

Abstract
The prevalence of hepatitis C virus (HCV) infection in Asian countries is high. This study assessed the efficacy and safety of elbasvir/grazoprevir (EBR/GZR) in participants with HCV infection from Asia‐Pacific countries and Russia. In this phase 3, randomized, placebo‐controlled, double‐blind study, treatment‐naive participants with HCV genotype (GT) 1, 4, or 6 infection were randomized to EBR 50 mg/GZR 100 mg (immediate‐treatment group [ITG]) or placebo (deferred‐treatment group [DTG]) once daily for 12 weeks (Protocol PN‐5172‐067, NCT02251990). The primary efficacy variable was a nonrandomized comparison of sustained virologic response at 12 weeks after the end of therapy (SVR12) for the ITG with a historical control. The primary safety outcome was a randomized comparison between the ITG and DTG. Three hundred thirty‐seven participants were randomized to the ITG (n = 251) or DTG (n = 86); 199 (59.2%) participants were Asian, and 250 (74.4%) had HCV GT1b infection. Overall, 232/250 (92.8%) participants in the ITG achieved SVR12 (97.5% confidence interval, 89.1, 96.5). Of the 18 participants who failed to attain SVR12, 1 was lost to follow‐up and 17 had virologic failure, 13 of whom had HCV GT6 infection. The incidence of adverse events was similar between participants receiving EBR/GZR and placebo (50.8% versus 51.2%; difference, −0.3%; 95% confidence interval, −12.3, 11.9). Conclusion: EBR/GZR for 12 weeks provides an effective and well‐tolerated regimen for chronic HCV GT1 infection in treatment‐naive people from Asia‐Pacific countries and Russia, particularly for the large population with GT1b infection. EBR/GZR is not recommended for the treatment of individuals with HCV GT6 infection. (Hepatology Communications 2018;2:595‐606)
Abbreviations
- AE
adverse event
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CI
confidence interval
- DTG
deferred‐treatment group
- EBR
elbasvir
- ECI
event of clinical interest
- GT
genotype
- GZR
grazoprevir
- HCV
hepatitis C virus
- IFN
interferon
- ITG
immediate‐treatment group
- LLoQ
lower limit of quantitation
- RAS
resistance‐associated substitution
- SAE
serious adverse event
- SVR12
sustained virologic response 12 weeks after completion of treatment
- ULN
upper limit of normal
The prevalence of hepatitis C virus (HCV) infection in Asian countries is high. China alone has a larger infected population than Europe and the Americas combined, with an estimated 13 million infected people.1 Prevalence estimates for Australia, South Korea, Taiwan, Thailand, and Vietnam range from 1.3% in Australia and South Korea to 4.4% in Taiwan. 1 In contrast to the Americas, where the HCV genotype (GT) 1a subtype predominates, the most prevalent genotype in the Asia‐Pacific region is GT1b.2, 3 In addition, GT6 is endemic in Southeast Asia and is highly prevalent in southern China, Vietnam, and Laos.3 GT1b is also the predominant subtype of HCV in Russia, where the prevalence of infection is 4.1%.2
All‐oral interferon (IFN)‐free treatment regimens consisting of direct‐acting antiviral agents are highly effective for the majority of people with chronic HCV infection. These treatments are generally well tolerated compared with older IFN‐based treatments and are well established as standard of care treatment throughout many Western countries.4, 5 However, in the Asia‐Pacific region, the high cost of these medications within an often resource‐constrained environment has limited the uptake of direct‐acting antiviral agent regimens, and consequently IFN‐containing regimens are still being used in many countries.6, 7
The combination of elbasvir (EBR), an NS5A inhibitor, and grazoprevir (GZR), an NS3/4a inhibitor, is approved in many Western countries and in Japan for treatment of HCV GT1 and 4 infection.8, 9, 10 This combination has broad and potent antiviral activity in vitro 11, 12, 13, 14 and has shown high efficacy in phase 2 and 3 studies when administered for 12 weeks across a wide spectrum of people, including those with cirrhosis, chronic kidney disease, or human immunodeficiency virus coinfection, and in previously treated people in whom IFN‐based therapy failed.15, 16, 17, 18, 19, 20, 21, 22, 23 Rates of sustained virologic response 12 weeks after completion of treatment (SVR12) in these predominantly Western populations generally exceeded 90%, and high rates of SVR12 have also been reported in Japanese individuals.24 The safety profile of EBR/GZR has been established in placebo‐controlled studies that included a deferred‐treatment group (DTG). In these studies, EBR/GZR was well tolerated and had a similar safety profile in participants with and without cirrhosis and when compared with placebo‐treated participants.17, 18, 23
The objective of the phase 3 C‐CORAL trial was to evaluate the efficacy and safety of a once‐daily, all‐oral, fixed‐dose combination of EBR/GZR for 12 weeks in treatment‐naive participants with or without cirrhosis and with HCV GT1, 4, or 6 infection from Asia‐Pacific countries and Russia. In Western countries, EBR/GZR is not currently indicated for the treatment of people with GT6 infection. Given the small number of people with GT6 infection who had been treated with EBR/GZR at the time the C‐CORAL protocol was written, the efficacy of EBR/GZR in this population was not well established. This publication describes the results of a preplanned interim analysis of SVR12 data from all participating countries except China. This interim analysis was prespecified in the study protocol for the purposes of supporting regulatory submission outside China.
Patients and Methods
STUDY DESIGN
C‐CORAL (Merck protocol PN‐5172‐067, https://clinicaltrials.gov/ct2/results?cond=-term=NCT02251990-cntry=-state=-city=-dist=) was a phase 3, randomized, international, parallel‐group, placebo‐controlled, double‐blind study conducted at 36 study centers in Australia (two), South Korea (six), Taiwan (seven), Thailand (three), Vietnam (three), and Russia (15). A second cohort of participants was enrolled later in 13 centers across mainland China, and study results from these participants will be reported separately. The study was conducted in accordance with the Declaration of Helsinki and good clinical practice guidelines. Independent institutional review boards or ethics committees reviewed and approved the protocol and applicable amendments for each institution, and all participants gave written informed consent.
