

Abstract
NI‐0801 is a fully human monoclonal antibody against chemokine (C‐X‐C motif) ligand 10 (CXCL10), which is involved in the recruitment of inflammatory T cells into the liver. The safety and efficacy of NI‐0801 was assessed in patients with primary biliary cholangitis. In this open‐label phase 2a study, patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid received six consecutive intravenous administrations of NI‐0801 (10 mg/kg) every 2 weeks. Patients were followed up for 3 months after the last infusion. Liver function tests, safety assessments, as well as pharmacokinetic and pharmacodynamic parameters were evaluated at different time points throughout the dosing period and the safety follow‐up period. Twenty‐nine patients were enrolled in the study and were treated with NI‐0801. The most frequently reported adverse events included headaches (52%), pruritus (34%), fatigue (24%), and diarrhea (21%). No study drug‐related serious adverse events were reported. NI‐0801 administration did not lead to a significant reduction in any of the liver function tests assessed at the end of the treatment period (i.e., 2 weeks after final NI‐0801 administration) compared to baseline. Conclusion: Despite clear pharmacologic responses in the blood, no therapeutic benefit of multiple administrations of NI‐0801 could be demonstrated. The high production rate of CXCL10 makes it difficult to achieve drug levels that lead to sustained neutralization of the chemokine, thus limiting its targetability. (Hepatology Communications 2018;2:492‐503)
Abbreviations
- AE
adverse event
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AMA
anti‐mitochondrial autoantibody
- APC
allophycocyanin
- AST
aspartate aminotransferase
- CCL
chemokine (C‐C motif) ligand
- CD
clusters of differentiation
- CXCL
chemokine (C‐X‐C motif) ligand
- CXCR
chemokine (C‐X‐C motif) receptor
- D
day
- ELF
enhanced liver fibrosis
- γGT
gamma glutamyl transpeptidase
- HV
healthy volunteer
- Ig
immunoglobulin
- mAb
monoclonal antibody
- NK
natural killer
- PBC
primary biliary cholangitis
- PBMC
peripheral blood mononuclear cell
- PD
pharmacodynamic
- PE
phycoerythrin
- PK
pharmacokinetic
- SAE
serious adverse event
- TMDD
target‐mediated drug disposition
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis,1 is a slowly progressive autoimmune disease characterized by a gradual destruction of the small intrahepatic bile ducts.2, 3 PBC is considered a multifactorial disease presumably arising from a combination of environmental and genetic factors.4, 5 The disease primarily affects women, and incidence is highest in the fifth decade of life. The immune‐mediated loss of bile ducts leads to decreased bile secretion and the retention of toxic substances within the liver. This inflammatory process can result in further hepatic damage, fibrosis, cirrhosis, and eventually liver failure.2, 3 The mainstay of treatment for PBC is ursodeoxycholic acid (UDCA). UDCA is an anticholestatic agent that improves liver biochemistry by promoting bile‐acid secretion and reducing the effect of accumulation of toxic bile acids in the liver. More recently, obeticholic acid has been approved as a second‐line therapy in combination with UDCA for patients with PBC with an inadequate response to UDCA.6 However, neither UDCA nor obeticholic acid address the underlying cause of the disease, i.e., autoimmune inflammation of the bile ducts.
A serologic hallmark of PBC is the presence of autoantibodies reactive with both mitochondrial and nuclear antigens.7 Most commonly, these anti‐mitochondrial autoantibodies (AMA) target the pyruvate dehydrogenase E2 complex, which is located in the inner mitochondrial matrix. Although AMAs are present in the serum of 95% of patients with PBC and represent a key diagnostic finding, they are unlikely to play a direct role in the induction of target cell damage.3 Histopathologically, PBC is characterized by a lymphocytic infiltrate of portal tracts and segmental inflammatory destruction of intrahepatic bile ducts, resulting in progressive loss of these ducts with associated periportal fibrosis and eventually cirrhosis. T cells infiltrating the liver in PBC were shown to include populations specific for the pyruvate dehydrogenase E2 complex, and there is evidence that the destruction of bile ducts is mediated by these liver‐infiltrating autoreactive T cells.8
Chemokine (C‐X‐C motif) ligand 10 (CXCL10) is a chemokine that is secreted by several cell types, including monocytes, endothelial cells, fibroblasts, and epithelial cells, including cholangiocytes and hepatocytes, in response to interferon‐γ. CXCL10 elicits its effects by binding to chemokine (C‐X‐C motif) receptor 3 (CXCR3), which is highly expressed on effector T cells and plays an important role in T‐cell trafficking and function.9 CXCL10 and CXCR3 levels are consistently found to be up‐regulated in serum and the liver of patients suffering from inflammatory hepatic disease, including PBC,10, 11, 12, 13, 14, 15, 16 and CXCR3 is considered the main chemokine receptor driving infiltration of inflammatory lymphocytes into the liver.17 In animal models of liver disease, it has been shown that CXCL10 neutralization reduces inflammation and fibrosis.18, 19 Altogether, these data suggest a potential role for CXCL10 as a therapeutic target in PBC.
NI‐0801 is a fully human immunoglobulin (Ig) G1 monoclonal antibody (mAb). This antibody binds with high affinity to human CXCL10 (Kd = 140 pM) but not to the other CXCR3 ligands CXCL9 and CXCL11. In vitro, NI‐0801 efficiently inhibits CXCL10‐induced calcium flux and chemotaxis as well as lymphocyte transendothelial migration across human sinusoidal endothelial cells under conditions of physiologic flow. In addition, NI‐0801 is capable of binding CXCL10 sequestered on glycosaminoglycans, assuring neutralization of CXCL10 in its physiologic context.
Phase 1 single‐dose studies in healthy volunteers (HVs) demonstrated that NI‐0801 was well tolerated and that the terminal half‐life of 18.5 days observed at the highest dose tested in HVs (i.e., 20 mg/kg) was consistent with the expected half‐life of an IgG. This open‐label phase 2a study was conducted to investigate the safety and efficacy of NI‐0801 in patients with PBC with an inadequate response to UDCA.
