

Abstract
The mechanisms by which the liver fails in end‐stage liver disease remain elusive. Disruption of the transcription factor network in hepatocytes has been suggested to mediate terminal liver failure in animals. However, this hypothesis remains unexplored in human subjects. To study the relevance of transcription factor expression in terminal stages of chronic liver failure in humans, we analyzed the expression of liver‐enriched transcription factors (LETFs) hepatocyte nuclear factor (HNF)4α, HNF1α, forkhead box protein A2 (FOXA2), CCAAT/enhancer‐binding protein (CEBP)α, and CEBPβ. We then selected downstream genes responsible for some hepatic functions (ornithine transcarbamylase [OTC], cytochrome P450 3A4 [CYP3A4], coagulation factor VII [F7], cadherin 1 [CDH1], phospho‐ezrin (Thr567)/radixin (Thr564)/moesin (Thr558) [p‐ERM], phospho‐myosin light chain [p‐MLC], low‐density lipoprotein receptor‐related protein 1 [LRP1]) in liver tissue from patients at different stages of decompensated liver function based upon Child‐Pugh classification, Model for End‐Stage Liver Disease score, and degree of inflammatory activity/fibrosis. We first examined differential expression of LETF and determined whether a relationship exists between transcript and protein expression, and liver function. We found HNF4α expression was down‐regulated and correlated well with the extent of liver dysfunction (P = 0.001), stage of fibrosis (P = 0.0005), and serum levels of total bilirubin (P = 0.009; r = 0.35), albumin (P < 0.001; r = 0.52), and prothrombin time activity (P = 0.002; r = 0.41). HNF4α expression also correlated with CYP3A4, OTC, and F7 as well as CDH1 RNA levels. The Rho/Rho‐associated protein kinase pathways, which have been implicated in the regulation of HNF4α, were also differentially expressed, in concert with LRP1, a reported upstream regulator of RhoA function. Conclusion: HNF4α and other members of the LETFs appear to be important regulators of hepatocyte function in patients with chronic hepatic failure. (Hepatology Communications 2018;2:582‐594)
Abbreviations
- CEBP
CCAAT/enhancer‐binding protein
- Ct
comparative threshold
- CYP3A4
cytochrome P450 3A4
- F7
coagulation factor VII
- FOXA2
forkhead box protein A2
- HNF
hepatocyte nuclear factor
- LETF
liver‐enriched transcription factor
- LRP1
low‐density lipoprotein receptor‐related protein 1
- MELD
Model for End‐Stage Liver Disease
- mRNA
messenger RNA
- NASH
nonalcoholic steatohepatitis
- OTC
ornithine transcarbamylase
- p‐ERM
phospho‐ezrin (Thr567)/radixin (Thr564)/moesin (Thr558)
- ROCK
Rho/Rho‐associated protein kinase
Cirrhosis of the liver is characterized by diffuse fibrosis, disruption of the normal lobular architecture of the liver with formation of regenerative nodules, and severe disruption of the vascular organization of the liver that can also result in portal hypertension.1 These profound structural and vascular changes can be accompanied by hepatocellular failure and the inability of hepatocytes to perform their normal synthetic and metabolic functions.2, 3, 4, 5 The causes of cirrhosis include hepatitis B virus infection, hepatitis C virus infection, alcohol‐mediated Laennec's cirrhosis, and nonalcoholic steatohepatitis (NASH)/metabolic syndrome, among others. When advanced liver cirrhosis progresses to terminal liver failure, the only definitive therapy is orthotopic liver transplantation.6 The worldwide prevalence of liver cirrhosis is roughly 25 to 400 per 100,000 subjects.1 In 2015, liver disease represented the eleventh leading cause of death, with an estimated 15.8 deaths per 100,000 globally. From 2000 to 2015, mortality increased nearly 30%,7 with the majority of cases occurring during the most productive years of life, between 25 and 64 years of age.7, 8
Although the etiologic agents and events that lead to cirrhosis may be known, the mechanisms responsible for deterioration of hepatocyte function and ultimately hepatic failure are largely unknown. Chronic injury to the liver is characterized by a decrease in hepatocyte mass, ongoing oxidative stress,9, 10 impaired mitochondrial function,11, 12 and limited regenerative capacity.13, 14 We have previously investigated the abnormal microenvironment associated with cirrhosis and the hepatocytes that reside within the cirrhotic liver in an experimental animal model of irreversible cirrhosis and fatal chronic liver failure that greatly resembles human disease.13, 14, 15, 16 We found that hepatocytes early in the development of cirrhosis suffer an adaptive metabolic and energy shift that involves a conversion from using oxidative phosphorylation to glycolysis and that dysfunctional hepatocytes during the terminal stages of chronic liver disease are unable to sustain the needed high levels of energy produced from glycolysis.15 Interestingly, we found that liver‐enriched transcription factors (LETFs) are stably down‐regulated in hepatocytes from animals with end‐stage cirrhosis and terminal hepatic failure. We showed that forced re‐expression of hepatocyte nuclear factor 4 alpha (HNF4α) can reprogram dysfunctional hepatocytes from terminally cirrhotic livers to function again, both in culture and in vivo, through expansion of new hepatocytes or stem cells and without the need for regeneration.16
While available animal models have been extremely useful for elucidating many aspects of hepatocyte dysfunction within the cirrhotic microenvironment, the relative expression of LETF pathways and downstream genes have yet to be examined in human degenerative liver disease and correlated with clinical hepatic function. Here, we examine expression of these LETF genes from explanted human liver specimens and correlate the results with clinical parameters of liver function and stage of liver decompensation. We demonstrate that the transcription factor HNF4α is significantly down‐regulated in association with the extent of hepatic dysfunction, in concert with select related genes. We thus corroborate previous findings from an animal model of irreversible cirrhosis with terminal liver failure.
Materials and Methods
HUMAN TISSUE SAMPLES
Between January 2009 and March 2013, 85 liver samples were obtained from patients who underwent partial hepatectomy or liver transplantation in the Department of Surgery and Science, Kyushu University Hospital. Diseases included liver metastasis, primary hepatocellular carcinoma, and decompensated cirrhosis. The patients with liver metastasis that did not undergo preoperative chemotherapy were classified as the normal liver group control. Specimens were analyzed histologically and for gene expression. Patient demographics and clinical status are shown in Table 1. The study was conducted with the approval of the institutional ethics review boards of Kyushu University (27‐245) and the University of Pittsburgh (PRO13010075). The work undertaken conforms to the provisions of the Declaration of Helsinki.
