the immune system are believed to play an important role in NASH progression.42 Hence, future investigations employing littermate controls in the analysis of the NOX2–ROS axis in obesity‐associated alterations of the intestinal microbiome and its contributions to NAFLD development and progression are of obvious importance.
NOX2‐mediated effects on ROS production were not specific to a single immune cell type (e.g., macrophages, neutrophils), suggesting that NOX2 broadly affects immune responsiveness and, as such, likely contributes to the pathogenesis of a variety of diseases, including obesity. Specifically, our data demonstrated that prolonged culture of primary cells outside the obesogenic environment, as seen with BMDMs and BMDCs, resulted in similar baseline ROS levels in NOX2−/− and WT cells. Challenge with an inflammatory stimulant was sufficient to uncover the differences in ROS production in such cells. These data suggest that the obesity‐associated inflammatory environment plays an important role in NOX2 activation. However, specific pathways and cellular mechanisms directly associated with NOX2‐dependent effects in disease pathogenesis remain underdefined.
Our data demonstrated that diminished ROS production in NOX2−/− mice did not affect HFD‐induced obesity, adiposity, and food consumption. Thermogenic capacity, which in part is dependent on BAT size and UCP‐1 expression/function, is central to whole body metabolism in both mice and humans.43 Furthermore, augmented mitochondrial ROS production represents a key step in Ucp1 activation and thermogenesis.44 In agreement with similar total body weight gain between obese WT and NOX2−/− mice, NOX2−/− mice exhibited similar BAT weight and Ucp1 expression. These findings suggest that NOX2‐dependent ROS does not regulate the thermogenic state in an experimental model of HFD‐induced obesity and that such effects are likely dependent on other, and possibly redundant, mechanisms of ROS induction.
Additionally, WAT redistribution and adipocyte death during weight gain is associated with obesity‐associated disease progression.24 An inverse proportion of eWAT to iWAT size coupled with an increased presence of apoptotic adipocytes in eWAT adipocytes were supportive of advanced adiposity in HFD‐fed WT mice compared to NOX2−/− mice.24 Our findings suggest that NOX2 does not modulate HFD‐induced body weight gain or thermogenic capacity but plays an important role in WAT redistribution and apoptosis. As genetic ablation of NOX2 modulates WAT distribution, adipocyte death, and the type of hepatic steatosis, a better understanding of NOX2 contribution in WAT physiology, adipocyte function, and lipid partitioning is required.
Glucose dysmetabolism, a consequence of excessive systemic nutrient load, WAT redistribution and inflammation, and imbalance in the ROS axis, represents one of the most common end‐organ sequelae of diet‐induced obesity.33 Our data suggest that despite similar obesity, systemic insulin production, and insulin tolerance in NOX2−/− mice compared to WT controls, NOX2−/− mice were protected from HFD‐induced glucose dysmetabolism. These findings suggest that divergent mechanisms,45 NOX2‐dependent and independent, are likely to impact glucose metabolism and insulin sensitivity in HFD‐fed mice. Further, the divergence between NOX2 deficiency and insulin sensitivity may have connections with a largely underappreciated role of ROS in stimulating tyrosine phosphorylation‐dependent insulin signaling.46 In fact, the observed impact on glucose dysmetabolism, a well‐known attribute of patients with NAFLD, warrants a further, detailed, mechanistic analysis on the role of NOX2 in glucose metabolisms and its interplay with NAFLD development.