Patients and Methods
PATIENTS
Male and female patients aged ≥18 years with proven PBC as demonstrated by the presence of at least two of the following three diagnostic factors were enrolled in this multicenter study: history of increased alkaline phosphatase (ALP) for at least 6 months; positive serum AMA titer (>1:40); liver biopsy consistent with PBC. Patients had an incomplete response to UDCA if they showed a failure to normalize ALP levels or to reduce ALP levels by >40% after 1 year of therapy with UDCA (Barcelona criteria20) and were receiving stable doses of UDCA for at least 6 months prior to screening. In addition to the above criteria, patients were included if they had elevated ALP levels (>1.5 upper limit of normal [ULN]) and elevated alanine aminotransferase (ALT) (>1.5 ULN) or aspartate aminotransferase (AST) (>1.5 ULN) levels.
Patients were excluded if at screening total bilirubin was >2.9 mg/dL (50 μmol/L) or creatinine clearance was <80 mL/minute. Patients with a history of or an ongoing hepatic decompensation or with a positive serology result for human immunodeficiency virus, hepatitis B or C, known or previous malignancy, presence of any active infection, or previous history of active tuberculosis within 12 months of screening were excluded. In addition, patients were excluded if the average alcohol ingestion exceeded 21 units/week (for male patients) or 14 units/week (for female patients). The use of immunosuppressants, steroids, or peroxisome proliferator activation receptor activators within 3 months prior to study treatment was prohibited.
Screening according to the inclusion and exclusion criteria occurred within 21 days prior to the first NI‐0801 dosing. All patients provided written informed consent before any study‐related procedures were undertaken.
STUDY DESIGN
This study was an open‐label, single‐arm, phase 2a, proof‐of‐concept, multicenter study in male and female patients with PBC, aged ≥18 years, with an incomplete response to UDCA; it was designed to provide safety, efficacy, pharmacokinetic (PK) and pharmacodynamic (PD) data on multiple administrations of NI‐0801 in PBC. Patients received 10 mg/kg NI‐0801 as a 1‐hour intravenous infusion every 2 weeks for a total of six doses in addition to UDCA. Patients were followed up for 3 months after the last infusion.
This study was registered on https://clinicaltrials.gov/ct2/show/NCT01430429 (NCT01430429). The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the institution's human research committee.
SAFETY ASSESSMENTS
Adverse events (AEs), defined as any undesirable experience occurring during the trial regardless of relationship to the study drug, were recorded and classified based on the intensity and relationship to the study drug. Events that were fatal, life threatening, required inpatient hospitalization or prolonged hospitalization, or surgical intervention were classified as serious AEs (SAEs). Patients were monitored for 3 months after final infusion for safety purposes. Physical examination, vital signs, hematology, serum chemistries, urinalyses, and electrocardiograms were performed for all patients at baseline (day [D] 1 predose) and at scheduled time points postdose. The presence of NI‐0801 anti‐drug antibodies was analyzed by ICON Development Solutions (Manchester, United Kingdom) using an electrochemiluminescence‐based bridging assay.
EFFICACY ASSESSMENTS
Effects on disease activity were assessed by measurement of liver function tests (including ALT, ALP, AST, gamma glutamyl transpeptidase (γGT), and total bilirubin) and IgG and IgM levels predose and postdose on the dosing days (i.e., D1, D15, D29, D43, D57, D71) and on D5, D75, D85, week 18, and week 24. An enhanced liver fibrosis (ELF) test was performed at D1 predose, D85, and week 18. The main outcome measurement consisted of the percentage reduction in liver function tests from baseline to D85 (i.e., 2 weeks after final NI‐0801 administration).
PK AND PD EVALUATION
Serum concentrations of NI‐0801 and total CXCL10 were assessed at predose and postdose on dosing days (i.e., D1, D15, D29, D43, D57, D71) and on D5, D75, D85, week 18, and week 24. NI‐0801 concentrations were quantified using an enzyme‐linked immunosorbent assay‐based assay developed to detect free (or partially bound) NI‐0801 in human serum. For measurement of total CXCL10 (i.e., free and antibody‐bound CXCL10), serum samples were first subjected to an acid–alcohol dissociation step. Subsequently, CXCL10 levels were analyzed by enzyme‐linked immunosorbent assay (Quantikine kit, R&D Systems).
CHEMOTAXIS ASSAY
The chemotactic activity of sera collected from patients with PBC was monitored using murine preB lymphoma L1.2 cells, which were stably transfected to express human CXCR3. Assays of chemotactic responsiveness were carried out using 96‐well ChemoTx plates with 5 μm pores (Neuroprobe, Gaithersburg, MD). Briefly, samples were added in triplicate to the lower chamber in the presence or absence of exogenously added recombinant human CXCL10 (R&D Systems). L1.2 transfected cells were stimulated with butyric acid (0.6 mg/mL) for 24 hours to induce expression of CXCR3 prior to the chemotaxis assay. Activated cells (2,000 cells per well) were seeded on top of the membrane. The culture plate was incubated at 37°C for 3‐4 hours. Cells having migrated across the membrane in the lower chamber were recovered by centrifugation (600g; 3 minutes) and transferred to FMAT optical black plates (Applied Biosystems). Harvested cells were stained with 1, 5‐bis{[2‐(di‐methylamino)ethyl]amino}‐4, 8‐dihydroxyanthracene‐9, 10‐dione (DRAQ5; Alexis Corporation, Biostatus Limited) and counted using the fluorometric microvolume assay technology (FMAT 8100 HTS system; Applied Biosystems).
ISOLATION OF PERIPHERAL BLOOD MONONUCLEAR CELLS AND FLOW CYTOMETRY
For flow cytometric analysis, matched samples collected at baseline (i.e., D1 predose) and 2 weeks following the final infusion of NI‐0801 (D85) were available for 7 patients. Peripheral blood was collected in ethylene diamine tetraacetic acid tubes, and mononuclear cells were isolated by overlaying Ficoll before spinning in a benchtop centrifuge for 30 minutes at 650g without a brake. The mononuclear band at the interface of the plasma and Ficoll was collected, washed twice, and resuspended to 10 × 106/mL or cryopreserved for future analysis.