Table 1.
Clinical Parameters of the Patients in This Study
| Factors |
All patients (n = 84) |
Normal Liver (n = 20) |
Child‐Pugh A (n = 25) |
Child‐Pugh B (n = 9) |
Child‐Pugh C (n = 30) |
|---|---|---|---|---|---|
| Age, years | 61 ± 11 | 59 ± 2 | 67 ± 12 | 63 ± 4 | 57 ± 7 |
| Sex, male, N (%) | 47 (56) | 13 (65) | 17 (68) | 4 (44) | 13 (43) |
| Etiology, N (%) | |||||
| Hepatitis B virus | 11 (13) | 0 (0.0) | 4 (16) | 3 (33) | 4 (13) |
| Hepatitis C virus | 36 (43) | 0 (0.0) | 15 (60) | 6 (67) | 15 (50) |
| Alcohol | 7 (8.3) | 0 (0.0) | 3 (12) | 0 (0.0) | 4 (13) |
| NASH | 7 (8.3) | 0 (0.0) | 2 (8.0) | 0 (0.0) | 5 (17) |
| Cryptogenic | 3 (3.6) | 0 (0.0) | 1 (4.0) | 0 (0.0) | 2 (6.7) |
| Metastasis | 20 (24) | 20 (100) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Hepatocellular carcinoma positive (%) | 40 (48) | 0 (0.0) | 24 (96) | 7 (78) | 9 (30) |
| Histologic cirrhosis, N (%) | 39 (46) | 0 (0.0) | 0 (0.0) | 9 (100) | 30 (100) |
| Total bilirubin (mg/dL) | 2.1 ± 2.6 | 0.74 ± 0.46 | 0.68 ± 0.26 | 2.4 ± 1.2 | 4.1 ± 3.3 |
| Albumin (g/dL) | 3.5 ± 0.8 | 4.3 ± 0.1 | 3.9 ± 0.5 | 3.1 ± 0.4 | 2.7 ± 0.4 |
| Prothrombin time activity (%) | 75 ± 25 | 102 ± 2.7 | 88 ± 14 | 68 ± 12 | 49 ± 9 |
| Ascites, N (%) | 33 (39) | 0 (0.0) | 0 (0.0) | 6 (67) | 27 (90) |
| MELD score | 11 ± 6 | 6.0 ± 0.5 | 6.2 ± 0.4 | 12 ± 2 | 17 ± 4 |
HISTOLOGIC EXAMINATION
All specimens were cut into serial slices of 5‐10 mm thickness and fixed in 10% formalin. After macroscopic examination, noncancerous tissue was trimmed, embedded in a paraffin block, and cut into 4‐μm microscopic sections. Sections were stained with hematoxylin and eosin, and pathologic findings were assessed according to the Liver Cancer Study Group of Japan.7, 8 Necroinflammatory activity and the diagnosis of liver cirrhosis were performed according to the New Inuyama Classification17 and the Ishak scoring system for histologic fibrosis staging.18, 19
QUANTITATIVE REAL‐TIME POLYMERASE CHAIN REACTION
Total RNA was extracted from noncancerous liver tissue using RNeasy Mini kits (QIAGEN, Hilden, Germany) and reverse transcribed using SuperScriptIII (Invitrogen, Carlsbad, CA) following the manufacturers' instructions. We performed quantitative polymerase chain reaction with a StepOnePlus system (Applied Biosystems, Foster City, CA) using TaqMan Fast Advanced Master Mix (Life Technologies, Waltham, MA). The probes used (all from Applied Biosystems) were HNF4α (Hs00604431_m1), forkhead box protein A2 (FOXA2) (Hs00232764_m1), CCAAT/enhancer binding protein alpha (CEBPα) (Hs00269972_s1), CEBPβ (Hs00270923_s1), HNF1α (Hs00167041_m1), ornithine transcarbamylase (OTC) (Hs00166892_m1), cytochrome P450 3A4 (CYP3A4) (Hs00604506_m1), coagulation factor VII (F7) (Hs01551992; _m1), and β‐actin (Hs01060665_g1). Each sample was examined in duplicate. Gene expression levels were analyzed according to the comparative threshold cycle (Ct) method, where the amount of target was normalized to β‐actin and relative to a control sample. As a relative control expression reference, we used normal human liver tissue from a liver resection performed on a patient (18 years old) without liver dysfunction, who underwent hepatectomy for a benign liver tumor. The sample reference control value was given by 2‐ΔΔCt. Ct indicates the polymerase chain reaction cycle number at which the amount of amplified target reaches a fixed threshold. The ΔCt value is determined by subtracting the average reference Ct value from the average target Ct value. The ΔΔCt value involves subtraction by the ΔCt experimental control value.
IMMUNOHISTOCHEMISTRY
Paraffin‐embedded liver tissue was deparaffinized with xylenes and dehydrated with ethanol. Antigen unmasking was performed by boiling in citrate buffer, pH 6.0. The slides were then incubated in 3% hydrogen peroxide, blocked with normal animal serum, and subsequently left incubating overnight at 4°C with primary antibodies; these included mouse anti‐HNF4α (41898; Abcam, Cambridge, MA), rabbit anti‐FOXA2 (108422; Abcam), rabbit anti‐low‐density lipoprotein receptor‐related protein 1 (anti‐LRP1) (92544; Abcam), rabbit anti‐phospho‐ezrin (Thr567)/radixin (Thr564)/moesin (Thr558) (p‐ERM) (3726; Cell Signaling, Beverly, MA), and rabbit anti‐myosin light chain (phosphor S20) (2480; Abcam). Tissue sections were then incubated with the secondary biotinylated antibody corresponding to the animal species of the primary antibody (BA‐1000; Vector Laboratories, Burlingame, CA) and exposed to 3,3'‐diaminobenzidine (SK‐4105; Vector Laboratories) to visualize the peroxidase activity. Counterstaining was performed with Richard‐Allan Scientific Signature Series Hematoxylin (Thermo Scientific, Waltham, MA). Control tissues were used for validation of antibodies used in this study (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1172/full). For quantification, immunoreactivities of HNF4α and FOXA2 were independently graded by two liver pathologists, with 1,000 hepatocytes in three high‐power fields being counted per sample. Normal livers (n = 5), Child‐Pugh A (n = 5), Child‐Pugh B (n = 5), and Child‐Pugh C (n = 13) were included for these analyses.