NOX2 is highly expressed in the liver, and a serum‐soluble NOX2‐derived peptide (sNOX2‐dp) is a known marker of liver steatosis.47 However, how NOX2 regulates immune responses in NAFLD progression is principally underdefined. Here, we demonstrated that NOX2−/− mice are protected from HFD‐induced macrosteatosis, lipid peroxidation, and hepatocellular damage despite similar liver weights and TG content. The reduced disease severity of hepatocellular damage correlated with attenuated hepatic immune cell infiltration, including macrophages, neutrophils, and T cells. Infact, Diet‐induced hepatic neutrophil, macrophage, and T‐cell infiltration are trademarks of NAFLD progression and pathogenesis.1, 35, 48 The reduction in hepatic macrophages in livers of NOX2−/− mice is consistent with a recent report of attenuated macrophage infiltration into WAT in myeloid‐specific NOX2−/− mice.36 Altered number of liver‐infiltrating immune cells in NOX2−/− mice also suggest reduced activation of inflammatory axes in these mice. Proinflammatory cytokine production, by liver‐infiltrating immune cells, is central to NAFLD pathogenesis.14 Our findings demonstrated that NOX2 modulated the production of IL‐17A, IFN‐γ, and TNF‐α, which are all well‐known promoters of NAFLD pathogenesis,1, 14, 17, 48, 49 by liver‐infiltrating immune cells, thus denoting a pivotal role of the NOX2–ROS axis in the pathophysiology of NAFLD. We also demonstrated that NOX2‐dependent effects were not unique to liver‐infiltrating T cells as polarization of naive splenic CD4+ T cells to effector T‐cell subtype and cytokine production following polyclonal T‐cell receptor activation were similarly affected. These data imply that intrinsic NOX2 effects in T cells in combination with the well‐established extrinsic signals (e.g., bacterial colonization, lipid partitioning) and inflammatory hepatic microenvironment may play vital roles in the regulation of T‐cell polarization and responsiveness in NAFLD. Thus, a definition of cell‐intrinsic and cell‐extrinsic mechanisms underlying NOX2‐driven modulation of T‐cell polarization and inflammatory vigor in NAFLD is clearly warranted. Further confirmation of the immune NOX2 expression as the central locus of disease pathology, by use of bone marrow chimera studies or immune cell‐specific NOX2 deletion, should be performed in the context of obesogenic diet feeding.
In conclusion, our novel findings suggest that NOX2 modulates the inflammatory milieu in obesity (Fig. 7) and, as such, regulates NAFLD pathogenesis. Our findings are in agreement with the role of NOX2 in the modulation of alcoholic fatty liver disease.50 Importantly, such insights suggest that pharmacologic targeting of NOX2 activation and/or function in immune cells may represent a viable approach to reducing morbidity of obesity‐associated sequelae.
Figure 7.
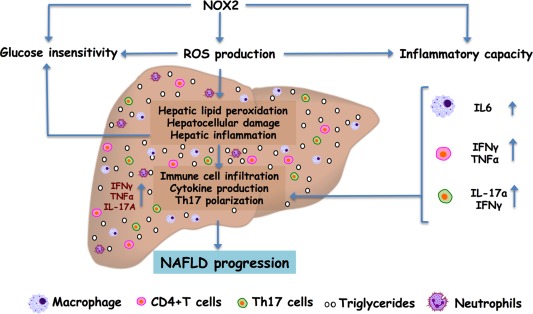
Summary figure depicting immunomodulatory roles of NOX2 in HFD‐induced NAFLD progression. NOX2 deficiency correlates with protection from HFD‐induced glucose insensitivity, hepatocellular damage, and hepatic immune cell infiltration, despite similar total body weight gain and hepatic steatosis. Mechanistically, protection from the pathogenesis of obesity‐associated sequelae correlated with NOX2‐driven modulation of ROS production and immune cell inflammatory capacity (e.g., macrophages, CD4+ T cells, and Th17 polarization). These data suggest a previously unappreciated role of the NOX2–ROS axis in the interplay between inflammatory vigor during NAFLD pathogenesis.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1162/full.
Supporting Information 1
Potential conflict of interest: Nothing to report.
Supported in part by National Institutes of Health awards R01DK099222 (to S.D.), T32AI118697 (to D.A.G. and C.C.C), and P30 DK078392 Pathology of the Digestive Disease Research Core Center at the Cincinnati Children's Hospital Medical Center; Cincinnati Children's Hospital Medical Center Pediatric Diabetes and Obesity Center (to S.D.); and American Heart Association 17POST33650045 (to M.E.M.F).