Peripheral blood mononuclear cells (PBMCs) were incubated with mouse anti‐human monoclonal fluorochrome‐conjugated antibodies for 30 minutes at 4°C prior to flow cytometry. All antibodies were purchased from BD Biosciences and were raised against clusters of differentiation (CD)3 (v500), CD4 (v450), CD8 (fluorescein isothiocyanate), CD25 (allophycocyanin [APC]‐Cy7), CD45RA (v500), CD45RO (APC‐H7), CD56 (peridinin chlorophyll protein complex‐Cy5.5), CD62L (phycoerythrin [PE]), CD69 (APC‐Cy7), CD127 (PE), CD154 (PE), CD183 APC, and CD197 (peridinin chlorophyll protein complex‐Cy5.5). Following incubation, cells were washed and resuspended in phosphate‐buffered saline supplemented with 0.5% fetal bovine serum for analysis. Previously cryopreserved samples were thawed rapidly at 37°C, washed in staining buffer, and resuspended in Roswell Park Memorial Institute medium–10% fetal bovine serum/50 IU interleukin‐2 before being allowed to rest overnight at 37°C, 5% CO2 in a humidified incubator. Labeling then proceeded as described above. Analysis was performed on a Beckman Coulter 9‐color CyAn.
Flow cytometric analysis was only performed on patient samples originating from the two study sites that had the possibility to perform PBMC isolation. Therefore, the results obtained from the flow cytometric analysis are not necessarily representative of the whole study population.
STATISTICAL ANALYSIS
For this phase 2 pilot study, a sample size calculation estimated that with 40 patients the study would have 80% power to detect (with a 0.05 significance level) reductions from baseline of 27% and 22% in ALP and AST, respectively. All statistical tests were two sided.
The safety analysis set includes all patients who received any part of an infusion of the study drug. The evaluable analysis set is defined as the safety analysis set excluding 3 patients who did not receive the scheduled six doses of NI‐0801.
Regular interim analyses were conducted during the study estimating the percentage reduction from baseline at D85 (i.e., 2 weeks after the final NI‐0801 administration) for each of the liver function tests. A two‐tailed one‐sample t test was used to test the null hypothesis that the percentage change from baseline at D85 was equal to 0. No imputations were made for missing data.
Statistical analysis was performed using SAS software (SAS Institute, Cary, NC) and GraphPad Prism 6.02 for Windows (GraphPad Software, San Diego, CA).
Results
PATIENT CHARACTERISTICS AT BASELINE AND PATIENT DISPOSITION
Patient demographics and disease characteristics at baseline for the 29 patients enrolled and treated at 10 investigational sites in Europe (eight in the United Kingdom, two in Italy) are displayed in Table 1. The majority of patients in the study were women (28/29). Patient age and weight ranged from 32 years to 72 years and from 48 kg to 92 kg, respectively. Median disease duration was 5.3 years. At baseline, the median value of ALP was 417 IU/L (range, 163‐834 IU/L) and the median values for the transaminases ALT and AST were 83 IU/L (range, 36‐269 IU/L) and 61 IU/L (range, 32‐171 IU/L), respectively.
Table 1.
Patient Characteristics at Baseline (Safety Analysis Set)
| Age (years) | 50 (32‐72) |
| Sex (M:F) | 1/28 |
| Weight (kg) | 63 (48‐92) |
| Disease duration (years) | 5.3 (1.2‐21.2) |
| Liver function tests | |
| ALP (IU/L) | 417 (163‐834) |
| ALT (IU/L) | 83 (36‐269) |
| AST (IU/L) | 61 (32‐171) |
| γGT (IU/L) | 292 (60‐1277) |
| Total bilirubin (μmol/L) | 12 (4‐45) |
Values are expressed as median (range). Normal ranges: ALP 40‐129 IU/L (men), 35‐104 IU/L (women); ALT 10‐50 IU/L (men), 10‐35 IU/L (women); AST 0‐37 IU/L (men), 0‐31 IUL/L (women); γGT 10‐71 IU/L (men), 6‐42 IU/L (women); total bilirubin 0‐20 μmol/L.
Twenty‐six patients completed the study, whereas 3 patients withdrew from the study after having received two to five infusions of NI‐0801. None of these patients discontinued due to safety concerns linked to the administration of NI‐0801.
Initially, the study aimed to recruit 40 patients; however, the trial was terminated due to lack of efficacy after an interim futility analysis conducted by the time 26 patients had reached D85.
SAFETY AND TOLERABILITY OF NI‐0801
One or more AEs were reported for all patients, and a total of 228 AEs were recorded during the study. Of these, 63 were of moderate/severe intensity. The most frequently reported AE was headache (by 52% of the patients), which was mostly of mild intensity (85%). Other frequently reported AEs included pruritus, fatigue, and diarrhea, reported by 34%, 24%, and 21% of the patients, respectively. The most frequently reported infection was urinary tract infection (24%), followed by nasopharingitis (21%), which mostly occurred during the winter season. None of the observed infections were opportunistic.
The only SAE reported was a domestic accident resulting in patient hospitalization. This SAE occurred 3 weeks after the final NI‐0801 administration and was considered by the investigator not related to the study drug administration. The AEs reported by 2 or more patients during the study are listed in Table 2. Immunogenicity analysis revealed trace positive antibody titers for 9 patients at the last follow‐up visit; these did not impact PK exposure (data not shown).
Table 2.