STATISTICAL ANALYSIS
The statistical software JMP 10J (SAS Institute, Cary, NC) was used for all analyses. Data are expressed as mean ± SEM. Continuous variables were compared using the Wilcoxon test and one‐way analysis of variance between two groups and among four groups, respectively. Additionally, the messenger RNA (mRNA) expression values in Fig. 1 were compared individually against control values using the Student t test. Regression analyses were performed to detect correlations between gene expression and clinical parameters and between relative expression of HNF4α and the other genes expressed. Values of P < 0.05 were considered statistically significant.
Figure 1.
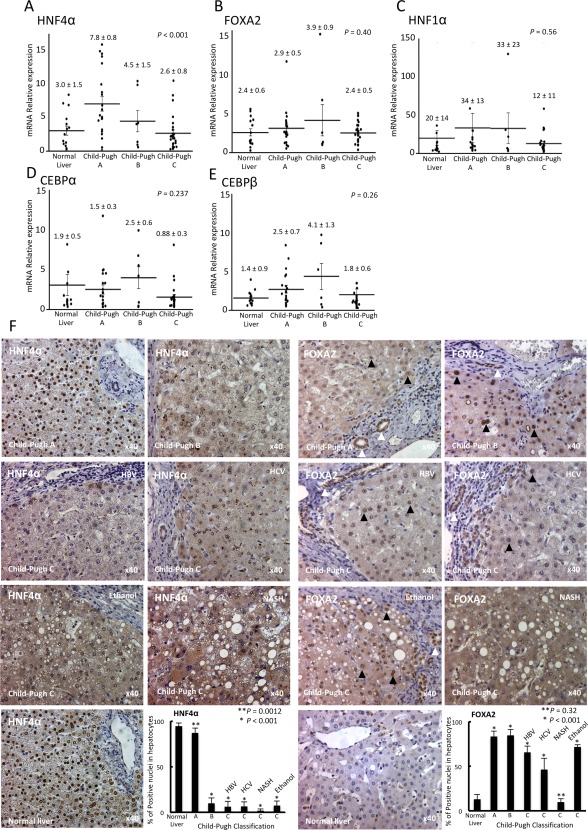
Expression levels of liver‐enriched transcription factors, grouped by the Child‐Pugh classification. Gene expression levels were analyzed according to the Ct method with the amount of target normalized to β‐actin and relative to a control sample. The relative expression of (A) HNF4α, (B) FOXA2, (C) HNF1α, (D) CEBPα, and (E) CEBPβ were analyzed using real‐time reverse‐transcription polymerase chain reaction. The expression values were compared using one‐way analysis of variance among four groups. To compare the mRNA expression values from each experimental group against controls, the Student t test was used. (F) HNF4α and FOXA2 expression in liver samples (normal livers [n = 5], Child‐Pugh A [n = 5], Child‐Pugh B [n = 5], and Child‐Pugh C [n = 13]) were analyzed by immunohistochemistry, and immunoreactivity was quantified in hepatocyte nuclei (magnification ×40). Abbreviations: HBV, hepatitis B virus; HCV, hepatitis C virus. Data are expressed as mean ± SEM.
Results
CLINICAL LIVER FUNCTION AND LETF EXPRESSION
Previous observations in animals with terminal chronic hepatic failure indicated that disruption of the transcription factor network and cellular dedifferentiation likely mediate hepatocyte decompensation. This observation led us to assess the relationship between LETF expression (HNF4α, FOXA2, HNF1α, CEBPα, and CEBPβ) and clinical evidence of liver dysfunction in human cirrhotic livers where gene expression was correlated with the Child‐Pugh score. There was a significant difference in expression of HNF4α among specimens from patients with normal liver and those with Child‐Pugh A, Child‐Pugh B, and Child‐Pugh C liver disease (3.0 ± 1.5 versus 7.8 ± 0.8 versus 4.5 ± 1.5 versus 2.6 ± 0.8; P < 0.001) using one‐way analysis of variance to determine differences among groups (Fig. 1A). Additionally, we performed individual comparisons of each group versus the normal liver control group. This analysis revealed that only mRNA expression of HNF4α in the Child‐Pugh A group was significantly higher than that in the normal liver group (7.8 ± 0.8 versus 3.0 ± 1.5; P = 0.008). However, the mRNA expression of HNF4A in the Child‐Pugh B or Child‐Pugh C group did not differ from that in normal liver (4.5 ± 1.5 or 2.6 ± 0.8 versus 3.0 ± 0.9; P = 0.32 or 0.65, respectively) (Fig. 1A). Moreover, HNF4α expression correlated significantly with serum total bilirubin level (r = –0.35, P = 0.009), serum albumin levels (r = 0.52, P < 0.001), and prothrombin time activity (r = 0.41, P = 0.002) among the patients analyzed (Fig. 2). In fact, HNF4α mRNA values correlated inversely and significantly with the Model for End‐Stage Liver Disease (MELD) score of disease severity (r = 0.42, P = 0.002). However, gene expression levels of FOXA2, HNF1α, CEBPα, and CEBPβ showed no significant difference in the livers among patients with Child‐Pugh A, B, and C scores (FOXA2, 2.4 ± 0.6 versus 2.9 ± 0.5 versus 3.9 ± 0.9 versus 2.4 ± 0.5, P = 0.40; HNF1α, 20 ± 14 versus 34 ± 13 versus 33 ± 23 versus 12 ± 11, P = 0.56; CEBPα, 1.9 ± 0.5 versus 1.5 ± 0.3 versus 2.5 ± 0.6 versus 0.88 ± 0.3, P = 0.237; CEBPβ, 1.4 ± 0.9 versus 2.5 ± 07 versus 4.1 ± 1.3 versus 1.8 ± 0.6, P = 0.26) (Fig. 3B‐E). In addition, FOXA2, HNF1α, CEBPα, and CEBPβ had no significant correlation with serum total bilirubin level, serum albumin levels, prothrombin time activity, or MELD score (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1172/full). These facts indicate that only loss of HNF4α expression is associated with the progression to terminal chronic hepatic failure in humans.
Figure 2.