Eligible participants were randomized 3:1 to one of two treatment arms. Participants in the immediate‐treatment group (ITG) received the fixed‐dose combination of EBR 50 mg/GZR 100 mg once daily for 12 weeks, followed by 24 weeks of follow‐up. Participants randomized to the DTG received placebo once daily for 12 weeks. Participants were unblinded after 12 weeks treatment plus 4 weeks follow‐up; participants in the DTG then received 12 weeks of open‐label treatment with EBR 50 mg/GZR 100 mg, with 24 weeks of follow‐up after dosing was completed. The blinded ITG/DTG grouping enabled safety assessment. Enrollment was managed to ensure that at least 20% of participants had compensated cirrhosis and approximately 15% had HCV GT4 or 6 infection.
STUDY PARTICIPANTS
Treatment‐naive adults (age >18 years) with chronic HCV GT1, 4, or 6 infection and baseline HCV RNA ≥10,000 IU/mL were enrolled. Race was self‐identified. Participants with cirrhosis or without cirrhosis were eligible, with cirrhosis defined as METAVIR F4 on liver biopsy within 24 months of enrollment, FibroScan >12.5 kPa within 12 months of enrollment, or a combination of FibroTest score >0.75 and aspartate aminotransferase (AST):platelet ratio index >2. Participants with evidence of decompensated liver disease, coinfection with hepatitis B virus (hepatitis B surface antigen positive) or human immunodeficiency virus, or a history of malignancy, clinically relevant drug or alcohol abuse within 12 months of screening, hepatocellular carcinoma, uncontrolled diabetes (hemoglobin A1c >10%), aminotransferase levels >10 times the upper limit of normal (ULN), albumin <3.0 g/dL, elevated prothrombin time unrelated to anticoagulation, creatinine clearance <50 mL/minute, hemoglobin level <9.5 g/dL, or platelet count <50 × 103 cells/μL were excluded.
RANDOMIZATION AND MASKING
Randomization was performed centrally using an interactive voice response system/integrated web response system and was stratified according to presence or absence of cirrhosis and GT subtype (GT1a versus non‐GT1a versus GT4/6). Participants, investigators, and sponsors were blinded to treatment assignment and HCV RNA results through week 16, after which time the assignments were unmasked and participants randomized to the DTG received active therapy. EBR, GZR, and placebo were manufactured to preserve masking (confirmed as visually identical) and packaged identically. All clinical supplies were provided by Merck & Co., Inc., Kenilworth, NJ.
PROCEDURES
Plasma HCV RNA levels were measured by the COBAS AmpliPrep/COBAS TaqMan HCV test, version 2.0 (Roche Molecular Diagnostics, Branchburg, NJ) with a lower limit of quantitation (LLoQ) of 15 IU/mL. Specimens for viral load measurements were collected at screening, baseline, and regular intervals during treatment and follow‐up. HCV genotype was determined using the Abbott HCV Real Time Genotype II assay, with the genotypes of all participants from outside China also confirmed by NS5B amplicon sequence analysis. Blood samples for viral resistance assays were collected at baseline and at the time of virologic failure. HCV NS3 and NS5A genes from plasma samples were amplified using reverse transcription polymerase chain reaction followed by population sequencing and comparison to HCV GT subtype‐specific reference sequences. The limit of viral detection was ≈25% of the viral population. For NS3, substitutions at amino acid positions 36, 54, 55, 56, 80, 107, 122, 132, 155, 156, 158, 168, 170, and 175 were analyzed for all GTs, whereas for NS5A, substitutions at amino acid positions 28, 30, 31, and 93 were assessed in participants with GT1 infection and at amino acid positions 24, 28, 30, 31, 32, 38, 58, 92, and 93 in participants with GT4 or 6 infection. Reference strains were H77 (https://www.ncbi.nlm.nih.gov/nuccore/NC_004102) for GT1a and Con1 (https://www.ncbi.nlm.nih.gov/nuccore/AJ238799) for GT1b. The clinical relevance of substitutions within the NS3 and NS5A regions in participants with GT4 or 6 infection is not well characterized. For this analysis, GT4 and 6 sequences were evaluated at the key loci noted above, as substitutions at these positions have been associated with in vitro or in vivo resistance to NS3 and NS5A inhibitors. While HCV GT4 and 6 have more subtype diversity than GT1 and likely have more naturally occurring polymorphisms at the key loci evaluated, only one reference strain was used for each of these genotypes in order to enable a comparison among participants infected with a particular genotype. For GT6, the 6a reference strain https://www.ncbi.nlm.nih.gov/nuccore/2326454) was used, while the GT4 analysis used a 4a reference strain (https://www.ncbi.nlm.nih.gov/nuccore/GU814265).
OUTCOME MEASURES
The primary efficacy outcome variable was SVR12, defined as the proportion of participants in the ITG with undetectable HCV RNA 12 weeks after the end of treatment. Virologic failure was defined as nonresponse (detectable HCV RNA at end of treatment with HCV RNA >LLoQ throughout treatment), rebound (>1 log10 increase in HCV RNA from nadir while on treatment), breakthrough (HCV RNA >LLoQ after previously being <LLoQ while on treatment), or relapse (HCV RNA >LLoQ during follow‐up after having undetectable HCV RNA at end of treatment). The primary safety outcome was a comparison between the two treatment arms during the 12‐week blinded period and up to 14 days after unblinding. Safety was assessed by monitoring adverse events (AEs), vital signs, and laboratory test results. Events of clinical interest (ECIs) were considered tier 1 safety events and included the first instances of alanine aminotransferase (ALT) or AST >500 IU/mL, ALT or AST >3 × baseline and >100 IU/mL, and alkaline phosphatase >3 × ULN. Tier 2 and 3 safety events consisted of AEs, drug‐related AEs, serious AEs (SAEs), serious drug‐related AEs, or an AE leading to discontinuation; an event was considered tier 2 if it occurred in ≥4 participants. Late elevations in ALT or AST were defined as ALT/AST elevations >5 × ULN in participants who had ALT/AST ≤ULN between weeks 2 and 4. Emergence of viral drug resistance was assessed in participants who met the criteria for virologic failure with HCV RNA >1,000 IU/mL.