REFERENCES
- 1. Giles DA, Moreno‐Fernandez ME, Divanovic S. IL‐17 axis driven inflammation in non‐alcoholic fatty liver disease progression. Curr Drug Targets 2015;16:1315‐1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehal WZ. The Gordian Knot of dysbiosis, obesity and NAFLD. Nat Rev Gastroenterol Hepatol 2013;10:637‐644. [DOI] [PubMed] [Google Scholar]
- 3. Polimeni L, Del Ben M, Baratta F, Perri L, Albanese F, Pastori D, et al. Oxidative stress: new insights on the association of non‐alcoholic fatty liver disease and atherosclerosis. World J Hepatol 2015;7:1325‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ganz M, Szabo G. Immune and inflammatory pathways in NASH. Hepatol Int 2013;7(Suppl. 2):771‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sumida Y, Niki E, Naito Y, Yoshikawa T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic Res 2013;47:869‐880. [DOI] [PubMed] [Google Scholar]
- 6. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012;24:981‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox‐dependent signalling. Nat Rev Mol Cell Biol 2014;15:411‐421. [DOI] [PubMed] [Google Scholar]
- 8. Paik YH, Iwaisako K, Seki E, Inokuchi S, Schnabl B, Osterreicher CH, et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011;53:1730‐1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sukumar P, Viswambharan H, Imrie H, Cubbon RM, Yuldasheva N, Gage M, et al. Nox2 NADPH oxidase has a critical role in insulin resistance‐related endothelial cell dysfunction. Diabetes 2013;62:2130‐2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Souto Padron de Figueiredo A, Salmon AB, Bruno F, Jimenez F, Martinez HG, Halade GV, et al. Nox2 mediates skeletal muscle insulin resistance induced by a high fat diet. J Biol Chem 2015;290:13427‐13439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pepping JK, Freeman LR, Gupta S, Keller JN, Bruce‐Keller AJ. NOX2 deficiency attenuates markers of adiposopathy and brain injury induced by high‐fat diet. Am J Physiol Endocrinol Metab 2013;304:E392‐E404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Divanovic S, Dalli J, Jorge‐Nebert LF, Flick LM, Galvez‐Peralta M, Boespflug ND, et al. Contributions of the three CYP1 monooxygenases to pro‐inflammatory and inflammation‐resolution lipid mediator pathways. J Immunol 2013;191:3347‐3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, et al. Negative regulation of Toll‐like receptor 4 signaling by the Toll‐like receptor homolog RP105. Nat Immunol 2005;6:571‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giles DA, Moreno‐Fernandez ME, Stankiewicz TE, Graspeuntner S, Cappelletti M, Wu D, et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex‐independent disease modeling. Nat Med 2017;23:829‐838. Erratum in: Nat Med 2017;23:1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cappelletti M, Presicce P, Lawson MJ, Chaturvedi V, Stankiewicz TE, Vanoni S, et al. Type I interferons regulate susceptibility to inflammation‐induced preterm birth. JCI Insight 2017;2:e91288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796‐1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harley IT, Stankiewicz TE, Giles DA, Softic S, Flick LM, Cappelletti M, et al. IL‐17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014;59:1830‐1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giles DA, Moreno‐Fernandez ME, Stankiewicz TE, Cappelletti M, Huppert SS, Iwakura Y, et al. Regulation of inflammation by IL‐17A and IL‐17F modulates non‐alcoholic fatty liver disease pathogenesis. PLoS One 2016;11:e0149783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giles DA, Ramkhelawon B, Donelan EM, Stankiewicz TE, Hutchison SB, Mukherjee R, et al. Modulation of ambient temperature promotes inflammation and initiates atherosclerosis in wild type C57BL/6 mice. Mol Metab 2016;5:1121‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tilg H, Moschen AR, Roden M. NAFLD and diabetes mellitus. Nat Rev Gastroenterol Hepatol 2017;14:32‐42. [DOI] [PubMed] [Google Scholar]
- 21. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007;56:1761‐1772. [DOI] [PubMed] [Google Scholar]
- 22. Mas E, Danjoux M, Garcia V, Carpentier S, Segui B, Levade T. IL‐6 deficiency attenuates murine diet‐induced non‐alcoholic steatohepatitis. PLoS One 2009;4:e7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Costford SR, Castro‐Alves J, Chan KL, Bailey LJ, Woo M, Belsham DD, et al. Mice lacking NOX2 are hyperphagic and store fat preferentially in the liver. Am J Physiol Endocrinol Metab 2014;306:E1341‐E1353. [DOI] [PubMed] [Google Scholar]
- 24. Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW 2nd, DeFuria J, Jick Z, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007;56:2910‐2918. [DOI] [PubMed] [Google Scholar]