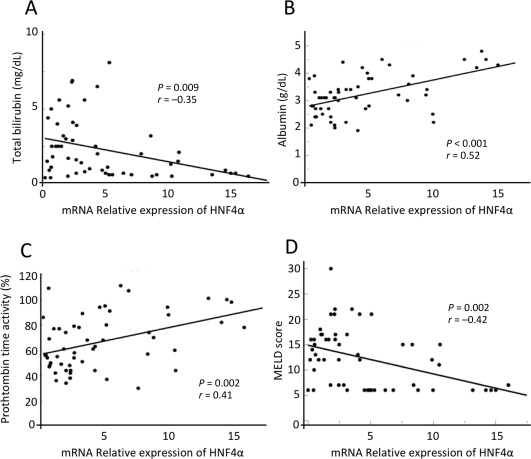
Regression analyses between HNF4α expression and clinical parameters. Gene expression levels in Child‐Pugh A, B, and C patients' samples were analyzed according to the Ct method with the amount of target normalized to β‐actin and relative to a control sample. Scatterplots are shown depicting the correlation between the expression of (A) HNF4α and total bilirubin level, (B) serum albumin, (C) prothrombin time activity, and (D) MELD score.
Figure 3.
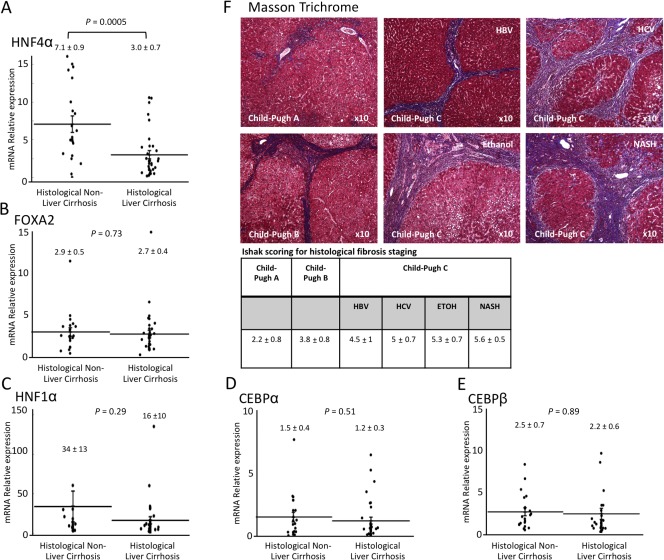
Comparison of liver‐enriched transcription factors and histologic cirrhosis. Livers were stained with Masson Trichrome and evaluated for fibrosis using the New Inuyama and Ishak classification. Data were analyzed according to the Ct method with the amount of target normalized to β‐actin and relative to a control sample. The relative expression of (A) HNF4α, (B) FOXA2, (C) HNF1α, (D) CEBPα, and (E) CEBPβ was compared between histologic liver cirrhosis and non‐liver cirrhosis. (F) Liver fibrosis in each Child‐Pugh classification (A, B, and C) and etiology was evaluated using Masson Trichrome staining and scored by Ishak scoring for histologic fibrosis (magnification ×10). Abbreviations: ETOH, ethanol; HBV, hepatitis B virus; HCV, hepatitis C virus.
HNF4α and FOXA2 expressions were then examined by immunohistochemistry. HNF4α immunoreactivity was observed in almost all nuclei from normal (n = 5) and Child‐Pugh A liver samples (n = 5), as expected. However, HNF4α was not observed in hepatocytes from Child‐Pugh B (n = 5) and C liver samples (n = 13) (Fig. 1F) independent of etiology (hepatitis B virus, hepatitis C virus, alcohol‐mediated Laennec's cirrhosis, and NASH). FOXA2, a LETF normally related to biliary epithelial cell specification in human adult normal livers but observed in hepatocytes of diseased human livers,20 was strongly expressed in duct structures from normal liver samples as expected, and Child‐Pugh A and B liver samples showed FOXA2 was confined mainly to duct‐like structures. However, weak expression was also observed in the majority of hepatocytes, and there was strong expression in a minority of hepatocytes. In Child‐Pugh C liver samples, FOXA2 was more variable and weak in hepatocytes; notably, NASH–Child‐Pugh C liver samples showed weak expression in a minority of hepatocytes (Fig. 1F). The findings related to specific transcription factor expression relate to nuclear localization (the nucleus is the site in which transcription factors exert their effects on gene expression); however, weak cytoplasmic staining was often observed, especially in patient samples with Child‐Pugh B and C.
HISTOLOGIC MEASURES OF LIVER CIRRHOSIS AND LETF EXPRESSION
We next evaluated whether the histologic findings and extent of fibrosis correlated with LETF expression. To accomplish this, we first evaluated the extent of fibrosis using Masson trichrome staining (Fig. 3F) and the New Inuyama classification.17 Cases were divided into two groups: those with histologic liver cirrhosis (n = 39) and those without liver cirrhosis (n = 25). We found that the level of HNF4α gene expression directly correlated with liver cirrhosis (P = 0.0005) (Fig. 3A). However, there was no significant difference in HNF1α, CEBPα, CEBPβ, or FOXA2 expression in cirrhotic and noncirrhotic liver specimens (FOXA2, 2.7 ± 0.4 versus 2.9 ± 0.5, P = 0.73; HNF1α, 16 ± 10 versus 34 ± 13, P = 0.29; CEBPα, 1.2 ± 0.3 versus 1.5 ± 0.4, P = 0.051; CEBPβ, 2.2 ± 0.6 versus 2.5 ± 0.7, P = 0.89) (Fig. 3A‐E). Additionally, patient liver specimens were also quantitatively scored using the Ishak scoring system for fibrosis and correlated with the Child‐Pugh score. An inverse relationship between the amount of fibrosis and hepatic function (Fig. 3F) was observed, indicating that the amount of liver fibrosis directly correlated with hepatocyte function and HNF4α gene expression.
