

Abstract
Acetaminophen (APAP)‐induced liver injury is closely associated with acute hepatic inflammation. Hypoxia‐inducible factor‐1 (HIF‐1) is activated during immunological processes and regulates gene expressions in various types of immune cells. Although HIF‐1 controls the differentiation and functions of conventional T cells in chronic inflammation, the pathological importance of HIF‐1 in innate‐like T cells during acute inflammation remains unknown. Here, we investigated the role of HIF‐1 in innate‐like γδ T cells during APAP‐induced acute liver injury. In response to APAP administration, T‐cell‐specific Hif‐1α gene knockout mice sustained severe liver damage compared to wild‐type control mice but without any impacts on the initial hepatic insult. This severe liver damage was accompanied by excessive neutrophil infiltration into the liver, increased serum interleukin (IL)‐17A levels, and increased hepatic expressions of C‐X‐C chemokine ligand (Cxcl) 1 and Cxcl2. Neutrophil depletion and IL‐17A neutralization completely abolished the aggravated phenotypes in T‐cell‐specific Hif‐1α gene knockout mice. Loss of the Hif‐1α gene enhanced the aberrant accumulation of IL‐17A‐producing innate‐like γδ T cells in the affected liver with no apparent effects on their IL‐17A‐producing ability. Adoptive transfer of Hif‐1α‐deficient splenic γδ T cells into recombination activating gene 2 (Rag2)‐deficient mice aggravated APAP‐induced liver injury with increased neutrophil accumulation in the liver compared to that of wild‐type γδ T cells. Furthermore, Hif‐1α‐deficient γδ T cells selectively showed aberrantly enhanced migratory ability. This ability was totally abolished by treatment with the mitochondrial adenosine triphosphate synthase inhibitor oligomycin. Conclusion: Deletion of Hif‐1α gene in T cells aggravates APAP‐induced acute inflammatory responses by enhancing aberrant innate‐like γδ T‐cell recruitment, thereby increasing excessive neutrophil infiltration into the liver. (Hepatology Communications 2018;2:571‐581)
Abbreviations
- AILI
acetaminophen‐induced liver injury
- ALT
alanine aminotransferase
- APAP
acetaminophen
- ATP
adenosine triphosphate
- CD
clusters of differentiation
- CXCL
C‐X‐C chemokine ligand
- FasL
Fas ligands
- HIF
hypoxia inducible factor
- IL
interleukin
- KO
knockout
- LPS
lipopolysaccharide
- NKT
natural killer T
- Th
T helper
- THIF‐1KO
T‐cell knockout of the Hif‐1α gene
- TNF‐α
tumor necrosis factor α
- Treg
regulatory T cell
- WT
wild type
T cells are a type of leukocytes that play an important role during immunologic processes. Conventional T cells require more than a week for their activation and maturation in adaptive immunity; they also contribute to chronic liver diseases, such as nonalcoholic fatty liver disease and hepatic fibrosis.1 In contrast, innate‐like T cells, such as natural killer T (NKT) cells and γδ T cells, control innate responses by directly killing infected or damaged cells and regulating functions of innate cells.2, 3 In addition, regulatory T (Treg) cells can suppress acute inflammatory responses.4, 5 Although the role of T cells in chronic liver diseases is well studied, information on how innate T cells are activated and contribute to inflammatory responses during acute liver injury remains elusive.
Acetaminophen (APAP) is used worldwide as an analgesic and antipyretic drug. However, accidental or intentional overdoses of APAP cause acute liver damage, known as acetaminophen‐induced liver injury (AILI), that is associated with high morbidity and mortality.6 APAP is metabolized mainly by hepatic cytochrome P450 2E1 (CYP2E1) into a toxic intermediate, N‐acetyl‐p‐benzoquinone‐imine, the excessive accumulation of which rapidly causes massive hepatocyte cell death. This toxic metabolite‐dependent liver injury evokes acute inflammatory responses mediated predominantly by macrophages and neutrophils. Activation of these innate immune cells further accelerates liver injury through the production of cytotoxic proteases and various cytokines, such as tumor necrosis factor α (TNF‐α) and interleukin (IL)‐1β.6, 7 These excessive inflammatory responses result in severe liver necrosis and can cause lethal hepatotoxicity.
In contrast to the information available for innate immune cells, little is known on the significance and subtypes of T cells involved in the pathogenesis of AILI. Overexpression of suppressor of cytokine signaling 3 in clusters of differentiation (CD)4+ T cells reportedly exacerbates APAP‐induced hepatotoxicity.8 Subsequent studies suggest that Treg cells and T helper (Th)17 cells, subsets of conventional CD4+ T cells, affect the level of APAP‐elicited liver injury. Treg cells suppress liver damage presumably by regulating the activities of other effector T cells.9 Furthermore, IL‐17/IL‐17A levels increase in patients with APAP‐induced liver failure,10 and a type of Th17 cells is considered to be the major source of IL‐17A.11 Among the various T‐cell subtypes, NKT cells and γδ T cells have been recognized as innate‐like T cells and play crucial roles in AILI pathogenesis. NKT cells protect or accelerate liver damage by regulating APAP metabolism,12, 13 whereas γδ T cells are activated by macrophage‐derived cytokines IL‐1β and IL‐23 and induce neutrophil‐mediated liver injury through IL‐17A production.9 These reports indicate the pathological significance of T cells in APAP hepatotoxicity. However, the types of T cells that are involved in the pathogenesis of AILI and the way in which their activation is controlled have not been fully elucidated.