Summary of AEs (Safety Analysis Set)
| Type of AE |
Number of Events (Number of Patients Reporting Event) |
|---|---|
| Total AEs | 228 (29) |
| Moderate AEs | 48 (19) |
| Severe AEs | 15 (7) |
| Serious AEs | 1 (1) |
| Serious adverse drug reactions | 0 (0) |
| Life‐threatening AEs/deaths | 0 (0) |
| AEs leading to drug withdrawal | 0 (0) |
| AEs reported in 2 or more subjects: | |
| Ear and labyrinth disorders | |
| Ear pain | 2 (2) |
| Gastrointestinal disorders | |
| Abdominal pain | 4 (3) |
| Diarrhea | 11 (6) |
| Nausea | 4 (4) |
| Toothache | 3 (2) |
| Vomiting | 3 (3) |
| General disorders and administration site conditions | |
| Fatigue | 13 (7) |
| Influenza‐like illness | 5 (4) |
| Injection‐site hematoma | 2 (2) |
| Edema peripheral | 3 (3) |
| Infections and infestations | |
| Candidiasis | 2 (2) |
| Gastroenteritis | 3 (3) |
| Nasopharyngitis | 7 (6) |
| Urinary tract infection | 9 (7) |
| Viral infection | 2 (2) |
| Musculoskeletal and connective tissue disorders | |
| Arthralgia | 8 (5) |
| Back pain | 2 (2) |
| Pain in extremity | 2 (2) |
| Nervous system disorders | |
| Dizziness | 5 (4) |
| Headache | 41 (15) |
| Lethargy | 3 (3) |
| Syncope | 3 (2) |
| Respiratory, thoracic, and mediastinal disorders | |
| Cough | 3 (3) |
| Oropharyngeal pain | 8 (5) |
| Skin and subcutaneous tissue disorders | |
| Pruritus | 14 (10) |
| Vascular disorder | |
| Hot flush | 2 (2) |
PKs AND PDs
NI‐0801 concentrations following six repeated intravenous infusions of 10 mg/kg NI‐0801 every 2 weeks in patients with PBC are shown in Fig. 1A. Mean maximum concentration was 242 ± 74.9 μg/mL, and mean area under the curve (AUC)0‐D85 was 201.5 ± 104.4 mg·hour/mL in the 26 treated patients.
Figure 1.
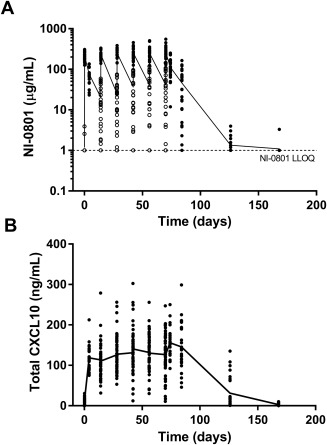
Serum concentration–time profile. (A) NI‐0801 and (B) total (antibody‐bound and free) CXCL10 in patients with PBC (evaluable analysis set) following six intravenous doses of NI‐0801. The solid line represents mean values, circles represent individual values. Empty circles in (A) represent predose NI‐0801 values on dosing days; the LLOQ of the PK assay is indicated by the dotted line. Abbreviation: LLOQ, lower limit of quantification.
Mean trough level was 32.8 ± 33.2 μg/mL and varied widely between patients, ranging from below the limit of detection (i.e., 1 μg/mL) to 144.5 μg/mL. At 2 months after the final NI‐0801 administration, drug levels were no longer detectable in any of the patients.
The serum concentration–time profile of total (i.e., antibody‐bound and free) CXCL10 in patients with PBC following six intravenous doses of NI‐0801 is shown in Fig. 1B. Following the administration of NI‐0801, a rapid increase in total CXCL10 levels from average baseline levels of 0.35 ng/mL to 14.3 ng/mL (1 hour after first dosing) was observed in the serum of patients with PBC, indicating the formation of NI‐0801/CXCL10 complexes. Four days after the first NI‐0801 administration, total CXCL10 levels reached a plateau and remained elevated throughout the dosing period (range of mean total CXCL10 levels, 111‐156 ng/mL). Maximal total CXCL10 levels varied considerably between patients, ranging from 93 ng/mL to 303 ng/mL. After the final NI‐0801 administration, total CXCL10 levels started decreasing again and had returned to baseline levels at the final safety follow‐up visit (week 24). The clearance of total CXCL10 in serum followed the kinetics of NI‐0801 clearance.
EFFECT OF NI‐0801 TREATMENT ON PBC
Multiple administrations of NI‐0801 at a dose of 10 mg/kg failed to reduce the liver biochemistry test values in patients with PBC. Values obtained for the various efficacy parameters measured at baseline and D85 (i.e., 2 weeks after the final NI‐0801 administration) in all 26 patients that completed the study are summarized in Table 3.
Table 3.
Comparison of Liver Function Tests, IgG, IgM, and ELF Test at Baseline and Study Day 85 (Evaluable Analysis Set)
| Laboratory Parameter | Baseline | D85 | D85 Change From Baseline (%) | P Value |
|---|---|---|---|---|
| ALP (IU/L) | 412 (163‐810) | 456 (171‐874) | 5.5 | 0.022* |
| ALT (IU/L) | 82 (36‐245) | 78 (33‐235) | –6.8 | 0.346 |
| AST (IU/L) | 61 (32‐171) | 57 (29‐165) | –1.9 | 0.909 |
| γGT (IU/L) | 292 (60‐1277) | 273 (62‐1051) | –4.6 | 0.501 |
| Total bilirubin (μmol/L) | 12 (4‐45) | 12 (6‐33) | 0 | 0.162 |
| IgG (g/L) | 11.71 (7.27‐18.01) | 12.65 (6.48‐21.66) | 4.3 | 0.014* |
| IgM (g/L) | 3.82 (0.84‐11.01) | 2.87 (0.9‐5.44) | –2.5 | 0.394 |
| ELF test | 10.06 (8.59‐11.96) | 10.29 (8.11‐12.70) | 3.1 | 0.109 |
Laboratory values are expressed as median (range); P values derived using two‐tailed one‐sample t test; *P ≤ 0.05.
At D85, a small but statistically significant increase in percentage change from baseline for ALP and IgG levels was noticeable (ALP median change from baseline, 5.5%, P = 0.022; IgG median change from baseline, 4.3%, P = 0.014), whereas no significant alterations in serum levels of ALT, AST, total bilirubin, γGT, IgM, or ELF were observed (median change from baseline, –6.8% [ALT], 1.9% [AST], −4.6% [γGT], 0% [total bilirubin], 3.1% [ELF]). No consistent changes were observed over time for any of the liver function tests (data not shown).