REGULATION OF HNF4α AND DOWNSTREAM EFFECTORS OF LIVER FUNCTION
To determine the extent to which HNF4α serves as a master regulator of liver function, HNF4α expression was correlated with the expression of three genes that are important indicators of hepatic function and known to be regulated by HNF4α: OTC, CYP3A4, and F7 (Fig. 4A‐D). The expression of HNF4α correlated with the expression of OTC (r = 0.37, P = 0.0064), CYP3A4 (r = 0.28, P = 0.040), and F7 (r = 0.29, P = 0.036). However, the expression of other LETFs did not correlate significantly with these hepatocyte‐specific genes (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1172/full). Indeed, these three hepatocyte‐specific genes were severely down‐regulated in association with low expression of HNF4α, indicating that HNF4α definitely affects the expression of many liver‐specific target genes involved in protein secretion, amino acid production, and xenobiotic and drug metabolism. However, the molecular changes that promote the loss of HNF4α in cirrhotic livers in chronic hepatic failure remain undefined. Thus, we evaluated the role of cell–cell and cell–matrix mechanotransduction cascade‐related pathways in this process. As fibrosis is a dominant feature in the cirrhotic liver and the scarring causes cell membrane changes, we evaluated the expression of cadherin 1 (CDH1), a gene that encodes the cell–cell adhesion protein E‐cadherin. Linear regression analysis showed that E‐cadherin significantly correlated with HNF4α expression (r = 0.41, P = 0.0024) (Fig. 4D). We further examined the activity of Rho/Rho‐associated protein kinase (ROCK), a known mechanotransducer, and its downstream target p‐ERM (Fig. 5A), using immunohistochemistry, and found no activity in hepatocytes from the Child‐Pugh A and Child‐Pugh B specimens. However, p‐ERM immunoreactivity was present in the cytoplasm of hepatocytes from Child‐Pugh C specimens. Moreover, we confirmed ROCK activation by examining the expression of phospho‐myosin light chain and found that immunoreactivity was present in the cytoplasm of hepatocytes from Child‐Pugh B and Child‐Pugh C specimens (Fig. 5B). Finally, as the loss of LRP1, a marker of mature hepatocytes that regulates surface urokinase receptor21 and integrins,22 also reportedly leads to increased RhoA/ROCK function,23 we examined the staining pattern of LRP1 in relation to Child‐Pugh status. LRP1 staining inversely correlated with that of p‐ERM as would be expected if LRP1 regulated RhoA/ROCK (Fig. 5F).
Figure 4.
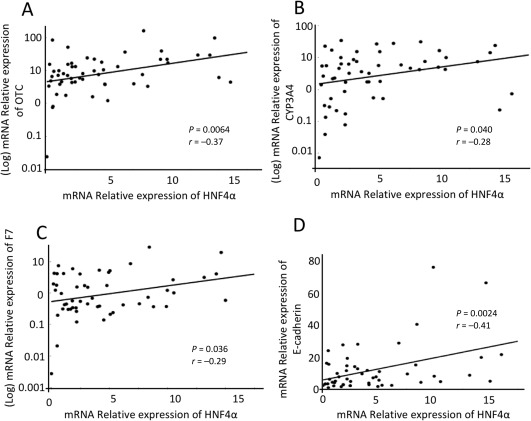
HNF4α expression and liver function genes. Gene expression levels in Child‐Pugh A, B, and C patients' samples were analyzed according to the Ct method with the amount of target normalized to β‐actin and relative to a control sample. Scatterplots are shown depicting the correlation between the expression of (A) HNF4α and OTC, (B) CYP3A4, (C) F7, and (D) E‐cadherin.
Figure 5.
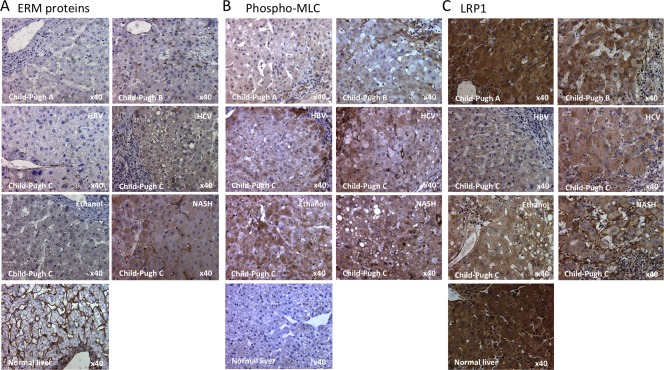
Immunohistochemistry of mechanosignaling molecules in human livers grouped by the Child‐Pugh classification. The expressions of (A) p‐ERM, (B) phospho‐MLT, and (C) LRP1 were analyzed by immunohistochemistry (magnification ×40). Abbreviations: HBV, hepatitis B virus; HCV, hepatitis C virus.
Discussion
Hepatic transcription factors are key regulators of normal liver function.24 Hepatocyte‐specific gene transcription is stimulated by the concerted action of HNF 1, CEBPα, CEBPβ (formerly called HNF2), FOXA (previously known as HNF3), HNF4, and HNF6.25 Current literature indicates that HNF4α is the master hepatic regulator and controls numerous functions that include maintenance of the hepatic epithelium and morphogenesis,26, 27 glucose and fatty acid metabolism, synthesis of blood coagulation factors, detoxification (CYPP450 activity),15, 28 preservation of hepatocyte polarity, and maintenance of differentiation.29, 30 HNF4α even controls the expression of other liver‐enriched factors, such as HNF1α,24 and can reestablish liver function and differentiated hepatocyte‐specific characteristics when reintroduced in models of liver cirrhosis and hepatocellular carcinoma.15, 16, 24, 28, 31
We now show levels of HNF4α, HNF1α, CEBPα, CEBPβ, and FOXA2 expression in human livers with and without cirrhosis and with varying degrees of liver failure as defined by the Child‐Pugh score. We previously demonstrated that levels of HNF4α decreased in end‐stage hepatocytes from animals with cirrhosis and terminal hepatic failure14 and showed that forced re‐expression of HNF4α can immediately convert senescent and irreversibly dysfunctional hepatocytes derived from terminal livers to reestablish normal function.16 In the present study, we show that patients with cirrhosis of the liver express significantly lower levels of HNF4α in their livers than those without cirrhosis.
In addition, we showed significant mRNA down‐regulation of HNF4α in Child‐Pugh C decompensated livers compared to mRNA expression in Child‐Pugh A compensated livers. Importantly, HNF4α mRNA expression in Child‐Pugh C decompensated livers was not significantly different when compared to normal liver samples. However, HNF4α localization to the nucleus was significantly reduced in Child‐Pugh B and Child‐Pugh C livers. These findings might indicate an essential role for HNF4α in preserving normal liver function. Its expression is significantly up‐regulated at the onset of injury (Child‐Pugh A) to potentially maintain liver homeostasis; however, as liver disease progresses, HNF4α protein expression is diminished in the nuclei of hepatocytes and/or rendered ineffective, leading to impaired regulation and hepatic function. Although several LETFs were down‐regulated in severe liver disease (Child‐Pugh C), only HNF4α expression directly correlated with liver function and disease progression. For instance, protein expression of FOXA2 in biliary endothelial cells remained consistent in all liver samples examined in this study. However, in liver samples from patients with Child‐Pugh A, B, and C, a substantial number of hepatocytes became positive for FOXA2, supporting that reprogramming of biliary endothelial cells to hepatocytes or vice versa is also operative in human liver.20, 32
Our findings indicate that failure of the hepatic transcription system appears to be a pathologic finding in liver disease progression. These findings suggest that any correlation between chronic liver failure in cirrhosis in humans and HNF4α‐dependent hepatic functions is not regulated at the level of HNF4α transcription but rather at the level of protein expression and/or nuclear localization.