STATISTICAL ANALYSIS
The target enrollment for the Asia‐Pacific region (including China) and Russia was 453 participants, with an interim analysis of the ITG from the ex‐China cohort prespecified in the protocol. For this interim analysis, with an assumed response rate of 85.5%, an overall sample size of 340 participants provided at least 99.9% power to show superiority to a historical reference SVR12 rate of 73% at a one‐sided 1.25% alpha level; this approximation was used as the historical reference rate to assess the primary endpoint of a study that enrolled a majority of participants with HCV GT1 in addition to participants with GT4 and GT6. The historical reference rate of 73% was based on data from studies of peginterferon/ribavirin in participants with HCV GT1 infection from Taiwan and South Korea, adjusted with a 5% decrease to correct for the expected improved safety profile with an IFN‐free regimen and shorter treatment duration.25, 26 The primary efficacy analysis was performed in the full analysis set population, which included all randomized participants who received at least one dose of study treatment. A two‐sided 97.5% asymptotic confidence interval (CI) was calculated for SVR12 in the ITG. The treatment effect was established if the lower bound of the two‐sided CI was >73%. Missing values were recorded as treatment failures unless the values immediately preceding and following the missing result were both successes, in which case the missing value was imputed as a success. Safety analyses were conducted in all participants who received at least one dose of study medication. For tier 1 safety parameters, P values and 95% CIs were calculated using the Miettinen and Nurminen method for between‐treatment differences in the percentage of participants with these events. Tier 2 safety parameters were assessed by point estimates and 95% CIs for between‐group comparisons, and tier 3 parameters were compared with point estimates.
Results
PARTICIPANT DISPOSITION AND BASELINE CHARACTERISTICS
A total of 371 participants were screened and 337 were randomized to the ITG (n = 251) or DTG (n = 86) (Fig. 1). The full analysis set included 250 participants in the ITG (1 randomized participant did not receive the study drug) and 86 participants in the DTG. Seven participants in the ITG discontinued treatment early. During the 12‐week blinded period, the first participant received the first dose of medication on March 2, 2015, and the final participant completed treatment on December 8, 2015.
Figure 1.
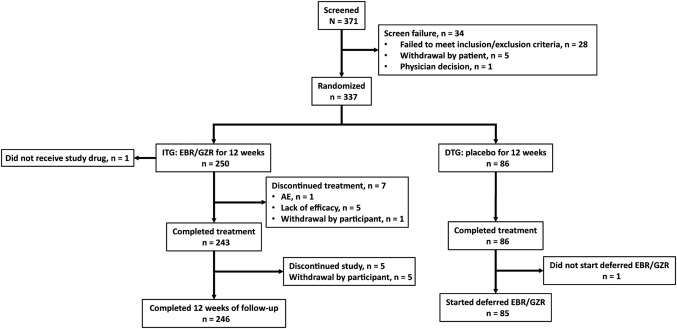
Study flow diagram.
The two treatment groups were generally balanced with respect to baseline characteristics (Table 1). The mean age was 50.1 years (range, 18, 76); 59.2% of participants were Asian and 40.5% were Caucasian; the majority were women (57.1%). Most participants had HCV GT1b infection (74.4%) and did not have cirrhosis (81.0%). Most participants were enrolled from study sites in Russia (35.4%), followed by Taiwan (25.3%), and South Korea (14.9%). Twelve of 249 participants had platelet counts <100,000 cells/μL (1 patient did not have a baseline sample). Nine of 16 (56.3%) participants from Thailand and 12 of 25 (48.0%) from Vietnam had HCV GT6 infection, as did 9 of 63 (14.3%) from Taiwan and 5 of 21 (23.8%) from Australia (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1177/full).
Table 1.
Baseline Participant Characteristics (Full Analysis Set)
|
Immediate‐Treatment Group (EBR/GZR for 12 Weeks) (n = 250) |
Deferred‐Treatment Group (Placebo for 12 Weeks) (n = 86) |
Total (N = 336) |
|
|---|---|---|---|
| Mean age, years (SD) | 49.9 (12.2) | 50.8 (11.7) | 50.1 (12.1) |
| Sex, n (%) | |||
| Male | 105 (42.0) | 39 (45.3) | 144 (42.9) |
| Female | 145 (580) | 47 (54.7) | 192 (57.1) |
| Race, n (%) | |||
| Caucasian | 101 (40.4) | 35 (40.7) | 136 (40.5) |
| Asian | 148 (59.2) | 51 (59.3) | 199 (59.2) |
| Other | 1 (0.4) | 0 | 1 (0.3) |
| Country of enrollment, n (%) | |||
| South Korea | 37 (14.8) | 13 (15.1) | 50 (14.9) |
| Taiwan | 63 (25.2) | 22 (25.6) | 85 (25.3) |
| Russia | 88 (35.2) | 31 (36.0) | 119 (35.4) |
| Australia | 21 (8.4) | 7 (8.1) | 28 (8.3) |
| Vietnam | 25 (10.0) | 8 (9.3) | 33 (9.8) |
| Thailand | 16 (64) | 5 (5.8) | 21 (6.3) |
| Body mass index, mean (SD), kg/m2 | 24.98 (3.71) | 24.86 (3.38) | 24.95 (3.62) |
| IL28B genotype, n (%) | |||
| CC | 158 (63.2) | 56 (65.1) | 214 (63.7) |
| Non‐CC | 88 (35.2) | 27 (31.4) | 115 (34.2) |
| Missing | 4 (1.6) | 3 (3.5) | 7 (2.1) |
| HCV genotype, n (%) | |||
| 1a | 26 (10.4) | 11 (12.8) | 37 (11.0) |
| 1b | 187 (74.8) | 63 (73.3) | 250 (74.4) |
| 4 | 2 (0.8) | 1 (1.2) | 3 (0.9) |
| 6 | 35 (14.0) | 11 (12.8) | 46 (13.7) |
| Baseline HCV RNA, n (%) | |||