Hypoxia‐inducible factors (HIFs) are key regulators of the transcriptional responses to low oxygen conditions (hypoxia) and have been reported to play crucial roles in the regulation of development, differentiation, and functions of immune cells.14 The accumulation of immune cells and vascular injury limit oxygen availability in the inflamed tissues, resulting in HIF activation in the inflammatory cells. Beside hypoxia, inflammatory stimuli, such as lipopolysaccharide (LPS),15 TNF‐α,16 and IL‐617 as well as T‐cell receptor ligation17, 18 can also activate HIF‐1α transcription and translation in an oxygen‐independent manner. HIF‐1 negatively regulates Th1 function19 and is required for transcriptional control of CD8+ T‐cell differentiation.20 HIF‐1 also determines the Th17/Treg balance in response to environmental cytokines.17, 21, 22, 23 Despite these intensive studies, the way in which HIF‐1 regulates the functions of T cells during acute APAP‐evoked inflammation is not known. Using T‐cell‐specific knockout mice (KO), we found that HIF‐1 inhibits excessive γδ T cell infiltration into the liver and thereby limits aberrant neutrophil‐mediated liver damage in APAP‐induced hepatotoxicity.
Materials and Methods
MICE AND EXPERIMENTAL PROCEDURES
Mice with T‐cell‐specific deletion of the Hif‐1α gene (THIF‐1KO) were generated by breeding Hif‐1α floxed mice (wild type [WT])24 with Lck‐cre transgenic mice that were kindly provided by Dr. Christopher Wilson (University of California San Francisco, San Francisco, CA). B6/recombination activating gene 2 (Rag2)‐deficient mice were purchased from Charles River Laboratories Japan, Inc. (Kanagawa, Japan). Mice were housed in cages containing irradiated sterile diet, autoclaved paper chips, and water under humidity‐ and temperature‐controlled specific pathogen‐free conditions in the animal facility of Tokyo Women's Medical University and Waseda University Institution. Littermate male mice aged 8‐10 weeks were used for experiments. All animal experiments were carried out in compliance with the Guideline for the Care and Use of Laboratory Animals and were approved by the Committee on Animal Care and Use of Waseda University (Protocol number, 2017‐A035). For detailed experimental procedures used in this study, see the http://onlinelibrary.wiley.com/doi/10.1002/hep4.1175/full.
STATISTICAL ANALYSIS
Results are expressed as mean ± SEM. Statistical analyses were performed using Mann‐Whitney U tests and one‐way analysis of variance followed by Tukey‐Kramer tests for all experiments. Survival rates were compared using the log rank test. P < 0.05 was considered significant.
Results
T‐CELL‐SPECIFIC DELETION OF THE Hif‐1α GENE EXACERBATED LIVER DAMAGE AND DECREASED SURVIVAL IN AILI
To explore the importance of HIF‐1 in T cells in the pathogenesis of AILI, we administered 250 mg/kg APAP intraperitoneally to mice fasted for 24 hours. Serum alanine aminotransferase (ALT) activity peaked at 6 hours after APAP injection and gradually decreased thereafter in WT mice (Fig. 1A). THIF‐1KO mice showed levels of initial liver damage comparable to those of WT mice. However, serum ALT levels were sustained at high levels until 24 hours and were significantly higher at 24 hours and 48 hours in THIF‐1KO mice than in WT mice (Fig. 1A). Hepatic glutathione levels further decreased at 6 hours by APAP treatment but were then gradually restored in WT mice. THIF‐1KO mice showed similar alterations in response to APAP, but the restoration was less in THIF‐1KO mice than in WT mice (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1175/full). The regression of hepatocyte necrosis (as evaluated by hematoxylin and eosin staining) was subsequently delayed in THIF‐1KO mice (Fig. 1B,C). However, the damage‐induced hepatic regeneration assessed by Ki67‐positive cell numbers and proliferating cell nuclear antigen (PCNA) expression occurred comparably, irrespective of Hif‐1α gene status in T cells (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1175/full). THIF‐1KO mice showed greater susceptibility to APAP (250 mg/kg) with decreased survival rates compared with WT mice (Fig. 1D). These results suggest that HIF‐1 in T cells inhibits aberrant APAP‐induced hepatotoxicity in the late phase of injury.
Figure 1.
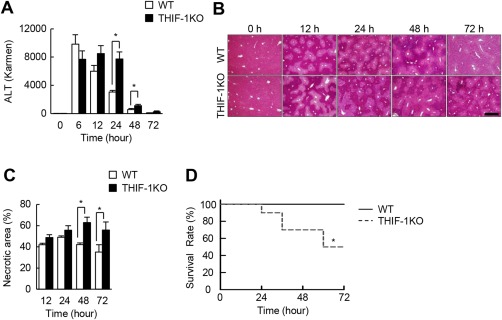
T‐cell‐specific Hif‐1α deficiency exacerbated hepatotoxicity and reduced survival rate during AILI. (A) Serum ALT levels in mice exposed to APAP (n = 6‐10). (B) Representative hematoxylin and eosin‐stained images of liver sections (scale, 500 μm). (C) Percentage of necrotic area in APAP‐treated liver (n = 5‐10). (D) Survival rate of mice administered APAP (n = 5‐10). *P < 0.05. Data represent mean ± SEM. One IU (international unit) / L = 1 Karmen × 0.482.