EXPLORATORY ASSESSMENT OF RELATIONSHIP BETWEEN EXPOSURE TO NI‐0801 AND CLINICAL EFFECT PARAMETERS
Based on their AUC0‐D85, patients were stratified into low‐ (63.4‐124.5 mg·hour/mL; n = 9), medium‐ (147.7‐224.2 mg·hour/mL; n = 8), and high‐exposure (228.4‐417.5 mg·hour/mL; n = 9) tertiles. The percentage change from baseline achieved at D85 (i.e., 2 weeks after the final NI‐0801 administration) for ALP, ALT, AST, γGT, IgG, and ELF by the level of NI‐0801 exposure is shown in Fig. 2.
Figure 2.
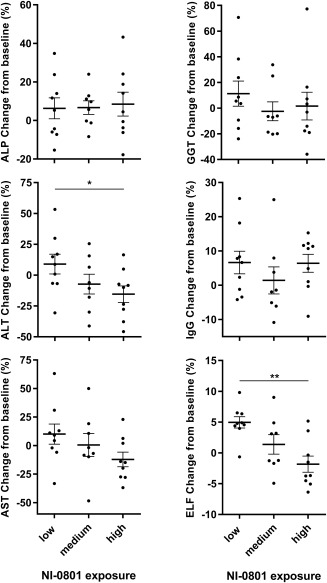
Change of baseline from clinical effect parameters (ALP, ALT, AST, γGT, IgG, and ELF) on D85 by the level of NI‐0801 exposure achieved. Bold horizontal bar indicates mean, error bars indicate SEM; P values were derived using two‐tailed unpaired t test (*P < 0.05, **P < 0.005).
The mean percentage change from baseline in ALT varied from 8.9% (range, –30.6% to +53.3%) for the group with low NI‐0801 exposure to –7.3% (range, −41.2% to +25.5%) and –15.4% (range, –45.7 to +16.42%; P value low versus high, 0.033) for the groups with medium and higher NI‐0801 exposures, respectively.
The mean percentage change from baseline in the ELF score varied from 5.0% (range, –0.6% to +9.8%) for the group with low NI‐0801 exposure to 1.4% (range, –4.9% to +9.0%) and –1.8% (range, –6.4% to +5.2%; P value poor versus adequate, 0.0006) for the groups with medium and higher NI‐0801 exposures, respectively.
Although not significant, a similar trend toward increased reduction in AST (mean of low reduction, +10.0% [–33.3% to +63.1%]; mean of medium reduction, +0.5% [–48.5% to +50.0%], mean of high reduction, –12.2% [–37.0% to + 23.0%]) was observed for the group achieving higher NI‐0801 exposure. No trends were apparent for the percentage change from baseline in ALP, γGT, or IgG levels.
CXCL10‐MEDIATED CHEMOTACTIC ACTIVITY IN PATIENT SERUM
The capacity of NI‐0801 in the patients' serum to inhibit CXCL10‐induced chemotaxis was investigated in a selected number of patients representing patients achieving high (>228.4 mg·hour/mL) or low (<124.5 mg·hour/mL) NI‐0801 exposure throughout the dosing period. For each of these patients, the predose sample (i.e., before first administration of NI‐0801) was used as baseline; 2 to 3 time points from the dosing and follow‐up period were analyzed.
Using murine L1.2 cells expressing human CXCR3, maximal chemotaxis was reached in the presence of CXCL10 levels ranging from 8 to 67 nM (corresponding to 70‐580 ng/mL). Endogenous levels of CXCL10 found in predose samples of patients with PBC were lower than 0.056 nM (corresponding to 0.5 ng/mL) and thus not sufficient to induce chemotaxis of CXCR3‐expressing cells. However, although the total CXCL10 levels measured in the majority of the postdosing samples were in the range expected to induce maximal chemotaxis, cells expressing CXCR3 did not migrate in response to endogenous CXCL10 in any of the postdosing serum samples tested (Fig. 3, empty bars). These findings indicate that endogenous CXCL10 present in the serum of patients with PBC exposed to NI‐0801 is neutralized by the antibody and not biologically active.
Figure 3.
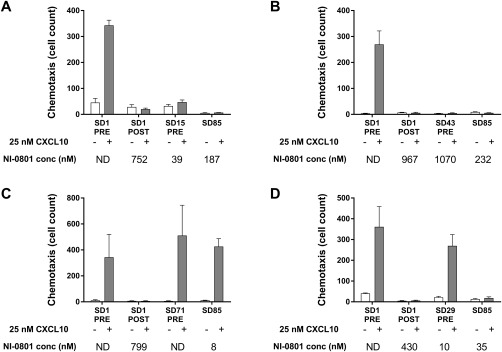
CXCL10 chemotactic activity in serum samples of representative NI‐0801‐treated patients with PBC. (A,B) High or (C,D) low NI‐0801 exposure in the absence (empty bars) or presence (solid bars) of 25 nM exogenous CXCL10. Data represent mean cell counts ± SEM. Abbreviations: ND, not detectable; SD, study day.
To mimic tissue conditions where high local concentrations of CXCL10 can be found, chemotaxis was also assessed after adding exogenous CXCL10 (25 nM) to the patient serum samples (Fig. 3). In all predose samples tested, CXCR3‐expressing cells migrated in response to the exogenously added CXCL10. For patients achieving high NI‐0801 exposure, no chemotaxis was observed for any of the postdosing samples in the presence of exogenous CXCL10. In contrast, chemotaxis to exogenously added CXCL10 was restored in patients with lower NI‐0801 exposure.
ANALYSIS OF CXCR3 EXPRESSION ON DIFFERENT LYMPHOCYTE SUBSETS
The impact of NI‐0801 dosing on the expression of CXCR3 on different lymphocyte subsets was investigated by flow cytometry on PBMCs from a subset of patients (n = 7) for whom matched pretreatment and posttreatment samples (i.e., D1 predose and D85) were available. The percentage of cells expressing CXCR3 was analyzed (Fig. 4). An increase in the percentage of CD3 cells expressing CXCR3 after treatment (mean, 45.73%; range, 28.72‐55.72) compared to pretreatment (mean, 37.22%; range, 29.77‐42.81) was recorded. Furthermore, this trend was repeated for both CD8+ T cells (pretreatment mean, 54.72% [34.8‐77.6]; posttreatment mean, 62.8% [35.35‐85.34]) and natural killer (NK) cells (pretreatment mean, 37.18% [22.68‐49.9]; posttreatment mean, 52.32% [30.58‐72.12]). Percentage expression of CXCR3 on CD4+ T cells followed a similar trend but did not reach significance (Fig. 4). The median channel fluorescence value for CXCR3 on each of the lymphocyte subset studied was also analyzed and revealed a consistent trend of greater expression posttreatment, although this did not reach significance (data not shown).