| ≤800,000 IU/mL | 83 (33.2) | 28 (32.6) | 111 (33.0) |
| >800,000 IU/mL | 167 (66.8) | 58 (67.4) | 225 (67.0) |
| ≤2,000,000 IU/mL | 140 (56.0) | 47 (54.7) | 187 (55.7) |
| >2,000,000 IU/mL | 110 (44.0) | 39 (45.3) | 149 (44.3) |
| Fibrosis stage | |||
| METAVIR F0‐F2 | 170 (68.0) | 61 (70.9) | 231 (68.8) |
| METAVIR F3 | 33 (13.2) | 8 (9.3) | 41 (12.2) |
| METAVIR F4 | 47 (18.8) | 17 (19.8) | 64 (19.0) |
| IFN‐eligible, n (%) | |||
| Yes | 250 (100.0) | 86 (100.0) | 336 (100.0) |
| Fibrosis stage by diagnosis, n (%) | |||
| Cirrhosis (yes) by biopsy | 6 (2.4) | 4 (4.7) | 10 (3.0) |
| Cirrhosis (yes) by FibroTest | 1 (0.4) | 2 (2.3) | 3 (0.9) |
| Cirrhosis (yes) by FibroScan | 40 (16.0) | 11 (12.8) | 51 (15.2) |
| Cirrhosis (no) by biopsy | 42 (16.8) | 10 (11.6) | 52 (15.5) |
| Cirrhosis (no) by FibroTest | 21 (8.4) | 5 (5.8) | 26 (7.7) |
| Cirrhosis (no) by FibroScan | 140 (56.0) | 54 (62.8) | 194 (57.7) |
| Baseline hemoglobin, g/dL mean (SD) | 14.0 (1.5) | 14.2 (1.3) | 14.1 (1.4) |
| Baseline albumin level, g/dL, mean (SD) | 4.48 (0.30) | 4.47 (0.27) | 4.48 (0.29) |
| Baseline ALT level, IU/L, mean (SD) | 66.52 (53.19) | 73.52 (48.54) | 68.32 (52.06) |
| Baseline AST level, IU/L, mean (SD) | 53.88 (36.81) | 61.98 (42.49) | 55.96 (38.44) |
| Baseline bilirubin level, mg/dL, mean (SD) | 0.60 (0.31) | 0.55 (0.25) | 0.59 (0.29) |
| Baseline platelet count, × 103/μL, mean (SD) | 197.74 (66.31) | 193.92 (69.53) | 196.76 (67.06) |
Abbreviation: IL, interleukin.
EFFICACY ANALYSES
Overall, 232/250 (92.8%) participants in the ITG achieved SVR12 (97.5% CI, 89.1, 96.5) (Fig. 2). As predefined in the protocol, the lower bound of the 97.5% CI was higher than the historical reference rate of 73%, and thus the treatment effect was considered established (P < 0.001). Of the 18 participants who failed to attain SVR12, 1 was lost to follow‐up and 17 had virologic failure (breakthrough, n = 3; rebound, n = 3; relapse, n = 11).
Figure 2.
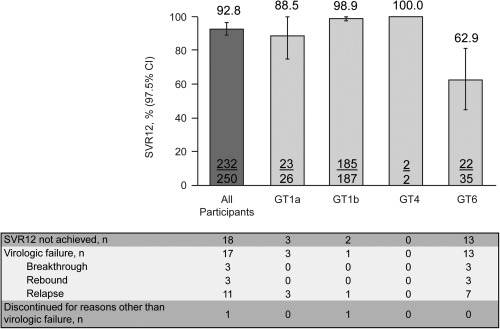
Rates of sustained virologic response at week 12 after cessation of study therapy. Data represent mean ± 97.5% CI.
SVR12 was higher among participants with HCV GT1b infection (185/187, 98.9%) than those with GT1a infection (23/26, 88.5%). Both participants with HCV GT4 infection achieved SVR12 (2/2, 100.0%), and responses were notably lower in those with GT6 infection (22/35, 62.9%; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1177/full). SVR12 remained high across most important participant subgroups (Fig. 3). SVR12 was high in participants with cirrhosis (43/47, 91.5%) and those aged ≥65 years (24/26, 92.3%) but was slightly lower in those with baseline viral load >2 million IU/mL (98/110, 89.1%) and those of Asian race (132/148, 89.2%), which included a high proportion of participants with GT6 infection. SVR12 rates were lower in participants from Thailand (8/16, 50.0%) and Vietnam (20/25, 80.0%) but remained above 90% in participants from Russia (87/88, 98.9%), Taiwan (62/63, 98.4%), South Korea (36/37, 97.3%), and Australia (19/21, 90.5%) (Table 2).
Figure 3.
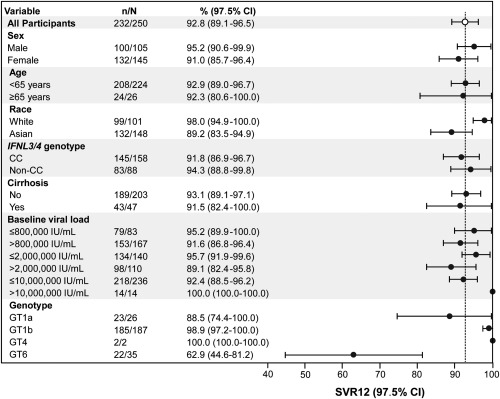
Subgroup analyses. Data represent mean ± 97.5% CI.
Table 2.
Rates of Sustained Virologic Response at Week 12 After Cessation of Study Therapy by Country and Genotype
|
All Genotypes n/N (%) [97.5% CI] |
GT1a n/N (%) |
GT1b n/N (%) |
GT4 n/N (%) |
GT6 n/N (%) |
|
|---|---|---|---|---|---|
| Country | |||||
| South Korea |
36/37 (97.3) [91.3‐100.0] |
1/1 (100.0) | 35/36 (97.2) | 0 (0) | 0 (0) |
| Taiwan |
62/63 (98.4) [94.9‐100.0] |
4/4 (100.0) | 50/50 (100.0) | 0 (0) | 8/9 (88.9) |
| Russia |
87/88 (98.9) [96.3‐100.0] |
1/1 (100.0) | 85/86 (98.8) | 1/1 (100.0) | 0 (0) |
| Australia |
19/21 (90.5) [76.1‐100.0] |
6/7 (85.7) | 8/8 (100.0) | 1/1 (100.0) | 4/5 (90.5) |
| Vietnam |
20/25 (80.0) [62.1‐97.9] |
9/9 (100.0) | 4/4 (100.0) | 0 (0) | 7/12 (58.3) |
| Thailand |
8/16 (50.0) [22.0‐78.0] |
2/4 (50.0) | 3/3 (100.0) | 0 (0) | 3/9 (33.3) |
RESISTANCE ANALYSES
NS3 resistance‐associated substitutions (RASs) were detected at baseline in 69.2% (18/26) and 78.9% (146/185) of participants with GT1a and 1b infection, respectively. The presence of NS3 RASs at baseline resulted in lower efficacy in participants with GT1a infection but not in those with GT1b infection. SVR12 rates were 99.3% (145/146) versus 100.0% (39/39) in participants with GT1b infection with and without baseline NS3 RASs and 83.3% (15/18) and 100.0% (8/8), respectively, in those with GT1a infection. Of the 3 participants with GT1a infection and NS3 RASs at baseline who failed to attain SVR12, 2 also had detectable NS5A RASs at baseline. All 10 participants with GT1a infection and the Q80K substitution at baseline achieved SVR12.