NEUTROPHIL INFILTRATION INCREASED IN THE LIVERS OF THIF‐1KO MICE DURING AILI
We next investigated the type of immune cells that contribute to aggravated liver damage in THIF‐1KO mice. Innate immune cells reportedly accelerate liver damage during AILI.6 To examine the possible involvement of innate immune cells in APAP hepatotoxicity in THIF‐1KO mice, isolated hepatic leukocytes were analyzed by flow cytometry. Macrophages were markedly recruited in the damaged liver 24 hours after APAP injection, but the percentage and absolute numbers of macrophages were not affected by T‐cell‐specific deletion of the Hif‐1α gene (Fig. 2A). Consistent with this finding, the expressions of Tnf‐α and Il‐12p40, hallmarks of inflammatory M1 macrophages, also increased with APAP challenge; however, expression levels were comparable in WT and THIF‐1KO mice (Fig. 2B). Moreover, death signaling mediated by Fas ligands (FasL) reportedly induces hepatocyte death in AILI,25 but the hepatic expressions of FasL did not differ between the two groups (Fig. 2B). In addition, flow cytometric analysis revealed that the expressions of CD69 and FasL on NKT cells were comparable between the groups, suggesting that NKT cells are not activated by deletion of Hif‐1α (data not shown). Neutrophils infiltrated substantially in the liver of APAP‐treated WT mice at 12 hours, and the number remained unchanged for the next 12 hours (Fig. 2C). In contrast, hepatic neutrophil infiltration further increased in THIF‐1KO mice compared to WT mice 24 hours after APAP treatment (Fig. 2C). This finding was further supported by immunohistochemical analysis showing that neutrophil infiltration was more evident in the necrotic regions in THIF‐1KO mice than in WT mice (Fig. 2D). Exposure to APAP has been reported to induce two major neutrophil chemoattractants, chemokine (C‐X‐C motif) ligand (Cxcl)1 and Cxcl2,26 and various types of liver cells, including hepatocytes, Kupffer cells, liver sinusoidal endothelial cells, and hepatic stellate cells secrete these chemokines in LPS‐induced liver inflammation.27 Consistently, the aggravated neutrophil infiltrations were closely associated with the aberrant increase in expressions of Cxcl1 and Cxcl2 at 24 hours in APAP‐treated THIF‐1KO liver compared with APAP‐treated WT liver (Fig. 2E). In addition, depletion of neutrophils with anti‐Gr‐1 antibody markedly reduced the levels of serum ALT in APAP‐treated WT mice. In contrast, the same treatment completely abolished the aggravated phenotype in THIF‐1KO mice (Fig. 2F). Collectively, these results suggest that the aggravated liver damage in THIF‐1KO mice is attributable to excessive neutrophil infiltration in the late phase of AILI.
Figure 2.
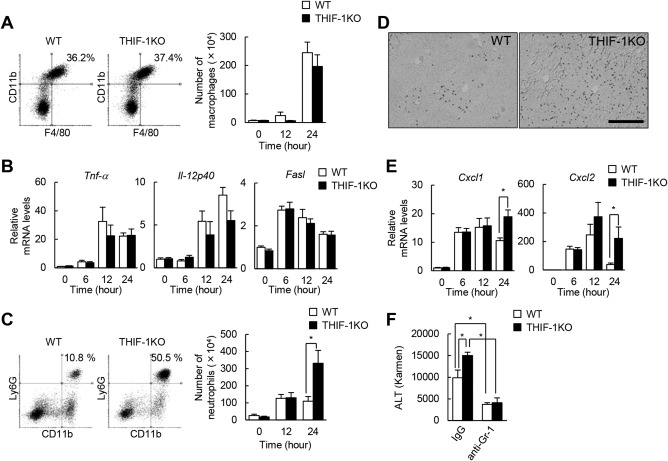
Neutrophil recruitment increased in THIF‐1KO liver exposed to APAP. (A) Flow cytometric analysis at 24 hours after APAP treatment and number of hepatic macrophages (CD45+Ly6G–F4/80+CD11b+) during the initial 24 hours (n = 4‐10). (B) Hepatic expressions of inflammatory genes (n = 8‐13). (C) Flow cytometric analysis at 24 hours after APAP treatment and number of hepatic neutrophils (CD45+CD11b+Ly6G+) (n = 4‐10). (D) Image of 24 hours APAP‐treated liver stained with Ly6G antibody (scale, 200 μm). (E) Hepatic mRNA levels of neutrophil‐chemotactic genes (n = 8‐13). (F) Serum ALT levels in 24 hours APAP‐treated mice pretreated with anti‐Gr‐1 antibody (n = 4‐5). *P < 0.05. Data represent mean ± SEM. Abbreviations: IgG, immunoglobulin G; mRNA, messenger RNA. One IU (international unit) / L = 1 Karmen × 0.482.