To confirm the role of LETFs in liver disease progression, we examined the possible correlation of HNF4α and other LETFs with clinical measures of hepatic function. We chose serum albumin, total bilirubin, and prothrombin time activity as indicators of overall hepatic function, although this only partially covers the many functions of the liver. These clinical markers, however, affect bile metabolism and indicate the capacity of the hepatocyte to synthesize proteins.33 Significant correlations were found between the levels of serum albumin and total bilirubin with expression of HNF4α.
Regulation of serum albumin has previously been noted to be associated with HNF4α as well as with the expression of HNF1α and the FOXA family of transcription factors.25 Biliary metabolism has also been linked to HNF4α expression as some enzymes involved in bile acid synthesis, such as 7α‐hydroxylase and 12α‐hydroxylase, have promoters that are bound directly by HNF4α34, 35 and the expression of bile acid transporter proteins, such as organic anion transporting polypeptide‐C and Na+‐taurocholate cotransporting polypeptide, is indirectly regulated by HNF4α‐mediated transactivation of HNF1α.36 Unfortunately, regulation of the LETFs is complex and has not been completely delineated in humans, although LETF regulatory networks that control the expression of liver function genes have been published based on microarray data.14
In our study, we evaluated hepatic function directly through expression of OTC, CYP3A4, and F7, which are three genes that loosely reflect metabolic, detoxification, and synthetic capacities of the liver. Interestingly, all three are downstream targets of HNF4α and their expression correlated strongly with HNF4α expression, although statistical significance was obtained only with OTC and CYP3A4. These findings emphasize the role of HNF4α in maintaining essential liver functions and, as described previously regarding its down‐regulation in decompensated liver disease, may explain the mechanism responsible for progression of cirrhosis to organ failure.
While the mechanism by which HNF4α and other LETFs are down‐regulated in cirrhosis has not yet been delineated, there are several possible candidate factors and pathways. For instance, there is evidence that nuclear localization of HNF4α can be regulated at the posttranslational level by phosphorylation37, 38 and acetylation,39 while SUMOylation can regulate HNF4α protein degradation and stability.40 At the transcriptional level, Desai et al.30 recently showed that hepatic HNF4α expression and its target genes are regulated by extracellular matrix rigidity; they found a threshold level of stiffness beyond which expression of HNF4α and liver function genes are lost. The extracellular matrix signals through a mechanotransduction cascade in which cells sense the stiffness of their environment through integrin clustering, activation of focal adhesion kinase, and further activation of intracellular signaling pathways involving the ROCK pathway.41, 42, 43 Activation of the ROCK pathway by mechanical cues leads to phosphorylation and activation of effector proteins, such as the ERM proteins, which induces actin stabilization, smooth muscle contraction, and transcriptional regulation.41 Our study shows a significant increase in the phosphorylated ERM proteins, indicative of ROCK activation, coincident with loss of E‐cadherin, HNF4α, and other LETFs in more severe liver failure. Although it is not clear how Rho/ROCK signaling affects gene expression, it has been found to interact with YAP/TAZ transcriptional regulators.44 Importantly, it was previously shown that loss of LRP1, a protein essential for normal hepatic development,45 a regulator of lipid metabolism in mature livers,46 and a known regulator of other mechanotransducers,21, 22 also activates the Rho/ROCK pathway in Schwann Cells.23 In this paper, we similarly show an inverse correlation between LRP1 levels and Rho/ROCK activation in hepatocytes. Interestingly, while little is known regarding the transcriptional regulation of LRP1, the https://doi.org/10.1093/database/baw100 indicates that LRP1 is a potential downstream transcriptional target of HNF4α. This suggests there may be a biological loop involving HNF4α, LRP1 expression, and suppression of Rho/ROCK that is disrupted in high‐grade liver failure.
E‐cadherin is a cell–cell junction protein that has many signaling partners in common with the integrin–focal adhesion kinase signaling pathway; it is involved in mechanical transduction and is greatly affected by fibrosis. Molecules involved in the genesis of liver fibrosis, such as transforming growth factor beta, are also implicated in the loss of E‐cadherin, and knockout of CDH1, the gene encoding E‐cadherin, has been shown to induce an inflammatory response and periductal fibrosis.47, 48 Furthermore, fibrocystin‐deficiency diseases, such as congenital hepatic fibrosis, down‐regulate E‐cadherin expression.49 Interestingly, E‐cadherin is a direct transcriptional target of HNF4α and loss of HNF4α results in loss of E‐cadherin expression.26, 27, 50 While there is no known link between mechanical transduction signaling and regulation of LETFs in fibrosis, two intracellular kinases involved in integrin‐ and E‐cadherin‐mediated signaling, e.g., proto‐oncogene tyrosine‐protein kinase Src (c‐Src) and protein kinase C, can phosphorylate HNF4α and other nuclear receptors, and their activation can lead to a decrease in stability and a loss of nuclear localization and transactivation functions.37, 51
In summary, gene and protein expression analysis of liver samples from patients with cirrhosis and worsening liver function demonstrate that liver disease disrupts the normal expression and activity of LETFs, especially HNF4α. Clinical data and MELD score correlations point to the loss of hepatic transcriptome expression with liver decompensation and suggest a possible mechanism responsible for disease progression in cirrhosis and terminal liver failure. Further studies are needed to more fully characterize how each LETF might affect liver function and to delineate molecules that directly regulate their expression levels.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1172/full.