At baseline, NS5A RASs were detected in 23.1% (6/26) of participants with GT1a infection and 21.1% (39/185) of those with GT1b infection (Fig. 4). SVR12 was impacted by the presence of baseline NS5A RASs in participants with GT1a infection but not in those with GT1b infection. SVR12 was achieved by 66.7% (4/6) of participants with GT1a infection and baseline NS5A RASs compared with 97.4% (38/39) of participants with GT1b infection and baseline NS5A RASs. SVR12 rates in participants with no baseline NS5A RASs were 95.0% (19/20) in those with GT1a infection and 100.0% (146/146) in those with GT1b infection.
Figure 4.
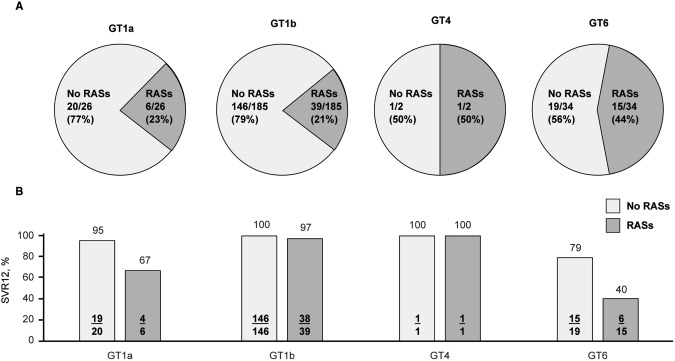
NS5A RASs at baseline in participants with GT1, 4, or 6 infection. (A) Prevalence of RASs; (B) effect on rates of SVR12.
All 35 participants with GT6 infection (including 10 different GT6 subtypes) had baseline NS3 RASs, and 15 participants had baseline polymorphisms in both NS3 and NS5A. SVR12 rates were 40.0% (6/15) in participants with dual baseline RASs compared with 78.9% (15/19) in those with only NS3 RASs (1 participant with GT6 infection did not have NS5A sequencing available; this participant achieved SVR12).
VIROLOGIC FAILURE
A total of 17 participants had virologic failure: 13 of these participants had GT6 infection, 3 had GT1a infection, and 1 had GT1b infection (see http://onlinelibrary.wiley.com/doi/10.1002/hep4.1177/full). All 3 participants with GT1a infection who relapsed had NS3 RASs detected both at baseline and at time of failure. NS5A RASs were also detected in 2 of these participants at baseline and in all 3 at relapse. The participant with GT1b infection who relapsed had NS5A L28M and Y93H RASs present at baseline and at time of failure and an NS3 Y56F RAS at baseline and a treatment‐emergent V132I variant at failure.
Of the 13 participants with GT6 infection who experienced virologic failure, 6 had on‐treatment failure (breakthrough, n = 3; rebound, n = 3) and 7 relapsed. Of the 6 participants with on‐treatment failure, 5 had subtype 6f infection. Notably, only 1 of 6 (16.7%) participants with GT6f infection achieved SVR12 (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1177/full); 5/6 had baseline viral load >2 million IU/mL, all 6 were enrolled in Thailand, and all had both NS3 and NS5A RASs present at baseline and failure. Of the 7 participants with GT6 infection who relapsed, 6 had both NS3 and NS5A RASs present at baseline and at failure and 1 had NS3 RASs only at baseline and time of failure. A complete listing of RASs present at baseline and at failure for the 17 participants with virologic failure is presented in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1177/full.
SAFETY
The incidence of AEs was similar between the ITG and DTG (50.8% versus 51.2%; difference, −0.3%; 95% CI, −12.3, 11.9) (Table 3). Drug‐related AEs occurred in 53 participants (21.2%) in the ITG and 17 (19.8%) participants in the DTG (difference, 1.5%; 95% CI, −9.3, 10.5), with headache the most commonly reported AE in the ITG (8.4%) and fatigue the most commonly reported drug‐related AE in the DTG (9.3%). SAEs were reported in 2 (0.8%) participants and 1 (0.2%) participant in the ITG and DTG, respectively, over the initial treatment period and 14 days of follow‐up. These included one case each of contusion and suicide in the ITG and one case of influenza in the DTG. None of the SAEs were considered related to treatment and, other than the suicide, all events resolved. A 49‐year‐old female Asian participant without cirrhosis in the ITG committed suicide on day 57 of the study after withdrawing consent. This participant had no known history of depression or psychiatric illness and had reported no prior or concomitant medications but had a history of suicidal ideation documented to have started 11 days earlier.
Table 3.