IL‐17A SECRETION INDUCED NEUTROPHIL‐MEDIATED LIVER INJURY IN THIF‐1KO MICE
Activated T cells predominantly produce IL‐17A, which induces the expressions of CXCL1 and CXCL2 and subsequently leads to neutrophil recruitment.28, 29 Neutrophil infiltration occurred in WT mice treated with APAP (Fig. 2C), but serum IL‐17A levels did not substantially change during AILI. In contrast, the loss of the Hif‐1α gene in T cells in THIF‐1KO mice did not have any impacts on serum IL‐17A levels for the first 12 hours after APAP injection; however, by 24 hours, serum IL‐17A levels had abruptly increased (Fig. 3A). To investigate whether increased IL‐17A caused the neutrophil‐mediated liver damage in THIF‐1KO mice, WT and THIF‐1KO mice were intravenously treated with anti‐IL‐17A antibody. Neutralization of IL‐17A completely abolished aberrantly elevated ALT levels and also abolished the increased neutrophil accumulation in THIF‐1KO mice (Fig. 3B,C). In contrast, the treatment showed little, if any, effect on liver damage and hepatic neutrophil numbers in WT mice exposed to APAP (Fig. 3B,C). These results suggest that HIF‐1 limits excessive neutrophil infiltration and the associated liver injury by suppressing IL‐17A secretion.
Figure 3.
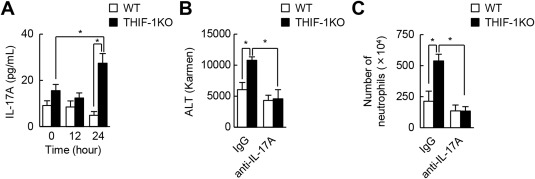
IL‐17A secretion induced neutrophil‐dependent hepatotoxicity in THIF‐1KO mice. (A) Serum IL‐17A levels in APAP‐treated mice (n = 11‐17). (B) Serum ALT levels and (C) number of hepatic neutrophils (CD45+CD11b+Ly6G+) of mice treated with either control IgG or anti‐IL‐17A antibody in AILI (n = 4‐6). *P < 0.05. Data represent mean ± SEM. Abbreviation: IgG, immunoglobulin G. One IU (international unit) / L = 1 Karmen × 0.482.
T‐CELL‐SPECIFIC Hif‐1α DEFICIENCY CONTRIBUTED TO INCREASED RECRUITMENT OF IL‐17A+ γδ T CELLS IN THE LIVER
We next investigated which types of T cells contributed to the aberrant increase in serum IL‐17A in THIF‐1KO mice. Isolated hepatic leukocytes were analyzed by flow cytometry after being restimulated with phorbol‐12‐myristial‐13‐acetate and ionomycin in the presence of GolgiStop. Hepatic accumulation of IL‐17A+ γδ T cells but not of other T cells expressing IL‐17A was significantly higher in THIF‐1KO mice than in WT mice when exposed to APAP (Fig. 4A,B). The mean fluorescence intensity of IL‐17A+ cells was notably higher in γδ T cells than in other T cells but was comparable in both T‐cell fractions between WT and THIF‐1KO mice (Fig. 4C). The expression of activation marker CD69 on γδ T cells did not differ between the two groups (Fig. 4D), suggesting that HIF‐1 has dispensable effects on the IL‐17A production of γδ T cells. In contrast, γδ T‐cell infiltration into the damaged liver substantially increased when exposed to APAP treatment, and these responses were markedly enhanced in THIF‐1KO mice compared to WT mice (Fig. 4E). To further assess the importance of HIF‐1 in γδ T cells in AILI, splenic γδ T cells of WT or THIF‐1KO mice were adoptively transferred to Rag2‐deficient mice, and mice were administrated 150 mg/kg APAP. Compared to mice receiving WT γδ T cells, those receiving Hif‐1α‐deficient γδ T cells showed elevated ALT levels and increased neutrophil accumulation in the liver (Fig. 4F). These findings further support our hypothesis that HIF‐1 prevents the accumulation of IL‐17A+ γδ T cells and the aberrant increase in serum IL‐17A levels exclusively by limiting the infiltration of γδ T cells into the liver.
Figure 4.
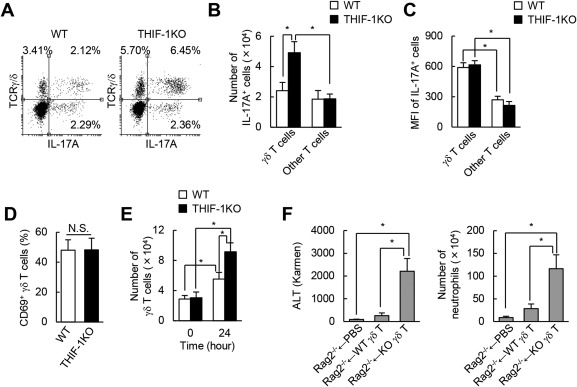
Loss of the Hif‐1α gene enhanced IL‐17A+ γδ T‐cell infiltration into APAP‐treated liver. (A‐C) Isolated hepatic leukocytes were restimulated with PMA and ionomycin. (A) Flow cytometric analysis of IL‐17A+ γδ T cells (CD45+CD3+TCRγ/δ+) isolated from 24‐hour APAP‐treated liver. (B) Numbers and (C) MFIs of hepatic IL‐17A+ γδ T (CD45+CD3+TCRγ/δ+) and other T cells (CD45+CD3+TCRγ/δ–) at 24 hours after APAP treatment (n = 5‐8). (D) Percentage of hepatic CD69+ γδ T cells at 24 hours after APAP treatment (n = 3). (E) Number of hepatic γδ T cells (n = 5‐8). (F) ALT levels and number of neutrophils in APAP (150 mg/kg)‐treated Rag2‐deficient mice adoptively transferred with splenic γδ T cells from WT or THIF‐1KO (n = 4‐6). *P < 0.05. Data represent mean ± SEM. Abbreviations: MFI, mean fluorescence intensity; N.S., not significant; PBS, phosphate‐buffered saline; PMA, phorbol‐12‐myristial‐13‐acetate; TCR, T‐cell receptor. One IU (international unit) / L = 1 Karmen × 0.482.