Figure 4.
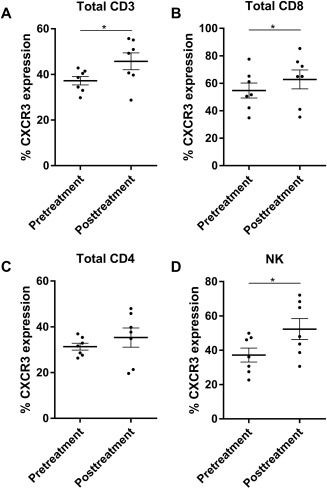
Flow cytometric analysis. The percentage of (A) CD3, (B) CD8, (C) CD4, and (D) NK cells expressing CXCR3 before and after treatment with NI‐0801. Bold horizontal bars indicate mean, error bars indicate SEM; *P < 0.05; P values derived using two‐tailed paired t test.
Discussion
CXCL10 and its receptor CXCR3 are involved in the recruitment of inflammatory T cells to the liver where these cells contribute to the destruction of the biliary tract in patients with PBC. NI‐0801 is a fully human mAb that binds to human CXCL10 with high affinity, inhibiting the interaction of CXCL10 with its receptor CXCR3.
This study was designed to investigate the efficacy, safety, PK, and PD profile of multiple doses of NI‐0801 in patients with PBC with an inadequate response to UDCA. NI‐0801 was administered at the dose of 10 mg/kg every 2 weeks.
NI‐0801 administration did not reveal any particular safety concerns. The most frequently occurring AEs reported in the NI‐0801‐03 study included symptoms commonly seen in patients with PBC, i.e., pruritus and fatigue,21, 22, 23, 24 or symptoms associated with UDCA therapy, e.g., diarrhea. Similarly, the most frequent infection reported was urinary tract infection, which is consistent with the increased frequency of this infection seen in patients suffering from PBC.25, 26 No SAE attributed to the drug was reported.
With NI‐0801 administration, total CXCL10 concentrations increased to peak levels that were approximately 450‐fold higher than baseline CXCL10 levels. This was most likely due to the formation of NI‐0801/CXCL10 complexes because CXCL10 present in the patient sera after NI‐0801 administration had no chemotactic activity, suggesting it was complexed to NI‐0801 and biologically inactive. The rise in total CXCL10 levels could be explained by several mechanisms. Chemokines bind rapidly to proteoglycans in the endothelial glycocalyx, which provides a mechanism to display and retain them at sites of inflammation. The antibody might bind to this sequestered chemokine and strip it off the endothelium into blood. In addition, binding of antibody to the chemokine interferes with its normal clearance pathway, thereby prolonging the half‐life of the chemokine. Complementary to the impact of NI‐0801 on the CXCL10 half‐life, the rate of neutralization of free NI‐0801 will be influenced by the levels of CXCL10. Rapid elimination of antibody in conjunction with an increase in antibody/target complex is characteristic for antibodies undergoing target‐mediated drug disposition (TMDD) and has been described for other mAbs targeting cytokines and chemokines.27, 28, 29
Flow cytometric analysis of CXCR3 expression on different lymphocyte subsets demonstrated a subtle but reproducible increase in the number of CXCR3‐expressing T cells and NK cells. Receptor–ligand interaction causes the internalization and degradation of CXCR3.30, 31 Thus, neutralization of CXCL10 in the blood by NI‐0801 could lead to reduced engagement of CXCR3, decreased receptor internalization, and an apparent increase in CXCR3 surface expression. Alternatively, the significant increase in CXCR3‐expressing T cells and in particular NK cells may be the result of reduced CXCL10‐mediated recruitment of these cells into the inflamed liver. However, the scope of the current study does not allow any conclusions to be drawn as to which mechanism underlies the observed increase in CXCR3 expressing cells in blood.
Despite these clear PD responses indicating modulation of the CXCR3/CXCL10 pathway in the blood, treatment with NI‐0801 did not translate into a clinical benefit for patients with PBC, as evidenced by the lack of improvement in any of the tests of liver damage and function measured. At D85, ALP levels were found to be modestly but significantly higher compared to baseline (ALP median change from baseline, 5.5%). Of note, ALP values at baseline were lower than the ALP screening values measured within 21 days prior to first drug administration (median screening versus baseline values for evaluable analysis set, 468 versus 412 IU/L, representing a reduction of 4.3% in ALP levels between screening and baseline). This suggests that the observed differences may simply reflect the normal fluctuations of ALP levels in patients with PBC. Of note, stratification of patients by level of NI‐0801 exposure suggests that higher drug exposure did lead to a reduction in ALT, AST, and ELF but not in ALP.
There are different possible explanations why treatment with NI‐0801 did not lead to clinical improvement in patients with PBC. The lack of improvement in patients with PBC following NI‐0801 treatment could be due to the high degree of redundancy present in the chemokine network. Apart from CXCL10, CXCR3 can also be activated by the chemokines CXCL9 and CXCL11.9 It has been shown that the neutralization of CXCL10 in animal models of liver disease reduces inflammation and fibrosis18, 19 and that CXCR3 deficiency delays and reduces progression of cellular inflammation in an autoimmune cholangitis animal model.32 However, existing animal models for PBC only have limited predictability, and we cannot exclude that interfering with CXCL10 alone might not be sufficient to reduce the entry of inflammatory cells into the liver to an extent that a clinical response can be achieved in patients with PBC.