Safety of Participants Randomly Assigned to Immediate or Deferred Therapy With EBR/GZR During the Initial Treatment Period and First 14 Days of Follow‐Up (Full Analysis Set)
| Variable |
Immediate‐Treatment Group (n = 250) n (%) |
Deferred‐Treatment Group (n = 86) n (%) |
Difference in % (Immediate − Deferred) (95% CI) |
|---|---|---|---|
| At least one AE | 127 (50.8) | 44 (51.2) | −0.3 (−12.3, 11.9) |
| SAEs | 2 (0.8) | 1 (1.2) | −0.4 (−5.5, 1.8) |
| Influenza | 0 | 1 (1.2) | ‐ |
| Contusion | 1 (0.4) | 0 | ‐ |
| Suicide | 1 (0.4) | 0 | ‐ |
| Deaths | 1 (0.4) | 0 | ‐ |
| Drug‐related AEs | 53 (21.2) | 17 (19.8) | 1.5 (−9.3, 10.5) |
| Drug‐related SAEs | 0 | 0 | 0 (−4.3, 1.5) |
| Discontinuation due to AEs | 1 (0.4) | 0 | 0.4 (−3.9, 2.3) |
| Discontinuation due to drug‐related AEs | 0 | 0 | ‐ |
| AEs occurring in ≥5% of participants | |||
| Diarrhea | 14 (5.6) | 4 (4.7) | 1.0 (−6.1, 5.5) |
| Fatigue | 13 (5.2) | 8 (9.3) | −4.1 (−12.4, 1.6) |
| Upper respiratory tract infection | 13 (5.2) | 3 (3.5) | 1.7 (−4.9, 6.1) |
| Headache | 21 (8.4) | 5 (5.8) | 2.6 (−5.0, 7.9) |
| Events of clinical interest | |||
| ALT or AST >500 IU/mL | 0 | 0 |
0 (−4.3, −1.5) P > 0.009 |
| ALT or AST >3× ULN and >100 IU/mL | 4 (1.6) | 4 (4.7) |
−3.0 (−9.9, 0.7) P = 0.114 |
| AP >3× ULN | 0 | 0 |
0 (−4.3 to 1.5) P > 0.999 |
| ALT elevation | |||
| 1.1‐2.5× BL | 13 (5.2) | 43 (50.0) | ‐ |
| >2.5‐5.0× BL | 3 (1.2) | 2 (2.3) | ‐ |
| >5.0× BL | 2 (0.8) | 2 (2.3) | ‐ |
| AST elevation | |||
| 1.1‐2.5× BL | 14 (5.6) | 36 (41.9) | ‐ |
| >2.5‐5.0× BL | 2 (0.8) | 4 (4.7) | ‐ |
| >5.0× BL | 0 (0.0) | 0 (0.0) | ‐ |
| Late ALT/ASTa | |||
| >2.0‐5.0× ULN | 5 (2.0) | 3 (3.5) | ‐ |
| >5.0× ULN | 2 (0.8) | 0 (0.0) | ‐ |
| Bilirubin elevation | |||
| >2.5‐5.0× BL | 2 (0.8) | 0 (0.0) | ‐ |
| >5.0‐10.0× BL | 1 (0.4) | 0 (0.0) | ‐ |
| >10.0× BL | 0 (0.0) | 0 (0.0) | ‐ |
| Hemoglobin | |||
| Grade 1: 10.0‐10.9 g/dL | 8 (3.2) | 1 (1.2) | ‐ |
| Grade 2: 9.0‐9.9 g/dL | 4 (1.6) | 0 (0.0) | ‐ |
| Grade 3: 7.0‐8.9 g/dL | 0 (0.0) | 0 (0.0) | ‐ |
| Grade 4: <7.0 g/dL | 0 (0.0) | 0 (0.0) | ‐ |
Defined as ALT/AST elevation occurring after treatment week 4 in patients who had normal ALT/AST levels between treatment weeks 2 and 4.
Abbreviations: AP, alkaline phosphatase; BL, baseline.
Eight participants reported ECIs (ITG, n = 4; DTG, n = 4); two ECIs in the ITG and two in the DTG were considered treatment related. Of the four ECIs reported in the ITG, two fulfilled the protocol‐specified criteria for a late ALT/AST elevation. One participant was discontinued from the study with an ALT/AST increase to >3 × ULN, with a simultaneous increase in total bilirubin to >2 × ULN that occurred 45 days after drug initiation. On the day prior to these laboratory abnormalities, the participant reportedly ingested two bottles (unknown size) of soju, an alcoholic beverage that varies in alcoholic content from 15% to 45% by volume. This participant achieved SVR12. Narratives for these participants are included in the http://onlinelibrary.wiley.com/doi/10.1002/hep4.1177/full.
Discussion
This study demonstrated that a 12‐week regimen of a fixed‐dose combination of EBR 50 mg/GZR 100 mg is highly effective and well tolerated in a treatment‐naive predominantly Asian population with HCV GT1 or 4 infection. The overall SVR12 rate was 92.8%, which exceeded the 73% SVR12 target based on historical data in Asian people treated with an IFN‐based regimen. Noteworthy is the SVR12 rate of 99% among participants with HCV GT1b infection, who comprise a majority of HCV‐infected individuals in Russia and the Asia‐Pacific region. The overall SVR12 rate with EBR/GZR in this predominantly Asian population is similar to rates of 95% to 98% reported in phase 3 studies of North American and European participants17, 18, 19, 20 and is also consistent with the SVR12 of 96.5% in a phase 3 study in Japanese participants.24 As in studies in Western populations, EBR/GZR was equally effective in participants with or without cirrhosis.17
Importantly, the incidence of virologic failures in the present study was driven largely by the proportion of participants with HCV GT6 infection who failed to attain SVR12. Among this population, SVR12 was 62.9%, with 13/35 participants (37.1%) experiencing virologic failure. EBR/GZR is not approved for the treatment of participants with GT6 infection in Western countries,8, 9, 10 and the low SVR12 rate in this population is consistent with these data.17, 20 Although there was marked variability in response across HCV GT6 subtypes (including no virologic failures among participants with GT6h, k, l, or w), few of these participants were enrolled in the study. Therefore, where available, treatment options other than EBR/GZR should be considered for people with HCV GT6 infection. Based on current U.S. treatment guidelines, regimens of glecaprevir/pibrentasvir, sofosbuvir/velpatasvir, or ledipasvir/sofosbuvir may be considered for treatment‐naive individuals with HCV GT6 infection.5 In contrast to the response profile in participants with GT6 infection, virologic failure was reported in only 1 of 187 participants with HCV GT1b infection (≈0.5%), also consistent with previous reports.17 Response rates among participants with HCV GT1b infection were ≥95%, regardless of the presence of baseline NS3 or NS5A RASs. These data are consistent with the current recommended use of EBR/GZR in participants with HCV GT1b infection that does not require testing for baseline RAS prior to initiation of therapy.