Supporting Information Figure 1A
Supporting Information Figure 1B
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Figure Caption
Potential conflict of interest: Dr. Soto‐Gutierrez., Dr. Bell, and Dr. Fox are inventors on a pending patent application that describes the use of transcription factors to treat chronic liver failure (US20140249209). Dr. Soto‐Gutierrez, Dr. Guzman‐Lepe, Dr. Takeishi, Dr. Collin de l'Hortet, Dr. Wang, and Dr. Fox are cofounders and have a financial interest in Von Baer Wolff, Inc., a company focused on biofabrication of autologous human hepatocytes from stem cell technology and programming liver failure; their interests are managed by the Conflict of Interest Office at the University of Pittsburgh in accordance with their policies. Dr. Fox, Dr. Soto‐Gutierrez, Dr. Takeishi, Dr. Guzman‐Lepe, Dr. Collin de l'Hortet, and Dr. Wang own stock in Von Baer Wolff. The other authors have nothing to report.
Supported by grants from the National Institutes of Health (DK099257 to A.S.‐G. and DK09932 to I.J.F.) and by the American Liver Foundation and the Uehara Memorial Foundation (to K.T.).
Contributor Information
Kazuki Takeishi, Email: ktake@surg2.med.kyushu-u.ac.jp.
Alejandro Soto‐Gutierrez, Email: als208@pitt.edu.
REFERENCES
- 1.Garcia‐Tsao G: Cirrhosis and its sequelae. In: Goldman L, Schafer AI, eds. Goldman‐Cecil Medicine. 25th ed. Philadelphia, PA: Elsevier; 2016:1023‐1031. [Google Scholar]
- 2. Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut 2015;64:830‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pessayre D, Lebrec D, Descatoire V, Peignoux M, Benhamou JP. Mechanism for reduced drug clearance in patients with cirrhosis. Gastroenterology 1978;74:566‐571. [PubMed] [Google Scholar]
- 4. Schuppan D, Afdhal NH. Liver cirrhosis. Lancet 2008;371:838‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hernaez R, Sola E, Moreau R, Gines P. Acute‐on‐chronic liver failure: an update. Gut 2017;66:541‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lopez PM, Martin P. Update on liver transplantation: indications, organ allocation, and long‐term care. Mt Sinai J Med 2006;73:1056‐1066. [PubMed] [Google Scholar]
- 7.[No authors listed]. The general rules for the clinical and pathological study of primary liver cancer. Liver Cancer Study Group of Japan . Jpn J Surg 1989;19:98‐129. [DOI] [PubMed] [Google Scholar]
- 8. Kudo M, Kitano M, Sakurai T, Nishida N. General rules for the clinical and pathological study of primary liver cancer, nationwide follow‐up survey and clinical practice guidelines: the outstanding achievements of the Liver Cancer Study Group of Japan. Dig Dis 2015;33:765‐770. [DOI] [PubMed] [Google Scholar]
- 9. Kitada T, Seki S, Iwai S, Yamada T, Sakaguchi H, Wakasa K. In situ detection of oxidative DNA damage, 8‐hydroxydeoxyguanosine, in chronic human liver disease. J Hepatol 2001;35:613‐618. [DOI] [PubMed] [Google Scholar]
- 10. Choi J, Ou JH. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am J Physiol Gastrointest Liver Physiol 2006;290:G847‐G851. [DOI] [PubMed] [Google Scholar]
- 11. Grattagliano I, Russmann S, Diogo C, Bonfrate L, Oliveira PJ, Wang DQ, et al. Mitochondria in chronic liver disease. Curr Drug Targets 2011;12:879‐893. [DOI] [PubMed] [Google Scholar]
- 12. Serviddio G, Bellanti F, Sastre J, Vendemiale G, Altomare E. Targeting mitochondria: a new promising approach for the treatment of liver diseases. Curr Med Chem 2010;17:2325‐2337. [DOI] [PubMed] [Google Scholar]
- 13. Kobayashi N, Ito M, Nakamura J, Cai J, Gao C, Hammel JM, et al. Hepatocyte transplantation in rats with decompensated cirrhosis. Hepatology 2000;31:851‐857. [DOI] [PubMed] [Google Scholar]
- 14. Liu L, Yannam GR, Nishikawa T, Yamamoto T, Basma H, Ito R, et al. The microenvironment in hepatocyte regeneration and function in rats with advanced cirrhosis. Hepatology 2012;55:1529‐1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nishikawa T, Bellance N, Damm A, Bing H, Zhu Z, Handa K, et al. A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J Hepatol 2014;60:1203‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nishikawa T, Bell A, Brooks JM, Setoyama K, Melis M, Han B, et al. Resetting the transcription factor network reverses terminal chronic hepatic failure. J Clin Invest 2015;125:1533‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fumihiro I, Takao T, Masao O, Takafumi I, Kyouichi I, Tomoteru K, et al. New Inuyama classification; new criteria for histological assessment of chronic hepatitis. Int Hepatol Commun 1996;6:112‐119. [Google Scholar]
- 18. Standish RA, Cholongitas E, Dhillon A, Burroughs AK, Dhillon AP. An appraisal of the histopathological assessment of liver fibrosis. Gut 2006;55:569‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, et al. Histological grading and staging of chronic hepatitis. J Hepatol 1995;22:696‐699. [DOI] [PubMed] [Google Scholar]
- 20. Limaye PB, Alarcon G, Walls AL, Nalesnik MA, Michalopoulos GK, Demetris AJ, et al. Expression of specific hepatocyte and cholangiocyte transcription factors in human liver disease and embryonic development. Lab Invest 2008;88:865‐872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonias SL, Gaultier A, Jo M. Regulation of the urokinase receptor (uPAR) by LDL receptor‐related protein‐1 (LRP1). Curr Pharm Des 2011;17:1962‐1969. [DOI] [PubMed] [Google Scholar]
- 22. Rabiej VK, Pflanzner T, Wagner T, Goetze K, Storck SE, Eble JA, et al. Low density lipoprotein receptor‐related protein 1 mediated endocytosis of beta1‐integrin influences cell adhesion and cell migration. Exp Cell Res 2016;340:102‐115. [DOI] [PubMed] [Google Scholar]
- 23. Mantuano E, Jo M, Gonias SL, Campana WM. Low density lipoprotein receptor‐related protein (LRP1) regulates Rac1 and RhoA reciprocally to control Schwann cell adhesion and migration. J Biol Chem 2010;285:14259‐14266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schrem H, Klempnauer J, Borlak J. Liver‐enriched transcription factors in liver function and development. Part I: the hepatocyte nuclear factor network and liver‐specific gene expression. Pharmacol Rev 2002;54:129‐158. [DOI] [PubMed] [Google Scholar]