- 25. Lynch CM, Kinzenbaw DA, Chen X, Zhan S, Mezzetti E, Filosa J, et al. Nox2‐derived superoxide contributes to cerebral vascular dysfunction in diet‐induced obesity. Stroke 2013;44:3195‐3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coats BR, Schoenfelt KQ, Barbosa‐Lorenzi VC, Peris E, Cui C, Hoffman A, et al. Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet‐induced obesity. Cell Rep 2017;20:3149‐3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Minokoshi Y, Kahn CR, Kahn BB. Tissue‐specific ablation of the GLUT4 glucose transporter or the insulin receptor challenges assumptions about insulin action and glucose homeostasis. J Biol Chem 2003;278:33609‐33612. [DOI] [PubMed] [Google Scholar]
- 28. Softic S, Gupta MK, Wang GX, Fujisaka S, O'Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest 2017;127:4059‐4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de Souza AH, Santos LRB, Roma LP, Bensellam M, Carpinelli AR, Jonas JC. NADPH oxidase‐2 does not contribute to beta‐cell glucotoxicity in cultured pancreatic islets from C57BL/6J mice. Mol Cell Endocrinol 2017;439:354‐362. [DOI] [PubMed] [Google Scholar]
- 30. Mofrad P, Contos MJ, Haque M, Sargeant C, Fisher RA, Luketic VA, et al. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology 2003;37:1286‐1292. [DOI] [PubMed] [Google Scholar]
- 31. Nalbantoglu IL, Brunt EM. Role of liver biopsy in nonalcoholic fatty liver disease. World J Gastroenterol 2014;20:9026‐9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nativ NI, Yarmush G, So A, Barminko J, Maguire TJ, Schloss R, et al. Elevated sensitivity of macrosteatotic hepatocytes to hypoxia/reoxygenation stress is reversed by a novel defatting protocol. Liver Transpl 2014;20:1000‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004;114:1752‐1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paquissi FC. Immune imbalances in non‐alcoholic fatty liver disease: from general biomarkers and neutrophils to interleukin‐17 axis activation and new therapeutic targets. Front Immunol 2016;7:490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014;59:1393‐1405. [DOI] [PubMed] [Google Scholar]
- 36. Pepping JK, Vandanmagsar B, Fernandez‐Kim SO, Zhang J, Mynatt RL, Bruce‐Keller AJ. Myeloid‐specific deletion of NOX2 prevents the metabolic and neurologic consequences of high fat diet. PLoS One 2017;12:e0181500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garcia‐Ruiz I, Solis‐Munoz P, Fernandez‐Moreira D, Grau M, Munoz‐Yague T, Solis‐Herruzo JA. NADPH oxidase is implicated in the pathogenesis of oxidative phosphorylation dysfunction in mice fed a high‐fat diet. Sci Rep 2016;6:23664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ye D, Li FY, Lam KS, Li H, Jia W, Wang Y, et al. Toll‐like receptor‐4 mediates obesity‐induced non‐alcoholic steatohepatitis through activation of X‐box binding protein‐1 in mice. Gut 2012;61:1058‐1067. [DOI] [PubMed] [Google Scholar]
- 39. Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll‐like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 2013;57:577‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang AM, Inamine T, Hochrath K, Chen P, Wang L, Llorente C, et al. Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest 2017;127:2829‐2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol 2016;13:412‐425. [DOI] [PubMed] [Google Scholar]
- 42. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 2013;57:601‐609. [DOI] [PubMed] [Google Scholar]
- 43. Kozak LP, Koza RA, Anunciado‐Koza R. Brown fat thermogenesis and body weight regulation in mice: relevance to humans. Int J Obes (Lond) 2010;34(Suppl. 1):S23‐S27. [DOI] [PubMed] [Google Scholar]
- 44. Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016;532:112‐116. Erratum in: Nature 2016;536:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol 2017;13:572‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab 2009;10:260‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Del Ben M, Polimeni L, Carnevale R, Bartimoccia S, Nocella C, Baratta F, et al. NOX2‐generated oxidative stress is associated with severity of ultrasound liver steatosis in patients with non‐alcoholic fatty liver disease. BMC Gastroenterol 2014;14:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alisi A, Carpino G, Oliveira FL, Panera N, Nobili V, Gaudio E. The role of tissue macrophage‐mediated inflammation on NAFLD pathogenesis and its clinical implications. Mediators Inflamm 2017;2017:8162421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Narayanan S, Surette FA, Hahn YS. The immune landscape in nonalcoholic steatohepatitis. Immune Netw 2016;16:147‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang M, Frasch SC, Li G, Feng D, Gao B, Xu L, et al. Role of gp91phox in hepatic macrophage programming and alcoholic liver disease. Hepatol Commun 2017;1:765‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1162/full.
Supporting Information 1