Among participants with GT1a infection, 11.5% (3/26) relapsed, all with both NS3 and NS5A RASs detected at the time of failure. The present analysis used population sequencing with a limit of detection of ≈25% of the viral species for the analysis of RASs. In participants with GT1a infection, RASs at amino acids 28, 30, 31, and 93 were assessed, as variants at these positions have been shown previously to have a clinically relevant impact on the efficacy of this regimen.8 Using this approach, NS5A RASs were found to be present in 23.1% (6/26) of participants with GT1a infection, and the SVR12 rate in this population was 66.7% (4/6). Studies in Western populations have shown that those with GT1a infection and baseline NS5A RASs can benefit from a 16‐week regimen of EBR/GZR in combination with ribavirin.8
The safety profiles of participants who received EBR/GZR or placebo in the present study were generally comparable, with similar frequencies of AEs, SAEs, and hepatic events. Only 1 of 250 participants in the ITG discontinued the study because of an AE, and there were no drug‐related SAEs in either treatment arm. Four tier 1 events were reported in participants receiving EBR/GZR (1.6%), all of which were elevated laboratory values reported as ECIs; of these, two met the criteria for late ALT/AST elevations >5 × ULN. This is consistent with the incidence of late ALT/AST elevations reported in an integrated safety analysis of data from the phase 2/3 clinical trials in 1,690 Western participants.27 In this retrospective analysis, there was a total of 13 participants (0.8%) who experienced ALT elevations from normal to >5 × ULN. These events generally occurred at or after treatment week 8 and were asymptomatic; most resolved with ongoing therapy. Cirrhosis was not a risk factor for these transaminase elevations, and they were not associated with clinically significant elevations in bilirubin or other changes in liver function. Only 3 of these 13 participants discontinued treatment early. Similarly, in the present study, transaminase increases were generally transient and normalized during or after completion of treatment, and no dose changes or discontinuations were required.
The present study was subject to several limitations. The historical comparator group was drawn from older studies of IFN‐/ribavirin‐based therapies, which are typically associated with lower response rates and poorer tolerability compared with all‐oral direct‐acting antiviral agent regimens. Few participants with GT4 were enrolled in this study, so conclusions regarding the efficacy of EBR/GZR in Asian people with HCV GT4 infection are limited. Several other studies have also assessed the efficacy and safety of all‐oral HCV treatments in people from the Asia‐Pacific region; however, comparisons with data from the present study cannot be drawn based on this study.
In conclusion, data from this study show that a once‐daily fixed‐dose combination of EBR/GZR for 12 weeks provides an effective and well‐tolerated regimen for chronic HCV GT1 infection in treatment‐naive individuals with or without cirrhosis from the Asia‐Pacific region and Russia, particularly the large population with HCV GT1b infection. EBR/GZR is not recommended for the treatment of individuals with HCV GT6 infection. This combination represents a safe and effective therapeutic option for Asian people with HCV GT1 infection.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1177/full.
Supporting Information 1
Acknowledgment
We thank the participants, their families, investigators, and site personnel who participated in this study. Medical writing and editorial assistance was funded by Merck & Co. and provided by Dr. Tim Ibbotson of ApotheCom.
Potential conflict of interest: Dr. Isakov has received grants from Merck, serves on advisory boards for Merck, AbbVie, R‐Pharm, and BMS, and is a speaker and consultant for Merck, AbbVie, R‐Pharm, BMS, and Gilead. Dr. Barr, Dr. B. Nguyen, Dr. Ginanni, Dr. Ingravallo, Dr. Liang, Dr. Lindore, Dr. Wahl, Dr. Talwani, and Dr. Robertson are employed by Merck; Dr. Barr, Dr. B. Nguyen, Dr. Ingravallo, Dr. Lindore, Dr. Wahl, and Dr. Robertson also own stock in Merck. Dr. George serves on advisory boards for AbbVie, Gilead, MSD, and BMS and is on the speakers' bureau for MSD. Dr. Tak advises Gilead, AbbVie, and Ono and received grants from Samil. Dr. Tanwandee has received grants from Merck. Dr. Kizhlo has received grants from Merck and personal fees from Merck, Janssen, and AbbVie. Dr. Zhdanov has received personal fees from Merck, AbbVie, Gilead, Janssen, R‐Pharm, and Biocad. The other authors have nothing to report.
Supported by Merck & Co., Inc., Kenilworth, NJ.
Clinical trials identifier https://clinicaltrials.gov/ct2/results?cond=-term=NCT02251990-cntry=-state=-city=-dist=
REFERENCES
- 1. Sievert W, Altraif I, Razavi HA, Abdo A, Ahmed EA, AlOmair A, et al. A systematic review of hepatitis C virus epidemiology in Asia, Australia and Egypt. Liver Int 2011;31(Suppl. 2):61‐80. [DOI] [PubMed] [Google Scholar]
- 2. Gower E, Estes C, Blach S, Razavi‐Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol 2014;61(Suppl.):S45‐S57. [DOI] [PubMed] [Google Scholar]
- 3. Hajarizadeh B, Grebely J, Dore GJ. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol 2013;10:553‐562. [DOI] [PubMed] [Google Scholar]
- 4. AASLD/IDSA . HCV Guidance: recommendations for testing, managing, and treating hepatitis C virus. https://www.hcvguidelines.org/. Published September 21, 2017. Accessed December 2017.
- 5. European Association for the Study of the Liver . EASL recommendations on treatment of hepatitis C 2016. J Hepatol 2017;66:153‐194. [DOI] [PubMed] [Google Scholar]
- 6. Lim SG, Dan YY. A 2015 roadmap for the management of hepatitis C virus infections in Asia. Korean J Intern Med 2015;30:423‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Omata M, Kanda T, Wei L, Yu M‐L, Chuang W‐L, Ibrahim A, et al. APASL consensus statements and recommendation on treatment of hepatitis C. Hepatol Int 2016;10:702‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zepatier (elbasvir and grazoprevir) prescribing information. Whitehouse Station, NJ: Merck Sharp & Dohme Corp; 2017. [Google Scholar]
- 9. European Medicines Agency . EPAR summary for the public. Zepatier: elbasvir/grazoprevir. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/004126/WC500211238.pdf. Accessed August 2017.