Alternatively, we cannot exclude that the lack of clinical effect is due to insufficient target coverage. The dose and dosage schedule of NI‐0801 in patients with PBC were selected based on PK data from two previous HV studies conducted with NI‐0801. Although the peak levels of NI‐0801 after each infusion were similar between patients, the trough levels varied considerably between individual patients; for 5/26 (19%) patients, NI‐0801 trough levels were below the limit of detection at 1 or more time points during the dosing period. Overall, NI‐0801 exposure in the patients with PBC was much lower than expected, suggesting an increased clearance of the drug in patients compared to HVs.
For antibodies undergoing TMDD, the extent of target expression and the turnover rate of the target have a major impact on the clearance of the antibody. CXCL10 is elevated in hepatocytes and cholangiocytes isolated from patients with inflammatory liver diseases, including autoimmune hepatitis and PBC, leading to higher CXCL10 serum levels in these patients than in HVs.10, 14, 17 PK/PD modeling of the clinical trial data from HVs and patients with PBC estimated a CXCL10 production rate of 0.17 nM/hour and 0.99 nM/hour in HVs and patients with PBC, respectively (data on file). Thus, the 5‐fold to 6‐fold increased production rate of CXCL10 in the patients with PBC probably accounted for the faster clearance of NI‐0801 by TMDD, resulting in the reduced exposure observed in the patients compared to HVs. High turnover rates have also been reported for other chemokines, including chemokine (C‐C motif) ligand 2 (CCL2) and CCL21.28, 29
Thus, the NI‐0801 levels achieved in the patients might not have been sufficient to neutralize the high CXCL10 levels present in the inflamed peripheral tissues in addition to the levels of CXCL10 circulating in the blood. This is supported by results from the chemotaxis assay that showed that trough serum samples taken from patients showing low exposure caused by rapid clearance of the antibody do not contain enough NI‐0801 to neutralize chemotaxis induced by an excess of exogenously added CXCL10.
Clinical trials conducted with other anti‐chemokine mAbs, including eldelumab (anti‐CXCL10 mAb for treatment of ulcerative colitis and Crohn's disease) and carlumab (anti‐CCL2 mAb for treatment of prostate cancer), suggested that very high drug levels requiring multiple administrations of mAb doses of at least 25 mg/kg would be needed to achieve a clinical response. These results provide further support to the hypothesis that higher NI‐0801 doses would be required to obtain prolonged suppression of CXCL10.29, 33, 34, 35
Finally, both treatment duration as well as patient characteristics might have limited the potential of observing clinical benefit in patients with PBC after NI‐0801 administration. Thus, it is possible that the duration of NI‐0801 treatment was not long enough to show a target injury effect in what is a chronic immune‐mediated disease, and we might have reduced liver infiltration by lymphocytes but not seen the benefit translate into less liver injury over the relatively short time frame studied. In addition, as indicated by the relatively high baseline ALP levels in a significant proportion of patients, the study population included patients that were far down the cholestatic pathway of injury. Immunotherapy‐based approaches might actually need to be used much earlier in the disease course in high‐risk patients to prevent the consequences of cholestasis becoming the more dominant pathologic feature.
In conclusion, this open‐label single‐arm study failed to demonstrate the therapeutic benefit of multiple administrations of NI‐0801 at a dose of 10 mg/kg in patients with PBC with an incomplete response to UDCA. Given the possible insufficient neutralization of the target in the tissue, the current study does not provide a clear answer regarding the potential role of CXCL10 in the pathogenesis of PBC. Nevertheless, PK/PD results indicate that achieving clinical benefit by neutralizing CXCL10 might be challenging as the high production rate of the chemokine might limit its targetability.
Author names in bold designate shared first co‐authorship.
Potential conflict of interest: Dr. de Graaf, Dr. de Min, Dr. Ferlin, and Dr. Lapeyre are employees of Novimmune; Dr. Guilhot is a former employee of Novimmune.
Supported by Novimmune SA, Switzerland.
European Clinical Trials Database (EudraCT) trial number https://www.clinicaltrialsregister.eu/ctr-search/trial/2011-001326-26/GB.
REFERENCES
- 1. Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from “cirrhosis” to “cholangitis.” J Hepatol 2015;63:1285‐1287. [DOI] [PubMed] [Google Scholar]
- 2. Invernizzi P. Primary biliary cirrhosis. Semin Liver Dis 2014;34:253‐254. [DOI] [PubMed] [Google Scholar]
- 3. Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med 2005;353:1261‐1273. [DOI] [PubMed] [Google Scholar]
- 4. Invernizzi P. Human leukocyte antigen in primary biliary cirrhosis: an old story now reviving. Hepatology 2011;54:714‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bianchi I, Carbone M, Lleo A, Invernizzi P. Genetics and epigenetics of primary biliary cirrhosis. Semin Liver Dis 2014;34:255‐264. [DOI] [PubMed] [Google Scholar]
- 6. European Association for the Study of the Liver . EASL Clinical Practice Guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol 2017;67:145‐172. [DOI] [PubMed] [Google Scholar]
- 7. Invernizzi P, Lleo A, Podda M. Interpreting serological tests in diagnosing autoimmune liver diseases. Semin Liver Dis 2007;27:161‐172. [DOI] [PubMed] [Google Scholar]