HIF‐1 SUPPRESSED THE MIGRATION CAPACITY OF γδ T CELLS BY INHIBITING MITOCHONDRIAL ADENOSINE TRIPHOSPHATE PRODUCTION
To examine whether the loss of the Hif‐1α gene selectively promotes γδ T‐cell migration, Boyden chamber assays were performed using splenocytes. Because C‐C chemokine ligand 20 and its chemokine receptor 6 play crucial roles in the infiltration of IL‐17A+ γδ T cells into the liver,30, 31 C‐C chemokine ligand 20 was used as a chemoattractant to induce γδ T‐cell migration. Hif‐1α‐deficient γδ T cells migrated to greater extents than WT γδ T cells (Fig. 5A), whereas non‐γδ T cells showed comparable abilities to move irrespective of Hif‐1α gene status (Fig. 5B). T cells require, in part, mitochondria‐derived energy for their migration because oligomycin, an inhibitor of mitochondrial adenosine triphosphate (ATP) synthase, inhibits the migration of T cells.32 Consistent with this report, oligomycin substantially decreased migration of either WT or THI‐1KO non‐γδ T cells (Fig. 5B). In contrast, the treatment slightly but not significantly reduced the migration of WT γδ T cells (Fig. 5A). On the other hand, the enhanced migration of Hif‐1α‐deficient γδ T cells was completely abrogated to levels of WT cells by oligomycin treatment (Fig. 5A). Furthermore, HIF‐1 did not affect the total mitochondrial content of γδ T cells (Fig. 5C), suggesting that enhanced migration in Hif‐1α‐deficient γδ T cells was not caused by increased mitochondrial biogenesis evoked by Hif‐1α gene inactivation. Collectively, these results suggest that HIF‐1 suppresses the migration of γδ T cells, presumably by limiting mitochondrial energy metabolism.
Figure 5.
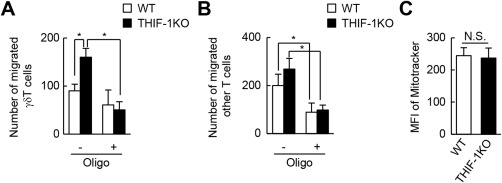
Deletion of the Hif‐1α gene enhanced migration capacity of γδ T cells. Migration assay of T cells from WT and THIF‐1KO mice. Cells were stimulated by CCL20 with or without oligomycin, and the number of migrated (A) γδ T cells (CD45+CD3+TCRγ/δ+) and (B) other T cells (CD45+CD3+TCRγ/δ–) in lower chambers was determined by flow cytometry (n = 6‐7). (C) Mitochondrial content of splenic CD45+CD3+TCRγ/δ+ γδ T cells (n = 3). *P < 0.05. Data represent mean ± SEM. Abbreviations: CCL, C‐C chemokine ligand; MFI, mean fluorescence intensity; N.S., not significant; Oligo, oligomycin; TCR, T‐cell receptor.
Discussion
Tight regulation of inflammation is important to limit excessive tissue damage and to restore functional integrity rapidly after inflammation has resolved. Inflammatory responses are governed, in part, by the controlled infiltration of different types of immune cells in the affected tissue. Moreover, appropriate numbers of T cells accumulated in inflamed tissues have been implicated in the resolution of inflammation, whereas their excessive infiltration in several diseases, including drug‐induced hepatitis, exacerbates inflammation. HIF‐1, a key transcription factor that mediates cellular responsiveness to hypoxia, is a critical regulator of the development, differentiation, and function of various types of immune cells. However, its pathological significance in the function of T cells during acute inflammation has not been fully elucidated. Here, we showed that T‐cell HIF‐1 plays a critical role in regulating the infiltration of innate‐like γδ T cells in APAP‐induced hepatitis, presumably by switching energy reliance from mitochondria to glycolysis. This switch subsequently inhibits excessive neutrophil accumulation in the damaged liver. HIF‐1 in T cells prevents excessive inflammatory responses by preventing aberrant migration of γδ T cells.