- 25. Costa RH, Kalinichenko VV, Holterman AX, Wang X. Transcription factors in liver development, differentiation, and regeneration. Hepatology 2003;38:1331‐1347. [DOI] [PubMed] [Google Scholar]
- 26. Battle MA, Konopka G, Parviz F, Gaggl AL, Yang C, Sladek FM, et al. Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc Natl Acad Sci U S A 2006;103:8419‐8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet 2003;34:292‐296. [DOI] [PubMed] [Google Scholar]
- 28. Shih DQ, Dansky HM, Fleisher M, Assmann G, Fajans SS, Stoffel M. Genotype/phenotype relationships in HNF‐4alpha/MODY1: haploinsufficiency is associated with reduced apolipoprotein (AII), apolipoprotein (CIII), lipoprotein(a), and triglyceride levels. Diabetes 2000;49:832‐837. [DOI] [PubMed] [Google Scholar]
- 29. Santangelo L, Marchetti A, Cicchini C, Conigliaro A, Conti B, Mancone C, et al. The stable repression of mesenchymal program is required for hepatocyte identity: a novel role for hepatocyte nuclear factor 4alpha. Hepatology 2011;53:2063‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Desai SS, Tung JC, Zhou VX, Grenert JP, Malato Y, Rezvani M, et al. Physiological ranges of matrix rigidity modulate primary mouse hepatocyte function in part through hepatocyte nuclear factor 4 alpha. Hepatology 2016;64:261‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lazarevich NL, Cheremnova OA, Varga EV, Ovchinnikov DA, Kudrjavtseva EI, Morozova OV, et al. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology 2004;39:1038‐1047. [DOI] [PubMed] [Google Scholar]
- 32. Rogler CE, Bebawee R, Matarlo J, Locker J, Pattamanuch N, Gupta S, et al. Triple staining including FOXA2 identifies stem cell lineages undergoing hepatic and biliary differentiation in cirrhotic human liver. J Histochem Cytochem 2017;65:33‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwartz RE, Fleming HE, Khetani SR, Bhatia SN. Pluripotent stem cell‐derived hepatocyte‐like cells. Biotechnol Adv 2014;32:504‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stroup D, Chiang JY. HNF4 and COUP‐TFII interact to modulate transcription of the cholesterol 7alpha‐hydroxylase gene (CYP7A1). J Lipid Res 2000;41:1‐11. [PubMed] [Google Scholar]
- 35. del Castillo‐Olivares A, Gil G. Suppression of sterol 12alpha‐hydroxylase transcription by the short heterodimer partner: insights into the repression mechanism. Nucleic Acids Res 2001;29:4035‐4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jung D, Kullak‐Ublick GA. Hepatocyte nuclear factor 1 alpha: a key mediator of the effect of bile acids on gene expression. Hepatology 2003;37:622‐631. [DOI] [PubMed] [Google Scholar]
- 37. Sun K, Montana V, Chellappa K, Brelivet Y, Moras D, Maeda Y, et al. Phosphorylation of a conserved serine in the deoxyribonucleic acid binding domain of nuclear receptors alters intracellular localization. Mol Endocrinol 2007;21:1297‐1311. [DOI] [PubMed] [Google Scholar]
- 38. Yu D, Chen G, Pan M, Zhang J, He W, Liu Y, et al. High fat diet‐induced oxidative stress blocks hepatocyte nuclear factor 4alpha and leads to hepatic steatosis in mice. J Cell Physiol 2018;233:4770‐4782. [DOI] [PubMed] [Google Scholar]
- 39. Soutoglou E, Katrakili N, Talianidis I. Acetylation regulates transcription factor activity at multiple levels. Mol Cell 2000;5:745‐751. [DOI] [PubMed] [Google Scholar]
- 40. Zhou W, Hannoun Z, Jaffray E, Medine CN, Black JR, Greenhough S, et al. SUMOylation of HNF4alpha regulates protein stability and hepatocyte function. J Cell Sci 2012;125:3630‐3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lessey EC, Guilluy C, Burridge K. From mechanical force to RhoA activation. Biochemistry 2012;51:7420‐7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005;8:241‐254. [DOI] [PubMed] [Google Scholar]
- 43. Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density‐induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK‐ERK linkage. Oncogene 2009;28:4326‐4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature 2011;474:179‐183. [DOI] [PubMed] [Google Scholar]
- 45. Roebroek AJ, Reekmans S, Lauwers A, Feyaerts N, Smeijers L, Hartmann D. Mutant Lrp1 knock‐in mice generated by recombinase‐mediated cassette exchange reveal differential importance of the NPXY motifs in the intracellular domain of LRP1 for normal fetal development. Mol Cell Biol 2006;26:605‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Basford JE, Wancata L, Hofmann SM, Silva RA, Davidson WS, Howles PN, et al. Hepatic deficiency of low density lipoprotein receptor‐related protein‐1 reduces high density lipoprotein secretion and plasma levels in mice. J Biol Chem 2011;286:13079‐13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shrestha N, Chand L, Han MK, Lee SO, Kim CY, Jeong YJ. Glutamine inhibits CCl4 induced liver fibrosis in mice and TGF‐beta1 mediated epithelial‐mesenchymal transition in mouse hepatocytes. Food Chem Toxicol 2016;93:129‐137. [DOI] [PubMed] [Google Scholar]
- 48. Nakagawa H, Hikiba Y, Hirata Y, Font‐Burgada J, Sakamoto K, Hayakawa Y, et al. Loss of liver E‐cadherin induces sclerosing cholangitis and promotes carcinogenesis. Proc Natl Acad Sci U S A 2014;111:1090‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Spirli C, Locatelli L, Morell CM, Fiorotto R, Morton SD, Cadamuro M, et al. Protein kinase A‐dependent pSer(675) ‐beta‐catenin, a novel signaling defect in a mouse model of congenital hepatic fibrosis. Hepatology 2013;58:1713‐1723. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50. Spath GF, Weiss MC. Hepatocyte nuclear factor 4 provokes expression of epithelial marker genes, acting as a morphogen in dedifferentiated hepatoma cells. J Cell Biol 1998;140:935‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chellappa K, Jankova L, Schnabl JM, Pan S, Brelivet Y, Fung CL, et al. Src tyrosine kinase phosphorylation of nuclear receptor HNF4alpha correlates with isoform‐specific loss of HNF4alpha in human colon cancer. Proc Natl Acad Sci U S A 2012;109:2302‐2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1172/full.
Supporting Information Figure 1A
Supporting Information Figure 1B
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Figure Caption