- 8. Shimoda S, Van De Water J, Ansari A, Nakamura M, Ishibashi H, Coppel RL, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest 1998;102:1831‐1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 2011;89:207‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nishioji K, Okanoue T, Itoh Y, Narumi S, Sakamoto M, Nakamura H, et al. Increase of chemokine interferon‐inducible protein‐10 (IP‐10) in the serum of patients with autoimmune liver diseases and increase of its mRNA expression in hepatocytes. Clin Exp Immunol 2001;123:271‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harvey CE, Post JJ, Palladinetti P, Freeman AJ, Ffrench RA, Kumar RK, et al. Expression of the chemokine IP‐10 (CXCL10) by hepatocytes in chronic hepatitis C virus infection correlates with histological severity and lobular inflammation. J Leukoc Biol 2003;74:360‐369. [DOI] [PubMed] [Google Scholar]
- 12. Chuang Y‐H, Lian Z‐X, Cheng C‐M, Lan RY, Yang G‐X, Moritoki Y, et al. Increased levels of chemokine receptor CXCR3 and chemokines IP‐10 and MIG in patients with primary biliary cirrhosis and their first degree relatives. J Autoimmun 2005;25:126‐132. [DOI] [PubMed] [Google Scholar]
- 13. Zeremski M, Petrovic LM, Chiriboga L, Brown QB, Yee HT, Kinkhabwala M, et al. Intrahepatic levels of CXCR3‐associated chemokines correlate with liver inflammatioin and fibrosis in chronic hepatitis C. Hepatology 2008;48:1440‐1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tacke F, Zimmermann HW, Berres M‐L, Trautwein C, Wasmuth HE. Serum chemokine receptor CXCR3 ligands are associated with progression, organ dysfunction and complications of chronic liver diseases. Liver Int 2011;31:840‐849. [DOI] [PubMed] [Google Scholar]
- 15. Manousou P, Kolios G, Drygiannakis I, Koulentaki M, Pyrovolaki K, Voumvouraki A, et al. CXCR3 axis in patients with primary biliary cirrhosis: a possible novel mechanism of the effect of ursodeoxycholic acid. Clin Exp Immunol 2013;172:9‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lleo A, Zhang W, Zhao M, Tan Y, Bernuzzi F, Zhu B, et al.; PBC Epigenetic Study Group . DNA methylation profiling of the X chromosome reveals an aberrant demethylation on CXCR3 promoter in primary biliary cirrhosis. Clin Epigenetics 2015;7:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Curbishley SM, Eksteen B, Gladue RP, Lalor P, Adams DH. CXCR 3 activation promotes lymphocyte transendothelial migration across human hepatic endothelium under fluid flow. Am J Pathol 2005;167:887‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoneyama H, Kai Y, Koyama J, Suzuki K, Kawachi H, Narumi S, et al. Neutralization of CXCL10 accelerates liver regeneration in carbon tetrachloride‐induced acute liver injury. Med Mol Morphol 2007;40:191‐197. [DOI] [PubMed] [Google Scholar]
- 19. Hintermann E, Bayer M, Pfeilschifter JM, Luster AD, Christen U. CXCL10 promotes liver fibrosis by prevention of NK cell mediated hepatic stellate cell inactivation. J Autoimmun 2010;35:424‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parés A, Caballería L, Rodés J. Excellent long‐term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology 2006;130:715‐720. [DOI] [PubMed] [Google Scholar]
- 21. Cauch‐Dudek K, Abbey S, Stewart DE, Heathcote EJ. Fatigue in primary biliary cirrhosis. Gut 1998;43:705‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jopson L, Jones DE. Fatigue in primary biliary cirrhosis: prevalence, pathogenesis and management. Dig Dis 2015;33(Suppl. 2):109‐114. [DOI] [PubMed] [Google Scholar]
- 23. Bergasa NV. The pruritus of cholestasis. J Hepatol 2005;43:1078‐1088. [DOI] [PubMed] [Google Scholar]
- 24. Beuers U, Kremer AE, Bolier R, Elferink RP. Pruritus in cholestasis: facts and fiction. Hepatology 2014;60:399‐407. [DOI] [PubMed] [Google Scholar]
- 25. Corpechot C, Chrétien Y, Chazouillères O, Poupon R. Demographic, lifestyle, medical and familial factors associated with primary biliary cirrhosis. J Hepatol 2010;53:162‐169. [DOI] [PubMed] [Google Scholar]
- 26. Varyani FK, West J, Card TR. An increased risk of urinary tract infection precedes development of primary biliary cirrhosis. BMC Gastroenterol 2011;11:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen P, Vu T, Narayanan A, Sohn W, Wang J, Boedigheimer M, et al. Pharmacokinetic and pharmacodynamic relationship of AMG 811, an anti‐IFN‐γ IgG1 monoclonal antibody, in patients with systemic lupus erythematosus. Pharm Res 2015;32:640‐653. [DOI] [PubMed] [Google Scholar]
- 28. Dudal S, Subramanian K, Flandre T, Law WS, Lowe PJ, Skerjanec A, et al. Integrated pharmacokinetic, pharmacodynamic and immunogenicity profiling of an anti‐CCL21 monoclonal antibody in cynomolgus monkeys. MAbs 2015;7:829‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fetterly GJ, Aras U, Meholick PD, Takimoto C, Seetharam S, Mcintosh T, et al. Utilizing pharmacokinetics/pharmacodynamics modeling to simultaneously examine free CCL2, Total CCL2 and Carlumab (CNTO 888) concentration time data. J Clin Pharmacol 2013;53:1020‐1027. [DOI] [PubMed] [Google Scholar]
- 30. Sauty A, Colvin RA, Wagner L, Rochat S, Spertini F, Luster AD. CXCR3 internalization following T cell‐endothelial cell contact: preferential role of IFN‐inducible T cell alpha chemoattractant (CXCL11). J Immunol 2001;167:7084‐7093. [DOI] [PubMed] [Google Scholar]
- 31. Meiser A, Mueller A, Wise EL, McDonagh EM, Petit SJ, Saran N, et al. The chemokine receptor CXCR3 is degraded following internalization and is replenished at the cell surface by de novo synthesis of receptor. J Immunol 2008;180:6713‐6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang W, Fei Y, Gao J, Liu B, Zhang F. The role of CXCR3 in the induction of primary biliary cirrhosis. Clin Dev Immunol 2011;2011:564062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mayer L, Sandborn WJ, Stepanov Y, Geboes K, Hardi R, Yellin M, et al. Anti‐IP‐10 antibody (BMS‐936557) for ulcerative colitis: a phase II randomised study. Gut 2014;63:442‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sandborn WJ, Colombel J‐F, Ghosh S, Sands BE, Dryden G, Hébuterne X, et al. Eldelumab [Anti‐IP‐10] induction therapy for ulcerative colitis: a randomised, placebo‐controlled, phase 2b study. J Crohn's Colitis 2016;10:418‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC‐chemokine ligand 2 (CCL2), in metastatic castration‐resistant prostate cancer. Invest New Drugs 2013;31:760‐768. [DOI] [PubMed] [Google Scholar]