Accumulating evidence shows that neutrophils play an important role during AILI, although there is controversy on how infiltrated neutrophils contribute to hepatocellular damage.33 Here, we found that THIF‐1KO mice aggravated APAP hepatotoxicity with increased neutrophil infiltration into the damaged liver. These findings are consistent with those of other reports demonstrating the pathological importance of neutrophil‐evoked inflammation in AILI,7, 34, 35 and this strongly supports our hypothesis that HIF‐1 in T cells inhibits excessive neutrophil accumulation and the subsequent expansion of the associated liver injury. High doses of APAP cause massive neutrophil infiltration in the necrotic region of the liver as early as 6 hours after administration.35 The administration of neutrophil‐depleting antibody or the deletion of Cxcr2, a receptor of neutrophil‐chemotactic chemokines CXCL1 and CXCL2, ameliorates APAP‐induced liver damage and enhances survival.9, 34, 35 In addition, various types of liver cells, such as hepatocytes, Kupffer cells, liver sinusoidal endothelial cells, and hepatic stellate cells, secrete these chemokines to promote neutrophil infiltration in LPS‐induced liver inflammation.27 We consistently found notable increases in the hepatic expression of Cxcl1 and Cxcl2 in THIF‐1KO mice, providing further evidence for crucial roles of T‐cell HIF‐1‐regulated neutrophil recruitment in aggravated APAP hepatotoxicity. However, such inhibitory effects of HIF‐1 seem to be limited to late‐onset and inflammation‐associated liver injury. Indeed, differences between WT and THIF‐1KO mice in serum ALT levels and areas of necrosis in the affected liver became evident only after 24 hours of APAP treatment. Considering that neutrophil depletion suppresses elevation of ALT levels even in the early phase of liver insults,35 HIF‐1 in T cells displays dispensable roles in toxic metabolite‐mediated early liver damage. Consistent with this hypothesis, 12 hours after APAP challenge, we observed that neutrophil accumulation occurred to similar extents irrespective of Hif‐1α gene status in T cells. Although we cannot rule out the possibility that the aggravated liver injury in THIF‐1KO is attributable to delayed glutathione recovery 24 hours after APAP challenge, HIF‐1 in T cells indeed suppresses neutrophil‐mediated liver injury because neutrophil depletion completely abolished the aggravated liver injury in THIF‐1KO mice. Furthermore, the aggravated hepatic injury that occurred in THIF‐1KO mice could not be explained by the difference in macrophage‐mediated inflammatory responses because hepatic macrophage accumulation and expression of its associated proinflammatory mediators were induced to similar extents in WT and THIF‐1KO mice. Collectively, this evidence suggests that HIF‐1 in T cells protects against the aberrant expansion of inflammation‐evoked liver damage resulting from APAP hepatotoxicity by limiting excessive neutrophil infiltration.
IL‐17A, a proinflammatory cytokine primarily produced by activated T cells, stimulates neutrophil recruitment to inflamed tissues.28, 29 Increased serum IL‐17 levels have been reported in human and mouse AILI.9, 10, 11 In contrast with the findings of these reports, we did not observe any changes in serum IL‐17A levels in WT mice exposed to APAP, suggesting dispensable roles for IL‐17A in our disease model. We cannot currently explain this discrepancy, although the smaller amounts of APAP used in our experiments might have failed to elicit substantial production of IL‐17A. However, our hypothesis was further confirmed when we found no apparent effects of IL‐17A neutralization on liver damage and neutrophil infiltration into the affected WT liver. In contrast, the aggravated APAP toxicity in THIF‐1KO mice can be attributed predominantly to an aberrant increase in serum IL‐17A at 24 hours after APAP challenge because anti‐IL‐17A neutralizing antibody completely abolished the aggravated liver damage and neutrophil recruitment in THIF‐1KO mice to the levels of APAP‐treated WT mice. This hypothesis is further supported by our findings showing much higher expressions of IL‐17A‐inducible neutrophil chemoattractants, such as Cxcl1 and Cxcl2, in THIF‐1KO liver at 24 hours after APAP treatment. Although IL‐17A+ γδ T cells have been reported to stimulate liver regeneration in partial hepatectomized mice,31 IL‐17A played dispensable roles in APAP‐induced liver regeneration in our experiments because hepatocyte proliferation rates were not significantly different between WT and THIF‐1KO mice. APAP‐evoked hepatic inflammation can occur in both an IL‐17A‐dependent and an IL‐17A‐independent manner, but tight regulation of local IL‐17A production by HIF‐1 in T cells in the immune‐mediated injury phase seems to be a prerequisite for the normal inflammatory response and its resolution in the liver.
Among the various types of T cells, Th17 cells were initially recognized as the primary source of IL‐17A. However, when considering the role of IL‐17A in the acute inflammatory response of AILI, innate immune cells, such as NK cells, NKT cells, and γδ T cells, seem to play crucial roles in the IL‐17A production in AILI. Wang et al.9 have clearly shown the importance of γδ T cells as a major source of IL‐17A. They also found increased accumulations of γδ T cells in liver exposed to APAP. Consistent with that report, we also found a moderate but not significant increase in the number of γδ T cells in APAP‐challenged WT liver. Deletion of the Hif‐1α gene in T cells stimulated a marked accumulation of hepatic IL‐17A+ γδ T cells without any impact on their IL‐17A‐producing ability. Given that APAP treatment tended to reduce the numbers of hepatic NK and NKT cells irrespective of the Hif‐1α gene status (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1175/full) and that adoptive transfer of Hif‐1α‐deficient γδ T cells into immunodeficient mice deteriorated AILI with enhanced neutrophil infiltrations compared to that of WT γδ T cells, present findings strongly suggest that HIF‐1 in T cells limits excessive infiltration of γδ T cells with high IL‐17A‐producing ability and thus abolishes aberrant neutrophil accumulation in APAP‐induced liver injury.