- 10. CATIE . Zepatier for hepatitis C approved in Canada. http://www.catie.ca/en/catienews/2016-01-29/zepatier-hepatitis-c-approved-canada. Accessed August 2017.
- 11. Harper S, McCauley JA, Rudd MT, Ferrara M, DiFilippo M, Crescenzi B, et al. Discovery of MK‐5172, a macrocyclic hepatitis C virus NS3/4a protease inhibitor. ACS Med Chem Lett 2012;3:332‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Summa V, Ludmerer SW, McCauley JA, Fandozzi C, Burlein C, Claudio G, et al. MK‐5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother 2012;56:4161‐4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lahser FC, Bystol K, Curry S, McMonagle P, Xia E, Ingravallo P, et al. The combination of grazoprevir, a hepatitis C virus (HCV) NS3/4A protease inhibitor, and elbasvir, an HCV NS5A inhibitor, demonstrates a high genetic barrier to resistance in HCV genotype 1a replicons. Antimicrob Agents Chemother 2016;60:2954‐2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu R, Curry S, McMonagle P, Yeh WW, Ludmerer SW, Jumes PA, et al. Susceptibilities of genotype 1a, 1b, and 3 hepatitis C virus variants to the NS5A inhibitor elbasvir. Antimicrob Agents Chemother 2015;59:6922‐6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lawitz E, Gane E, Pearlman B, Tam E, Ghesquiere W, Guyader D, et al. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK‐5172) and elbasvir (MK‐8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C‐WORTHY): a randomised, open‐label phase 2 trial. Lancet 2015;385:1075‐1086. [DOI] [PubMed] [Google Scholar]
- 16. Sulkowski M, Hezode C, Gerstoft J, Vierling J, Mallolas J, Pol S, et al. Efficacy and safety of 8 weeks versus 12 weeks of treatment with grazoprevir (MK‐5172) and elbasvir (MK‐8742) with or without ribavirin in patients with hepatitis C virus genotype 1 mono‐infection and HIV/hepatitis C virus co‐infection (C‐WORTHY): a randomised, open‐label phase 2 trial. Lancet 2015;385:1087‐1097. [DOI] [PubMed] [Google Scholar]
- 17. Zeuzem S, Ghalib R, Reddy KR, Pockros PJ, Ben Ari Z, Zhao Y, et al. Grazoprevir‐elbasvir combination therapy for treatment‐naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: a randomized trial. Ann Intern Med 2015;163:1‐13. [DOI] [PubMed] [Google Scholar]
- 18. Roth D, Nelson DR, Bruchfeld A, Liapakis A, Silva M, Monsour HM Jr, et al. Grazoprevir plus elbasvir in treatment‐naive and treatment‐experienced patients with hepatitis C virus genotype 1 infection and stage 4‐5 chronic kidney disease (the C‐SURFER study): a combination phase 3 study. Lancet 2015;386:1537‐1545. Erratum in: Lancet 2015;386:1824. [DOI] [PubMed] [Google Scholar]
- 19. Rockstroh JK, Nelson M, Katlama C, Lalezari J, Mallolas J, Bloch M, et al. Efficacy and safety of grazoprevir (MK‐5172) and elbasvir (MK‐8742) in patients with hepatitis C virus and HIV co‐infection (C‐EDGE CO‐INFECTION): a non‐randomised, open‐label trial. Lancet HIV 2015;2:e319‐e327. [DOI] [PubMed] [Google Scholar]
- 20. Kwo P, Gane E, Peng C‐Y, Pearlman B, Vierling JM, Serfaty L, et al. Effectiveness of elbasvir and grazoprevir combination, with or without ribavirin, for treatment‐experienced patients with chronic hepatitis C infection. Gastroenterology 2017;152:164‐175.e4. [DOI] [PubMed] [Google Scholar]
- 21. Sperl J, Horvath G, Halota W, Ruiz‐Tapiador JA, Streinu‐Cercel A, Jancoriene L, et al. Efficacy and safety of elbasvir/grazoprevir and sofosbuvir/pegylated interferon/ribavirin: a phase III randomized controlled trial. J Hepatol 2016;65:1112‐1119. [DOI] [PubMed] [Google Scholar]
- 22. Forns X, Gordon SC, Zuckerman E, Lawitz E, Calleja JL, Hofer H, et al. Grazoprevir and elbasvir plus ribavirin for chronic HCV genotype‐1 infection after failure of combination therapy containing a direct‐acting antiviral agent. J Hepatol 2015;63:564‐572. [DOI] [PubMed] [Google Scholar]
- 23. Dore GJ, Altice F, Litwin AH, Dalgard O, Gane EJ, Shibolet O, et al; C‐EDGE CO‐STAR Study Group . Elbasvir‐grazoprevir to treat hepatitis C virus infection in persons receiving opioid agonist therapy: a randomized trial. Ann Intern Med 2016;165:625‐634. [DOI] [PubMed] [Google Scholar]
- 24. Kumada H, Suzuki Y, Karino Y, Chayama K, Kawada N, Okanoue T, et al. The combination of elbasvir and grazoprevir for the treatment of chronic HCV infection in Japanese patients: a randomized phase II/III study. J Gastroenterol 2017;52:520‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jin Y‐J, Lee J‐W, Lee JI, Park SH, Park CK, Kim YS, et al. Multicenter comparison of PEG‐IFN α2a or α2b plus ribavirin for treatment‐naive HCV patient in Korean population. BMC Gastroenterol 2013;13:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu C‐H, Liu C‐J, Lin C‐L, Liang C‐C, Hsu S‐J, Yang S‐S, et al. Pegylated interferon‐α‐2a plus ribavirin for treatment‐naive Asian patients with hepatitis C virus genotype 1 infection: a multicenter, randomized controlled trial. Clin Infect Dis 2008;47:1260‐1269. [DOI] [PubMed] [Google Scholar]
- 27. Dusheiko GM, Manns MP, Vierling JM, Reddy KR, Sulkowski MS, Kwo PY, et al. Safety and tolerability of grazoprevir/elbasvir in patients with chronic hepatitis C (HCV) infection: integrated analysis of phase 2‐3 trials [Abstract 712]. Hepatology 2015;62(Suppl. 1):562A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1177/full.
Supporting Information 1