A coordinated regulation of immune cell migration is essential for normal inflammatory responses.36 Our findings of a higher migratory ability of γδ T cells lacking the Hif‐1α gene strongly suggest that HIF‐1 in T cells prevents aberrant inflammatory responses by inhibiting excessive infiltration of γδ T cells into APAP‐challenged liver. Furthermore, because oligomycin treatment completely abrogated the enhanced migration of Hif‐1α‐deficient γδ T cells to the levels of WT γδ T cells, this inhibitory regulation seems to occur by suppressing mitochondrial ATP generation. This finding is in sharp contrast to the stimulatory effects of HIF‐1 on the migratory ability of proinflammatory macrophages and cancerous cells.37, 38 In these cells, HIF‐1 promotes cell migration exclusively by shifting cells from oxidative phosphorylation toward glycolysis to provide energy for various cell migratory processes, such as actin remodeling. Once activated, T cells also reportedly undergo a rapid metabolic reprogramming with increased glycolysis, achieved in part by activating HIF‐1.39 However, activated T cells are still likely to rely on mitochondria‐associated metabolism to exert their functions. In fact, the redistribution of mitochondria and local mitochondrial ATP production occur in the uropod during T‐cell migration.32 Considering that oligomycin treatment greatly but not completely inhibits T cell chemotaxis, both mitochondria‐dependent and mitochondria‐independent metabolic processes are prerequisites for efficient T‐cell migration. This hypothesis is further supported by our observation of a partial inhibition of Hif‐1α‐deficient γδ T‐cell migration by oligomycin. Interestingly, this HIF‐1‐mediated regulation of T‐cell migration was limited to γδ T cells and was not found in other T cells in our experiments, suggesting cell‐type‐specific effects of HIF‐1 on cell migration. Furthermore, HIF‐1 reportedly inhibits mitochondrial biogenesis,40 but the mitochondrial content of γδ T cells was not affected by deletion of the Hif‐1α gene in our experiments, suggesting the importance of an HIF‐1‐dependent reduction of metabolic fluxes into mitochondria in the regulation of γδ T‐cell migration. Therefore, HIF‐1 appears to selectively limit excessive accumulation of γδ T cells in liver exposed to APAP by suppressing their mitochondrial energy production (Fig. 6).
Figure 6.
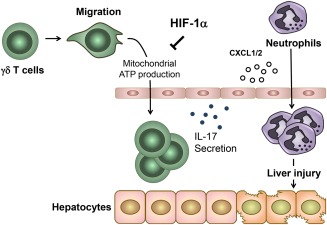
Schematic illustration on the role of HIF‐1 in γδ T cells during AILI. HIF‐1 inhibits aberrant migration of γδ T cells into APAP‐treated liver, presumably by suppressing mitochondrial metabolism and thereby limiting excessive neutrophil recruitment and subsequent liver injury.
In conclusion, our study provides evidence that HIF‐1 in T cells prevents aberrant APAP‐induced liver injury by suppressing aberrant recruitment of proinflammatory γδ T cells, thereby preventing the excessive accumulation of neutrophils. Because this newly proposed concept may extend to γδ T‐cell‐mediated acute sterile inflammation in conditions other than APAP‐induced liver injury, further study is necessary to explore the scope of this new mechanism and to develop new therapies targeting HIF‐1 in T cells.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1175/full.
Supporting Information 1
Acknowledgment
We thank the members of the Goda Laboratory for stimulating discussions.
Potential conflict of interest: Nothing to report.
Supported in part by the Japanese Government MEXT Program for the Strategic Research Foundation at Private Universities (No. S1201006 to N.G.), Grant‐in‐Aid for Scientific Research (C) (No. 17570119 to N.G.), and Grant‐in‐Aid for Scientific Research on Innovative Areas “Oxygen biology: a new criterion for integrated understanding of life” (No. 2611103 to N.G.).
REFERENCES
- 1. Heymann F, Tacke F. Immunology in the liver‐‐from homeostasis to disease. Nat Rev Gastroenterol Hepatol 2016;13:88‐110. [DOI] [PubMed] [Google Scholar]
- 2. Vantourout P, Hayday A. Six‐of‐the‐best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol 2013;13:88‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Van Kaer L, Parekh VV, Wu L. Invariant natural killer T cells: bridging innate and adaptive immunity. Cell Tissue Res 2011;343:43‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Okeke EB, Okwor I, Mou Z, Jia P, Uzonna JE. CD4+CD25+ regulatory T cells attenuate lipopolysaccharide‐induced systemic inflammatory responses and promotes survival in murine Escherichia coli infection. Shock 2013;40:65‐73. [DOI] [PubMed] [Google Scholar]
- 5. Carney EF. Acute kidney injury: targeting Treg cells to protect the kidney. Nat Rev Nephrol 2017;13:444. [DOI] [PubMed] [Google Scholar]
- 6. Krenkel O, Mossanen JC, Tacke F. Immune mechanisms in acetaminophen‐induced acute liver failure. Hepatobiliary Surg Nutr 2014;3:331‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil‐mediated injury amplification following necrosis. J Clin Invest 2015;125:539‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Numata K, Kubo M, Watanabe H, Takagi K, Mizuta H, Okada S, et al. Overexpression of suppressor of cytokine signaling‐3 in T cells exacerbates acetaminophen‐induced hepatotoxicity. J Immunol 2007;178:3777‐3785. [DOI] [PubMed] [Google Scholar]
- 9. Wang X, Sun R, Wei H, Tian Z. High‐mobility group box 1 (HMGB1)‐Toll‐like receptor (TLR)4‐interleukin (IL)‐23‐IL‐17A axis in drug‐induced damage‐associated lethal hepatitis: interaction of gammadelta T cells with macrophages. Hepatology 2013;57:373‐384. [DOI] [PubMed] [Google Scholar]
- 10. Li J, Zhu X, Liu F, Cai P, Sanders C, Lee WM, et al. Cytokine and autoantibody patterns in acute liver failure. J Immunotoxicol 2010;7:157‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu X, Uetrecht J. A novel T(H)17‐type cell is rapidly increased in the liver in response to acetaminophen‐induced liver injury: T(H)17 cells and the innate immune response. J Immunotoxicol 2013;10:287‐291. [DOI] [PubMed] [Google Scholar]
- 12. Downs I, Aw TY, Liu J, Adegboyega P, Ajuebor MN. Valpha14iNKT cell deficiency prevents acetaminophen‐induced acute liver failure by enhancing hepatic glutathione and altering APAP metabolism. Biochem Biophys Res Commun 2012;428:245‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin‐Murphy BV, Kominsky DJ, Orlicky DJ, Donohue TM, Jr. , Ju C. Increased susceptibility of natural killer T‐cell‐deficient mice to acetaminophen‐induced liver injury. Hepatology 2013;57:1575‐1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity 2014;41:518‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blouin CC, Page EL, Soucy GM, Richard DE. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia‐inducible factor 1alpha. Blood 2004;103:1124‐1130. [DOI] [PubMed] [Google Scholar]
- 16. Albina JE, Mastrofrancesco B, Vessella JA, Louis CA, Henry WL Jr, Reichner JS. HIF‐1 expression in healing wounds: HIF‐1alpha induction in primary inflammatory cells by TNF‐alpha. Am J Physiol Cell Physiol 2001;281:C1971‐C1977. [DOI] [PubMed] [Google Scholar]
- 17. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia‐inducible factor 1. Cell 2011;146:772‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ikejiri A, Nagai S, Goda N, Kurebayashi Y, Osada‐Oka M, Takubo K, et al. Dynamic regulation of Th17 differentiation by oxygen concentrations. Int Immunol 2012;24:137‐146. [DOI] [PubMed] [Google Scholar]
- 19. Shehade H, Acolty V, Moser M, Oldenhove G. Cutting edge: hypoxia‐inducible factor 1 negatively regulates Th1 function. J Immunol 2015;195:1372‐1376. [DOI] [PubMed] [Google Scholar]
- 20. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo‐Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia‐inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med 2012;209:2441‐2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha‐dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 2011;208:1367‐1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Higashiyama M, Hokari R, Hozumi H, Kurihara C, Ueda T, Watanabe C, et al. HIF‐1 in T cells ameliorated dextran sodium sulfate‐induced murine colitis. J Leukoc Biol 2012;91:901‐909. [DOI] [PubMed] [Google Scholar]
- 23. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia‐inducible factor‐1 alpha‐dependent induction of FoxP3 drives regulatory T‐cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A 2012;109:E2784‐E2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, et al. Hypoxia‐inducible factor‐1alpha is a positive factor in solid tumor growth. Cancer Res 2000;60:4010‐4015. [PubMed] [Google Scholar]
- 25. Knight TR, Jaeschke H. Acetaminophen‐induced inhibition of Fas receptor‐mediated liver cell apoptosis: mitochondrial dysfunction versus glutathione depletion. Toxicol Appl Pharmacol 2002;181:133‐141. [DOI] [PubMed] [Google Scholar]
- 26. Saiman Y, Friedman SL. The role of chemokines in acute liver injury. Front Physiol 2012;3:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bigorgne AE, John B, Ebrahimkhani MR, Shimizu‐Albergine M, Campbell JS, Crispe IN. TLR4‐dependent secretion by hepatic stellate cells of the neutrophil‐chemoattractant CXCL1 mediates liver response to gut microbiota. PLoS One 2016;11:e0151063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Onishi RM, Gaffen SL. Interleukin‐17 and its target genes: mechanisms of interleukin‐17 function in disease. Immunology 2010;129:311‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin‐17 family members. Immunity 2011;34:149‐162. [DOI] [PubMed] [Google Scholar]
- 30. Hammerich L, Bangen JM, Govaere O, Zimmermann HW, Gassler N, Huss S, et al. Chemokine receptor CCR6‐dependent accumulation of gammadelta T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology 2014;59:630‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rao R, Graffeo CS, Gulati R, Jamal M, Narayan S, Zambirinis CP, et al. Interleukin 17‐producing gammadeltaT cells promote hepatic regeneration in mice. Gastroenterology 2014;147:473‐484.e472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Campello S, Lacalle RA, Bettella M, Manes S, Scorrano L, Viola A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med 2006;203:2879‐2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jaeschke H. How relevant are neutrophils for acetaminophen hepatotoxicity? Hepatology 2006;43:1191‐1194. [DOI] [PubMed] [Google Scholar]
- 34. Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 2006;43:1220‐1230. [DOI] [PubMed] [Google Scholar]
- 35. Ishida Y, Kondo T, Kimura A, Tsuneyama K, Takayasu T, Mukaida N. Opposite roles of neutrophils and macrophages in the pathogenesis of acetaminophen‐induced acute liver injury. Eur J Immunol 2006;36:1028‐1038. [DOI] [PubMed] [Google Scholar]
- 36. Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol 2005;6:1182‐1190. [DOI] [PubMed] [Google Scholar]
- 37. Mendez O, Zavadil J, Esencay M, Lukyanov Y, Santovasi D, Wang SC, et al. Knock down of HIF‐1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol Cancer 2010;9:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, et al. HIF‐1alpha‐PDK1 axis‐induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun 2016;7:11635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med 2015;212:1345‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF‐1 inhibits mitochondrial biogenesis and cellular respiration in VHL‐deficient renal cell carcinoma by repression of C‐MYC activity. Cancer Cell 2007;11:407‐420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1175/full.
Supporting Information 1